
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePigments with antimicrobial and cytotoxic activities from the coprophilous fungus Fusarium solani isolated from horse dung†
Bel Youssouf G. Mountessou a,
Iliassou L. Mouafonb,
Rukesh Maharjanc,
Joseph Tchamgoued,
Gesquiere Laure M. Tianie,
Patrick Dibouloula,
Muhammad I. Choudharyc and
Simeon F. Kouam*a
a,
Iliassou L. Mouafonb,
Rukesh Maharjanc,
Joseph Tchamgoued,
Gesquiere Laure M. Tianie,
Patrick Dibouloula,
Muhammad I. Choudharyc and
Simeon F. Kouam*a
aDepartment of Chemistry, Higher Teacher Training College, University of Yaoundé I, P. O. Box 47, Yaoundé, Cameroon. E-mail: kfogue@yahoo.com
bDepartment of Pharmaceutical Sciences, Tshwane University of Technology, P. O. Box 0083, Pretoria, South Africa
cH.E.J. Research Institute of Chemistry, International Centre for Chemical and Biological Sciences (ICCBS), University of Karachi, Karachi-75270, Pakistan
dDepartment of Organic Chemistry, Faculty of Science, University of Yaoundé I, P. O. Box 812, Yaoundé, Cameroon
eDivision of Wood Chemistry, University Institute of Wood Technology Mbalmayo, P. O. Box 306, Mbalmayo, Cameroon
First published on 17th July 2025
Abstract
Four previously undescribed phenazine-derived pigments (1–4), along with seventeen known compounds, were isolated from a Fusarium solani strain cultured on solid rice medium. Structural elucidation of the new compounds was achieved through extensive spectroscopic analysis, while known compounds were identified by comparison with literature data. The antimicrobial and cytotoxic activities of the isolated compounds were assessed. Compounds 2, 5, and 11 exhibited weak antifungal activity against Candida albicans at 200 μM, while compounds 5, 6, 11, 12, 15, and 20 demonstrated moderate antibacterial effects against Staphylococcus aureus at the same concentration. Notably, compound 4 showed significant cytotoxicity against breast cancer cells (MCF-7, IC50 = 6.8 μM), and moderate cytotoxicity against cervical cancer cells (HeLa, IC50 = 25.0 μM). Moreover, the known quinone-derived pigments 10, 12, and 13 displayed potent cytotoxic activities against the three tested cancer cell lines (MCF-7, PC3, and HeLa). These results highlight the potential of these compounds as promising bioactive agents for cancer treatment.
1. Introduction
Coprophilous fungi, which thrive in the dung of herbivores or decaying organic matter, play a crucial ecological role in nutrient cycling and have emerged as promising sources of bioactive metabolites.1–5 Among these fungi, Fusarium solani is a ubiquitous species with both pathogenic and biotechnological significance. While primarily studied for its role in plant diseases,6 F. solani also produces a diverse range of secondary metabolites, including water-soluble pigments such as phenazines, quinones, and furans, as well as alkaloids, terpenoids, and polyketides.7,8 These compounds exhibit a broad spectrum of bioactivities—ranging from antimicrobial and anticancer to antioxidant and immunomodulatory effects—making them attractive candidates for pharmaceutical and industrial applications.7–9 Despite their potential, only a limited number of these fungal pigments have been thoroughly investigated for their biological properties.7Given the ecological adaptability of F. solani10,11 and its underexplored chemical diversity, we sought to characterise its metabolic potential as part of our ongoing search for bioactive fungal natural products.12–14 In this study, a strain of F. solani was isolated from horse dung, leading to the identification of four new fungal metabolites (1–4, Fig. 1) alongside seventeen known derivatives (5–21) (Fig. S1†). Structural elucidation was conducted using spectroscopic techniques, and the antimicrobial and cytotoxic activities of the compounds were assessed. Our findings not only expand the known chemical repertoire of F. solani but also provide insights into the therapeutic potential of fungal pigments, paving the way for future drug discovery efforts.
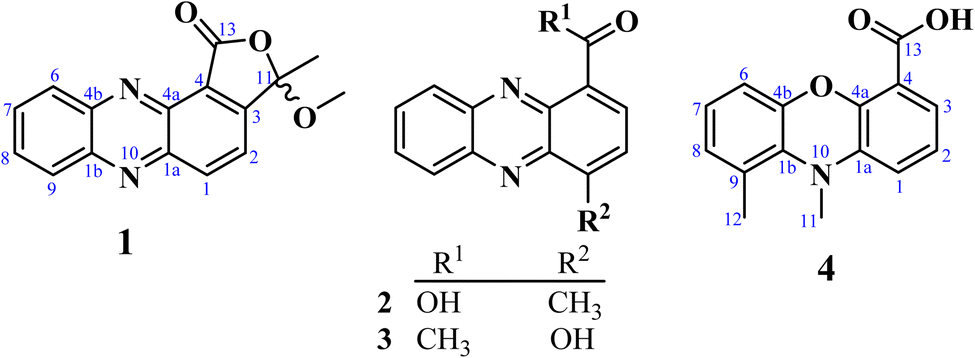 | ||
| Fig. 1 Chemical structures of compounds 1–4. | ||
2. Results and discussion
2.1. Structural elucidation of compounds
Well-grown Fusarium solani cultures on Yeast-Mold-Agar (YMA) plates were fermented in solid rice medium and extracted with ethyl acetate, yielding 4.2 g of organic extract. The extract was subjected to repeated silica gel and Sephadex LH-20 column chromatography, which resulted in several fractions. These fractions were further purified by normal- and reversed-phase preparative HPLC, leading to the isolation of twenty-one compounds, of which compounds 1–4 were hitherto unknown.Fusaphenazinone (1) was obtained as a yellowish pigment. Its molecular formula, C16H12N2O3, was determined by high-resolution electron ionization mass spectrometry (HREIMS), which revealed the molecular ion peak [M]˙+ (Fig. S2†) at m/z 280.0846 (calcd for 280.0848), corresponding to twelve double bond equivalents (DBE). The IR spectrum showed strong absorption bands for a conjugated lactone carbonyl, C–O, and C–H stretching vibrations at 1769, 1188, and 2923–2852 cm−1, respectively (Fig. S3†). The UV spectrum (Fig. S4†) exhibited absorption maxima at λmax 256, 267, and 371 nm characteristic of phenazine derivatives15 and related compounds.16 Analysis of the NMR (Table 1) and HSQC data (Fig. S8†) revealed the presence of a methyl group (δH/C 1.95/24.7), a methoxy group (δH/C 3.17/51.6), six aromatic methines (δH/C 8.60/138.4, 8.50/130.7, 8.28/129.9, 7.94/132.2, 7.94/131.9, 7.87/122.5), and seven quaternary carbons (δC 165.3, 154.7, 144.5, 144.0, 139.0, 123.0, 106.0) (Fig. S5–S7†). The 13C chemical shifts observed in the range δC 144.0–139.0 further confirmed the phenazine core structure in compound 1 (Fig. 1). This structure was identified as a 1,2-disubstituted scaffold, as the 1H NMR spectrum (Fig. S5†) revealed two sets of proton signals: (a) two sharp doublets of ortho-coupled protons at δH 8.60 and 7.87 (1H each, d, 8.5 Hz, H-2, H-1), and (b) a pair of doublets of doublets at δH 8.50 and 8.28 (1H each, dd, 2.0; 8.5 Hz, H-9, H-6), along with a signal for two overlapping protons at δH 7.95 (H-7 and H-8). The 1-proton sharp doublet at δH 7.87 showed HMBC cross-peaks with carbon resonances at δC 143.6 (C-1a), 123.0 (C-4), and the hemiketal carbon signal at δC 106.7 (C-11). These key HMBC correlations helped establish the attachment of the hemiketal carbon at C-3 of the phenazine skeleton, to an acid-derived carbonyl group at C-4. Consistent with the molecular weight of 280 and the thirteen degrees of unsaturation, the acid-derived carbonyl group was cyclized with the hydroxyl of the hemiketal group to form a lactone. Additional HMBC correlations were observed between the hemiketal carbon signal (δC 106.7) and the proton signals of both the methyl (δH 1.95) and the methoxy (δH 3.17) groups (Fig. S9†). These findings were further supported by COSY correlations (Fig. 2 and S10†) and by electron ionization mass spectrometry (EI-MS), which showed important ion peaks at m/z 265 [M−15]˙+, m/z 249 [M−31]˙+, and m/z 178 [C12H6N2]˙+ (Fig. S2†). The proposed structure of compound 1 was fully confirmed by HMBC, DEPT, and COSY spectra. Fig. 2 illustrates key HMBC and COSY correlations of compound 1. However, the absence of optical rotation indicates that compound 1 exists as a racemic mixture at C-11. While chiral HPLC analysis could have enabled the separation of the enantiomers and definitive determination of their absolute configurations, this approach was precluded by the unavailability of an appropriate chiral column and the insufficient amount of material (1.3 mg).
| No. | 1a | 2b | 3c | 4b | ||||
|---|---|---|---|---|---|---|---|---|
| δH mult. (J in Hz) | δC, type | δH mult. (J in Hz) | δC, type | δH mult. (J in Hz) | dδC, type | δH mult. (J in Hz) | dδC, type | |
| a Data were recorded in CDCl3 at 500 MHz for 1H NMR and 150 MHz for 13C NMR.b Data were recorded in CDCl3 at 800 MHz for 1H NMR and 200 MHz for 13C NMR.c Data were recorded in CD3OD at 500 MHz for 1H NMR and 125 MHz for 13C NMR.d Chemical shifts for 13C were retrieved from the HSQC-DEPT spectra, as the 13C NMR spectra were not recorded. | ||||||||
| 1 | 8.60 d (8.5) | 138.4 (CH) | — | 142.9 (C) | — | 157.1 (C) | 8.52 dd (1.6; 8.8) | 135.0 (CH) |
| 1a | — | 143.6 (C) | — | 145.0 (C) | — | 142.2 (C) | — | 143.1 (C) |
| 1b | — | 144.0 (C) | — | 143.1 (C) | — | 144.7 (C) | — | 139.7 (C) |
| 2 | 7.87 d (8.5) | 122.5 (CH) | 7.85 d (7.2) | 129.5 (CH) | 8.13 d (8.8) | 128.3 (CH) | 8.03 dd (6.4; 8.8) | 130.4 (CH) |
| 3 | — | 154.6 (C) | 8.86 d (7.2) | 137.5 (CH) | 8.23 d (8.8) | 130.9 (CH) | 8.97 dd (1.6; 6.4) | 137.3 (CH) |
| 4 | — | 123.0 (C) | — | 122.7 (C) | — | 133.9 (C) | — | 125.0 (C) |
| 4a | — | 139.0 (C) | — | 140.3 (C) | — | 144.8 (C) | — | 139.0 (C) |
| 4b | — | 144.5 (C) | — | 139.4 (C) | — | 145.3 (C) | — | 144.5 (C) |
| 6 | 8.50 dd (2.0; 8.5) | 130.7 (CH) | 8.26 d (8.8) | 127.8 (CH) | 8.33 d (8.8) | 131.4 (CH) | 8.18 d (8.8) | 128.2 (CH) |
| 7 | 7.95 m | 132.2 (CH) | 7.99 ddd (8.8; 6.4; 1.6) | 133.0 (CH) | 7.91 pseudo t (8.0) | 132.0 (CH) | 7.87 dd (7.2; 8.8) | 131.9 (CH) |
| 8 | 7.95 m | 131.9 (CH) | 7.95 ddd (8.8; 6.4; 1.6) | 131.2 (CH) | 7.96 pseudo t (8.0) | 133.3 (CH) | 7.83 d (7.2) | 132.6 (CH) |
| 9 | 8.28 dd (2.0; 8.5) | 129.9 (CH) | 8.37 d (8.8) | 130.4 (CH) | 8.21 d (8.8) | 129.9 (CH) | — | 135.8 (C) |
| 11 | — | 106.7 (C) | — | — | — | — | 3.47 s | 50.8 (CH3) |
| 12 | — | — | — | — | — | — | 2.94 s | 18.4 (CH3) |
| 13 | — | 165.3 (C) | — | 166.4 (C) | — | 201.3 (C) | — | 166.1 (C) |
| 11-CH3 | 1.95 | 24.7 (CH3) | — | — | — | — | — | — |
| 11-OCH3 | 3.17 | 51.6 (CH3) | — | — | — | — | — | — |
| 1-CH3 | — | — | 3.01 s | 18.3 (CH3) | — | — | — | — |
| 13-CH3 | — | — | — | — | 2.85 s | 30.8 (CH3) | — | — |
| 13-OH | — | — | 15.65 s | — | — | — | 15.81 s | — |
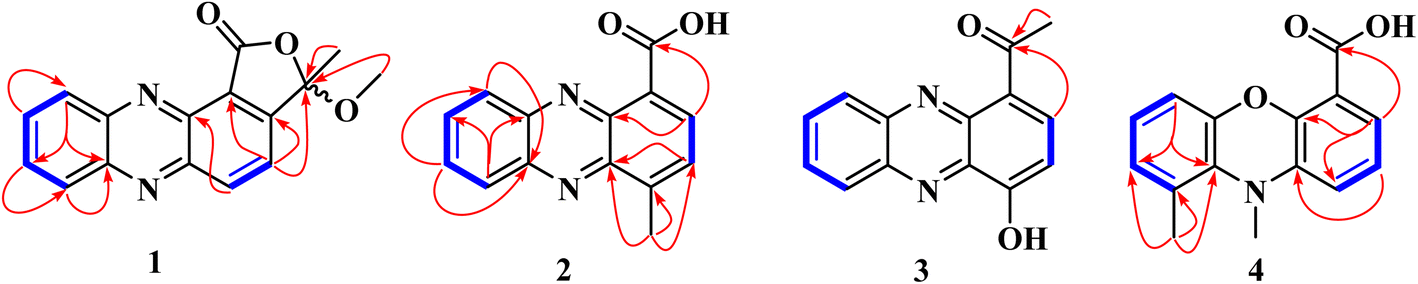 | ||
Fig. 2 Key COSY ( ) and HMBC ( ) and HMBC ( ) correlations of compounds 1–4. ) correlations of compounds 1–4. | ||
4-Methylphenazine-1-carboxylic acid (2) was obtained as a yellowish, gummy pigment. The molecular formula C14H10N2O2 was assigned to the compound based on HREI-MS data (m/z 238.0744; calcd for 238.0742) (Fig. S11†), corresponding to 11° of unsaturation. The broadband decoupled 13C NMR spectrum of 2 (Fig. S15†) showed 14 carbon signals, which were sorted by DEPT and HSQC techniques into four methyl, six methine, and seven quaternary carbons, including a carbonyl signal (δC 166.4) associated with the acid group, and four nitrogenized carbons from phenazine moiety (δC 145.0, 143.1, 140.3, 139.4). The IR spectrum of 2 (Fig. S12†) displayed characteristic vibration bands due to a methyl group (2922–2852 cm−1 and 760–741 cm−1), a conjugated acid carbonyl group (1715 cm−1), and benzene rings (1569–1532 cm−1). The UV-vis spectrum of 2 (Fig. S13†) showed absorption maxima at λmax 257, 266, and 372 nm, similar to those of phenazine derivatives, as observed in compound 1. In the 1H NMR spectrum of 2 (Fig. S14†), the deshielded singlet signal at δH 15.65 in the downfield region indicated the presence of a carboxylic acid group, with its OH group chelated to the nitrogen of the phenazine moiety. Similar to 1, the 1H NMR spectrum of 2 displayed a pair of doublets at δH 8.86 and 7.85 (1H each, d, 7.2 Hz, H-3, H-2), assignable to two ortho-coupled protons, along with another pair of doublets at δH 8.26 and 8.37 (1H each, d, 8.8 Hz, H-6, H-9), and proton signals at δH 7.99 and 7.95 (1H each, ddd, 8.8; 6.4; 1.6 Hz, H-7, H-8) (Table 1). HMBC cross-peaks observed between the 3-proton signal at δH 3.01 (3H, s) and the carbon signals at δC 145.0 (C-1a), 142.9 (C-1), and 129.5 (C-2) permitted to attach the methyl group at C-1 of the phenazine moiety. Additional HMBC correlations supporting the structure of 2 include the correlation between the proton signal at δH 8.86 (H-3) and the carbon signals at δC 166.4 (COOH), 145.0 (C-1a), and 140.3 (C-4a) (Fig. 2). The structure of 2 was further confirmed by DEPT, HSQC, and COSY spectra (Fig. S16–S19†), and its spectroscopic data were consistent with those of 4-methylphenazine-1-carboxylic acid, a synthetic compound.17 To the best of our knowledge, this is the first report of its spectroscopic data as a naturally occurring secondary metabolite.
1-Acetyl-4-hydroxyphenazine (3) was obtained as a gummy, yellow pigment. Its molecular formula, C14H10N2O2, with 11 double bond equivalents, was determined from HREI-MS (Fig. S20†), which showed the molecular weight at m/z 238.0740, matching the calculated value of 238.0742. The IR spectrum exhibited strong absorption bands for hydroxyl (3400 cm−1) and conjugated carbonyl (1590 cm−1) groups, along with characteristic absorption bands for aliphatic C–H stretching vibrations at 2919 and 2851 cm−1 (Fig. S21†). Similar to compounds 1 and 2, the UV spectrum of compound 3 displayed absorption maxima characteristic of phenazines at λmax 247, 264, and 368 nm (Fig. S22†). The 1H NMR spectrum of 3, recorded in CD3OD (Fig. S23†), displayed similarities to the spectra of compounds 1 and 2, with more distinct multiplicities for the aromatic protons. The structural elucidation of 3 was straightforward, as it was inferred from the structures of 1 and 2. In the 1H NMR spectrum of 3, two sets of proton signals on the phenazine core were observed: (a) a pair of doublets for ortho-coupled protons at δH 8.33 and 8.21 (1H each, d, 8.8 Hz, H-12, H-9), each coupling with a pair of pseudo-triplets at δH 7.96 and 7.91 (1H each, pseudo t, 8.0 Hz, H-10, H-11), and (b) a pair of doublets at δH 8.23 and 8.13 (1H each, d, 8.8 Hz, H-2, H-3) (Table 1 and Fig. S23†). HMBC cross-peaks between the 1-proton doublet at δH 8.23 and the carbon signals at δC 201.3 and 144.8 confirmed the presence of a phenazine moiety in 3, with an acetyl group at position C-1. This acetyl group was further confirmed by HMBC cross-peaks between the 3-proton singlet at δH 2.85 and the deshielded carbon signal at δC 201.3 (Fig. S25†). Additional HMBC correlations were observed between the proton signal at δH 8.13 and the carbon signals at δC 142.2 (C-5) and 133.9 (C-1), as well as between the proton signal at δH 7.96 and the carbon signals at δC 144.8 (C-8) and 131.4 (C-12). The structure of 3 was fully supported by other spectra, including HSQC (Fig. S24†) and COSY (Fig. S26†) spectra, with key HMBC and COSY correlations illustrated in Fig. 2.
Compound 4, named 9,10-dimethylphenoxazine-4-carboxylic acid, was obtained as a yellowish pigment. Its molecular formula, C15H13NO3, was established from HREI-MS (Fig. S27†), which showed the molecular ion peak [M]˙+ at m/z 255.0893 (calcd for 255.0895). The IR spectrum revealed absorption bands for hydroxyl, methyl, and conjugated carbonyl groups at 3366, 2923–2853, and 1737 cm−1, respectively (Fig. S28†), while absorption maxima at λmax 262, and 374 nm observed in the UV spectrum (Fig. S29†) were also suggestive of a phenazine derivative. The 1H NMR spectrum of compound 4 (Fig. S30†) exhibited similarities to those of compounds 1–3, with additional signals of two deshielded methyl groups at δH 3.47 and 2.94, along with a 1-proton singlet at δH 15.81, assigned to a chelated OH group of carboxylic acid. The spectrum also revealed six aromatic proton signals, sorted into two sets: (a) δH 8.97 (dd, 1.6; 6.4 Hz), 8.03 (dd, 6.4; 8.8 Hz), and 8.52 (dd, 1.6; 8.8 Hz), and (b) δH 8.18 (d, 8.8 Hz), 7.87 (dd, 7.2; 8.8 Hz), and 7.83 (d, 7.2 Hz), further supporting the phenazine derivative structure. However, the 13C NMR and DEPT spectra, recorded on an 800 MHz NMR CryoProbe, did not show any peaks due to the low sample amount (0.2 mg). Approximate 13C chemical shift values as listed in Table 1, were obtained from the HSQC and HMBC spectra (Fig. S31 and S32†). The structural elucidation of this compound was also facilitated by its similarities to compound 2. In the HMBC spectrum, cross-peaks were observed between the proton signal at δH 8.97 and the carbon resonances at δC 166.1, 139.1, and 134.8, also between the proton signal at δH 8.52 and the carbon signal at δC 137.2. Further HMBC correlations were observed between the proton signal at δH 8.18 and the carbon signals at δC 139.5, and 132.6, and between the proton signal at δH 8.03 and the carbon signals at δC 143.1 and 125.0 (Fig. 2). Additionally, the methyl proton signals at δH 2.94 correlated with the carbon signals at δC 139.5, 135.8, and 132.6. The deshielded proton signal at δH 3.47 was found to be attached to the nitrogen atom. Consistent with the molecular mass and formula, another nitrogen atom in the phenazine core was replaced by an oxygen atom. The structure of compound 4 was also supported by the COSY spectrum (Fig. S33†), and all protons and carbons were assigned using HSQC cross-peaks. The structure was further validated by important ion fragments at m/z 210 [M–45]˙+ (i.e., M–COOH), m/z 180 [M–75]˙+ (i.e., M–COOH–2 × CH3), m/z 120 [C6H3–COOH]˙+, and m/z 90 [C6H3–CH3]˙+ (Fig. S2†).
The known compounds (Fig. S1†) were identified as phenazine-1-carboxylic acid (5),18 phenazine-1-carboxamide (6),15,19 lumichrome (7),20 anhydrofusarubin (8),21,22 3-methyl ether-fusarubin (9),21,23 fusarubin (10),21,24,25 bostrycoidin (11),26 javanicin (12),21,24,27 5,8-dihydroxy-2-methoxy-6-hydroxymethyl-7-(2-hydroxypropyl)-1,4-naphthalenedione (13),28 1,3,6-trihydroxy-7-(1-hydroxyethyl)anthracene-9,10-dione (14),29,30 2,3-dihydro-5-hydroxy-4-hydroxymethyl-8-methoxy-2-methylnaphtho[1,2-b]furan-6,9-dione (15),31 5,7-dimethoxyisobenzofuran-1(3H)-one (16),32 apocynin (17),33 2-heptyl-3-hydroxy-4(1H)-quinolone (18),34 haplacutine F (19),35,36 ganodermaside D (20),37 cerevisterol (21).38,39
It is worth noting that, although nitrogen-containing heterocyclic compounds such as phenazines are often associated with the secondary metabolism of certain bacteria and fungi, they have been reported to be produced by some Fusarium species.7 However, to the best of our knowledge, no phenazine has been isolated and reported from Fusarium. Phenazine production in Fusarium is influenced by environmental factors like temperature, pH, oxygen availability, and the composition of the culture medium used during fermentation.40,41
2.2. Antimicrobial and cytotoxic activities
Water-soluble pigments, including phenazines, quinones, furans, flavins, pyrans, and others, are known in the food industry to exhibit antimicrobial, cytotoxic, and antioxidant activities.7 Thus, the isolated compounds, sorted into phenazine-derived pigments (1–4), flavins (7), quinones (8–15), furanone (16), quinolones (18 and 19), the steroidal compounds 20 and 21, and others were assessed for their antimicrobial and cytotoxic activities.| Compounds | Antimicrobial (MIC, μM) | Cytotoxicityb (IC50, μM) | |||||
|---|---|---|---|---|---|---|---|
| SA1 | SA2 | EC | CA | MCF-7 | PC3 | HeLa cell | |
| a MIC = Minimum inhibitory concentration.b Tested in triplicate at each concentration, with IC50 values representing mean ± SD from three independent experiments; — = inactive at MIC > 400 μM; nt = not tested; nd = not determined (as the dose-dependent inhibition curves did not reach 50% inhibition within the tested range of 6.25–100 μM); ✗ = inactive; SA1 = Staphylococcus aureus (NCTC 13277); SA2= Staphylococcus aureus biofilms (ATCC 6538); EC = Escherichia coli (ATCC 25922); CA = Candida albicans (ATCC 36082).c Standard antibacterial drug.d Standard antifungal drug.e Standard drug for cytotoxicity assay. | |||||||
| 1 | — | — | nt | nt | nd | nt | nd |
| 2 | — | — | — | 200 | nd | nt | nd |
| 3 | — | — | — | — | ✗ | nt | nd |
| 4 | nt | nt | nt | nt | 6.8 | nd | 25.0 |
| 5 | 400 | 200 | — | 200 | nd | nt | 36.2 |
| 6 | 200 | 200 | — | — | 37.1 | 35.0 | 8.9 |
| 7 | — | — | nt | — | ✗ | nt | nt |
| 8 | — | — | — | nt | nd | 27.6 | 49.0 |
| 9 | — | — | — | — | nd | nt | nt |
| 10 | — | — | — | — | 9.7 | 8.6 | 9.8 |
| 11 | 400 | 200 | — | 200 | nd | 14.7 | ✗ |
| 12 | 200 | 200 | — | — | 7.2 | 7.3 | 8.3 |
| 13 | — | — | — | — | 9.4 | 7.3 | 7.1 |
| 14 | — | — | — | — | nt | nt | nt |
| 15 | 400 | 400 | — | — | 19.4 | 7.1 | 29.4 |
| 16 | — | — | — | — | nd | 17.9 | ✗ |
| 18 | — | — | — | — | 10.8 | 14.3 | nd |
| 19 | — | — | nt | — | ✗ | nt | ✗ |
| 20 | 400 | 200 | — | nt | 12.2 | 6.4 | 91.2 |
| 21 | — | — | — | — | ✗ | nd | nt |
| Ofloxacinc | 25 | 25 | 1–2 | ✗ | nt | nt | nt |
| Amphotericind | ✗ | ✗ | ✗ | 0.4–1.0 | nt | nt | nt |
| Doxorubicine | nt | nt | nt | nt | 0.10 | 0.50 | 0.36 |
In contrast to the previous work of Trisuwan and collaborators (2010),21 who reported moderate activity for compound 12 (javanicin) against MCF-7 breast cancer cells, our findings revealed a more potent cytotoxic profile, with an IC50 value of 7.2 μM. To validate these results, we conducted additional dose–response experiments with compounds 10 and 12 across a concentration range of 6.25–100 μM. Compound 12 showed clear dose-dependent inhibition of MCF-7 cell growth, achieving 91.8% inhibition at 50 μM, 83.6% at 25 μM, 69.4% at 12.5 μM, and 45.2% at 6.25 μM (Fig. S34†). These differences in activity may stem from variations in experimental conditions or cell line characteristics. Further investigation using quantum structure–activity relationship (QSAR) analysis could help elucidate the molecular features underlying these divergent results. Additionally, ganodermaside D (20), a steroidal compound previously reported as an anti-aging agent by Weng and co-workers (2011),37 exhibited moderate inhibitory activities against MCF-7 breast (IC50 = 12.2 μM) and HeLa cervical (IC50 = 91.2 μM) cancer cell lines. Its potency was good against PC3 prostate cancer cells (IC50 = 6.4 μM).
In cytotoxicity assays, several compounds showed promising activity against breast (MCF-7), cervical (HeLa), and prostate (PC-3) cancer cell lines, with derivatives such as 4 and 6 exhibiting strong inhibition, possibly through ROS generation or DNA intercalation. Quinone derivatives (10 and 12) displayed dose-dependent cytotoxicity, suggesting redox-mediated apoptosis, while steroidal ganodermaside D (20) showed moderate and potent effects against hormone-related cancers (MCF-7 and PC-3), potentially via steroid receptor modulation. However, inconsistencies in MCF-7 inhibition between studies indicate that cell line-specific metabolic differences or compound stability may influence activity. Further mechanistic investigations, including transcriptomic profiling, ROS detection assays, and in vivo tumour models, are needed to validate these hypotheses. Quantum structure–activity relationship (QSAR) analyses could also guide the rational design of more selective derivatives.
3. Experimental
3.1. General experimental procedures
For large-scale fermentation, the fungus was grown on 4 mm petri dishes containing Yeast-Mold-Agar (YMA) and fermented using a solid rice medium, locally known as “tota chawal” (broken rice) in Karachi. For column chromatography, silica gel 60 (230–400 mesh, Merck, Germany) was used. Semi-pure compounds were further purified using a recycling preparative HPLC-LC-908 system (Japan) with a JAIGEL-ODS-M-80 column (250 mm × 20 mm, 4 μm, 80 Å). The eluent for reverse-phase chromatography was a 50% MeCN–50% H2O mixture. For normal-phase chromatography, a JAIGEL-SIL D-60–10 column was used.The purity of isolated compounds was assessed using Merck TLC plates (silica gel 60 F254). A JASCO P-2000 polarimeter was used to measure optical rotations. UV spectra were recorded on a Hitachi U-3200 spectrophotometer, and IR spectra were obtained on a Bruker Alpha Platinum-ATR spectrometer using the ATR technique (4000–400 cm−1, 4 cm−1 resolution, 16 scans). Electron ionization mass spectra (EI-MS) were acquired using a JEOL JMS600H-1 mass spectrometer. NMR experiments were performed on Bruker Ascend 500, 600, and 800 MHz spectrometers in deuterated solvents, with chemical shifts referenced to TMS at 0.00 ppm for both 1H and 13C NMR. Coupling constants were reported in hertz (Hz).
3.2. Fungal material
Fresh horse dung was collected at Ecopark in Ahala, Yaoundé, Cameroon. Fungal isolation was carried out using the moist chamber protocol49 with slight modifications. Briefly, approximately 0.5 g of faeces was placed in four petri dishes containing filter paper. The petri dishes were pre-treated with a chloramphenicol solution (0.5 μM) to prevent bacterial growth. After 4 days of incubation, the first series of spores were observed, with subsequent series appearing after 8 and 15 days. The first series of spores was transferred to Malt Extract Agar (MEA) for further purification, resulting in the isolation of seven fungal strains, which were subjected to chemical pre-screenings. One of these strains showed potential for large-scale fermentation and was identified as Fusarium solani by Inqaba Biotec (https://inqababiotec.co.za/), through ITS nrDNA sequencing (Table S1†). A voucher specimen of the selected strain was stored at −80 °C in our laboratory under the number CECA065.For identification, genomic DNA was extracted using the Quick-DNA™ Fungal/Bacterial kit (Zymo Research, Catalogue No. D6005). The ITS target region was amplified with OneTaq® Quick-Load® 2X Master Mix (NEB, Catalogue No. M0486) using ITS-1 and ITS-4 primers. The PCR product was analysed by gel electrophoresis and cleaned up enzymatically using the EXOSAP method. The extracted fragments were sequenced in both the forward and reverse directions (Nimagen, BrilliantDye™ Terminator Cycle Sequencing Kit V3.1, BRD3-100/1000) and subsequently purified (Zymo Research, ZR-96 DNA Sequencing Clean-up Kit™, Catalogue No. D4050). The purified fragments were analysed on the ABI 3500xl Genetic Analyser (Applied Biosystems, ThermoFisher Scientific). The resulting sequence data (*.ab1 files) were processed using DNASTAR software and identified through a BLAST search (NCBI).
3.3. Fermentation and extraction
Ten 500 mL-Erlenmeyer flasks, each containing 80 g of broken rice (tota chawal) and 100 mL distilled water, were autoclaved and inoculated with well-grown Fusarium solani cultures from Yeast-Mold-Agar (YMA) plates. The flasks were incubated at 20 °C in an air-conditioned room. After 15 days, the cultures were harvested, and the fungal mycelia were extracted with ethyl acetate. The filtrate was concentrated under reduced pressure, yielding 4.2 g of purple crude extract.3.4. Isolation
The organic extract (4 g) was subjected to silica gel column chromatography and eluted with mixtures of n-hexane/acetone (0–100% acetone, v/v) of increasing polarity, followed by acetone/methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 8:2, v/v), resulting in 100 fractions (fr1–fr100, ∼400 mL each). Fractions fr51–fr54 formed a precipitate, which was washed with n-Hex/acetone (95:5, v/v) to yield compound 5 (15 mg, yellow amorphous powder, tR: 22 min). Based on TLC profiles, the fractions were grouped into 7 main series (I–VII). Series I (200 mg) and VII (1.2 g), eluted with pure n-hexane and 10–20% methanol in acetone (v/v), were complex mixtures and were not further investigated.
:1 to 8:2, v/v), resulting in 100 fractions (fr1–fr100, ∼400 mL each). Fractions fr51–fr54 formed a precipitate, which was washed with n-Hex/acetone (95:5, v/v) to yield compound 5 (15 mg, yellow amorphous powder, tR: 22 min). Based on TLC profiles, the fractions were grouped into 7 main series (I–VII). Series I (200 mg) and VII (1.2 g), eluted with pure n-hexane and 10–20% methanol in acetone (v/v), were complex mixtures and were not further investigated.
Series II (20 mg), obtained with 5% acetone in hexane, was further purified by normal-phase preparative HPLC equipped with a JAIGEL-SIL (D-60-10) column, using a mixture of 1% methanol in chloroform (CHCl3) as the eluent. This purification yielded compound 8 (15 mg), a pigment with an intense purple colour (tR: 22 min). Series III (37 mg), obtained with n-Hex/acetone (90:10, v/v), was purified by normal-phase preparative HPLC, using 1% methanol in chloroform as the eluent, to afford compound 9 (5.2 mg), an orange pigment (tR: 18 min).
Series IV (1.2 g), obtained with n-Hex/acetone (80:20, v/v), was separated by silica gel column chromatography using a CHCl3/MeOH (99:1, v/v) mixture, resulting in compounds 16 (7.2 mg, white gum, tR: 12 min), 6 (105 mg, yellow needle crystals, tR: 18 min), and sub-fractions IVA and IVB. Sub-fraction IVA (8.3 mg) was further purified on a Sephadex LH-20 column using 50% MeOH in CH2Cl2, yielding compound 20 (4 mg, yellow oil). Sub-fraction IVB (88 mg) was purified by reverse phase preparative HPLC using 50% MeCN in H2O, yielding two pink pigments: compounds 12 (3.8 mg, tR: 32 min) and 11 (9.2 mg, tR: 36 min).
Series V (300 mg), eluted from the main column with n-Hex/acetone (70:30, v/v) was further subjected to column chromatography over silica gel using a CH2Cl2/MeOH (96:4, v/v) mixture to give sub-fractions VA–VC. Sub-fraction VA (45 mg) was purified by reverse phase preparative HPLC (50% MeCN–50% H2O), to afford compounds 3 (3.1 mg, yellow pigment, tR: 18 min), and 1 (1.3 mg, yellow pigment, tR: 22 min). Using the same HPLC method and equipment, sub-fraction VB (12 mg) yielded compounds 7 (0.5 mg, white amorphous powder, tR: 25 min) and 2 (1.1 mg, yellow pigment, tR: 28 min). Lastly, sub-fraction VC (30 mg) afforded compounds 19 (6 mg, white gum, tR: 30 min), and 18 (10 mg, white gum, tR: 36 min).
Series VI (102 mg), which was previously eluted from the main column with n-Hex/acetone (60:40, v/v) was further subjected to silica gel column chromatography using an isocratic mixture of CH2Cl2/acetone (85:15, v/v) to afford compounds 21 (7.2 mg, white neat solid), and sub-fractions VIA and VIB. Both sub-fractions were separately purified by reverse preparative HPLC using MeCN/H2O (1:1, v/v). Sub-fractions VIA provided orange pigments of 10 (1.5 mg, tR: 30 min), and 17 (1.3 mg, tR: 32 min). Sub-fractions VIB yielded orange pigments, including 15 (2.3 mg, tR: 28 min), 14 (4.3 mg, tR: 34 min), and 13 (5.2 mg, tR: 38 min).
3.5. Physico-chemical properties of compounds 1–4
Fusaphenazinone (1): yellowish pigment; [α]D25 0 (c 0.10, CHCl3); UV (CHCl3) λmax (Abs.) 256 (1.55), 267 (1.52), 371 (1.70) nm; IR (ATR) νmax 2923, 2852, 1769, 1557, 1525, 1305, 1188, 1158, 957, 901, 759, 671 cm−1; 1H and 13C NMR data, Table 1; EI-MS m/z (rel. int., %): 280 (11), 265 (26), 249 (32), 235 (6), 220 (13), 205 (6), 193 (9), 179 (5), 149 (8), 111 (4), 85 (5), 71 (9), 57 (12), 44 (100); HREI-MS m/z 280.0846 [M]˙+ (mol. formula C16H12N2O3, calcd value 280.0848).4-Methylphenazine-1-carboxylic acid (2): yellowish pigment; UV (CHCl3) λmax (Abs.) 257 (1.51), 266 (1.50), 372 (2.09) nm; IR (ATR) νmax 2922, 2852, 1715, 1569, 1531, 1464, 1264, 1293, 800, 760, 741 cm−1; 1H and 13C NMR data, Table 1; EI-MS m/z (rel. int., %): 238 (0.8), 194 (100), 193 (32.7), 102 (2.6), 179 (1.1), 128 (0.6); HREI-MS m/z 238.0744 [M]˙+ (mol. formula C14H10N2O2, calcd value 238.0742).
1-Acetyl-4-hydroxyphenazine (3): yellowish pigment; UV (CHCl3) λmax (Abs.) 247 (2.14), 264 (1.99), 368 (1.38) nm; IR (ATR) νmax 3400, 2929, 2851, 1684, 1590, 1398, 1364, 1246, 667, 494 cm−1; 1H and 13C NMR data, Table 1; EI-MS m/z (rel. int., %): 238 (13), 221 (60), 195 (100), 178 (42), 162 (29), 145 (12), 119 (35), 102 (29); HREI-MS m/z 238.0740 [M]˙+ (mol. formula C14H10N2O2, calcd value 238.0742).
9,10-dimethylphenoxazine-4-carboxylic acid (4): yellowish pigment; UV (CHCl3) λmax (Abs.) 262 (0.50), 374 (0.13) nm; IR (ATR) νmax 3366, 2923, 2853, 1737, 1575, 1540, 1464, 1416, 1377, 1174, 1113, 768 cm−1; 1H and 13C NMR data, Table 1; EI-MS m/z (rel. int., %): 255 (8), 210 (100), 180 (55), 150 (8), 120 (21), 105 (6), 90 (12); HREI-MS m/z 255.0893 [M]˙+ (mol. formula C15H13NO3, calcd value 255.0895).
3.6. Biological assays
4. Conclusions
This study reports the isolation of four new phenazine-derived pigments and seventeen known compounds from Fusarium solani, along with an evaluation of their antimicrobial and cytotoxic activities. None of the compounds exhibited significant antimicrobial effects against Staphylococcus aureus, Candida albicans, and Escherichia coli, although compounds 5 and 11 showed weak to moderate activity against C. albicans and S. aureus. In contrast, the new compound, 9,10-dimethylphenoxazine-4-carboxylic acid (4), showed strong cytotoxicity against MCF-7 human breast cancer cell lines. Some known quinone-derived compounds, including 10, 12, and 13, exhibited potent activity against the tested cancer cell lines. These findings suggest that the primary bioactivity of phenazine- and quinone-derived pigments is cytotoxic, highlighting their strong potential for anticancer drug development. However, our findings regarding the potencies of quinones 8 and 12 on MCF-7 breast cancer cells differ from those reported by Trisuwan and collaborators (2010).21 Further research, including structure–activity relationship (SAR) studies, is necessary to optimise these compounds and the new derivative 4 for enhanced chemotherapeutic efficacy.Data availability
The data generated and analysed during this study are available from the corresponding author upon reasonable request. Data related to the chemical structures and spectroscopic analysis of the isolated compounds are available in the ESI.†Author contributions
B. Y. G. M., P. D., and J. T.: isolation, identification, and cultivation of the fungus. B. Y. G. M.: methodology, large scale fermentation, purification and isolation of compounds, spectroscopic analysis, and data curation. B. Y. G. M. and R. M.: bioassays, data curation, and preparing the original draft manuscript. B. Y. G. M., I. L. M., and S. F. K.: structure elucidation. G. L. M. T., M. I. C., and S. F. K.: reviewing, editing, and correcting the draft. B. Y. G. M., J. T., G. L. M. T., and S. F. K.: conceptualization. M. I. C. and S. F. K.: supervision, funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
B. Y. G. M. gratefully acknowledges The World Academy of Sciences (TWAS) for an eight-month Postdoctoral Fellowship (FR number: 3240325064) under the ICCBS-UNESCO-TWAS programme, which supported his research visit to the H.E.J. Research Institute of Chemistry, ICCBS, University of Karachi, Pakistan. He also expresses his gratitude to the Ministerial Standing Committee on Scientific and Technological Cooperation (COMSTECH) of the Organization of Islamic Cooperation (OIC) for partially supporting his living expenses at the ICCBS. This work was conducted as part of the Humboldt Research Hub-CECANAPROF (3.4-CMR-Hub) on antimicrobial natural products from fungi (https://cecanaprof.com/), funded by the Alexander von Humboldt Foundation. The authors further acknowledge the German Academic Exchange Service (DAAD) for providing equipment grants (PKZ 308665 - Ref. ST 42) to their laboratory at the Higher Teacher Training College, University of Yaoundé I, Cameroon.References
- A. A. Farouq, D. K. Abdullah, F. Hooi-Ling, N. Abdullah, S. S. Malaysia and J. Biol, J. Agric. Saf. Health., 2012, 2, 44–51 Search PubMed.
- A. Mumpuni, A. Amurwanto and D. J. Wahyono, Biodivers. J., 2021, 22, 1550–1557 Search PubMed.
- A. S. Jasim, B. A. Abass and I. M. Al-Rubayae, Arch. Razi. Inst., 2021, 76, 1333–1341 CAS.
- M. J. Richardson, Mycol. Res., 2001, 105, 387–402 CrossRef.
- M. A. Moghalles and S. M. Al-Bader, YJAVS, 2014, 1, 22–26 CrossRef.
- V. Edel-Hermann and C. Lecomte, Phyopathology, 2019, 109, 512–530 CrossRef CAS PubMed.
- R. Poorniammal, S. Prabhu, L. Dufossé and J. Kannan, J. Fungi, 2021, 7, 692 CrossRef CAS PubMed.
- S. R. M. Ibrahim, A. Sirwi, B. G. Eid, S. G. A. Mohamed and G. A. Mohamed, J. Fungi, 2021, 7, 943 CrossRef CAS PubMed.
- B. S. Menezes, L. S. Solidade, A. A. Conceição, M. N. Santos Junior, P. L. Leal, E. S. de Brito, K. M. Canuto, S. Mendonça, F. G. de Siqueira and L. M. Marques, Appl. Microbiol. Biotechnol., 2020, 10, 117 CAS.
- S. Sarrocco, Pest. Manag. Sci., 2016, 72, 643–652 CrossRef CAS PubMed.
- M. Vašutová, P. Mleczko, A. López-García, I. Macek, G. Boros, J. Ševcík, S. Fujii, D. Hackenberger, I. H. Tuf, E. Hornung, B. Páll-Gergely and R. Kjøller, Mycorrhiza, 2019, 29, 413–434 CrossRef PubMed.
- B. Y. G. Mountessou, M. E. G. Anoumedem, B. M. Kemkuignou, Y. Marin-Felix, F. Surup, M. Stadler and F. S. Kouam, Beilstein J. Org. Chem., 2023, 19, 1555–1561 CrossRef CAS PubMed.
- M. E. G. Anoumedem, B. Y. G. Mountessou, F. S. Kouam, A. Narmani and F. Surup, Antibiotics, 2020, 9, 753 CrossRef PubMed.
- M. G. Happi, S. F. Kouam, F. M. Talontsi, C. Nkenfou, F. Longo, B. T. Ngadjui and M. Spiteller, Z. Naturforsch., 2013, 70, 625–630 CrossRef.
- V. Shanmugaiah, N. Mathivanan and B. Varghese, J. Appl. Microbiol., 2010, 108, 703–711 CrossRef CAS PubMed.
- J. A. VanAllan, G. A. Reynolds and R. E. Adel, J. Org. Chem., 1962, 27, 1659–1664 CrossRef CAS.
- G. W. Rewcastle, W. A. Denny and B. C. Baguley, J. Med. Chem., 1987, 30, 843–851 CrossRef CAS PubMed.
- A. Cimmino, Z. Bahmani, S. Castaldi, M. Masi, R. Isticato, J. Abdollahzadeh, J. Amini and A. Evidente, J. Agric. Food Chem., 2021, 69, 12143–12147 CrossRef CAS PubMed.
- G. P. Jones, D. G. Lewis and M. E. Tate, Acta Cryst., 1988, C44, 2220–2222 CAS.
- K. Ströch, A. Zeeck, N. Antal and H. P. Fiedler, J. Antibiot., 2005, 58, 103–110 CrossRef PubMed.
- K. Trisuwan, N. Khamthong, V. Rukachaisirikul, S. Phongpaichit, S. Preedanon and J. Sakayaroj, J. Nat. Prod., 2010, 73, 1507–1511 CrossRef CAS PubMed.
- I. Kurobane, L. C. Vining, A. G. McInnes and J. A. Walter, Can. J. Chem., 1980, 58, 1380–1385 CrossRef CAS.
- J. H. Tatum and R. A. Baker, Phytochemistry, 1983, 22, 543–547 CrossRef CAS.
- Y. Wen, Y. Lv, J. Hao, H. Chen, Y. Huang, C. Liu, H. Huang, Y. Ma and X. Yang, Nat. Prod. Res., 2020, 34, 1879–1883 CrossRef CAS PubMed.
- T. Kurobane, L. C. Vining, A. G. McInnes and D. G. Smith, Can. J. Chem., 1978, 56, 1593 CrossRef.
- N. Khan, F. Afroz, M. N. Begum, S. R. Rony, S. Sharmin, F. Moni, C. M. Hasan, K. Shaha and M. H. Sohrab, Toxicol Rep, 2018, 5, 970–976 CrossRef CAS PubMed.
- D. Bergeron, B. Caron and P. Brassard, J. Org. Chem., 1993, 58, 509–511 CrossRef CAS.
- J. H. Tatum, R. A. Baker and R. E. Berr, Phytochemistry, 1985, 24, 3019–3021 CrossRef CAS.
- D.-L. Zhao, D. Wang, X.-Y. Tian, F. Cao, Y.-Q. Li and C.-S. Zhang, Mar. Drugs, 2018, 16, 36 CrossRef PubMed.
- P. Wang, Y. Cui, C. Cai, H. Chen, Y. Dai, P. Chen, F. Kong, J. Yuan, X. Song and W. Mei, Mar. Drugs, 2018, 17, 4 CrossRef PubMed.
- H. Liu, C. Yan, C. Li, T. You and Z. She, Molecules, 2020, 25, 576 CrossRef CAS PubMed.
- S.-J. Wang, H.-W. Liu, Y.-Q. Wang, L. Bao, X.-L. Yang and H.-A. Wen, Mycosystema, 2013, 32, 1028–1033 CAS.
- H. Finnemore, J. Chem. Soc., 1908, 93, 1513–1519 RSC.
- S. P. Diggle, S. Matthijs, V. J. Wright, M. P. Fletcher, S. R. Chhabra, I. L. Lamont, X. Kong, R. C. Hider, P. Cornelis, M. Cámara and P. Williams, Chem. Biol., 2007, 14, 87–96 CrossRef CAS PubMed.
- D. Li, N. Oku, A. Hasnada, M. Shimizu and Y. Igarashi, Beilstein J. Org. Chem., 2018, 14, 1446–1451 CrossRef CAS PubMed.
- D. Staerk, J. R. Kesting, M. Sairafianpour, M. Witt, J. Asili, S. A. Emami and J. W. Jaroszewski, Phytochemistry, 2009, 70, 1055–1061 CrossRef CAS PubMed.
- Y. Weng, J. Lu, L. Xiang, A. Matsuura, Y. Zhang, Q. Huang and J. Qi, Biosci., Biotechnol., Biochem., 2011, 75, 800–803 CrossRef CAS PubMed.
- Q.-X. Wang, S.-F. Li, F. Zhao, H.-Q. Dai, L. Bao, R. Ding, H. Gao, L.-X. Zhang, H.-A. Wen and H.-W. Liu, Fitoterapia, 2011, 82, 777–781 CrossRef CAS PubMed.
- H. Kawagishi, R. Katsumi, T. Sazawa, T. Mizuno, T. Hagiwara and T. Nakamura, Phytochemistry, 1988, 21, 2111–2119 Search PubMed.
- S. Kumaria, V. Khanna and N. Sharma, Int. J. Pest Manag., 2024, 70, 1086–1099 CrossRef.
- A. Upadhyay and S. Srivastava, Microbiol. Res., 2011, 166, 323–335 CrossRef CAS PubMed.
- A. H. Mussa, A. F. Abdulkareem, H. F. Abbas and H. Farhan, Syst. Rev. Pharm., 2020, 11, 408 CAS.
- V. F. Cardozo, A. G. Oliveira, E. K. Nishio, M. R. E. Perugini, C. G. T. J. Andrade, W. D. Silveira, N. Durán, G. Andrade, R. K. T. Kobayashi and G. Nakazato, Ann. Clin. Microbiol. Antimicrob., 2013, 12, 1–8 CrossRef PubMed.
- O. P. Martínez-Rodríguez, R. García-Contreras, R. Aguayo-Ortiz and M. Figueroa, Biofouling, 2023, 39, 830–837 CrossRef PubMed.
- H. Satriawan, T. C. Teoh, M. Rizman-Idid, A. Krishnan, N. A. Bakar and S. A. Alias, Chiang Mai J. Sci., 2024, 51, e2024043 Search PubMed.
- D. K. Morales, N. J. Jacobs, S. Rajamani, M. Krishnamurthy, J. R. Cubillos-Ruiz and D. A. Hogan, Mol. Microbiol., 2010, 78, 1379–1392 CrossRef CAS PubMed.
- W. Xun, B. Gong, X. Liu, X. Yang, X. Zhou and L. Jin, Int. J. Mol. Sci., 2023, 24, 11274 CrossRef CAS PubMed.
- R. J. N. Frandsen, S. A. Rasmussen, P. B. Knudsen, S. Uhlig, D. Petersen, E. Lysøe, C. H. Gotfredsen, H. Giese and T. O. Larsen, Sci. Rep., 2016, 6, 26206 CrossRef CAS PubMed.
- D. L. Hawksworth, Mycologist's Handbook, Commonwealth Mycological Institute, Kew, Hawksworth, UK, 1974 Search PubMed.
- R. Maharjan, A. I. Khan, M. Nadeem-ul-Haque, M. Maresca, M. I. Choudhary, F. Shaheen and S. U. Simjee, Probiotics. Antimicrob. Proteins, 2022, 14, 391–405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra04180e |
| This journal is © The Royal Society of Chemistry 2025 |