
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn NMR insight into the solvatochromic behaviour of Brooker's merocyanine dyes†
Michal Afria,
Hugo E. Gottlieba,
Natalia Fridmanb and
Abed Saady *a
*a
aDepartment of Chemistry, Bar-Ilan University, Ramat-Gan, 52900, Israel. E-mail: abed.saady@biu.ac.il
bSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa City 3200003, Israel
First published on 17th July 2025
Abstract
Systematic NMR/UV-Vis analysis of Brooker's merocyanine dyes reveals solvent polarity governs electronic structure and spectroscopy. Absorption maxima 13C and 15N NMR shifts correlate with ET(30), probing zwitterionic/vinylogous amide. Substituent and solvent effects modulate these interactions, directly linking colour changes to electronic structure for rational design of environment-sensitive probes.
Introduction
Nuclear magnetic resonance (NMR) spectroscopy is a fundamental tool for probing the structure, dynamics, and interactions of organic molecules, providing detailed insights that are indispensable in material science, drug development, and biochemical research.1–3 NMR enables chemists to elucidate molecular properties by revealing subtle changes in electronic environments, molecular dynamics and molecular conformations.4,5Brooker's merocyanine (BM),6 is a highly conjugated dye known for its strong solvatochromic behaviour, namely, its colour changes depending on the solvent polarity (Fig. 1A). Numerous seminal studies have established the theoretical and experimental foundations of solvatochromism, including key reports on spectroscopic correlations—particularly between UV/Vis and NMR spectroscopy.7–14
 | ||
| Fig. 1 (A) BM in solution in various solvents. (B) The structural formula of BM. | ||
The unique electronic structure of BM makes it valuable in developing advanced materials for sensors, photonic devices, and molecular electronics.15,16 Its well-defined electronic transitions and ability to exist in multiple resonance forms make BM an ideal model for exploring fundamental mechanisms of nonlinear optical behaviour, such as intramolecular charge transfer and polarizability, which are central to the development of advanced photonic and optoelectronic materials.17–19
Structurally, BM, 1a can be drawn in two canonical forms, as shown in Fig. 1B. The form on the left preserves the aromaticity of the two rings, and is zwitterionic, involving charge separation, and would, therefore, be expected to be dominant in polar solvents. Conversely, the form on the right is a vinylogous amide and is neutral, eliminating the charges on the heteroatoms expected to be dominant in non-polar solvents.
In our previous work on the synthesis and applications of BM derivatives,20,21 we observed substantial solvent-dependent variations in their NMR spectra of various derivatives of BM's consistent with literature reports.22,23
This is reminiscent of the well-known solvatochromic material, Reichardt's dye, from which C. Reichardt derived the UV/Vis-based ET(30) index of solvent polarity (Reichardt himself mentions Brooker as an early proposer of a dye-based scale of solvent polarity).24 Extending this concept further, Frimer et al. reported an NMR study of Reichardt's dye, in which they showed that 13C chemical shifts correlate very well with solvent polarity and, therefore, with colour.25
Although basic NMR study of BM laid the foundation, here we significantly expand this scope by systematically investigating the correlation of ET(30) index and the photophysical properties of nine BM derivatives. Our results reveal a direct correlation between solvent polarity and both NMR chemical shifts (1H, 13C and 15N) and UV-Vis absorption, demonstrating that changes in the local environment are consistently reflected in both spectroscopic signatures. These findings provide new insights into the structure–property relationships in BM dyes and underscore the broader applicability of NMR and UV-Vis methods for understanding solvatochromism in this important class of materials.
Results and discussion
Compounds 1a–i, 2g and 2i (Fig. 2) were synthesized according to a previously reported procedure.20,21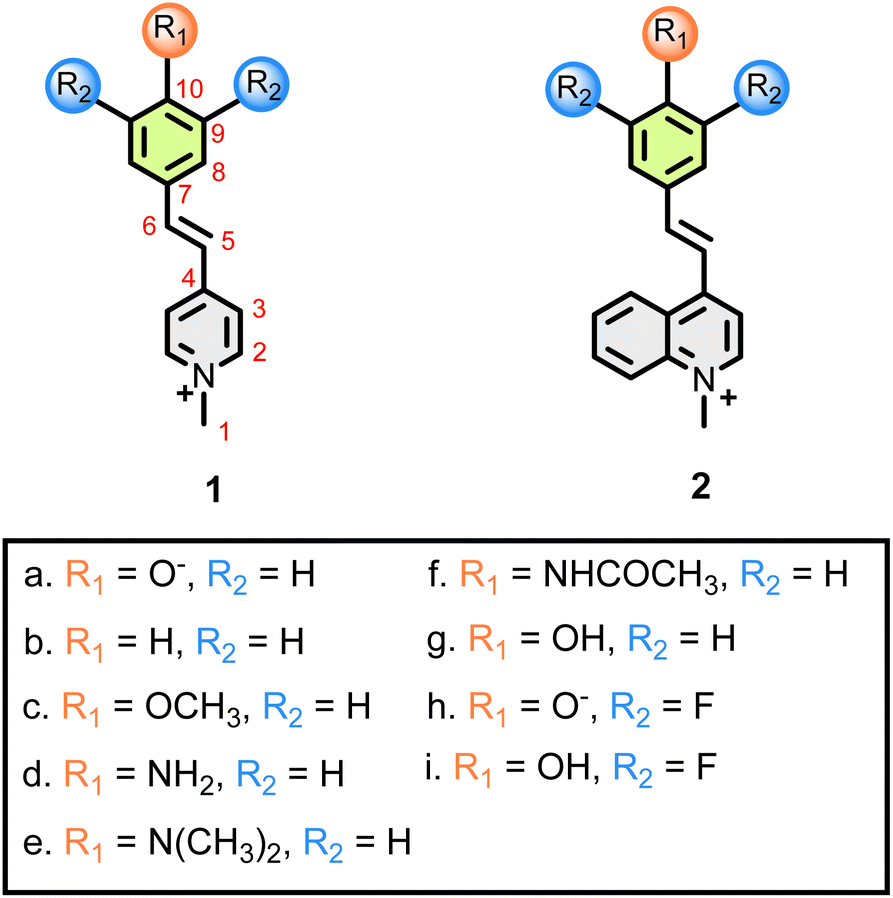 | ||
| Fig. 2 BM derivatives synthesized and evaluated in this study. | ||
The UV/Vis spectrum of parent merocyanine 1a shows two absorption maxima, one in the yellow (λmax = ca. 600 nm), and one in the violet region (λmax = ca. 400 nm) of the spectrum. We measured UV/Vis spectra for 1a in a variety of solvents ranging from THF (least polar) to water (most polar) (see; ESI†).
A plot of the wavelengths of the absorption maxima in nm and eV, as a function of the ET(30) index of the polarity of the solvent is shown in Fig. 3. As can be seen, there is a good correlation between ET(30) and the λmax values, which suggests that the ability of the solvents to induce separation of charges is similar for both the Brooker and the Reichardt dyes.
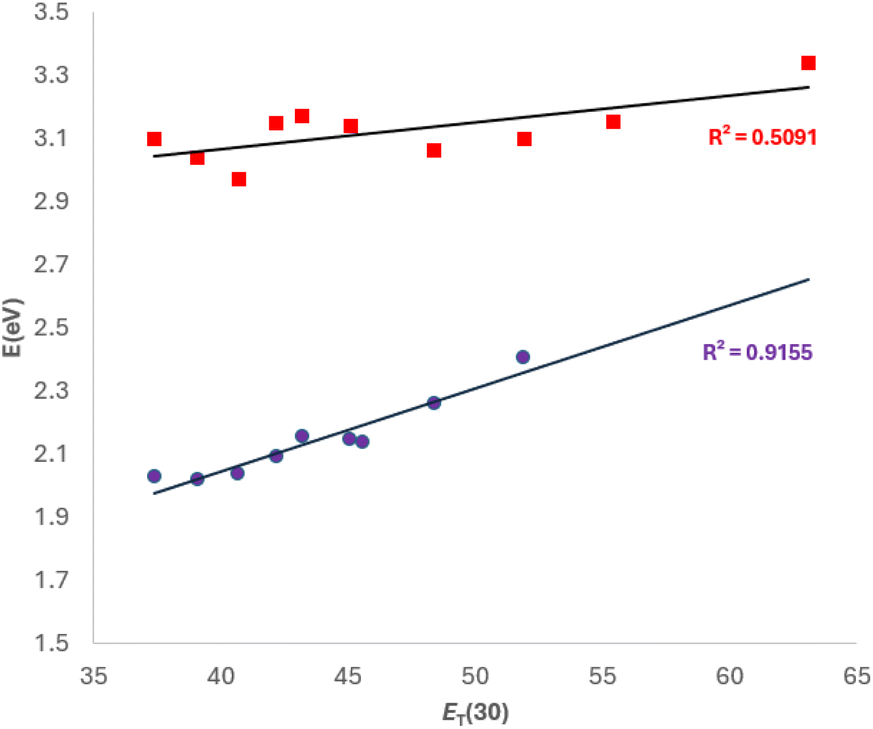 | ||
| Fig. 3 λmax (eV) values for 1a, as a function of the polarity of the solvent [ET(30) values, in kcal mol−1] ranging from THF (least polar) to water (most polar). | ||
With a view to compare the UV/Vis and NMR spectral properties we then took 1H and 13C NMR spectra of 1a in a variety of solvents. We unambiguously assigned all the NMR signals by using several 2D-NMR techniques (COSY – 1H × 1H correlation, HMQC – 1H × 13C one-bond correlation and HMBC – 1H × 13C long-range correlation). In addition, we indirectly obtained 15N chemical shifts by 1H × 15N HMBC spectra (see; ESI, Table S1†).
As the polarity increases, the high-wavelength peak becomes less intense and approaches the lower wavelength, and for the most polar solvents (methanol and water) it cannot be observed. (Full data in the ESI, Table S12†).
Inspection of the data indicates the chemical shifts of carbons 5, 7 and 10 are the most solvent-dependent – (Fig. 4). C-10 is deshielded in non-polar solvents, suggesting a more carbonyl-like nature, and shielded in polar solvents – more resembling an aromatic-carbon. Following this trend, even numbered carbons (C-8, C-6), also have negative slopes (albeit smaller in absolute terms) in the δ × ET(30) graph. Conversely, odd-number carbons (C-3, C-5, C-7) have positive slopes. Especially striking are the chemical shift of the N-1 nitrogen, with a strongly positive slope (Fig. 4). In non-polar solvents, this nitrogen is more like a VA and therefore deshielded relative to polar solvents, where it is more resembles a typical pyridinium salt.
 | ||
| Fig. 4 Solvent dependence of the chemical shifts for selected carbon and nitrogen atoms of 1a. | ||
All the compounds examined dissolve well in CD3OD and DMSO-D6; however, compounds 1a–i, 2g and 2i can be dissolved in less polar solvents (e.g. CDCl3) or in D2O, but this often resulted in more dilute samples which required overnight runs. Also, methanol and DMSO are representative more and less polar solvents, [ET(30) = 55.5 and 45.1, respectively].
Therefore, for the sake of brevity and comparison purposes, we decided to consistently report in the body of this paper only the differences of the chemical shifts in these two solvents of (δmethanol − δDMSO) for carbons 5, 7 and 10 and for N-1 (Table 1).
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | 2g | 1h | 1i | 2i | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C-5 | 8.24 | 0.71 | 0.72 | 1.44 | 0.75 | 1.24 | 2.57 | 3.81 | 6.09 | 0.61 | 6.00 |
| C-7 | 6.41 | 1.53 | 1.56 | 2.72 | 1.83 | 2.40 | 3.46 | 2.09 | 8.45 | 2.36 | 6.25 |
| C-10 | −4.58 | 1.48 | 2.47 | 1.55 | 2.27 | 1.25 | −0.04 | −1.76 | −4.20 | 2.19 | −4.84 |
| N-1 | 13.7 | −1.9 | −2.2 | −1.8 | −2.6 | −1.5 | 0.8 | 6.4 | 6.6 | −1.8 | 12.1 |
The complete data we have obtained is available in the ESI.†
Unsurprisingly, compound 1b is not solvatochromic – all solutions we examined were orange, this finding is expected, as no zwitterionic canonical form can exist. The observed uniformity of the UV/Vis spectra is mirrored in the NMR results – much smaller absolute δmethanol – δDMSO values are observed (Table 1).
Compounds 1b, was analysed by single crystal X-ray diffraction (Fig. 5).
 | ||
| Fig. 5 Solid-state structure of 1b, shown in ORTEP mode. Ellipsoids are shown at 50% probability. | ||
Similarly, dyes 1c–1f are also not solvatochromic, as seen both by the consistent orange colour of their solutions as by their small absolute δmethanol − δDMSO values (Table 1), as compared to 1a. It turns out that, for all compounds 1b–1f, for each specific carbon/nitrogen atom the Δδ values are not zero but are very similar (if not quite identical) across the sequence of dyes, and are likely due to unrelated specific solvent interactions – and we can use these trends as a baseline to distinguish other, more subtle changes, as will be shown below.
Interestingly, 1g is the protonated (phenol rather than phenolate) form of 1a – and, unlike 1b–1f, it was found to be solvatochromic. Solutions of 1g show the characteristic colour change of phenolate 1a (red in methanol, purple in DMSO, Fig. 1A) – even though to obtain a vinylogous amide form a carbonyl oxygen would have to carry a positive charge and would likely deprotonate, with the departing H+ attaching itself either to the nitrogen or to a solvent molecule.
In either case, we could envisage an equilibrium between an acid and its conjugate base or between tautomers, but not a resonance hybrid. Inspection of the Δδ data as indicated in Table 1, indeed suggests an equilibrium of this type. Δδ values may be used to estimate the degree of deprotonation for the phenol – specifically, the Δδ value (δmethanol − δDMSO) for N-1 is +13.7 for the phenolate, but ca. −2 for the non-solvatochromic substances (1b–1f); this means the corrected Δδ for the nitrogen atom is ca. +16 ppm.
For phenol 1g, the corrected Δδ would be ca. +3 ppm, indicating ca. 20% deprotonation to the phenolate 1a. This is supported by the observation of small broadenings of some carbon signals (in DMSO, 20 and 7 Hz for C-5 and C-7, respectively), indicating that there is an equilibrium which, at 300 K, is not fast enough for the extreme narrowing of the lines. It is worthwhile noticing that in 1d and 1f, we see no indication of an equivalent deprotonation of a less acidic NH-containing substituent.
Compound 2g is related to 1g, having a quinoline system rather than pyridine. The behaviour of the two phenols is very similar – 2g is also solvatochromic, and the Δδ values in this Table 1 indicate that also in this case, the OH group is partially deprotonated. The degree of deprotonation for this phenol, estimated as in item 2.4, indicates an even larger value – ca. 50%.
Compounds 1h and 1i are the phenolate/phenol pair, having difluoro substitution ortho to the OH group.26 As expected, 1h is indeed solvatochromic and the NMR data (ESI, Table S2†) clearly indicates the contribution of the vinylogous amide form as in the parent 1a. To our surprise, phenol 1h, however, does not deprotonate and is not solvatochromic, even though the ortho fluorine substituents are expected to increase the acidity of the phenol function.
Compound 2i is the quinoline version of the ortho-difluorinated phenol. 2i, however, is solvatochromic and is >80% deprotonated in solution (ESI, Table S9†). An equilibrium between protonated and deprotonated forms is also indicated by extensive broadening (the kinetics of this process seem to be slightly slower than what we observed for 1g) of several of the NMR lines (see ESI† file) – the broadest carbon signal (linewidth ca. 40 Hz) is C-10.
In order to estimate the contribution of the VA canonical form to the structure of 1a, we used 15N chemical shifts as the most sensitive tool for this purpose. We thus measured the NMR spectra for 1,4-dimethylpyridinium iodide, 3, and 1-methyl-4(1H)-pyridinone, 4, as model compounds (Fig. 6).
 | ||
| Fig. 6 Structure of model compounds 3 and 4 and solid-state structure of 3, shown in ORTEP mode. Ellipsoids are shown at 50% probability. | ||
These compounds were selected as reference structures for the two principal resonance forms of BM: 3 as a representative of the charge-separated pyridinium form, and 4 as a model for the vinylogous amide form. This allows a direct comparison of 15N chemical shifts, facilitating a quantitative assessment of resonance contributions in the studied dyes.
The 15N chemical shift for salt 3 is indeed quite solvent-independent, at ca. −183 ppm, and this value is a good reference for this type of quaternary nitrogen atom. Conversely, we use the 15N chemical shift – ca. −246 ppm – of 4, in CDCl3, as a model for the uncharged, vinylogous amide form. This chemical shift is quite typical for amide nitrogen.
We thus get a Δδ value of 63 ppm between the two chemical shifts, which allows us to estimate the contribution of the VA form of 1a as 44% in DMSO, 22% in methanol and 17% in water. Compound 4 can be also described as a resonance hybrid between the vinylogous amide form shown here and a charge-separated zwitterion. Using the same Δδ value, we estimate that the canonical form depicted in 4 is the dominant contributor to the resonance hybrid (78%) even in methanol. Of course, in all amides there is some contribution of a charge-separated (zwitterionic) form, and this is especially true for a vinylogous amide, even in a non-polar solvent, so that these values are obviously overestimates, but we still feel that they add to our understanding of this type of chemical function.
Conclusions
In conclusion, we have demonstrated that solvent polarity and molecular structure play key roles in dictating the electronic properties and resonance equilibria of merocyanine dyes. Our combined UV/Vis and NMR studies reveal that only dyes capable of adopting zwitterionic resonance forms, such as 1a, exhibit strong solvatochromism, with both absorption maxima and NMR chemical shifts shifting systematically with solvent environment. Through analysis of structural analogues and 15N chemical shifts, we show that substituents finely tune the extent of charge separation and deprotonation equilibria. These findings provide a clear framework for designing merocyanine-based dyes and probes with tailored, environment-sensitive properties.Experimental
General information
Reagents and solvents were purchased from commercial sources and were used without further purification. Progress of reactions was monitored by TLC on precoated Merck silica gel plates (60F-254), with visualization by UV light. Compounds were characterized by nuclear magnetic resonance using a Bruker Avance-III-700 spectrometer (700.45, 176.13 and 70.98 MHz for 1H, 13C and 15N, respectively). Chemical shifts are reported in ppm units (δ) relative to TMS (δH and δC = 0 ppm). The residual signals of under-deuterated solvents (δH = 7.26 ppm in CDCl3, 4.79 ppm in D2O and 2.50 ppm in (CD3)2SO) in 1H NMR spectra, and the central peaks of CDCl3 (δC = 77.00 ppm), (CD3)2SO (δC = 39.52 ppm) or TMS in 13C NMR spectra were used as internal standards. The coupling constants (J), calculated according to the first order NMR spectra, are reported in Hz. Standard abbreviations indicating multiplicity were used as follows: m = multiplet, quint = quintet, q = quartet, t = triplet, d = doublet, s = singlet, br = broad, sept = septet. 1H signal assignment was carried out using 2D NMR methods (COSY, HMQC, HMBC) as appropriate. Chemical shifts for 15N NMR spectra were referenced to neat nitromethane (an external standard). Absorption spectra were measured on a UV-2401PC UV-VIS recording spectrophotometer (Shimadzu, Kyoto, Japan).Compounds 1a–1i, 2g and 2i were synthetized according to literature procedures.20,21 Typically, 1,4-dimethylpyridinium iodide, 3 (0.1 mmol), the appropriate substituted benzaldehyde (0.1 mmol), and a catalytic amount of piperidine were dissolved in 2 mL of absolute ethanol in a 10 mL microwave CEM vial. The reaction mixture was irradiated in a microwave reactor at 80 °C for 2 hours. After cooling to room temperature, 10 mL of diethyl ether was added to precipitate the product, which was collected by vacuum filtration and washed three times with 3 mL portions of diethyl ether to afford the pure compound. The products were characterized by NMR and UV/Vis spectroscopy (see ESI† for details).
Data availability
The data supporting this article have been included as part of the Electronic supplementary information (ESI): Detailed spectroscopic data; NMR, UV/Vis spectra and SCXRD as appropriate of the reported compounds. CCDC 2468658 and 2468660. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra03758aAuthor contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Abed Saady thanks Bar-Ilan University (Start-up grant) and the Council for Higher Education (MAOF scholarship).Notes and references
- Y. Hu, K. Cheng, L. He, X. Zhang, B. Jiang, L. Jiang, C. Li, G. Wang, Y. Yang and M. Liu, Anal. Chem., 2021, 93, 1866–1879 CrossRef CAS PubMed.
- S. W. Homans, Angew. Chem., Int. Ed. Engl., 2004, 43, 290–300 CrossRef CAS PubMed.
- H. Zhu and L. A. O'Dell, Commun. Chem., 2021, 57, 5609–5625 RSC.
- E. E. Kwan and S. G. Huang, Eur. J. Org Chem., 2008, 2008, 2671–2688 CrossRef.
- T. D. Claridge, High-resolution NMR Techniques in Organic Chemistry, Elsevier, 2016 Search PubMed.
- L. Brooker, G. Keyes and D. Heseltine, J. Am. Chem. Soc., 1951, 73, 5350–5356 CrossRef CAS.
- N. K. Karmakar, S. Pandey, R. K. Pandey and S. S. Shukla, Appl. Spectrosc. Rev., 2021, 56, 513–529 CrossRef CAS.
- N. Boens, L. Wang, V. Leen, P. Yuan, B. Verbelen, W. Dehaen, M. Van der Auweraer, W. D. De Borggraeve, L. Van Meervelt and J. Jacobs, J. Phys. Chem. A, 2014, 118, 1576–1594 CrossRef CAS PubMed.
- D. Avcı, Ö. Tamer and Y. Atalay, J. Mol. Liq., 2016, 220, 495–503 CrossRef.
- W. Kuznik, M. N. Kopylovich, G. I. Amanullayeva, A. J. Pombeiro, A. H. Reshak, K. T. Mahmudov and I. Kityk, J. Mol. Liq., 2012, 171, 11–15 CrossRef CAS.
- J. M. Marković, N. P. Trišović, D. Mutavdžić, K. Radotić, I. O. Juranić, B. J. Drakulić and A. D. Marinković, Spectrochim. Acta Mol. Biomol. Spectrosc., 2015, 135, 435–446 CrossRef PubMed.
- A. V. Kulinich, N. A. Derevyanko and A. A. Ishchenko, J. Photochem. Photobiol., A, 2007, 188, 207–217 CrossRef CAS.
- D. Avcı, S. Altürk, Ö. Tamer, M. Kuşbazoğlu and Y. Atalay, J. Mol. Struct., 2017, 1143, 116–126 CrossRef.
- L. Zhang, J. M. Cole and X. Liu, J. Phys. Chem. C, 2013, 117, 26316–26323 CrossRef CAS.
- L. G. Nandi, C. R. Nicoleti, I. C. Bellettini and V. G. Machado, Anal. Chem., 2014, 86, 4653–4656 CrossRef CAS PubMed.
- J. Nicolini, F. M. Testoni, S. M. Schuhmacher and V. G. Machado, Tetrahedron Lett., 2007, 48, 3467–3470 CrossRef CAS.
- M. Dekhtyar, W. Rettig, A. Rothe, V. Kurdyukov and A. Tolmachev, J. Phys. Chem. A, 2019, 123, 2694–2708 CrossRef CAS PubMed.
- V. Cavalli, D. C. da Silva, C. Machado, V. G. Machado and V. Soldi, J. Fluoresc., 2006, 16, 77–86 CrossRef CAS PubMed.
- H. Mustroph, Phys. Sci. Rev., 2022, 7, 143–158 Search PubMed.
- A. Saady, E. Varon, A. Jacob, Y. Shav-Tal and B. Fischer, Dyes Pigm., 2020, 174, 107986 CrossRef CAS.
- A. Saady, P. Sudhakar, M. Nassir and A. Gedanken, Ultrason. Sonochem., 2020, 67, 105182 CrossRef CAS PubMed.
- J. O. Morley, R. M. Morley, R. Docherty and M. H. Charlton, Am. Chem. Soc., 1997, 119, 10192–10202 CrossRef CAS.
- J. O. Morley, R. M. Morley and A. L. Fitton, Am. Chem. Soc., 1998, 120, 11479–11488 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- M. Afri, H. E. Gottlieb and A. A. Frimer, Can. J. Chem., 2014, 92, 128–134 CrossRef CAS.
- Compound 1h was obtained after treatment of compound 1i with aqueous KOH according to the general protocol appears on ref. 6.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03758a |
| This journal is © The Royal Society of Chemistry 2025 |