
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall-molecule strategies to combat antibiotic resistance: mechanisms, modifications, and contemporary approaches
Hazem Elkady
 *a,
Ibtehal Nasser Salman
b and
Mohamed M. Khalifa
*ab
*a,
Ibtehal Nasser Salman
b and
Mohamed M. Khalifa
*ab
aPharmaceutical Medicinal Chemistry, Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt. E-mail: hazemelkady@azhar.edu.eg; MohamedKhalifa2321.el@azhar.edu.eg
bDepartment of Pharmacy, Alamal College for Specialized Medical Sciences, Karbala, 56001, Iraq. E-mail: ibtihal.naseer@alamal.edu.iq
First published on 14th July 2025
Abstract
Antibiotic resistance poses a formidable threat to human health, representing a critical challenge that demands urgent attention. Without decisive feat, we confront the alarming prospect of a world where effective antibiotics are no longer available. Bacteria employ various mechanisms to elude antibiotics, including modifying antibiotic targets, utilizing efflux pumps to avoid antibiotics, and inactivating antibiotics. This review focuses on small-molecule-based approaches to overcoming resistance, with emphasis on chemical adjuvants (such as β-lactamase inhibitors, efflux pump inhibitors, and membrane permeabilizers), synergistic combination therapies, repurposed non-antibiotic drugs, and structural modifications of known antibiotics like ciprofloxacin. We critically analyze structure–activity relationships (SAR), biochemical mechanisms, and clinical barriers associated with each strategy. By addressing antibiotic resistance, we aim to fortify our ability to combat bacterial infections effectively and sustain the efficacy of existing antibiotics in the face of evolving resistance.
 Hazem Elkady | Hazem Osama Radwan Elkady is an Associate Professor of Medicinal Chemistry at the Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt. He earned his PhD in pharmaceutical sciences in 2018 and has published over 75 peer-reviewed articles. His research focuses on the design and synthesis of novel anticancer and immunomodulatory agents targeting VEGFR-2, EGFR, topoisomerase II, and other therapeutic targets. Dr Elkady is a recognized reviewer for over 25 international journals and was ranked among the top 2% of scientists worldwide by the Stanford/Elsevier list in 2024. |
 Ibtehal Nasser Salman | Ibtehal Naseer Salman is a pharmacist who graduated in 2011 from the College of Pharmacy at Al-Mustansiriya University. She worked at the Iraqi Ministry of Health from 2012 to 2019. She was then accepted into the College of Pharmacy at Al-Mustansiriya University to study for a master's degree in pharmacology and toxicology. In 2023, she worked as a lecturer at Al-Amal College of Medical Sciences. Below are her published research. |
 Mohamed M. Khalifa | Mohamed M. M. Khalifa is an Associate Professor of Medicinal and Pharmaceutical Chemistry and Drug Design at Al-Azhar University, Cairo, Egypt. He received his PhD in Pharmaceutical Chemistry in 2018. His research focuses on anticancer and antiviral drug discovery, molecular modeling, and repurposing FDA-approved drugs. He has authored over 25 peer-reviewed publications in reputed journals and currently holds a research position at Al-Amal College for Specialized Medical Sciences in Iraq. Dr Khalifa is proficient in several molecular modeling platforms and has experience in both academic teaching and scientific research. |
1. Introduction
Antibiotics have been widely recognized as crucial medications in combating infectious diseases for the last century. Paul Ehrlich, a pioneer in modern chemotherapy, is credited with discovering the first antibiotic, Salvarsan, in 1909. This groundbreaking synthetic antibiotic, derived from arsenic, was utilized for treating syphilis, caused by Treponema pallidum.1,2 In 1928, Alexander Fleming fortuitously discovered that the fungus Penicillium notatum could inhibit the growth of Staphylococcus aureus colonies. In 1930, Gerhard Domagk discovered the sulfa drugs.3 Following World War II, semi-synthetic antibiotics like amoxicillin and quinolones were generated to improve stability and broaden antibacterial effectiveness. Macrolides, third-generation cephalosporins, and linezolid are among the most recent developments, which are designed to improve the pharmacokinetics of antibiotics and combat Gram-negative bacterial resistance.4,5 Notwithstanding these advancements, the pervasiveness of antibiotic-resistant bacterial strains has escalated in current eras and necessitating a reevaluation of antibiotic utilization.6,7Antibiotic resistance denotes to bacteria's capability to withstand the impact of antibiotic agents, categorized as natural or acquired resistance. Natural resistance implies inherent resistance within bacteria. Acquired resistance transpires when a bacterium develops resistance to an antimicrobial agent to which it was before susceptible. Antibiotic resistance is often acquired and may arise via gene changes during normal physiological processes, modifications in cellular structures, the gaining of exogenous resistance genes, or a blend of these tactics.8
In 1945, Alexander Fleming issued a public admonition against the perils of antibiotic misuse, acknowledging the hazards linked to its improper application. Excessive or unwarranted use of antibiotics might result in bacterial adaptations that render the medicines ineffective against them.9 This review focuses specifically on small-molecule strategies to combat bacterial resistance. We provide a chemistry-driven perspective on the mechanisms of resistance and explore the medicinal chemistry innovations aimed at restoring antibiotic efficacy. Strategies covered include the development of adjuvants, structural modifications of existing drugs, rational combinations, drug repurposing, and novel chemical scaffolds.
2. Key mechanisms underlying antibiotic resistance
Knowing the processes that cause antibiotic resistance is a crucial step in finding a solution to this tricky problem. Various parts of the mechanisms of antimicrobial resistance have been reviewed in pertinent literature. We will go over the mechanisms that have been found in human infections that have been isolated from clinical settings. Typically, these strategies may be grouped into four main types: changing the drug's target, activating the drug efflux pump, inhibiting the absorption of the drug, and inactivating by enzymes.2.1. Alteration of drug target
An explicit illustration of this process is the resistance to β-lactam drugs noted in Gram-positive bacteria. These bacteria modify the configuration of penicillin-binding proteins (PBPs). PBPs are transpeptidases that play a crucial role in the synthesis of peptidoglycan within the cell wall. Structural modifications of PBPs, such as PBP2a in S. aureus due to the acquisition of the mecA gene, will diminish or entirely obstruct the drug's binding capability.10,11Additionally, certain Gram-positive bacteria, including enterococci and staphylococci, exhibit resistance to glycopeptides like vancomycin, which functions as a cell wall synthesis inhibitor. Resistance emerges via the acquisition of van genes, resulting in structural modifications of peptidoglycan precursors that reduce vancomycin binding efficacy.10,12 vancomycin-susceptible staphylococci produce cell-wall precursors that terminate in D-Ala-D-Ala. Subsequent to their transfer from the cytoplasm to the cell membrane, these precursors exhibit a high affinity for vancomycin; upon binding, they are rendered incapable of participating in cell-wall synthesis. Vancomycin-resistant staphylococci, when exposed to an inducer such as vancomycin, produce substrates with varying termini (D-Ala-D-Lac, or D-Ala-D-Ser) that exhibit low affinity for vancomycin, enabling continued synthesis of the cell wall.13
2.2. Activation of drug efflux pump
Antibiotic efflux is a primary method by which bacteria eject antibiotics from their cytoplasm to the exterior environs utilizing specialized transporter proteins known as efflux pumps.14 These pumps afford a protective function by extruding the antibiotic from the bacterial cell.15,16 Certain entities are selective, expelling just certain substrates, while others are non-selective, accommodating a broad spectrum of structurally varied substances, including colors, organic solvents, detergents, and many families of antibiotics.17 Efflux pump genes are encoded in the bacterial chromosome. Some forms of expression are constitutive, whilst others are induced or overexpressed in reaction to specific environmental stimuli or the availability of a proper substrate.18The classification of bacterial multidrug efflux systems is based on their construction and energy source, dividing them into five primary families: resistance nodulation cell division (RND), small multidrug resistance (SMR), major facilitators (MFs), multidrug and toxic compound extrusion (MATE), and ATP-binding cassette (ABC).19
2.3. Inhibition of drug uptake
Bacteria have evolved methods to inhibit the antibiotic from accessing its intracellular target by reducing the absorption of the antimicrobial compound. This process is especially significant in Gram-negative bacteria because their outer membrane consists of glycolipids, primarily lipopolysaccharide (LPS). This membrane decreases permeability and serves as a barrier to many antimicrobial drugs.20Hydrophilic molecules, including β-lactams and definite fluoroquinolones, are significantly influenced by alterations in the penetrability of the outer membrane, as they typically utilize water-filled diffusion ducts, referred to as porins, to traverse this fence.21 The efficiency of this natural fence is exemplified by vancomycin which is ineffective against Gram-negative germs due to its inability to invade the outer membrane. Likewise, Pseudomonas naturally shows lower sensitivity to β-lactams compared to Enterobacteriaceae, which is partly due to a decreased quantity and/or different patterns of porin expression.22
Numerous porin classes have been recognized and may be categorized based on their construction (trimeric or monomeric), selectivity, and regulatory mechanisms of expression. The three extensively studied porins generated by E. coli, specifically OmpF, OmpC, and PhoE, in conjunction with P. aeruginosa's OprD (protein D2), exemplify porin-mediated antibiotic resistance. Porin modifications can transpire via three principal mechanisms: (i) a change in the kind of porins expressed, (ii) an adjustment in the degree of porin expression, and (iii) a disruption of porin functionality. Alterations in permeability via these pathways frequently lead to minimal resistance and are generally linked with additional resistance mechanisms, including heightened expression of efflux pumps.23
2.4. Inactivation of drug by enzymes
The majority of antibiotics work by attaching themselves to their targets in a certain way, which stops these targets' physiological functions. Nevertheless, due to prolonged struggle with antibiotics, several bacteria have developed resistance enzymes that render clinically significant drugs like aminoglycosides, carbapenems, and β-lactams inactive.24 These resistance enzymes are primarily classified into two categories: modifying enzymes and hydrolytic enzymes.25Two classes of β-lactamases can be recognized grounded on their structure and mechanism: serine-β-lactamases (SBLs) and metallo-β-lactamases (MBLs). The SBLs employ a nucleophilic serine residue for the process of hydrolysis, which holds significant clinical relevance. The MBLs utilize zinc ions to trigger water molecules for the hydrolysis of β-lactam antibiotics, garnering significant attention in recent years due to their exceptionally broad substrate range and robust carbapenemase activity (Fig. 1).29,30
 | ||
| Fig. 1 β-Lactams are hydrolyzed by (i) SBLs and (ii) MBLs. | ||
MBLs can be categorized into three distinct subclasses (B1, B2, and B3), which are primarily discriminated by their metal composition and the unique characteristics of their active sites. The majority of metallo-β-lactamases identified to date are classified within subclass B1, with the imipenemase (IMP), Verona imipenemase (VIM), and New Delhi metallo-β-lactamase (NDM) families being the three most prevalent metallo-β-lactamases observed in clinical isolates.31–34
Macrolides rely on a pivotal cyclization process mediated by ester bonds to achieve their antibacterial efficacy. However, macrolide esterases possess the remarkable ability to disrupt this cyclic framework,35 cleaving the ring and initiating a sequence of internal cyclization and dehydration through intramolecular condensation.36,37 Accordingly, the open-ring macrolides are rendered inactive, forfeiting their antimicrobial properties.
Enzymatic hydrolysis of fosfomycin 1, an epoxide antibiotic, transpires via the decomposition of its reactive epoxide ring. This process is arbitrated either by a thiol-containing co-substrate or through water-induced ring breakdown, orchestrated by three discrete fosfomycin-resistance enzymes: FosA, FosB, and FosX (Fig. 2). FosA, found in Gram-negative bacteria, functions as a glutathione transferase that depends on Mn2+ and K+ ions. FosB acts as an L-cysteine thioltransferase requiring Mg2+, whereas FosX is a Mn2+-dependent epoxide hydrolase that specifically targets fosfomycin.38,39
 | ||
| Fig. 2 Enzymatic ring-opening of fosfomycin 1 mediated by FosA, FosB, and FosX. | ||
2.4.2.1. Modification on antibiotics. Aminoglycoside antibiotics have three basic classes of aminoglycoside-modifying enzymes: nucleotidyltransferases (ANTs), phosphotransferases (APHs), and acetyltransferases (AACs).40 In addition, chlormamphenicol 2 is susceptible to modification by acetyl transferase (Fig. 3).41
 | ||
| Fig. 3 Acetylation of chlormamphenicol 2. | ||
2.4.2.2. Modification of antibiotic targets. Alterations in the conformation and/or extent of penicillin-binding proteins (PBPs) represent one of the key bacterial strategies to resist β-lactam antibiotics.42 Variations in the number of PBPs affect the amount of medication that can bind to the target.43 The erythromycin ribosome methylase (erm) gene family methylates 16S ribosomal ribonucleic acid (rRNA), changing the drug-binding site and stopping the binding of macrolides and lincosamines.44
3. Strategies to tackle the problem of antibiotic resistance
3.1. Use of antibiotic adjuvants
Antibiotic adjuvants are substances that exhibit minimal or no antimicrobial activity independently. They act harmonically with antibiotics to reduce or inhibit bacterial resistance, thereby restoring and preserving the efficacy of antibiotics. Three primary categories of adjuvants have been investigated: β-lactamase inhibitors, efflux pump inhibitors (EPIs), and membrane permeabilizers.3.1.2.1. Classical SBLs inhibitors. Clavulanic acid 3, the first β-lactamase inhibitor approved for therapeutic utilization, was insulated from Streptomyces clavuligerus in the early seventies.46 Sulbactam 4 and Tazobactam 5 are penicillinate sulfones that were developed later by drug developers in 1978 and 1980, respectively.47 These three approved β-lactam-ring containing β-lactamase inhibitors can irreversibly acylate β-lactamases by targeting the enzyme's serine residue (Fig. 4)48 meaning that serine-β-lactamases are unable to hydrolyze the β-lactam antibiotics that are given with them (SBLs). Nonetheless, these inhibitors possess the same β-lactam core structure characteristic of β-lactam antibiotics. Bacteria swiftly develop resistance to these structurally similar compounds by utilizing or altering pre-existing mechanisms.27
 | ||
| Fig. 4 Acylation of serine-β-lactamases (SBLs) by tazobactam 5 resulting in the formation of an ester bond.49 | ||
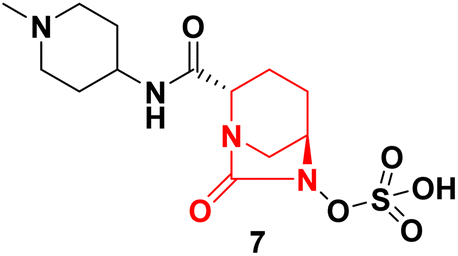
3.1.2.2. Diazabicyclooctanes (DBOs) based SBL inhibitor. Second-generation β-lactamase inhibitors with diazabicyclooctane (DBO) scaffolds have shown significant promise in restoring the activity of β-lactam antibiotics against resistant Gram-negative bacteria. These compounds overcome limitations of traditional inhibitors by targeting a broader range of β-lactamases, including Class A, C, and some D serine β-lactamases. This section summarizes four key DBO-based inhibitors including Avibactam 6, Relebactam 7, Durlobactam 8, and Nacubactam 9 highlighting their structures, co-administered antibiotics, mechanisms, and clinical relevance.50 Table 1 below presents a comparative analysis between the four DBO inhibitors, whereas Fig. 5 and 6 depict the structural and mechanistic attributes of Avibactam and Durlobactam, respectively as a representative examples of DBO-based β-lactamase inhibitors.
| Feature | Avibactam | Relebactam | Durlobactam | Nacubactam |
|---|---|---|---|---|
| Chemical structure |  |
 |
 |
 |
| Core structure | Diazabicyclooctane (DBO) | DBO derivative with piperidine tail | Rigidified DBO (3,4-double bond + methyl) | DBO with PBP2 inhibition |
| Target enzymes | Class A, class C, some class D β-lactamases | Class A & C β-lactamases | Class A, C, D carbapenemases | Class A, C, some D + PBP2 |
| Co-administered β-lactams | Ceftazidime | Imipenem-cilastatin | Sulbactam | Meropenem, Aztreonam, piperacillin |
| FDA status | Approved (2014)51 | Approved (2019)52–54 | Approved (2023)55 | Phase III trials56–58 |
| Key features/advantages | Reversible acylation; avoids β-lactam ring resistance | Used for pyelonephritis and complicated intra-abdominal infections | Dual inhibition; effective vs. MDR Acinetobacter | Dual action; improves antibacterial activity |
 | ||
| Fig. 5 Acylation of SBLs by avibactam 6 to form a carbamoyl linkage.49 | ||
 | ||
| Fig. 6 Mechanism of action of durlobactam 8. | ||
3.1.2.3. Boronate β-lactamase inhibitors.
3.1.2.3.1. Varborbactam.
Varborbactam 10 represents the inaugural non-β-lactam boronic acid β-lactamase inhibitor. In 2017, the FDA confirmed a mixture of meropenem and vaborbactam to manage complicated urinary tract infections in adult patients.59,60 Varborbactam demonstrated a diverse array of effectiveness against several serine β-lactamases, encompassing class A carbapenemases and class C cephalosporinases. However, it exhibited minimal activity against class B metallo-β-lactamases and class D carbapenemases.61,62 The combination of meropenem and vaborbactam safeguards meropenem from being degraded by serine carbapenemases through a novel enzyme inhibition mechanism. This process involves vaborbactam binding covalently to the catalytic serine residue within β-lactamase enzymes. The boron atom in vaborbactam mimics the tetrahedral intermediate that naturally forms during the enzyme's acylation and deacylation steps in β-lactam hydrolysis. By stabilizing this transition state analogue, vaborbactam effectively blocks the enzyme's activity, leading to rapid and irreversible inactivation of β-lactamases, thereby protecting meropenem from degradation.63,64

(4) Biological versatility: in contrast to serine β-lactamases, which are exclusively bacterial enzymes, metallo-β-lactamases (MBLs) are part of a more extensive superfamily of metalloproteins that engage in a variety of biological functions that extend beyond the hydrolysis of β-lactams.65 Consequently, there are now no effective MBL inhibitors utilized in clinical practice. The challenges encountered have hindered the advancement of clinically approved MBL inhibitors, highlighting the necessity for novel strategies in inhibitor design.
MBLs are categorized into three subclasses: B1, B2, and B3, based on the quantity of zinc atoms present. The B1 subtype is the most clinically significant and encompasses New Delhi. Metallo-β-lactamase-1 (NDM-1) is an enzyme that poses a significant hazard to human health due to its substrate promiscuity, broad-spectrum activity, emergence of variations, and capacity for transferability. The pursuit of an effective NDM-1 inhibitor continues, and despite extensive research over the years, an optimal treatment remains elusive. Significant advancements in NDM-1 research are underway, with several molecular structures having been altered and evaluated against NDM-1. In the past decade, a substantial volume of research on NDM-1 inhibitors has been published.
3.1.3.1. Natural MBLs inhibitors. Aspergillomarasmine A (AMA) 11 is a polyamino acid synthesized by Aspergillus versicolor that effectively inhibits antibiotic resistance enzymes in Gram-negative pathogenic bacteria, including Pseudomonas spp, Acinetobacter spp, and Enterobacteriaceae.66,67 Despite never achieving clinical significance as a therapy, it has been reassessed for NDM-1 inhibition. This molecule is particularly noteworthy due to its capacity to bind zinc while exhibiting benign effects in vivo, as demonstrated in mice. A 95% survival rate was observed post-infection with NDM-1 positive K. pneumoniae, but meropenem monotherapy resulted in significantly higher death.67

3.1.3.2. Chelating agents. Initial compounds recognized as NDM-1 inhibitors were chelating drugs like ethylenediamine-N,N,N′,N′-tetracetic acid (EDTA) 12 and trispicolylamine (TPA) 13.68,69 These compounds bind zinc at the active site of MBL enzymes, therefore limiting their action and facilitating the efficacy of antibiotics. Regrettably, the prevalence of metal-containing enzymes in the human system renders chelating drugs unsuitable for clinical use due to their ubiquitous cytotoxicity.

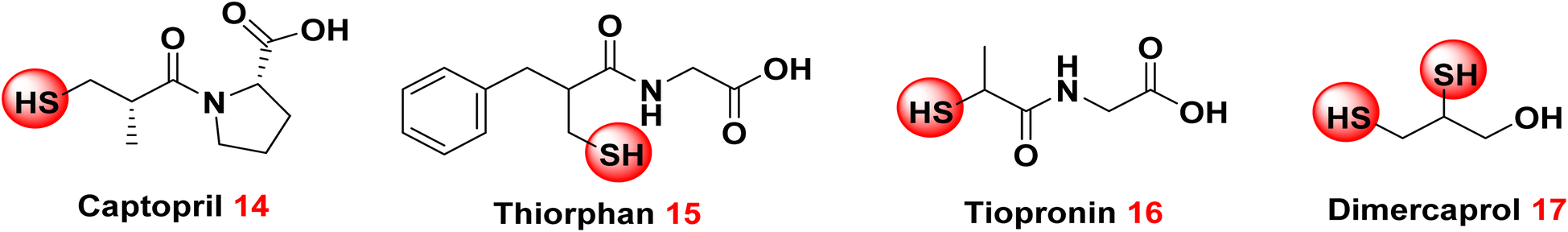
3.1.3.3. Thiol comprising congeners. The thiol group is renowned as a zinc chelator, making its presence in initiatives focused on discovering new inhibitors for class B β-lactamases unsurprising.70–72 In 2015, Klingler et al. evaluated eleven authorized pharmaceuticals or their bioactive metabolites featuring a free thiol group, identifying four medicines that inhibit the clinically significant metallo-β-lactamases NDM-1, Verona integron-encoded metallo-β-lactamase (VIM-1), and imipenemase-7 (IMP-7).73 These inhibitors include captopril 14, a clinically approved angiotensin-converting enzyme (ACE) inhibitor commonly used for the treatment of hypertension; thiorphan 15, functioning as an enkephalinase inhibitor and is used primarily as an antidiarrheal agent; tiopronin 16, a therapeutic agent utilized in managing severe cases of homozygous cystinuria by reducing cystine levels in the urine, and dimercaprol 17, an established chelating agent employed in the management of heavy metal toxicity. These compounds exhibited IC50 values in the low micromolar range for all evaluated metallo-β-lactamases and shown significant synergy with imipenem.
The crystal structure of NDM-1 complexed with captopril reveals that the thiolate group inhabits the fourth coordination site for each ion, facilitating intercalation between the two Zn2+ ions. The hydroxide anion positioned between the two Zn2+ ions is substituted by the thiolate group, therefore initiating the hydrolytic process (Fig. 7).

3.1.3.4. Bisthiazolidines (BTZs). Bisthiazolidines are thiol-containing bicyclic compounds that interact with the dizinc cores of metallo-β-lactamases (MBLs) via free thiol groups, therefore substituting hydroxide ions in the dizinc clusters during hydrolysis (Fig. 8).75 These compounds have two principal advantages: extensive action against all class B enzymes and effortlessness of production. Further refinement of the bisthiazolidine framework to establish additional binding interactions with active site residues could enhance their inhibitory efficacy, potentially reaching the desired nanomolar range.
 | ||
| Fig. 8 Mechanism of action of bisthiazolidines as MBLs inhibitors.49 | ||
In 2015, González et al. elucidated the identification of bisthiazolidines 18a,b and 19a,b as novel metallo-β-lactamase inhibitors capable of reinstating the efficacy of imipenem.76 The compounds in question exhibit certain traits reminiscent of β-lactam compounds, which serve as effective substrates for these enzymes. They possess a bicyclic configuration characterized by a bridging nitrogen atom and a carboxylate group situated in the α-position relative to the nitrogen atom. Furthermore, they possess the thiol group essential for coordination with the catalytic Zn2+ ion. Bisthiazolidines 18a,b and 19a,b have demonstrated remarkable versatility as inhibitors, effectively targeting a broad spectrum of class B enzymes, including NDM-1, VIM-2, IMP-1, and BcII, all exhibiting values within the low micromolar range.75

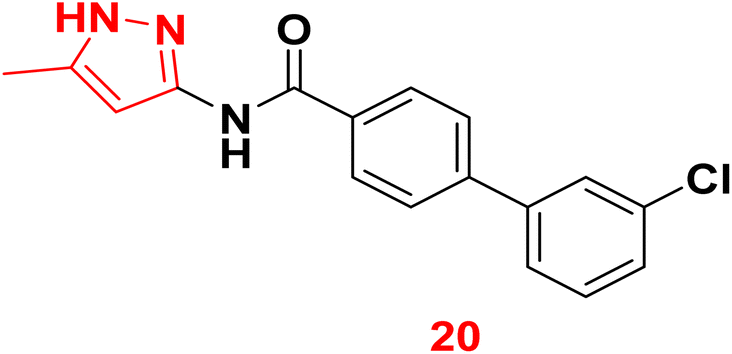
3.1.3.5. Pyrazole derivatives as NDM-1 inhibitors. In 2021, Ahmad et al., reported the design and synthesis of a series of pyrazole-based derivatives specifically developed as potent inhibitors of the NDM-1 enzyme, addressing the critical challenge of resistance posed by metallo-β-lactamases. The in vitro antibacterial effect against NDM-1-positive Acinetobacter baumannii and Klebsiella pneumoniae of the target derivatives were determined. Furthermore, a molecular docking analysis of the candidate molecules against NDM-1-producing A. baumannii was achieved to explore their binding interactions. Among the compounds, compound 20 exhibited the strongest binding affinity to the receptor, highlighting its potential as a remarkable antibacterial agent (Fig. 9).77
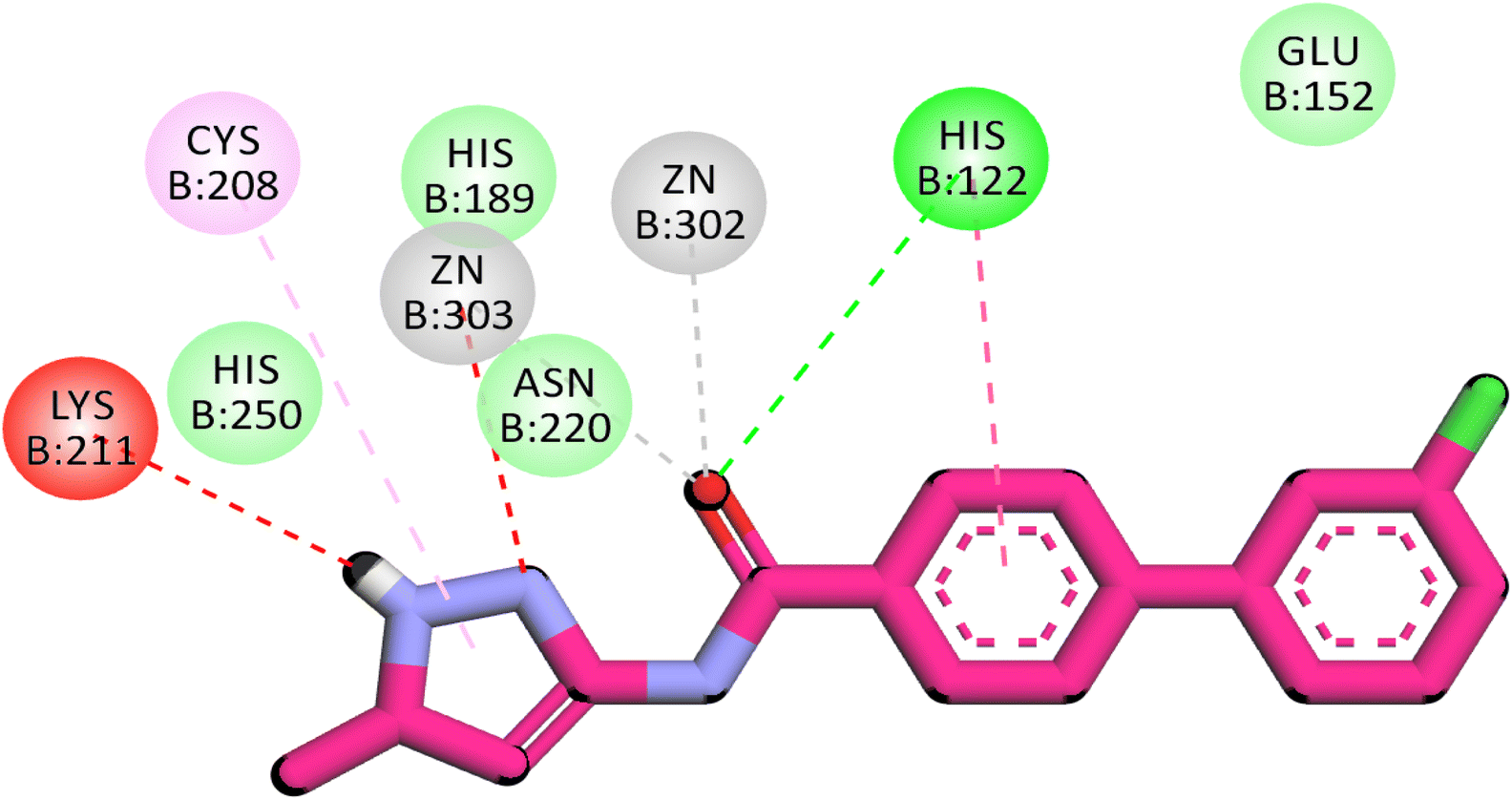 | ||
| Fig. 9 Two-dimensional diagram of compound 20 with NDM1 protein (Pdb: 4EXS). | ||
3.1.3.6. Isatine as NDM-1 inhibitor. In 2023, compound 21 (Zndm19) was identified to treat drug-resistant bacterial infections through evaluation of NDM-1 enzyme activity suppression. The biological in vitro outcomes verified the compound 21 suppressed NDM-1 activity and reinstating the bactericidal activity of MEM against NDM-1-positive E. coli. The mice peritonitis infection model study revealed that the Zndm19-meropenem combination therapy exhibited synergistic activity leading to nearly a 60% rise in survival rates along with lowered bacterial load in tissue. Furthermore, molecular docking revealed the ability of Zndm19 to chelate the two zinc centers and interact with the key amino acid residues within the active site of NDM-1 (Fig. 10).78
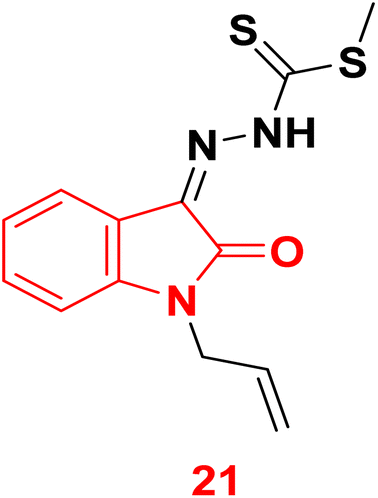
 | ||
| Fig. 10 Binding mode of compound 21 with NDM-1protein (Pdb: 5ZIO). | ||
3.1.3.7. Quinoline derivatives as NDM-1 inhibitors. In 2022, thirty-one quinolinyl sulfonamides and sulfonyl esters were synthesized and evaluated for their inhibitory activity against metallo-β-lactamase NDM-1.79 Among these, compounds 22 and 23 revealed the highest inhibitory potency, with IC50 values of 0.02 μM using meropenem (MEP) as the substrate. Structure–activity relationship analysis revealed that halogen substitution on the phenyl group significantly enhances the inhibitory effects of quinoline derivatives on NDM-1. Minimum inhibitory concentration (MIC) assays showed that quinolinyl sulfonamides improved the antibacterial effectiveness of MEP against Escherichia coli strains expressing NDM-1 (EC01 and EC08), resulting in a 2-64-fold reduction in MICs. In vivo mouse studies demonstrated that compound 22 synergized with MEP, significantly reducing the bacterial load of EC08 in the liver and spleen following a one intraperitoneal dose. Molecular docking investigation showed that the endocyclic nitrogen atom of the quinoline ring and the exocyclic nitrogen of the sulfonamide group directly synchronize with the Zn2+ ions in the active site of NDM-1, as depicted in Fig. 11.


3.1.4.1. Phenylalanine–arginine β-naphthylamide (PAβN).
It is worth highlighting that phenylalanine–arginine β-naphthylamide (PAβN) 25 might have a dual antibiotic adjuvant action.83–85 As it is one of the most studied EPIs through competitive inhibition mechanism, where the efflux pumps recognize it as a substrate instead of the target antibiotics (quinolones mainly ciprofloxacin and levofloxacin). Moreover, it has recently been shown to permeabilize bacterial membranes and enhanced the efficacy of β-lactams against overexpressing strains of P. aeruginosa.83

The upregulation of genes producing NorA and related efflux pumps significantly contributes to drug and biocide resistance in S. aureus. Consequently, NorA represents a compelling target in medicinal chemistry, since a chemical capable of inhibiting its actions might reinstate the efficacy of substrate antibiotics like fluoroquinolones. No NorA EPI has received approval for clinical application to yet.
3.1.4.2. Indole based weapons as promising NorA efflux pump inhibitors. The indole moiety has demonstrated potential regarding the EPI action of a drug. Compound 26 is among the initial indole-based inhibitors of NorA and can enhance the sensitivity of S. aureus to ciprofloxacin by four-fold.86 Subsequent structural changes were implemented at the C2, C3, and C5 locations of compound 26 (Fig. 12). In the primary identified members, an electron-withdrawing group was consistently retained (a NO2 group in 28, 29, 30 or a CN group in 27). Moreover, an aromatic moiety with various substituents was consistently maintained across all compounds to optimize their efficacy. Recently, indoles containing halogens at the C5 position and a distinctive nitrone moiety at the C3 position (compounds 31a–c) have garnered significant attention due to their potential as pharmacologically active agents, demonstrating promising inhibitory activity against key bacterial enzymes. All the engineered members had favorable NorA EPI characteristics, equal to or exceeding those of the lead compound 26.87–89
 | ||
| Fig. 12 Chemical structures of indole-based candidates observed to bind to NorA. | ||
Next, Lepri et al., have synthesized 48 indole derivatives via substitutions at the C5 and N1 positions of indole.90 The synthesized members have been tested against norA-overexpressing S. aureus. Compound 32 was identified as the lead and subsequently refined through modifications to four essential structural elements: the terminal dimethylamine moiety, the ethyl linker, the benzyl group, and the ethyl carboxylate (Fig. 13). Among the synthesized indole derivatives, compound 33 distinguished itself as the most formidable candidate. The analysis of the structure–activity relationship (SAR) indicated that the presence of a propoxyl chain featuring terminal cyclic amino groups is essential for the effective inhibition of the NorA pump at low micromolar concentrations. Furthermore, the N-benzyl moiety was observed to not only preserve inhibitory activity but also to affect biological effects and ADME properties depending on its substituents.
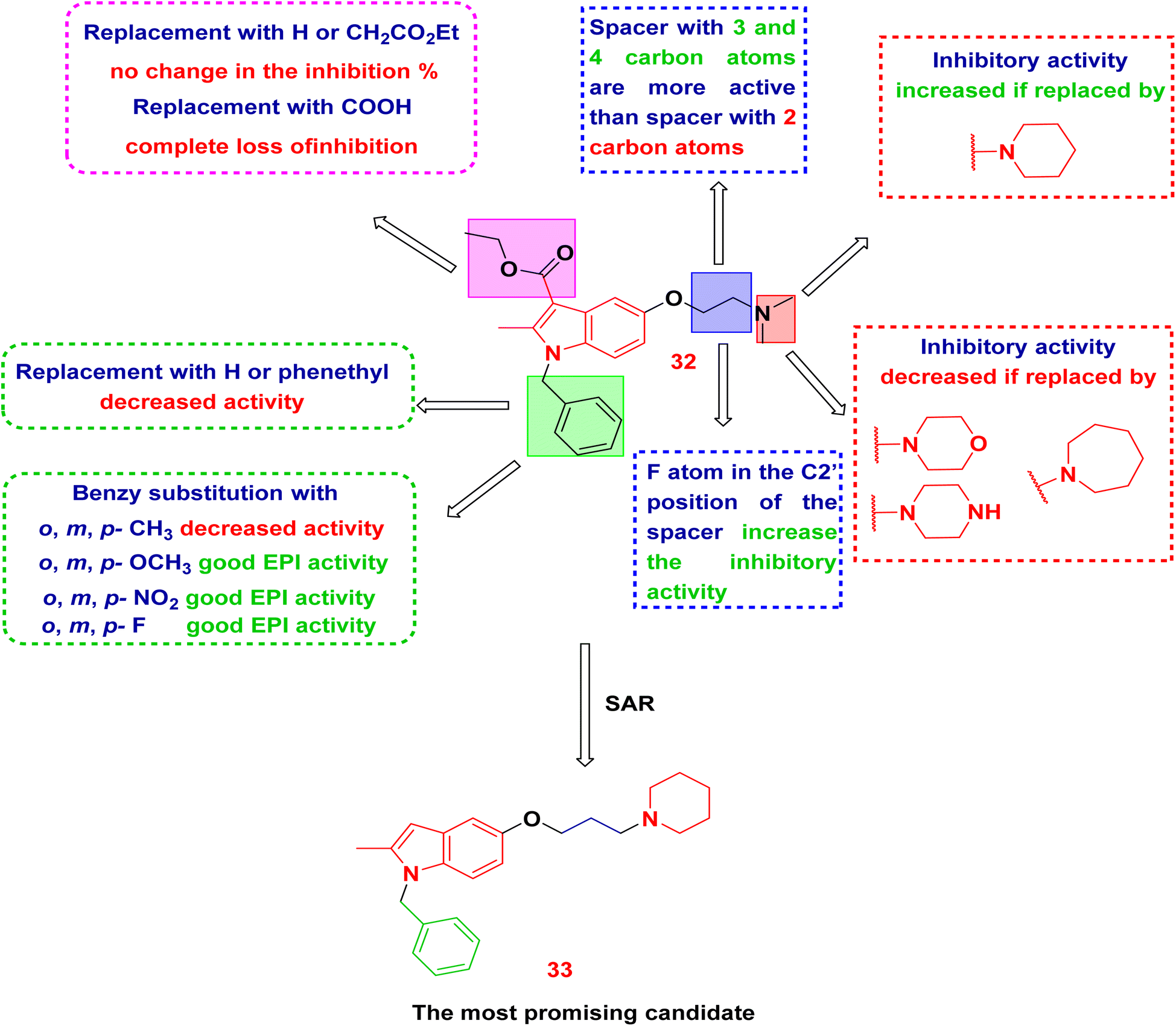 | ||
| Fig. 13 Detailed structural modification of lead compound 32 and SAR study. | ||
3.1.4.3. Pyranopyridines as NorA efflux pump inhibitors. Sjuts et al.91 developed a series of pyranopyridines that demonstrated greater potency than PAβN in sensitizing Enterobacteriaceae to antibiotics. The two pyranopyridine compounds, MBX3132 34 and MBX3135 35, demonstrated potency at a concentration as low as 0.1 μM, which is 500-fold lower than that of PAβN. Given the significant steric hindrance, 34 and 35 exhibited a stronger binding affinity to the binding pocket of the AcrB transporter in E. coli compared to PAβN. Consequently, 34 and 35 demonstrated effective inhibition at remarkably low concentrations. To improve solubility and biocompatibility, the terminal acetyl group was modified to a polar tetrazole as in MBX4191 36.92 Compound 36 exhibited significant water solubility and minimal cytotoxic effects.

The challenge in targeting efflux pumps arises from their diverse physiological functions, which may lead to unforeseen toxicities upon inhibition. Research is thus directed towards identifying agents that specifically stop pumps working exclusively in prokaryotes.17 In response to this need, numerous scientific trials have been conducted to ascertain the blockers of these pumps, as well as to explore the application of efflux pump inhibition strategies. Currently, no efflux pump suppressor have been ratified for the treatment of bacterial infections affecting humans and animals. The only reported inhibitor is MP-601, 37, which is supplied as an aerosol in combination with ciprofloxacin for treating respiratory infections in patients with ventilator-associated pneumonia caused by a multidrug-resistant P. aeruginosa.73,93

3.1.4.4. Oxadiazoles as NorA efflux pump inhibitors. In 2021, a new series of 1,3,4-oxadiazole conjugates linked to capsaicin was synthesized. The evaluation of ciprofloxacin activity potentiation was conducted for the entire set of synthesized members. Among them, compound 38 demonstrated significant activity. The results obtained indicated that compound 38 exhibited inhibition of the NorA efflux pump in the ethidium bromide (EtBr) fluorescence assay, significantly reducing the efflux of ethidium bromide.94

3.1.5.1. Colistin.
Colistin 40 is a naturally derived polymyxin antibiotic isolated from Bacillus polymyxa, composed of a decapeptide with six amino acids forming a cyclic peptide ring linked to a fatty acid side chain. It belongs to the class of antimicrobial peptides, characterized by cationic and amphiphilic properties, which have been extensively investigated as antibiotic adjuvants.96,97 Colistin primarily targets the negatively charged lipid A moieties within lipopolysaccharides (LPS). Due to its stronger affinity for LPS compared to divalent cations such as Mg2+ and Ca2+, colistin competitively displaces these ions, destabilizing the outer membrane by releasing LPS molecules and creating permeabilized pores. The ability of colistin to form physical pores in the outer membrane enables synergistic effects with various antibiotic classes, including rifampicin and carbapenems.98,99 Although colistin is an older antimicrobial agent that was previously withdrawn because of nephrotoxicity and neurotoxicity concerns,100 it has re-emerged as a last-resort therapy for infections caused by carbapenem-resistant Gram-negative bacteria. Consequently, clinical administration of colistin necessitates vigilant monitoring of key biomarkers to manage potential toxicities during treatment.101
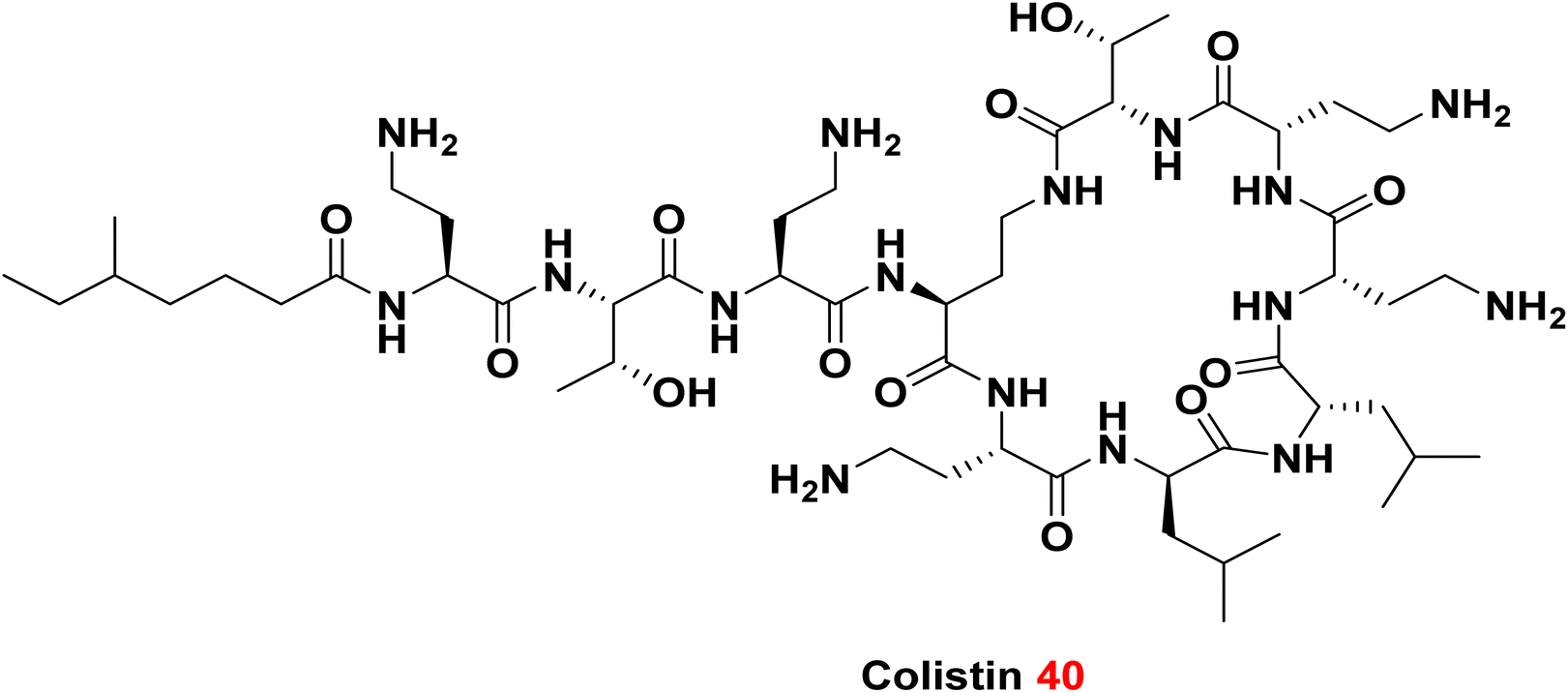
3.1.5.2. Polymyxin B nonapeptide (PMBN).
This derivative of polymyxin-B is characterized by a shorter fatty acid lipid tail deficiency while maintaining the outer membrane-permeabilizing activity of polymyxin B.102 PMBN binds to LPS, leading to the release of divalent cross-linkers that enhance outer membrane permeability. PMBN enhances the sensitivity of E. coli to hydrophobic antibiotics by augmenting outer membrane permeability.103 The enantiomer of PMBN exhibits no activity, highlighting the significance of stereochemical configuration.104 Despite being less toxic than colistin, PMBN exhibited nephrotoxicity in preclinical studies, hindering its advancement as a therapeutic adjuvant.105

3.2. Combination therapies and small molecule optimization
3.2.1.1. Combinations of trimethoprim-sulfamethoxazole (TMP-SMX). The combination therapy of trimethoprim 42 and sulfamethoxazole 43 targets bacterial folate production by hindering two critical enzymes: trimethoprim blocks dihydrofolate reductase, while sulfamethoxazole inhibits dihydropteroate synthase. By interfering with these sequential steps, the duo effectively halts folate production, essential for bacterial growth and replication. This synergistic antibiotic combination is widely used to treat several infections such as urinary tract infections, respiratory tract infections, and opportunistic infections in patients with weakened immune systems.111 It is especially important for managing infections triggered by methicillin-resistant Staphylococcus aureus (MRSA) and for treating pneumocystis pneumonia.112

3.2.1.2. Combinations of aminoglycosides with β-lactam antibiotics. Aminoglycosides, including gentamicin 44, can enhance clinical treatment efficacy, expedite bacterial clearance, and bolster antibiotic resistance, particularly when used in association with β-lactam antibiotics like ampicillin 45.113,114 β-Lactam antibiotics possess the capability to disrupt the bacterial cell wall, thereby facilitating the entry of aminoglycosides into bacteria and augmenting their bactericidal efficacy. Aminoglycosides combined with β-lactam antibiotics are frequently employed in the treatment of severe hospital-acquired infections caused by multidrug-resistant organisms, including acquired pneumonia, ventilator-associated pneumonia, and sepsis.115

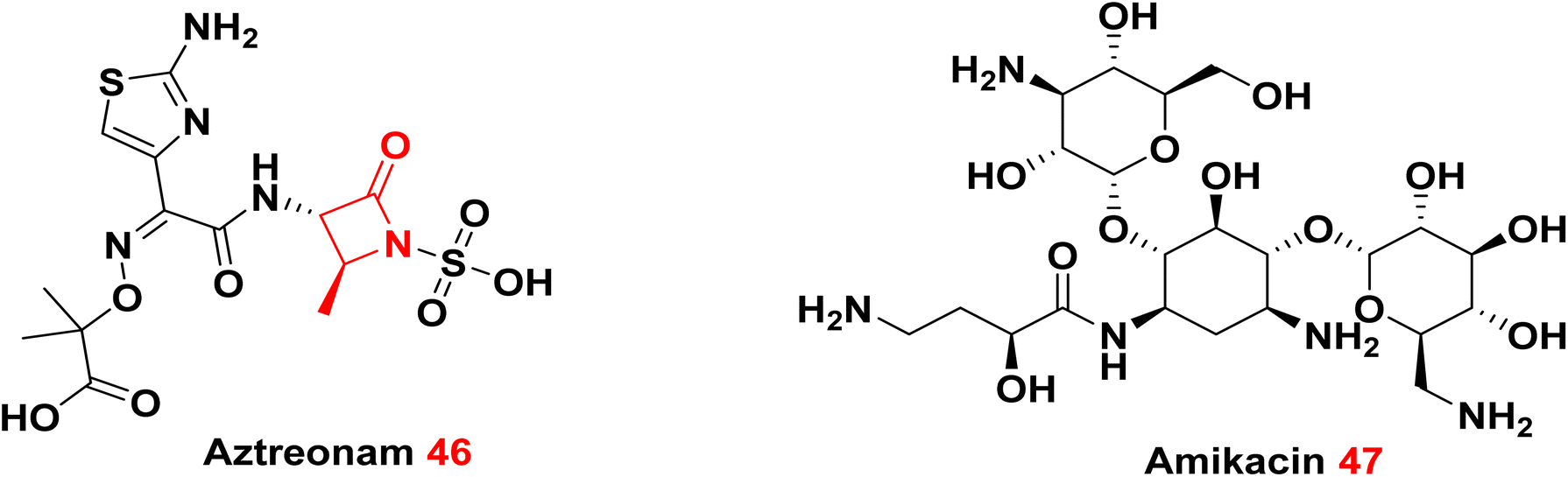
3.2.1.3. Combination of monobactams and aminoglycoside. It was reported that combination of monobactam such as Aztreonam 46 and aminoglycoside such as amikacin 47 gave promising results against metallo-β-lactamase-producing multidrug-resistant P. aeruginosa.116
Novel ciprofloxacin derivatives were synthesized in 2021 by reacting ciprofloxacin 48 with various organic reagents. The synthesized compounds were identified through elemental analysis, X-ray analysis, and spectral data. The new derivative, 49, was exceedingly effective against all the tested organisms. The newly synthesized compounds exhibited excellent efficacy against ciprofloxacin-resistant K. pneumoniae DF72F in comparison to the commercial ciprofloxacin disc (5 μg). However, they exhibited moderate efficacy against E. coli U65M and P. aeruginosa TA74F, which are clinical isolates (Fig. 14).117
 | ||
| Fig. 14 Structural optimization of ciprofloxacin. | ||
In 2023, six analogs of ciprofloxacin have been synthesized by introducing new functional groups at C-3 and C-7 positions. The antibacterial activity of the synthesized derivatives was assessed against a resistant series of Gram-positive and Gram-negative bacteria using ciprofloxacin as a reference. Among all, the synthesized derivatives 50, 51, and 52 showed better activity in comparison to the ciprofloxacin.

3.3. Repurposing nonantibiotic drugs as antibacterials
Examining current pharmaceuticals for possible efficacy against drug-resistant bacteria is a viable approach in combating antibiotic resistance. This method entails reassessing existing drugs to see if they exhibit antibacterial capabilities, regardless of their original intent. The repurposing of pharmaceuticals can yield useful therapies for drug-resistant illnesses.118 An essential benefit of examining current pharmaceuticals is the abundance of safety and effectiveness data accessible. These pharmaceuticals have previously undergone comprehensive testing in human subjects, which can facilitate their repurposing for antimicrobial uses. Identifying non-traditional antimicrobial compounds inside current pharmaceuticals can augment the antibiotic arsenal and provide novel therapies for drug-resistant microbes.119It has been established that atorvastatin 53 and simvastatin 54 were more effective against MRSA, vancomycin-resistant enterococcus (VRE), and Staphylococcus epidermidis, when compared to rosuvastatin 55. Conversely, Enterobacter cloacae and E. coli were more susceptible to Atorvastatin 53 than to simvastatin 54 and rosuvastatin 55.120





3.4. Improving the profile of existing antibiotic classes
Monocyclic β-lactams, including aztreonam, exhibit greater stability against hydrolysis by β-lactamases in comparison to other β-lactams.116,131 The monobactams are classified as a subclass of monocyclic beta-lactam moieties.132 Consequently, concentrating on these monobactam compounds presents a significant opportunity to discover new and innovative β-lactamase inhibitors.In 2021, a series of monobactam compounds were synthesized and assessed for their β-lactamase inhibitory activities. The MIC was calculated for the monobactam derivatives against four strains of β-lactamase Gram-positive and Gram-negative bacteria. The results obtained were compared with clavulanic acid as a co-inhibitor alongside amoxicillin against the same four strains of bacteria. The biological findings indicated that compounds 61, 62, and 63 exhibited a β-lactamase inhibitory effect against E. coli species similar to that of clavulanic acid.133

4. Discussion and future perspectives
The fight against antibiotic resistance is multidimensional, and while different small-molecule-based treatments have showed promise, each has unique benefits and drawbacks.4.1. Comparison of strategies
• Adjuvants (e.g., β-lactamase and efflux pump inhibitors) offer a rapid path to clinical utility by enhancing existing antibiotics. However, their effectiveness is often limited by target specificity and pharmacokinetic mismatches with partner drugs.• Structural modification of known antibiotics can restore activity against resistant strains while maintaining established safety profiles. However, such modifications may be insufficient to overcome complex resistance mechanisms like efflux pumps or porin loss.
• Drug repurposing provides a cost-effective route by leveraging known drugs, but many repurposed agents exhibit limited antibacterial potency at clinically relevant concentrations.
• Combination therapies can offer synergistic effects, yet they also pose challenges related to dosing, drug–drug interactions, and multi-component resistance.
4.2. Clinical translation bottlenecks
Despite encouraging advances in the design of small-molecule strategies, the successful translation of these candidates into clinical use remains limited. Several key obstacles hinder this process:• Toxicity of novel adjuvants or dual-target inhibitors (e.g., β-lactamase and PBP2 inhibitors).
• Poor outer membrane permeability, especially in Gram-negative species.
• Rapid emergence of secondary resistance under selective pressure.
• Regulatory complexity in approving combination or repurposed agents without new clinical trials.
4.3. Emerging and underexplored directions
Several areas hold untapped potential for innovative resistance-fighting solutions:• Hybrid molecules that chemically link an antibiotic and adjuvant into a single scaffold.
• AI-assisted antibiotic design, accelerating structure-based scaffold development and optimization.
• Membrane-targeting agents and synthetic antimicrobial peptides with non-traditional mechanisms.
• Non-classical adjuvants, such as quorum sensing inhibitors and antivirulence agents.
In summary, although current methodologies have shown encouraging results, a more profound combination of medicinal chemistry, microbiology, and translational research is essential to transform these techniques into sustainable therapeutic outcomes. The future of antibiotic development depends on adopting complexity through precision-targeted, multifunctional drugs.
5. Novel antibiotics targeting drug-resistant bacteria in the recent five years
The development of new antibiotics targeting drug-resistant bacteria represents a vital initiative in addressing the growing issue of antimicrobial resistance. Researchers are diligently investigating novel therapeutic approaches to combat these resilient pathogens.5.1. Lefamulin
Lefamulin 64 is a pleuromutilin that was approved by the FDA in 2019.134 Lefamulin is indicated for the treatment of community-acquired bacterial pneumonia, encompassing infections due to drug-resistant Streptococcus pneumoniae. This agent inhibits bacterial protein synthesis by binding to the peptidyl transferase center of the 50S bacterial ribosome, thereby preventing the binding of transfer RNA for peptide transfer. It serves as an alternative treatment in cases where resistance to older antibiotics has developed.135,136

5.2. Pretomanid
In 2019, the FDA approved use of Pretomanid 65 with Bedaquiline and Linezolid as a treatment regimen for pulmonary MDR TB. Pretomanid 65 is also used for treatment of extensive drug-resistant tuberculosis (XDR-TB) in adult patients.137 The mechanism of action involves the inhibition of mycolic acid biosynthesis (similar to isoniazid) to disrupt cell wall formation.138

5.3. Cefiderocol
Cefiderocol 66 is a broad-spectrum cephalosporin antibiotic. It has been approved in 2019 by FDA and has marketed in several countries. It is highly effective against many drug-resistant Gram-negative bacteria, including carbapenem-resistant Enterobacteriaceae.139 Cefiderocol works by disrupting bacterial cell walls and facilitating iron uptake, an innovative approach to addressing antibiotic resistance.140

6. Conclusion
Antibiotic resistance is becoming a bigger and bigger danger to global health, making therapies that save lives less effective. This study looked at a number of small-molecule-based tactics for fighting bacterial resistance, such as using adjuvants, synergistic drug combinations, repurposed non-antibiotic agents, and changing the structure of current antibiotics. Each of these methods has its own benefits, but they all have big problems when it comes to practical translation, such toxicity, low permeability, or the chance of resistance evolving quickly. We have spoken about structure activity relationships (SAR), mechanistic insights, and biological targets that might help with rational drug design from the point of view of medicinal chemistry. To come up with strong and long-lasting remedies, we need to know more about how resistance works at the molecular level. The use of new technologies like AI-driven compound design, hybrid molecule engineering, and innovative target identification will be highly important in speeding up the search for next-generation antibiotics. Continued interdisciplinary collaboration among chemists, microbiologists, and clinicians is crucial to overcome current bottlenecks and to preserve the efficacy of antimicrobial agents in the face of evolving bacterial threats.Data availability
This article is a review of existing literature, and no primary datasets were generated or analyzed during the preparation of this manuscript. All referenced data and materials are available in the cited sources.Conflicts of interest
There are no conflicts to declare.References
- R. I. Aminov, Front. Microbiol., 2010, 1, 134 CrossRef PubMed.
- K. Williams, J. R. Soc. Med., 2009, 102, 343–348 CrossRef CAS.
- C. Jeśman, A. Młudzik and M. Cybulska, Pol. Merkur. Lek., 2011, 30, 320–322 Search PubMed.
- K. Iskandar, J. Murugaiyan, D. Hammoudi Halat, S. E. Hage, V. Chibabhai, S. Adukkadukkam, C. Roques, L. Molinier, P. Salameh and M. Van Dongen, Antibiotics, 2022, 11, 182 CrossRef CAS PubMed.
- S. B. Christensen, Molecules, 2021, 26, 6057 CrossRef CAS.
- R. Chait, K. Vetsigian and R. Kishony, Nat. Chem. Biol., 2012, 8, 2–5 CrossRef CAS.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- F. C. Tenover, Am. J. Med., 2006, 119, S3–S10 CrossRef CAS PubMed.
- G. Sulis, B. Daniels, A. Kwan, S. Gandra, A. Daftary, J. Das and M. Pai, BMJ Glob. Health, 2020, 5, e003393 CrossRef PubMed.
- A. Beceiro, M. Tomás and G. Bou, Clin. Microbiol. Rev., 2013, 26, 185–230 CrossRef CAS.
- W. Reygaert, Clin. Lab. Sci., 2009, 22, 115 Search PubMed.
- G. Cox and G. D. Wright, Int. J. Med. Microbiol., 2013, 303, 287–292 CrossRef CAS PubMed.
- B. E. Murray, N. Engl. J. Med., 2000, 342, 710–721 CrossRef CAS PubMed.
- S. M. Soto, Virulence, 2013, 4, 223–229 CrossRef.
- F. Van Bambeke, Y. Glupczynski, P. Plesiat, J. Pechere and P. M. Tulkens, J. Antimicrob. Chemother., 2003, 51, 1055–1065 CrossRef CAS PubMed.
- G. P Tegos, M. Haynes, J. Jacob Strouse, M. Md T Khan, C. G Bologa, T. I Oprea and L. A. Sklar, Curr. Pharm. Des., 2011, 17, 1291–1302 CrossRef.
- B. Zechini and I. Versace, Recent Pat. Anti-Infect. Drug Discovery, 2009, 4, 37–50 CrossRef CAS.
- K. Nishino and A. Yamaguchi, J. Bacteriol., 2001, 183, 5803–5812 CrossRef CAS PubMed.
- P. Blanco, S. Hernando-Amado, J. A. Reales-Calderon, F. Corona, F. Lira, M. Alcalde-Rico, A. Bernardini, M. B. Sanchez and J. L. Martinez, Microorganisms, 2016, 4, 14 CrossRef.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- J.-M. Pagès, C. E. James and M. Winterhalter, Nat. Rev. Microbiol., 2008, 6, 893–903 CrossRef PubMed.
- R. E. Hancock and F. S. Brinkman, Annu. Rev. Microbiol., 2002, 56, 17–38 CrossRef CAS PubMed.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS PubMed.
- G. D. Wright, Adv. Drug Delivery Rev., 2005, 57, 1451–1470 CrossRef CAS PubMed.
- Y. Liu, R. Li, X. Xiao and Z. Wang, Molecules, 2018, 24, 43 CrossRef PubMed.
- G. A. Jacoby and L. S. Munoz-Price, N. Engl. J. Med., 2005, 352, 380–391 CrossRef CAS PubMed.
- K. Bush and G. A. Jacoby, Antimicrob. Agents Chemother., 2010, 54, 969–976 CrossRef CAS PubMed.
- S. Kumar, M. M. Mukherjee and M. F. Varela, Int. J. Bacteriol., 2013, 2013 Search PubMed.
- K. H. Tehrani and N. I. Martin, Medchemcomm, 2018, 9, 1439–1456 RSC.
- M. Mora-Ochomogo and C. T. Lohans, RSC Med. Chem., 2021, 12, 1623–1639 RSC.
- T. Naas, S. Oueslati, R. A. Bonnin, M. L. Dabos, A. Zavala, L. Dortet, P. Retailleau and B. I. Iorga, J. Enzyme Inhib. Med. Chem., 2017, 32, 917–919 CrossRef CAS PubMed.
- A. Kondratieva, K. Palica, C. Frøhlich, R. R. Hovd, H.-K. S. Leiros, M. Erdelyi and A. Bayer, Eur. J. Med. Chem., 2024, 116140 CrossRef CAS.
- G. Bahr, L. J. Gonzalez and A. J. Vila, Chem. Rev., 2021, 121, 7957–8094 CrossRef CAS PubMed.
- K. Bush, J. Infect. Chemother., 2013, 19, 549–559 CrossRef CAS.
- T. Golkar, M. Zieliński and A. M. Berghuis, Front. Microbiol., 2018, 9, 1942 CrossRef.
- M. Morar, K. Pengelly, K. Koteva and G. D. Wright, Biochemistry, 2012, 51, 1740–1751 CrossRef CAS PubMed.
- G. P. Dinos, Br. J. Pharmacol., 2017, 174, 2967–2983 CrossRef CAS.
- M. K. Thompson, M. E. Keithly, G. A. Sulikowski and R. N. Armstrong, Perspect. Sci., 2015, 4, 17–23 CrossRef.
- R. E. Rigsby, K. L. Fillgrove, L. A. Beihoffer and R. N. Armstrong, Methods Enzymol., 2005, 401, 367–379 CAS.
- K. Shaw, P. Rather, R. Hare and G. Miller, Microbiol. Rev., 1993, 57, 138–163 CrossRef CAS.
- A. Robicsek, J. Strahilevitz, G. A. Jacoby, M. Macielag, D. Abbanat, C. Hye Park, K. Bush and D. C. Hooper, Nat. Med., 2006, 12, 83–88 CrossRef CAS PubMed.
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482 CAS.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6(8), a025247 CrossRef.
- E. Peterson and P. Kaur, Front. Microbiol., 2018, 9, 2928 CrossRef PubMed.
- G. Dhanda, Y. Acharya and J. Haldar, ACS Omega, 2023, 8, 10757–10783 CrossRef CAS PubMed.
- C. Reading and M. Cole, Antimicrob. Agents Chemother., 1977, 11, 852–857 CrossRef CAS.
- J. Fisher, J. Belasco, R. Charnas, S. Khosla and J. R. Knowles, Philos. Trans. R. Soc. London, Ser. B, 1980, 289, 309–319 CAS.
- J.-D. Docquier and S. Mangani, Drug Resistance Updates, 2018, 36, 13–29 CrossRef.
- Z. Si, K. Pethe and M. B. Chan-Park, JACS Au, 2023, 3, 276–292 CrossRef CAS.
- K. Coleman, Curr. Opin. Microbiol., 2011, 14, 550–555 CrossRef CAS.
- P. Lagacé-Wiens, A. Walkty and J. A. Karlowsky, Core Evid., 2014, 13–25 CrossRef.
- B. Moya, I. M. Barcelo, S. Bhagwat, M. Patel, G. Bou, K. M. Papp-Wallace, R. A. Bonomo and A. Oliver, Antimicrob. Agents Chemother., 2017, 61(6) DOI:10.1128/aac.02529-16.
- M. A. Powles, A. Galgoci, A. Misura, L. Colwell, K. H. Dingley, W. Tang, J. Wu, T. Blizzard, M. Motyl and K. Young, Antimicrob. Agents Chemother., 2018, 62(8) DOI:10.1128/aac.02577-17.
- J. Wu, F. Racine, M. K. Wismer, K. Young, D. M. Carr, J. C. Xiao, R. Katwaru, Q. Si, P. Harradine and M. Motyl, Antimicrob. Agents Chemother., 2018, 62(5) DOI:10.1128/aac.02323-17.
- S. J. Keam, Drugs, 2023, 83, 1245–1252 CrossRef CAS PubMed.
- N. L. Mallalieu, E. Winter, S. Fettner, K. Patel, E. Zwanziger, G. Attley, I. Rodriguez, A. Kano, S. M. Salama and D. Bentley, Antimicrob. Agents Chemother., 2020, 64(5) DOI:10.1128/aac.02229-19.
- D. M. Livermore, M. Warner, S. Mushtaq and N. Woodford, Antimicrob. Agents Chemother., 2016, 60, 554–560 CrossRef CAS.
- A. Morinaka, Y. Tsutsumi, M. Yamada, K. Suzuki, T. Watanabe, T. Abe, T. Furuuchi, S. Inamura, Y. Sakamaki and N. Mitsuhashi, J. Antimicrob. Chemother., 2015, 70, 2779–2786 CrossRef CAS.
- M. McCarthy and T. Walsh, Drugs Today, 2017, 53, 521–530 CrossRef CAS PubMed.
- A. Krajnc, P. A. Lang, T. D. Panduwawala, J. Brem and C. J. Schofield, Curr. Opin. Chem. Biol., 2019, 50, 101–110 CrossRef CAS PubMed.
- O. Lomovskaya, D. Sun, D. Rubio-Aparicio, K. Nelson, R. Tsivkovski, D. C. Griffith and M. N. Dudley, Antimicrob. Agents Chemother., 2017, 61(11) DOI:10.1128/aac.01443-17.
- S. Dhillon, Drugs, 2018, 78, 1259–1270 CrossRef CAS PubMed.
- A. Marino, E. Campanella, S. Stracquadanio, M. Calvo, G. Migliorisi, A. Nicolosi, F. Cosentino, S. Marletta, S. Spampinato and P. Prestifilippo, Antibiotics, 2023, 12, 1521 CrossRef CAS.
- H. Fatima, N. Goel, R. Sinha and S. K. Khare, Colloids Surf., B, 2021, 205, 111901 CrossRef CAS.
- H. Daiyasu, K. Osaka, Y. Ishino and H. Toh, FEBS Lett., 2001, 503, 1–6 CrossRef CAS.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503 CrossRef CAS PubMed.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503–506 CrossRef CAS PubMed.
- C. Schnaars, G. Kildahl-Andersen, A. Prandina, R. Popal, S. Radix, M. Le Borgne, T. Gjøen, A. M. S. Andresen, A. Heikal and O. A. Økstad, ACS Infect. Dis., 2018, 4, 1407–1422 CrossRef CAS.
- R. Azumah, J. Dutta, A. Somboro, M. Ramtahal, L. Chonco, R. Parboosing, L. Bester, H. Kruger, T. Naicker and S. Essack, J. Appl. Microbiol., 2016, 120, 860–867 CrossRef CAS PubMed.
- B. M. Liénard, G. Garau, L. Horsfall, A. I. Karsisiotis, C. Damblon, P. Lassaux, C. Papamicael, G. C. Roberts, M. Galleni and O. Dideberg, Org. Biomol. Chem., 2008, 6, 2282–2294 RSC.
- N. Li, Y. Xu, Q. Xia, C. Bai, T. Wang, L. Wang, D. He, N. Xie, L. Li and J. Wang, Bioorg. Med. Chem. Lett., 2014, 24, 386–389 CrossRef CAS.
- P. Wang, J. Cheng, C.-C. Liu, K. Tang, F. Xu, Z. Yu, B. Yu and J. Chang, Curr. Top. Med. Chem., 2018, 18, 834–843 CrossRef CAS PubMed.
- F.-M. Klingler, T. A. Wichelhaus, D. Frank, J. Cuesta-Bernal, J. El-Delik, H. F. Müller, H. Sjuts, S. Göttig, A. Koenigs and K. M. Pos, J. Med. Chem., 2015, 58(8), 3626–3630 CrossRef CAS PubMed.
- D. T. King, L. J. Worrall, R. Gruninger and N. C. Strynadka, J. Am. Chem. Soc., 2012, 134, 11362–11365 CrossRef CAS.
- P. Hinchliffe, M. M. González, M. F. Mojica, J. M. González, V. Castillo, C. Saiz, M. Kosmopoulou, C. L. Tooke, L. I. Llarrull and G. Mahler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3745–E3754 CrossRef CAS.
- M. M. González, M. Kosmopoulou, M. F. Mojica, V. Castillo, P. Hinchliffe, I. Pettinati, J. r. Brem, C. J. Schofield, G. Mahler and R. A. Bonomo, ACS Infect. Dis., 2015, 1, 544–554 CrossRef PubMed.
- G. Ahmad, N. Rasool, M. U. Qamar, M. M. Alam, N. Kosar, T. Mahmood and M. Imran, Arabian J. Chem., 2021, 14, 103270 CrossRef CAS.
- H. Lv, Z. Zhu, C. Qian, T. Li, Z. Han, W. Zhang, X. Si, J. Wang, X. Deng and L. Li, Biomed. Pharmacother., 2023, 166, 115439 CrossRef CAS PubMed.
- J.-Z. Chigan, Z. Hu, L. Liu, Y.-S. Xu, H.-H. Ding and K.-W. Yang, Bioorg. Chem., 2022, 120, 105654 CrossRef CAS.
- A. K. Bhardwaj and P. Mohanty, Recent Pat. Anti-Infect. Drug Discovery, 2012, 7, 73–89 CrossRef CAS PubMed.
- J.-M. Bolla, S. Alibert-Franco, J. Handzlik, J. Chevalier, A. Mahamoud, G. Boyer, K. Kieć-Kononowicz and J.-M. Pagès, FEBS Lett., 2011, 585, 1682–1690 CrossRef CAS PubMed.
- F. Van Bambeke and V. J. Lee, Recent Pat. Anti-Infect. Drug Discovery, 2006, 1, 157–175 CrossRef CAS.
- R. P. Lamers, J. F. Cavallari and L. L. Burrows, PLoS One, 2013, 8, e60666 CrossRef CAS PubMed.
- E. E. Gill, O. L. Franco and R. E. Hancock, Chem. Biol. Drug Des., 2015, 85, 56–78 CrossRef CAS PubMed.
- E. Reading, Z. Ahdash, C. Fais, V. Ricci, X. Wang-Kan, E. Grimsey, J. Stone, G. Malloci, A. M. Lau and H. Findlay, Nat. Commun., 2020, 11, 5565 CrossRef CAS PubMed.
- P. N. Markham, Antimicrob. Agents Chemother., 1999, 43, 988–989 CrossRef CAS PubMed.
- S. Samosorn, J. B. Bremner, A. Ball and K. Lewis, Bioorg. Med. Chem., 2006, 14, 857–865 CrossRef CAS.
- J. I. Ambrus, M. J. Kelso, J. B. Bremner, A. R. Ball, G. Casadei and K. Lewis, Bioorg. Med. Chem. Lett., 2008, 18, 4294–4297 CrossRef CAS PubMed.
- A. Hequet, O. N. Burchak, M. Jeanty, X. Guinchard, E. Le Pihive, L. Maigre, P. Bouhours, D. Schneider, M. Maurin and J. M. Paris, ChemMedChem, 2014, 9, 1534–1545 CrossRef CAS.
- S. Lepri, F. Buonerba, L. Goracci, I. Velilla, R. Ruzziconi, B. D. Schindler, S. M. Seo, G. W. Kaatz and G. Cruciani, J. Med. Chem., 2016, 59(3), 867–891 CrossRef CAS PubMed.
- H. Sjuts, A. V. Vargiu, S. M. Kwasny, S. T. Nguyen, H.-S. Kim, X. Ding, A. R. Ornik, P. Ruggerone, T. L. Bowlin and H. Nikaido, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3509–3514 CrossRef CAS.
- M. Laws, A. Shaaban and K. M. Rahman, FEMS Microbiol. Rev., 2019, 43, 490–516 CrossRef CAS PubMed.
- K. Poole, K. Krebes, C. McNally and S. Neshat, J. Bacteriol., 1993, 175, 7363–7372 CrossRef CAS PubMed.
- F. Naaz, A. Khan, A. Kumari, I. Ali, F. Ahmad, B. A. Lone, N. Ahmad, I. A. Khan, V. S. Rajput and A. Grover, Bioorg. Chem., 2021, 113, 105031 CrossRef CAS.
- D. H. Kwon and C.-D. Lu, Antimicrob. Agents Chemother., 2006, 50, 1623–1627 CrossRef CAS.
- M. Cassone and L. Otvos Jr, Expert Rev. Anti-Infect. Ther., 2010, 8, 703–716 CrossRef CAS.
- B. P. Lazzaro, M. Zasloff and J. Rolff, Science, 2020, 368, eaau5480 CrossRef CAS.
- C. Tascini, G. Gemignani, S. Ferranti, E. Tagliaferri, A. Leonildi, A. Lucarini and F. Menichetti, J. Chemother., 2004, 16, 282–287 CrossRef CAS.
- N. Petrosillo, E. Ioannidou and M. Falagas, Clin. Microbiol. Infect., 2008, 14, 816–827 CrossRef CAS PubMed.
- L. M. Lim, N. Ly, D. Anderson, J. C. Yang, L. Macander, A. Jarkowski III, A. Forrest, J. B. Bulitta and B. T. Tsuji, Pharmacotherapy, 2010, 30, 1279–1291 CrossRef CAS PubMed.
- S. Biswas, J.-M. Brunel, J.-C. Dubus, M. Reynaud-Gaubert and J.-M. Rolain, Expert Rev. Anti-Infect. Ther., 2012, 10, 917–934 CrossRef CAS.
- H. Tsubery, I. Ofek, S. Cohen and M. Fridkin, J. Med. Chem., 2000, 43, 3085–3092 CrossRef CAS PubMed.
- M. Vaara and T. Vaara, Nature, 1983, 303, 526–528 CrossRef CAS PubMed.
- H. Tsubery, I. Ofek, S. Cohen and M. Fridkin, Biochemistry, 2000, 39, 11837–11844 CrossRef CAS PubMed.
- M. Vaara, J. Antimicrob. Chemother., 2013, 68, 1213–1219 CrossRef CAS.
- P. D. Tamma, S. E. Cosgrove and L. L. Maragakis, Clin. Microbiol. Rev., 2012, 25, 450–470 CrossRef CAS.
- I. Karaiskos, A. Antoniadou and H. Giamarellou, Expert Rev. Anti-Infect. Ther., 2017, 15, 1123–1140 CrossRef CAS PubMed.
- V. Defraine, M. Fauvart and J. Michiels, Drug Resistance Updates, 2018, 38, 12–26 CrossRef PubMed.
- G. J. Sullivan, N. N. Delgado, R. Maharjan and A. K. Cain, Curr. Opin. Microbiol., 2020, 57, 31–40 CrossRef CAS PubMed.
- M. J. Rybak and B. J. McGrath, Drugs, 1996, 52, 390–405 CrossRef CAS.
- A. Wróbel, K. Arciszewska, D. Maliszewski and D. Drozdowska, J. Antibiot., 2020, 73, 5–27 CrossRef.
- M. de Górgolas, P. Aviles, C. Verdejo and M. Fernandez Guerrero, Antimicrob. Agents Chemother., 1995, 39, 953–957 CrossRef.
- A. C. Sick, S. Tschudin-Sutter, A. E. Turnbull, S. J. Weissman and P. D. Tamma, Pediatrics, 2014, 133, e1148–e1155 CrossRef.
- G. D. Wright, Trends Microbiol., 2016, 24, 862–871 CrossRef CAS PubMed.
- N. Wang, J. Luo, F. Deng, Y. Huang and H. Zhou, Front. Pharmacol, 2022, 13, 839808 CrossRef CAS PubMed.
- H. Araoka, M. Baba, S. Takagi, N. Matsuno, K. Ishiwata, N. Nakano, M. Tsuji, H. Yamamoto, S. Seo and Y. Asano-Mori, Scand. J. Infect. Dis., 2010, 42, 231–233 CrossRef CAS PubMed.
- A. Alsughayer, A.-Z. A. Elassar, A. A. Hasan and F. Al Sagheer, Bioorg. Chem., 2021, 108, 104658 CrossRef CAS PubMed.
- N. K. Boyd, C. Teng and C. R. Frei, Front. Cell. Infect. Microbiol., 2021, 11, 684515 CrossRef CAS.
- Y. Liu, Z. Tong, J. Shi, R. Li, M. Upton and Z. Wang, Theranostics, 2021, 11, 4910 CrossRef CAS.
- M. Masadeh, N. Mhaidat, K. Alzoubi, S. Al-Azzam and Z. Alnasser, Ann. Clin. Microbiol. Antimicrob., 2012, 11, 1–5 CrossRef PubMed.
- A. Liappis, V. Kan, C. Rochester and G. Simon, Clin. Infect. Dis., 2001, 33, 1352–1357 CrossRef CAS.
- V. Chopra, M. A. Rogers, M. Buist, S. Govindan, P. K. Lindenauer, S. Saint and S. A. Flanders, Am. J. Med., 2012, 125, 1111–1123 CrossRef CAS PubMed.
- Y.-S. Cheng, W. Sun, M. Xu, M. Shen, M. Khraiwesh, R. J. Sciotti and W. Zheng, Front. Cell. Infect. Microbiol., 2019, 8, 438 CrossRef PubMed.
- B. Rácz and G. Spengler, Antibiotics, 2023, 12, 137 CrossRef.
- M. M. Kristiansen, C. Leandro, D. Ordway, M. Martins, M. Viveiros, T. Pacheco, J. E. Kristiansen and L. Amaral, Int. J. Antimicrob. Agents, 2003, 22, 250–253 CrossRef CAS.
- J. M. Stokes, C. R. MacNair, B. Ilyas, S. French, J.-P. Côté, C. Bouwman, M. A. Farha, A. O. Sieron, C. Whitfield and B. K. Coombes, Nat. Microbiol., 2017, 2, 1–8 Search PubMed.
- E. B. Breidenstein, C. de la Fuente-Núñez and R. E. Hancock, Trends Microbiol., 2011, 19, 419–426 CrossRef CAS.
- C. Vilaplana, E. Marzo, G. Tapia, J. Diaz, V. Garcia and P.-J. Cardona, J. Infect. Dis., 2013, 208, 199–202 CrossRef CAS.
- D. P. Eisen, E. S. McBryde and A. Walduck, J. Infect. Dis., 2013, 208, 1925–1927 CrossRef CAS.
- A. M. Kalle and A. Rizvi, Antimicrob. Agents Chemother., 2011, 55, 439–442 CrossRef CAS PubMed.
- P. Fernandes and E. Martens, Biochem. Pharmacol., 2017, 133, 152–163 CrossRef CAS PubMed.
- R. Li, R. A. Oliver and C. A. Townsend, Cell Chem. Biol., 2017, 24, 24–34 CrossRef CAS PubMed.
- A. A. Mahmood, M. A. Al-Iraqi and F. T. Abachi, Mater. Today: Proc., 2021, 42, 1860–1866 CAS.
- Y. R. Lee and K. L. Jacobs, Drugs, 2019, 79(17), 1867–1876 CrossRef CAS.
- M. P. Veve and J. L. Wagner, Pharmacotherapy, 2018, 38(9), 935–946 CrossRef CAS.
- N. Eraikhuemen, D. Julien, A. Kelly, T. Lindsay and D. Lazaridis, Infect. Dis. Ther., 2021, 10, 149–163 CrossRef PubMed.
- P. Nahid, S. R. Mase, G. B. Migliori, G. Sotgiu, G. H. Bothamley, J. L. Brozek, A. Cattamanchi, J. P. Cegielski, L. Chen and C. L. Daley, Am. J. Respir. Crit. Care Med., 2019, 200, e93–e142 CrossRef.
- K. A. Abrahams, S. M. Batt, S. S. Gurcha, N. Veerapen, G. Bashiri and G. S. Besra, Nat. Commun., 2023, 14, 3828 CrossRef CAS.
- D. R. Giacobbe, E. Ciacco, C. Girmenia, F. Pea, G. M. Rossolini, G. Sotgiu, C. Tascini, M. Tumbarello, P. Viale and M. Bassetti, Infect. Drug Resist., 2020, 4697–4711 CrossRef CAS.
- S. Domingues, T. Lima, M. J. Saavedra and G. J. Da Silva, Life, 2023, 13(7), 1427 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |