
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn search of a photoswitchable drug for serotonin receptors: a molecular dynamics simulation study†
Arshjot S. Dhaliwal,
Arunima Verma and
Padmabati Mondal *
*
Department of Chemistry, Indian Institute of Science Education and Research, Srinivasapuram, Yerpedu Mandal, Tirupati Dist., Andhra Pradesh 517619, India. E-mail: padmabati.mondal@iisertirupati.ac.in; Tel: +91 8772500926
First published on 23rd June 2025
Abstract
G-protein-coupled receptors are membrane-bound proteins that control physiological and psychological activities such as hormonal regulation, sensory signaling and neurotransmission in the human body. The photoisomerization capability of azobenzenes has enabled the development of a new generation of photoswitchable drugs that offer greater efficacy while reducing the side effects typical of conventional treatments. Serotonin binding to the serotonin receptor can be controlled by conjugating it with cis and trans photoswitchable isomers of azobenzene at the hydroxyl position. We have used a combination of molecular dynamics (MD) simulation and free-energy methods for receptor binding to trans-azobenzene-fused-serotonin (TAS) and cis-azobenzene-fused serotonin (CAS) ligand systems. The CAS ligand experienced greater conformational fluctuations in comparison to the TAS ligand. Binding free-energy values showed significant differences between the CAS–receptor and TAS–receptor complexes, indicating that the TAS ligand binds actively to the serotonin receptor as compared to the CAS ligand. It also revealed that the presence of a lipid membrane has a strong influence on the stability and dynamics of the receptor–ligand complex and, thus, on the differential binding free-energy values. The change in binding free energy between the CAS–receptor and TAS–receptor complexes is found to be entropically driven. A qualitative and quantitative analysis of the stacking interactions between aromatic residues of interest and each ligand indicates that T-type stacking interactions predominate and particularly Trp327 and Phe330 residues contribute notably to the stability of the TAS–receptor complex. The study indicates that by leveraging the photoswitchable properties of azobenzenes, a photoswitchable serotonin-based drug with tunable binding affinity can be designed, which may facilitate advanced drug discovery and therapeutic approaches for mental health treatments.
1 Introduction
Membrane-bound proteins are an essential family of biological macromolecules that function through interactions with other proteins or small molecules called ligands or drugs.1–3 These proteins interact with small molecules or each other via non-covalent interactions to create complexes, a process known as molecular recognition, which is fundamental to all biological processes.4,5 Computational drug discovery6–8 is vital to understanding the mechanism of protein and drug functioning, which requires a thorough understanding of the interactions associated with proteins and drugs. Hence, it is crucial to methodically decipher the atomistic and electronic levels of non-covalent protein–drug interactions, which can only be accomplished via computational research.9G-protein-coupled receptors (GPCRs)10–12 are the most prominent membrane-bound family of proteins and are responsible for controlling various psychological and physiological activities, such as neurotransmission, hormonal regulation, and sensory signaling, because they possess a signal transduction mechanism that transforms extracellular signals into intracellular responses.13–16 GPCRs are also marked by their participation in endocrine,17 alzheimer's,18 and cancer-like diseases.19 Serotonin, or 5-hydroxytryptamine (5-HT), is a chemical compound exclusively produced in the human body for several psychological and physiological functions.15,20 It is regulated at the functional level when it interacts with serotonin receptors, also called 5-HT receptors,21,22 which are a subtype of GPCRs. 5-HT receptors can modulate serotonin secretion based on their location in the neuronal network: when located presynaptically, they can inhibit serotonin release, while postsynaptically, they can enhance serotonin signaling.23 Proteins of the 5-HT receptor family are classified into seven subtypes, 5-HT1–7,24 according to their signaling pathways. Among these subfamilies, all except 5-HT3 are guanine nucleotide-binding proteins.20,25 This study is focused on the GPCR called 5-HT1B. The 5-HT1B receptor consists of seven alpha helices named TM1 to TM7 (where TM represents transmembrane), which are embedded in the lipid bilayer.26 Serotonin binds to the 5-HT1B receptor and initiates a series of functions, including receptor activation, G-protein signaling, and downstream effects that influence neural excitability and neurotransmitter release.27 Computational studies previously conducted in our research group focused on serotonin–receptor interactions, binding, stability, and signal transduction, providing a foundation for further research.20,28 Understanding the binding and unbinding mechanisms of serotonin with 5-HT1B and their control is important for further developing antidepressant therapeutic strategies with different drugs for disorders associated with serotonin dysregulation, such as depression and anxiety.
There are several known classes of drugs designed to interact with biological macromolecule targets. One of the ways to control the binding and unbinding of these drugs is via making them photoswitchable by attaching a photoswitchable molecule to them.7 These photoswitchable drugs possess the ability to reversibly photoisomerize between two conformations, i.e., a trans isomer (E form) and a cis isomer (Z form).29,30 The exposure to light with varied wavelengths triggers photoswitching activity, resulting in changes in drug polarity and stability and the affinity of the molecule to the target biomolecules.31 Among various known photoswitchable molecules, azobenzene-based molecules are one of the best-characterized photoswitches in terms of photochemistry because they have large spectra of possibilities for modification of the core structure of the compound and capability of isomerization in constrained environments.32–34 The dynamic properties of azobenzenes makes it possible to create a new generation of photoswitchable drugs that offer greater efficacy and reduced side effects. In this study, azobenzene is covalently attached to the serotonin molecule, resulting in the formation of two ligands, the E-isomer and Z-isomer, namely trans-azobenzene-fused-serotonin (TAS) and cis-azobenzene-fused-serotonin (CAS), respectively.15,35 By utilizing light of varied wavelengths, it is possible to manipulate binding affinities and minimize the side effects associated with the drugs.36 Similar azobenzene-based drugs for receptor proteins are already reported in previous literature by Matera et al.,37 López-Cano et al.,38 and Noev et al.39 Selective photoswitchable azobenzene-based allosteric agonists specifically for G protein-coupled receptors have also been studied and Donthamsetti et al.40 and by Saβmannshausen et al.7 These are studies for the potential biological or pathological significance of azobenzene-based drugs in general. Experimental studies conducted by Bahamonde et al.41 state that azobenzene moieties can be integrated into GPCR ligands, allowing their activity to be modulated by light that influences downstream signaling pathways. Photoswitchable drugs also help increase the receptor's selectivity and affinity towards drug–receptor binding, which plays a significant role in serotonin receptor signaling.40 The primary objective of conjugating serotonin with azobenzene is to enhance targeted drug delivery and provide an innovative therapeutic strategy for individuals suffering from serotonin-related disorders. Therefore, delineating how two isomers of azobenzene affect serotonin binding to the 5-HT1B receptor can help advance knowledge about the control of serotonin signaling. This approach will also advance the understanding of how the 5-HT1B receptor incorporates, adjusts and accommodates such a photoswitchable drug by exposing its more dynamic nature.
This study primarily focuses on the binding of azobenzene-based photoswitchable serotonin derivatives to the 5-HT1B receptor. The main motive here is to understand the non-covalent interactions responsible for the ligand binding to the 5-HT1B receptor and to estimate the difference in the binding affinity of two photoisomers to the receptor, which helps to assess the photocontrol of the ligand–receptor binding. However, since the 5-HT1B receptor is a membrane-bound protein, exploring the impact of a lipid membrane on these protein–ligand systems is also crucial to gain a more profound understanding of what changes in the functional dynamics and ligand interactions it can bring within these protein–ligand systems.
2 Computational details
2.1 Preparation of ligands
The azobenzene molecule undergoes a reversible photoisomerization process, in which a trans-to-cis isomerization occurs upon excitation with UV light of wavelength 365 nm and a cis-to-trans isomerization occurs with exposure to visible light of wavelength 420 nm.29 To prepare the azobenzene-fused-serotonin photoswitchable ligand, the 5-hydroxyl group of the 6-membered aromatic ring was removed from the serotonin molecule, and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N along with an aromatic ring was covalently attached at that position. The main reason behind replacing the hydroxyl group was to add the function of photoswitching of azobenzene (as shown in Fig. 1). Both isomers of the azobenzene-fused-serotonin drug were prepared and the energy was minimized based on universal force fields using the Avogadro molecular editor and visualizer software. Finally, the geometry of both TAS and CAS ligands was optimized separately at the B3LYP/6-31G(d,p) level of theory using Gaussian software.42 TDDFT calculations at the B3LYP/6-311+G(d,p) level of theory confirm that the TAS and CAS also absorb in the UV and visible regions, respectively.
N along with an aromatic ring was covalently attached at that position. The main reason behind replacing the hydroxyl group was to add the function of photoswitching of azobenzene (as shown in Fig. 1). Both isomers of the azobenzene-fused-serotonin drug were prepared and the energy was minimized based on universal force fields using the Avogadro molecular editor and visualizer software. Finally, the geometry of both TAS and CAS ligands was optimized separately at the B3LYP/6-31G(d,p) level of theory using Gaussian software.42 TDDFT calculations at the B3LYP/6-311+G(d,p) level of theory confirm that the TAS and CAS also absorb in the UV and visible regions, respectively.
 | ||
| Fig. 1 Chemical structures of trans-azo-serotonin (TAS) and cis-azo-serotonin (CAS) and their switching. | ||
2.2 Simulation setup
The initial 5-HT1B receptor structure was taken from the cryo-EM structure of the heterotrimeric Go protein coupled to the 5-HT1B receptor (chain S), PDB ID: 6G79.43 To prepare the apo-receptor, the heterotrimeric Go protein was removed along with all subunits and heteroatoms. The gas-phase optimized structures of the TAS and CAS ligands were docked to the 5-HT1B apo-receptor using Autodock4.2.6 software.44 The most stable protein–ligand binding free energy was ∼−8.4 kcal mol−1 for CAS and ∼−10 kcal mol−1 for TAS. The most stable and reasonable structures of the ligand-bound 5-HT1B receptor were taken as the initial structures of the ligand–receptor complexes. For the sake of simplicity, the CAS ligand docked to the receptor has been named a ‘CAS–receptor complex’, and the TAS ligand docked to the receptor a ‘TAS–receptor complex’. For molecular dynamics (MD) simulations, four different systems were prepared as described below:(i) The systems set up for MD simulations were created using the CHARMM-GUI solution builder.45 The initial docked structures of both the ligand–receptor complexes were separately solvated in rectangular water boxes of volume 85 Å × 85 Å × 85 Å. Then, both systems were separately neutralized by Na+ and Cl− ions.46 These two systems are labelled as ‘without POPC membrane’ in the following (Fig. 2(A)).
(ii) A 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)47,48 lipid-membrane model based on CHARMM36m force fields was constructed around the 5-HT1B receptor for each initial docked structure of the CAS–receptor and TAS–receptor complexes. The initial input files for MD simulations were prepared using the CHARMM-GUI membrane builder,45,49,50 ensuring the proper orientation of receptor structures within the lipid membrane. These complexes were then separately solvated in water boxes of volume 85 Å × 85 Å × 110 Å. These two systems are labelled as ‘with POPC membrane’ in the further sections (Fig. 2(B)).
(iii) Then, all four systems were energy-minimized using the steepest-descent method for 5000 steps. The systems were then gradually heated from 0 K to 300 K, and the periodic boundary conditions of all systems were equilibrated for 5 ns. Finally, 500 ns simulations were performed for all four systems using the GROMACS molecular dynamics simulation package51 in NPT ensembles with a pressure of 1 atm, controlled using the Parrinello–Rahman barostat.52 The CHARMM36m force field implemented in GROMACS was used for the POPC lipid membrane and receptor.53 The force fields for the ligands were obtained from CGENFF, which is included in the local CHARMM force field database of GROMACS.
 | ||
| Fig. 2 The initial structures of the 5-HT1B receptor (blue) docked to the photoswitchable azo-sero ligand (orange), without (A) and with (B) the POPC membrane. | ||
3 Analysis methods
3.1 Binding free-energy estimation and interaction-entropy calculations
The molecular mechanics/generalized born surface area (MM/GBSA)8,54 method, using a single molecular dynamic trajectory, was employed to calculate the binding free energy (ΔGMM/GBSA) using gmx_MMPBSA software.55 It is a tool based on AMBER's mmpbsa.py script56 that aims to perform end-state free-energy calculations57 with GROMACS output files. The ff14SB58 force field was used for all complexes to obtain the molecular mechanical energy. Here, for all systems, 1000 frames from 500 ns trajectories were extracted within equal intervals to perform calculations as follows:| ΔGMM/GBSA = ΔH − TΔS ≈ (ΔEMM + ΔGSolv) − TΔS. |
The entropic contribution (−TΔS) to the binding free energies is calculated using the interaction entropy (σIE) method,61,62 as implemented in gmx_MMPBSA, defined as follows:
−TΔS = kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln〈exp(βΔEintpl)〉. ln〈exp(βΔEintpl)〉. |
3.2 Structural and conformational changes of the ligands and receptor binding pocket
The conformational and positional changes in the ligands, along with the pocket volume and shape of receptors, play a key role in the molecular recognition process. Several theories, such as the induced-fit and lock-and-key models, explain the molecular recognition or protein–ligand interaction processes. The structural complementarity between protein and ligand plays an important role because the ligand will not bind actively to the binding pocket of the protein if the ligand cannot physically fit into it. Therefore, a quantitative assessment of the conformational and positional changes of the ligand and receptor, along with the calculation of binding pocket volume, is essential to understand the molecular recognition process systematically and quantitatively.3.3 Order parameters for qualitative description of stacking
Among non-covalent interactions, stacking interactions are among the primary interactions that biological systems with aromatic rings often encounter. These interactions play a crucial role in the stability and binding affinity of various protein–ligand complexes. Offset parallel (or antiparallel) stacking and T-type stacking are the two main kinds of stacking interactions found in macromolecular biological systems. Rcom and γ were defined to obtain a qualitative understanding of stacking interactions and determine the type and nature of stacking interactions between each of the primary aromatic residues and the ligand at the binding site.20,66 Rcom is the center-of-mass distance between the aromatic residue of interest and the ligand, while γ is the angle between the surface normals of the aromatic residue of interest and the ligand, which provides information on the orientation of the interacting residues (Fig. 3). For the γ angle, the coordinates of only three heavy atoms that form a triangle over the entire ring are considered when calculating the surface plane of the ring from which the surface normal is projecting out. In contrast, the coordinates of all the heavy carbon atoms in each aromatic ring are considered when calculating Rcom. | ||
| Fig. 3 Graphical representation of the order parameters Rcom and γ for (A) off-set parallel (or anti-parallel) stacking and (B) T-type stacking interactions. A similar approach was used in a previous study from our group.20 | ||
3.4 Quantitative estimation of per-residue non-bonded interaction energy
This study employed GROMACS2024 to estimate the molecular mechanical non-bonded interaction energy (based on CHARMM36m force fields) involving the ligands and residues of interest. The calculations were carried out using GROMACS's capability to break down short-range non-bonded energies between any two specified energy groups. It is important to note that these interaction energies differ from the free energy of binding. These energies represent the non-bonded enthalpic contributions from each residue of interest. CHARMM36m force fields have been parameterized to broadly capture molecular interactions, including those with solvent water, to give accurate interaction strength representation53 and therefore are a good choice for the calculation of MM interaction energies. By quantifying the MM non-bonded interaction energies, we can obtain insights into the contributions from each important aromatic residue of interest in the ligand binding process.3.5 Dynamic cross-correlation analysis
To understand the correlation between different parts of the proteins and the degree of conformational fluctuation due to change of ligand, dynamic cross-correlation (DCC) map analysis has been performed. The DCC matrix is calculated using the following equation:
 | (1) |
The difference dynamic cross-correlation (DDCC) was introduced and used as the difference in the DCC of the complex and apo-receptor in our previous studies to understand the effect of ligand binding.20,28
| DDCC(i,j) = DCC(i,j)complex − DCC(i,j)apo-receptor | (2) |
| DDCC(i,j) = DCC(i,j)CAS–receptor − DCC(i,j)TAS–receptor | (3) |
4 Results and discussion
4.1 Stability of the simulated systems
MD simulations were performed on the prepared input systems for 500 nanoseconds (ns). With superimposition of the C-alpha atoms, root-mean-square deviations (RMSDs) were calculated for the C-alpha carbon atoms (in each transmembrane helix) of receptors in both the ‘with and without the POPC membrane’ systems to monitor their stability over the simulation time.67As evident from the RMSD plots of the receptors, the TAS–receptor complexes (black) and the CAS–receptor complexes (red) exhibited conformational fluctuations (Fig. 4). These conformational fluctuations indicate that the initial docked conformations of all complexes were well-adjusted by MD samplings during the simulations. The relatively large fluctuations in C-alpha carbon atoms of the receptor in the CAS–receptor complexes compared to the TAS–receptor complexes can be interpreted as a lack of stabilization of the CAS ligand, consequently impacting the conformations of the receptor and ligand binding pocket. The RMSD values of the CAS ligand (green) exhibited more significant conformational fluctuations as compared to the TAS ligand (blue) (Fig. 4). Such fluctuations affect the stability of the binding site, indicating the disruption of non-covalent interactions between protein and ligand, and therefore the overall affinity and functionalities of the ligand are hindered. Based on the results from RMSD analysis, which indicate instability of the system till 50 ns, the initial 50 ns was excluded from the trajectories for binding free-energy and clustering analysis. To confirm convergence of the results, independent simulations have been performed for each system and results from those are discussed in Section S2 of the ESI.†
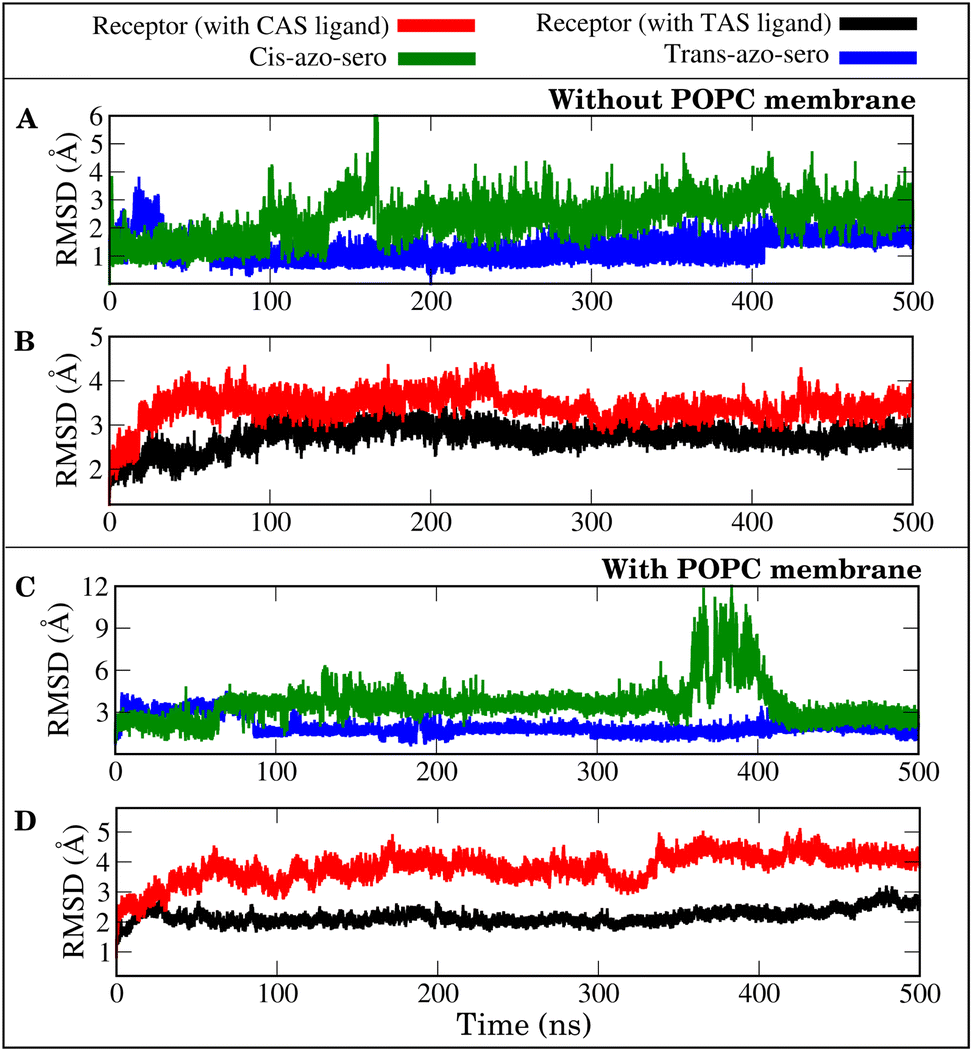 | ||
| Fig. 4 Root mean square deviations (RMSDs) of ligands and C-alpha carbon atoms of the receptor, plotted as a function of simulation time (ns): (A and B) without POPC membrane and (C and D) with POPC membrane. | ||
4.2 Binding free energy and per-residue energy decomposition
The binding free energies were computed using the MM/GBSA approach over the 500 ns MD trajectories and the initial 50 ns were excluded for relaxation. Fig. 5 shows the calculated values of enthalpy (ΔH), entropy (−TΔS) and binding free energy (ΔGMM/GBSA) for all the complexes.The small difference in ΔH values between the CAS–receptor and TAS–receptor complexes (ΔΔH ≅ 5.5 kcal mol−1) can be attributed to the similar structures of both ligands. However, the major differences seen in the entropy (ΔS) values between the TAS–receptor and CAS–receptor complexes influence the determination of binding free energies. This indicates that the CAS and TAS ligands introduce varied conformational changes to the receptor structures, resulting in the differences in binding free-energy values between the TAS–receptor and the CAS–receptor complexes. The relatively large ΔS value for the CAS–receptor complex can be attributed to large fluctuations exhibited by the CAS ligand (as also indicated by Fig. 4). Such fluctuations reflect the inherent complexity of protein–ligand interactions, which lead to high variability in binding affinities. The resulting relative binding free-energy differences clearly favour the TAS ligand, which stabilized the interactions with the 5-HT1B receptor, and the CAS ligand exhibited weaker binding affinity to the 5-HT1B receptor. The results from independent simulations are given in Table S1 of the ESI† and error bars for the binding free energies are shown in Table S2 of the ESI.†
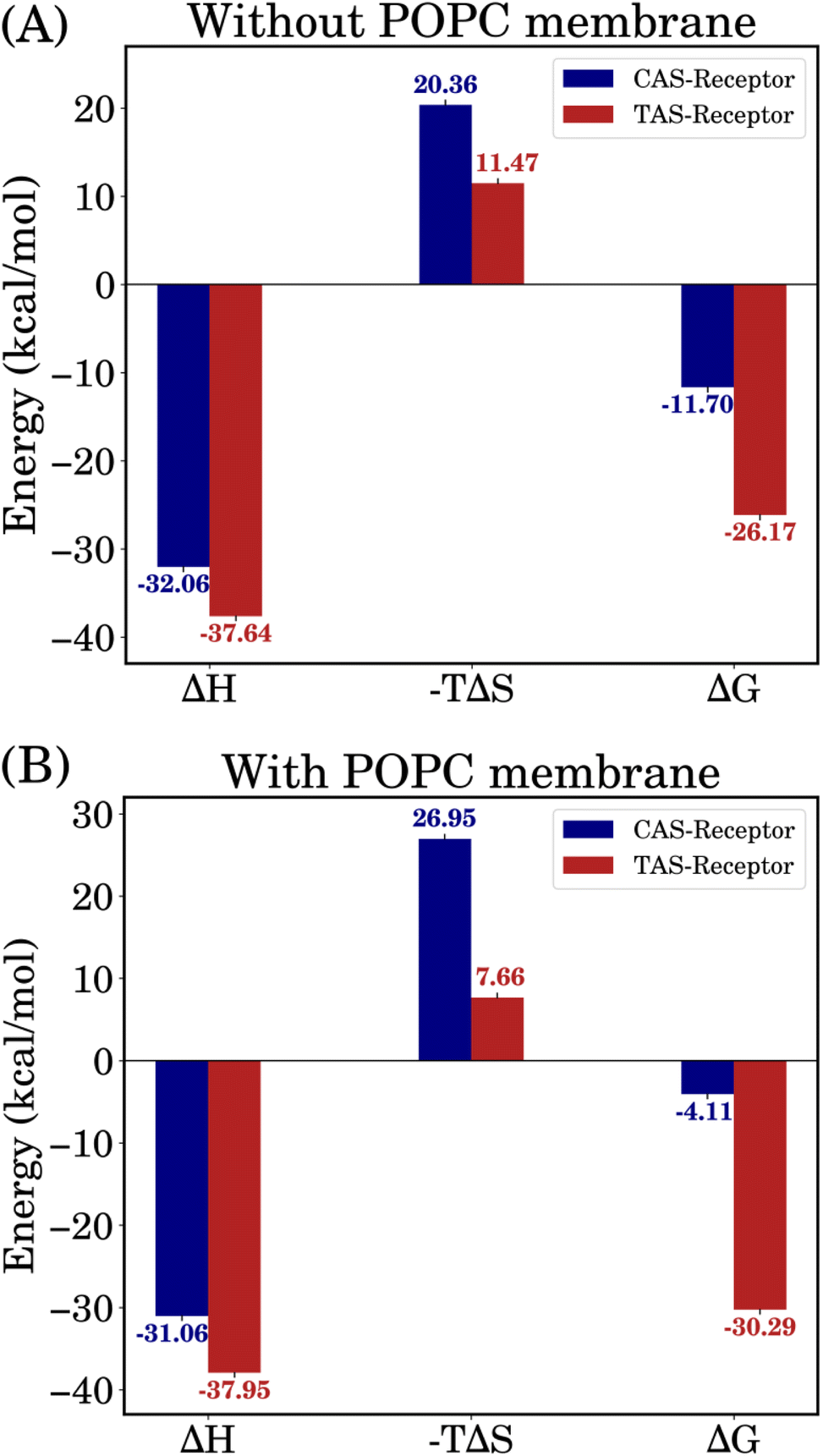 | ||
| Fig. 5 Binding free-energy results incorporating enthalpy and entropy contributions for both systems, i.e., (A) without and (B) with POPC membrane. | ||
 | ||
| Fig. 6 Comparison of per-residue binding free-energy contributions for TAS- (black) and CAS- (red) bound complexes, with and without POPC membrane. | ||
4.3 Comparative analysis of ligand conformations and their impact on binding-pocket volumes
The binding free-energy analysis revealed that the CAS ligand exhibited an entropic influence greater than that of the TAS ligand, indicating significant deviations in its molecular conformation throughout the simulations. To comprehend that, the ligand conformations were clustered using the clustering analysis method as described in the method section and representative conformations are shown in Fig. 7.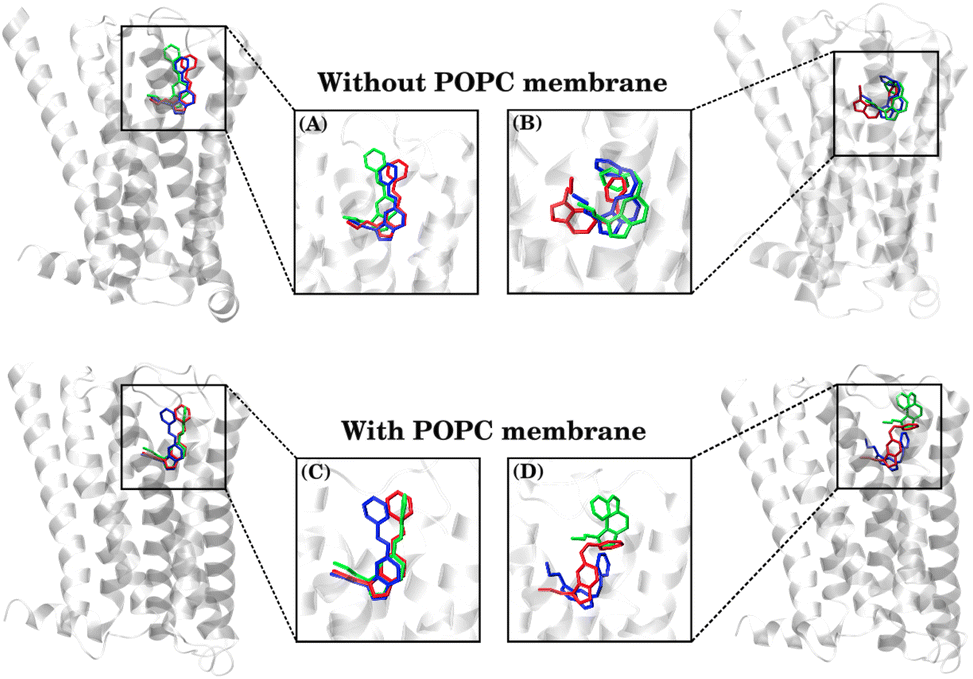 | ||
| Fig. 7 Comparison of MD-simulated conformations of the TAS ligand (A and C) and CAS ligand (B and D), representing the three most populated clusters, as obtained via VMD clustering analysis. | ||
Among these clusters, the blue conformation represents the most populated and initial structures, while the red conformation indicates the cluster observed towards the end of the simulation. Here, it is important to note that a significant number of conformations (especially in the CAS–receptor systems) could not be categorized as any of the three highest populated clusters due to the diverse conformational states populated by the ligands, as also indicated by the higher RMSDs of the CAS ligand.
The percentage of population for each cluster of each system is given as follows:
(1) Systems ‘without the POPC membrane’:
• (A) In the TAS–receptor complex, the blue conformation represents the most populated cluster at 91.28%, the red conformation indicates the cluster with a population of 3.85%, and the green indicates the cluster with a population of 2.61%. Only a negligible number of conformations remained non-clustered in both TAS–receptor complexes.
• (B) In the CAS–receptor complex, the blue-colored conformation represents the most populated cluster at 61.27%, the red conformation indicates the cluster with a population of 11.72%, and the green indicates the cluster with a population of 7.43%. The remaining 20% of structures could not be categorized as any of these three clusters.
(2) Systems ‘with the POPC membrane’:
• (C) In the TAS–receptor complex, the blue conformation represents the most populated cluster at 80.86%, the red conformation indicates the cluster with a population of 14.68% and the green indicates the cluster with a population of 2.44%.
• (D) In the CAS–receptor complex, the blue conformation represents the most populated cluster at 34.74%, the red conformation indicates the cluster with a population of 16.50% and the green indicates the cluster with a population of 9.25%. The remaining 40% structures could not be categorized into any of these three clusters.
This clustering analysis provides insights into the positional changes and different binding poses of the ligands, highlighting that the CAS ligand underwent higher positional changes (driven by entropy) compared to the TAS ligand within the binding pocket of the serotonin receptor.
 | ||
| Fig. 8 The volume changes in the binding pocket of the receptor due to changes in the conformations of the ligands as a function of time (ns): (A) systems without the POPC membrane and (B) systems with the POPC membrane. | ||
A larger pocket volume in the CAS–receptor complexes was observed for both the ‘with and without the POPC membrane’ systems. This analysis aligns with the increased conformational flexibility of the CAS ligand and thereby of the CAS-bound receptor. All of these analyses indicate that while the CAS ligand may initially bind to the receptor, the complex is overall less stable with the CAS ligand than with the TAS ligand.
The observed variations in the binding-pocket volumes of systems ‘with the POPC membrane’ can be interpreted as a result of non-covalent interactions between the receptors and the POPC membrane. However, unlike the CAS–receptor complex, these interactions do not influence the ligand–receptor binding in the TAS–receptor complex. The POPC membrane holds the receptor in such a way that it favours the interactions in the TAS–receptor complex compared to the CAS–receptor complex.
To further validate the results of the binding-pocket volume analysis, we performed a clustering analysis on receptor conformations using VMD in all MD trajectories. All receptor conformations were clustered into three clusters, and the representative conformations from the most populated clusters were selected for visualization of the changes in binding pocket volumes. The selected receptor structures were superimposed to visualize the differences in binding-pocket volumes across all MD systems (as shown in Fig. S4, ESI†). This analysis corroborates the quantitative data obtained from the calculations of the binding-pocket volumes.
4.4 Receptor–ligand local non-covalent interactions
 | ||
| Fig. 9 Aromatic residues of interest in the binding site (black) in a single snapshot. | ||
To obtain insight into the probable types of stacking interactions in these ligand–receptor complexes, a qualitative analysis is performed based on the two order parameters described in the methodology section.20,66 The order-parameter criteria for stacking interactions are considered as follows:
(i) Rcom: for parallel stacking interactions, 3 Å ≤ Rcom ≤ 5 Å, and for T-type stacking interactions, 5 Å ≤ Rcom ≤ 7.5 Å.
(ii) γ: for parallel stacking interactions, 0° ≤ γ ≤ 30°, and for T-type stacking interactions, 60° ≤ γ ≤ 120°.
The qualitative stacking-interaction analysis was conducted for all of the trajectories, i.e., protein–ligand complexes without and with the POPC membrane, as follows.
(I) Analysis for simulations without the POPC membrane. Fig. 10(A) and (B) show that while the Rcom values for residues Phe330 and Phe331 with the azo part of the CAS ligand lie between 5 Å and 7.5 Å, they are likely to have T-type stacking interactions, while the Rcom values for Tyr359, Phe217 and Trp327 are distributed around a mean value of 10 Å. However, multiple peaks for some residues are observed in the Rcom and γ plots, indicating that the azo part of the CAS ligand underwent significant fluctuations over time (ns).
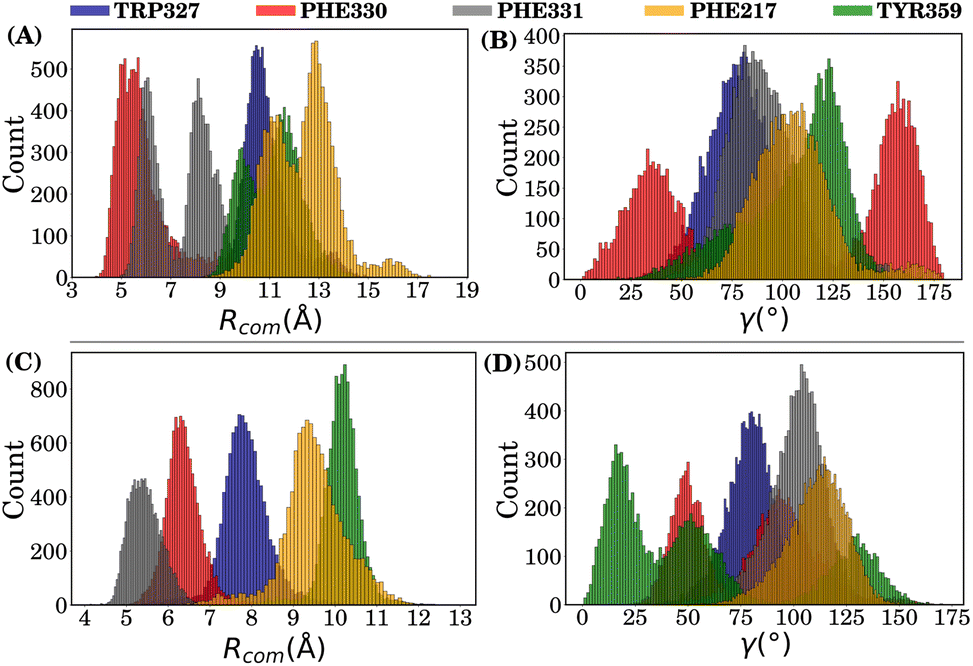 | ||
| Fig. 10 Distribution of order parameters in the CAS–receptor complex (without the POPC membrane): panels (A) and (B) represent the Rcom and γ, respectively, for the azo part, while panels (C) and (D) are the corresponding plots for the sero part. | ||
Fig. 10(C) and (D) show that while the Rcom values for residues Phe330, Phe331 and Trp327 with the sero part of the CAS ligand lie between 5 Å and 7.5 Å, indicating that they are likely to have T-type stacking interactions, the Rcom values for Tyr359 and Phe217 are distributed around a mean value of 9 Å–10 Å. However, here again, multiple peaks are visible for the γ angle distribution for the main aromatic residues Phe330 and Tyr359, indicating that the sero part of the CAS ligand also underwent fluctuations over time (ns).
Hence, order-parameter analysis suggests that the sero part of the CAS ligand forms partial T-type stacking interactions with Phe331 and Phe330, but with high fluctuations. Interestingly, the azo part of the CAS ligand forms a weak parallel off-set stacking interaction with Phe330 and partial T-type stacking with Phe331, as reflected by the γ angle distribution.
Fig. 11(A) and (B) show that the Rcom values for residue Phe330 with the azo part of TAS lie around a mean value of 6 Å, along with a mean γ value of ≈100°, indicating that it is likely to have T-type stacking interactions. The Rcom values for other residues are distributed beyond 8 Å.
 | ||
| Fig. 11 Distribution of order parameters in the TAS–receptor complex (without the POPC membrane): panels (A) and (B) represent Rcom and γ, respectively, for the azo part, while panels (C) and (D) correspond to the sero part. | ||
Fig. 11(C) and (D) show that the Rcom values for residues Phe331 and Phe330 with the sero part of the TAS ligand are 5 Å and 6 Å, respectively, indicating their strong T-type stacking interactions with the TAS ligand. Trp327 (considering statistical fluctuations) is distributed around a mean value of 7.8 Å and therefore partially contributes to T-type stacking interactions. The Rcom values for Phe217 and Tyr359 are distributed around mean values of 9 Å and 11 Å, respectively, which are beyond the stacking-interaction range.
(II) Analysis for simulations with the POPC membrane. The qualitative stacking interaction analysis of the ligand–receptor complexes ‘within the POPC membrane’ environment further supports the findings (Fig. S6 of the ESI†). The TAS–receptor complex demonstrated enhanced stability, possessing narrow distributions of order parameters and indicating consistent T-type stacking interactions. In contrast, the CAS–receptor complex exhibited broader distributions of order parameters (with multiple peaks) for each main aromatic residue of interest, suggesting significant conformational fluctuations of both the CAS ligand and the receptor in the CAS–receptor complex. The presence of the POPC membrane emphasizes the stability differences between the MD systems while maintaining the same qualitative stacking interaction patterns observed in non-membrane systems.
In overall conclusion, the narrower distributions (with higher probability count) of the main aromatic residues of interest, Phe331, Phe330 and Trp327, in the TAS–receptor complex indicate that they have strong T-type stacking interactions with the TAS ligand, which is not the case in the CAS–receptor complex, since the CAS ligand possesses much broader distributions of order parameters.
In the context of the analysis for complexes ‘without the POPC membrane’ (Fig. S7, ESI†), the TAS ligand demonstrates a stronger stabilizing effect on the residues of interest compared to the CAS ligand. Notably, Trp327, Phe331 and Phe330 stand out with mean values of −1.117 kcal mol−1, −2.908 kcal mol−1 and −3.414 kcal mol−1, respectively. They exhibited narrower mean distributions (lower standard deviations), indicating a strong affinity in terms of the MM non-bonded interaction energy in the TAS–receptor complex. The CAS ligand demonstrated a weaker stabilizing effect on the residues of interest. Although residues such as Phe331, Trp327 and Phe217 have more negative mean values for the non-bonded energy with the CAS ligand, they exhibited broader distributions and higher standard deviations, indicating a weak affinity in terms of the MM non-bonded interaction energy in the CAS–receptor complex. Tyr359 and Phe217 showed average quantitative contributions close to zero in both complexes, but their stability is still greater in the TAS–receptor complex than in the CAS–receptor complex.
In the analysis for complexes ‘with the POPC membrane’ (Fig. S8, ESI†), the obtained MM non-bonded energy values for both the TAS and the CAS ligand resemble the findings described previously. Introducing the POPC membrane brought further instability in the interaction energy between the residues of interest and the CAS ligand in the CAS–receptor complex. However, the TAS ligand illustrated similar results for MM non-bonded interaction energy between the residues of interest in the TAS–receptor complex.
This statistical analysis improves the interpretation of the contribution of each residue of interest to the total binding affinity and interaction energy in all the MD systems. By considering both the mean interaction energies and their associated standard deviations, a more comprehensive and nuanced understanding of the ligand–receptor binding dynamics has been achieved.
4.5 Dynamic cross-correlation map
To indicate the conformational correlations between different parts of the protein, dynamic cross-correlation maps have been plotted for the TAS–receptor and CAS–receptor complexes and are shown in Fig. 12(A) and (B), respectively. The difference in dynamic cross-correlation has also been plotted and included in Fig. S9, ESI.† It is clearly visible from the figures that different parts of the CAS–receptor are more strongly correlated than those of the TAS-bound protein. This observation also indicates that the conformational fluctuations in the CAS–receptor are significantly higher than in the TAS–receptor complex. | ||
| Fig. 12 Dynamic cross correlation maps for (A) the TAS–receptor complex and (B) the CAS–receptor complex. | ||
Moreover, to demonstrate how much significant conformational change each ligand can induce in the receptor, a DCC map for the apo-receptor is also plotted and shown in the ESI (Fig. S10 in Section S7).† All these results indicate that unlike the TAS ligand (which induces very little conformational change in the receptor), the CAS ligand destabilizes the receptor significantly, resulting in high conformational fluctuations and correlations between different parts of the CAS–receptor with respect to the apo-receptor.
4.6 Dynamic effects of the POPC membrane on the simulation systems
Serotonin receptors are membrane-bound proteins, so it was essential to evaluate how these ligand–receptor complexes behave upon the introduction of a lipid membrane environment. However, the primary focus of this research still remained on how these azobenzene-fused-serotonin drugs bind to the 5-HT1B receptor. The RMSD, binding free energy and supporting analysis revealed that the presence of a POPC lipid membrane has greater effects on the stability and dynamics of these receptor–ligand complexes. In the presence of the POPC membrane, the TAS–receptor system showed greater stabilization compared to the CAS–receptor system.The stabilization in the TAS–receptor system can be attributed to the favourable interactions between the receptor and the POPC lipid membrane. This aided in maintaining conformation of the receptor in such a way that non-covalent interactions between the TAS ligand and receptor remained intact throughout the simulation. These POPC–receptor interactions likely involve hydrophobic bonding between the receptor's transmembrane domains and lipid acyl chains, as well as electrostatic complementarity with lipid headgroups.68 In contrast, the CAS–receptor complex demonstrated greater instability. This instability is likely due to the already known unfavourable interactions within the CAS–receptor complex, which were further impacted by the interactions between the receptor and the POPC lipid membrane. These findings highlight the necessity of including native-like lipid environments in computational drug design to better predict the in vivo behavior of receptor–ligand complexes.
5 Conclusion
In present times, many drug-discovery researchers are using photoswitchable drugs to control the activity of biological macromolecules. However, there is less focus on potential research to critically examine photoswitchable-drug–receptor interactions in different environments. We used binding free energy, receptor binding pocket volume and other supporting analyses to study the stability of photoswitchable azobenzene-fused-serotonin drugs and receptor binding. We also introduced a POPC lipid membrane to analyze the change a lipid membrane can bring to these photoswitchable drug-receptor interactions.The analyses for simulation systems without the POPC membrane, revealed that the trans-azobenzene-fused-serotonin (TAS) ligand shows a more stable binding affinity to the 5-HT1B receptor than the cis-azobenzene-fused-serotonin (CAS) ligand. These differences in binding affinities highlight the impact of the ligand structure, shape and conformations on the receptor functionality. However, the structural similarities between the CAS and TAS ligands lead to a small difference in enthalpy (ΔH) values between the TAS–receptor and CAS–receptor complexes (ΔΔH ≅ 5.5 kcal mol−1), which can be attributed to the similar structures of both ligands. The major change in binding free energies emerged due to the entropy contributions. The interaction entropy (σIE) values indicate that entropy significantly contributes to the dynamics of these protein–ligand complexes.
The binding-pocket volume analysis of the receptor indicates a tighter binding pocket with the TAS ligand, suggesting more energetically favourable interactions compared to those of the CAS ligand. This is further corroborated by qualitative stacking analyses and molecular mechanics non-bonded energy calculations for key residues of interest with the ligands in all systems. The stacking interactions of aromatic residues of interest, Trp327, Phe330 and Phe331, with the TAS ligand played a critical role in stabilizing the TAS–receptor complex. These stacking interactions play a major role in enhancing binding affinity and receptor activation.
In the presence of the lipid membrane, the TAS–receptor system demonstrated further stabilization, whereas the CAS–receptor system exhibited further destabilization. The CAS ligand in the CAS–receptor system experienced conformational fluctuations, resulting in an increase in entropy and, thereby, an unfavourable binding free energy of −5.06 kcal mol−1 for the CAS–receptor complex. The TAS–receptor complex exhibited an enhanced binding free energy of −28.79 kcal mol−1, further supporting its higher stability in the lipid membrane environment. This difference in binding free energy highlights the profound impact of bilayer membranes in protein–ligand interactions.
This study achieves two key objectives. Firstly, the photoswitchable azobenzene-fused-serotonin molecule offers a proposed drug-like molecule for controlling serotonin receptor activity upon light exposure, where the trans isomer actively binds and the cis isomer exhibits inactive binding to the serotonin receptor. Secondly, this study accentuates the critical role of the membrane environment in governing ligand–receptor dynamics. This highlights the importance of the impact of lipid membranes in drug design strategies. Although there are several further steps to go to obtain an effective photoswitchable drug for 5HT1B receptors, we hope that these results will inspire researchers to carry out further studies on developing azobenzene-based photoswitchable serotonin derivatives to control the functions of the 5-HT1B receptor.
A common question about using an azobenzene-based molecule as a photoswitchable drug would be whether the cis-to-trans thermal back transformation hinders the use of the molecule as a photo-regulatory drug. However, it should be noted that in the absence of light, the cis-to-trans thermal back conversion is, in general, very slow and the rate is very much dependent on the molecular environment. Therefore, within the timescales we are interested in, it is unlikely that the cis-to-trans back conversion will happen. However, this needs further detailed investigation on the energetics and rate of cis-to-trans thermal conversion in the protein environment using a hybrid quantum mechanics molecular mechanics (QM/MM) method, which will be considered in the future extension of the present work. Furthermore, detailed understanding about the timescale, mechanism and energetics of the photoisomerisation of TAS in the binding site of the 5HT1B receptor is beyond the scope of the present work and will be performed in future using a hybrid quantum mechanics/molecular mechanics method.
Abbreviations
| 5-HT | 5-Hydroxytryptamine |
| CAS | Cis-azobenzene-fused-serotonin |
| TAS | Trans-azobenzene-fused-serotonin |
| MD | Molecular dynamics |
| VMD | Virtual molecular dynamics |
| TM | Transmembrane region |
| MM/GBSA | Molecular mechanics/generalized born surface area |
Data availability
The data supporting this article have been included as part of the ESI.† Further data can be made available by the author upon request.Author contributions
ASD performed the simulations, analyzed the results and wrote the original draft manuscript. AV assisted ASD in different phases of simulation and analyses. PM conceptualized the project, reviewed the analysis results, and corrected the manuscript draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PM acknowledges the SERB and ANRF for funding through grant no. CRG/2023/005494.References
- S. Holderbach, L. Adam, B. Jayaram, R. C. Wade and G. Mukherjee, Front. Mol. Biosci., 2020, 7, 601065 CrossRef CAS
.
- H. Kim, H. Shim, A. Ranganath, S. He, G. Stevenson and J. E. Allen, Front. Pharmacol, 2025, 15, 1518875 CrossRef PubMed
- D. Greene, W. M. Botello-Smith, A. Follmer, L. Xiao, E. Lambros and R. Luo, J. Phys. Chem. B, 2016, 120, 12293–12304 CrossRef CAS PubMed
- P. Mondal and M. Huix-Rotllant, Phys. Chem. Chem. Phys., 2019, 21, 8874–8882 RSC
- X. Du, Y. Li, Y.-L. Xia, S.-M. Ai, J. Liang, P. Sang, X.-L. Ji and S.-Q. Liu, Int. J. Mol. Sci., 2016, 17, 144 CrossRef PubMed
- A. V. Sadybekov and V. Katritch, Nature, 2023, 616, 673–685 CrossRef CAS PubMed
- T. Saßmannshausen, H. Glover, M. Trabuco, W. Neidhart, R. Cheng, M. Hennig, C. Slavov, J. Standfuss and J. Wachtveitl, J. Am. Chem. Soc., 2024, 47, 32670–32677 CrossRef
- Y. Zhang, G. Zheng, T. Fu, J. Hong, F. Li, X. Yao, W. Xue and F. Zhu, Phys. Chem. Chem. Phys., 2020, 22, 5132–5144 RSC
- W. J. Pichler, Allergy, 2022, 77, 404–415 CrossRef CAS PubMed
- H. B. Schiöth and R. Fredriksson, Gen. Comp. Endocrinol., 2005, 142, 94–101 CrossRef
- W. K. Kroeze, D. J. Sheffler and B. L. Roth, J. Cell Sci., 2003, 116, 4867–4869 CrossRef CAS
- D. M. Rosenbaum, S. G. F. Rasmussen and B. K. Kobilka, Nature, 2009, 459, 356–363 CrossRef CAS PubMed
- H. R. Kim, N. M. Duc and K. Y. Chung, Biomol. Ther., 2018, 26, 101–108 CrossRef CAS
- S. Basith, M. Cui, S. J. Y. Macalino, J. Park, N. A. B. Clavio, S. Kang and S. Choi, Front. Pharmacol, 2018, 9, 128 CrossRef PubMed
- B. L. Roth, S. M. Hanizavareh and A. E. Blum, Psychopharmacology, 2004, 174, 17–24 CrossRef CAS PubMed
- M. Berger, J. A. Gray and B. L. Roth, Annu. Rev. Med., 2009, 60, 355–366 CrossRef CAS PubMed
- G. Vassart and S. Costagliola, Nat. Rev. Endocrinol., 2011, 7, 362–372 CrossRef CAS
- A. Thathiah and B. De Strooper, Nat. Rev. Neurosci., 2011, 12, 73–87 CrossRef CAS PubMed
- R. T. Dorsam and J. S. Gutkind, Nat. Rev. Cancer, 2007, 7, 79–94 CrossRef CAS
- P. Mondal, RSC Adv., 2020, 10, 37995–38003 RSC
- D. Wacker, S. Wang, J. D. McCorvy, R. M. Betz, A. J. Venkatakrishnan, A. Levit, K. Lansu, Z. L. Schools, T. Che, D. E. Nichols, B. K. Shoichet, R. O. Dror and B. L. Roth, Cell, 2017, 168, 377–389e12 CrossRef CAS PubMed
- C. Wang, Y. Jiang, J. Ma, H. Wu, D. Wacker, V. Katritch, G. W. Han, W. Liu, X.-P. Huang, E. Vardy, J. D. McCorvy, X. Gao, X. E. Zhou, K. Melcher, C. Zhang, F. Bai, H. Yang, L. Yang, H. Jiang, B. L. Roth, V. Cherezov, R. C. Stevens and H. E. Xu, Science, 2013, 340, 610–614 CrossRef CAS
- K. B. Fink and M. Göthert, Pharmacol. Rev., 2007, 59, 360–417 CrossRef CAS PubMed
- D. Hoyer and G. Martin, Neuropharmacology, 1997, 36, 419–428 CrossRef CAS PubMed
- W. Yin, X. E. Zhou, D. Yang, P. W. de Waal, M. Wang, A. Dai, X. Cai, C.-Y. Huang, P. Liu, X. Wang, Y. Yin, B. Liu, Y. Zhou, J. Wang, H. Liu, M. Caffrey, K. Melcher, Y. Xu, M.-W. Wang, H. E. Xu and Y. Jiang, Cell Discovery, 2018, 4, 12 CrossRef PubMed
- K. Sriram and P. A. Insel, Mol. Pharmacol., 2018, 93, 251–258 CrossRef CAS
- A. L. Pehrson, D. Roberts, A. Khawaja and R. McNair, Psychopharmacology, 2022, 239, 1823–1838 CrossRef CAS
- A. Verma and P. Mondal, Biophys. Chem., 2025, 318, 107386 CrossRef CAS
- C. R. Crecca and A. E. Roitberg, J. Phys. Chem. A, 2006, 110, 8188–8203 CrossRef CAS
- I. C. Haskologlu, E. Erdag, O. Uludag and N. Abacioglu, Chronobiol. Med., 2024, 6, 194–204 CrossRef
- O. Bozovic, B. Jankovic and P. Hamm, Nat. Rev. Chem., 2022, 6, 112–124 CrossRef CAS PubMed
- D. Samanta, J. Gemen, Z. Chu, Y. Diskin-Posner, L. J. W. Shimon and R. Klajn, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9379–9384 CrossRef CAS PubMed
- J. Volarić, J. Buter, A. M. Schulte, K.-O. van den Berg, E. Santamaría-Aranda, W. Szymanski and B. L. Feringa, J. Org. Chem., 2022, 87, 14319–14333 CrossRef
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC
- B. Cheng, J. Morstein, L. K. Ladefoged, J. B. Maesen, B. Schiøtt, S. Sinning and D. Trauner, ACS Chem. Neurosci., 2020, 11, 1231–1237 CrossRef CAS PubMed
- M. Gao, D. Kwaria, Y. Norikane and Y. Yue, Nat. Sci., 2023, 3, e220020 CrossRef CAS
- C. Matera, A. M. J. Gomila, N. Camarero, M. Libergoli, C. Soler and P. Gorostiza, J. Am. Chem. Soc., 2018, 140, 15764–15773 CrossRef CAS
- M. López-Cano, M. Scortichini, D. K. Tosh, V. Salmaso, T. Ko, G. Salort, I. Filgaira, C. Soler, D. Trauner, J. Hernando, K. A. Jacobson and F. Ciruela, J. Am. Chem. Soc., 2025, 147, 874–879 CrossRef PubMed
- A. Noev, N. Kuznetsov, G. Korenev, N. Morozova, Y. Vasil'ev, N. Suvorov, E. Diachkova, M. Usachev, A. Pankratov and M. Grin, Int. J. Mol. Sci., 2022, 23, 5352 CrossRef CAS PubMed
- P. Donthamsetti, D. B. Konrad, B. Hetzler, Z. Fu, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2021, 143, 8951–8956 CrossRef CAS
- M. I. Bahamonde, J. Taura, S. Paoletta, A. A. Gakh, S. Chakraborty, J. Hernando, V. Fernández-Dueñas, K. A. Jacobson, P. Gorostiza and F. Ciruela, Bioconjugate Chem., 2014, 25, 1847–1854 CrossRef CAS
- M. Frisch, F. Clemente, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino and G. Zhe, Gaussian 09, 2009 Search PubMed
- J. García-Nafría, R. Nehmé, P. C. Edwards and C. G. Tate, Nature, 2018, 558, 620–623 CrossRef
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS
- D. Provasi, M. C. Artacho, A. Negri, J. C. Mobarec and M. Filizola, PLoS Comput. Biol., 2011, 7, e1002193 CrossRef CAS PubMed
- M. C. Owen, W. Kulig, T. Rog, I. Vattulainen and B. Strodel, J. Membr. Biol., 2018, 251, 521–534 CrossRef CAS
- M. Kurki, A. Poso, P. Bartos and M. S. Miettinen, J. Chem. Inf. Model., 2022, 62, 6462–6474 CrossRef CAS
- S. Jo, T. Kim and W. Im, PLoS One, 2007, 2, e880 CrossRef PubMed
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS PubMed
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed
- L. Dong, X. Qu, Y. Zhao and B. Wang, ACS Omega, 2021, 6, 32938–32947 CrossRef CAS
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef
- B. R. Miller3rd, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. H. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef CAS PubMed
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS
- A. Onufriev, D. Bashford and D. A. Case, J. Phys. Chem. B, 2000, 104, 3712–3720 CrossRef CAS
- J. Weiser, P. S. Shenkin and W. C. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS
- L. Duan, X. Liu and J. Z. H. Zhang, J. Am. Chem. Soc., 2016, 138, 5722–5728 CrossRef CAS
- V. Ekberg and U. Ryde, J. Chem. Theory Comput., 2021, 17, 5379–5391 CrossRef CAS PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS
- J. Li, A. L. Jonsson, T. Beuming, J. C. Shelley and G. A. Voth, J. Am. Chem. Soc., 2013, 135, 8749–8759 CrossRef CAS
- J. D. Durrant, L. Votapka, J. Sørensen and R. E. Amaro, J. Chem. Theory Comput., 2014, 10, 5047–5056 CrossRef CAS PubMed
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed
- B. A. Reva, A. V. Finkelstein and J. Skolnick, Folding Des., 1998, 3, 141–147 CrossRef CAS
- P. Sarkar, S. Mozumder, A. Bej, S. Mukherjee, J. Sengupta and A. Chattopadhyay, Biophys. Rev., 2021, 13, 101–122 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03789a |
| This journal is © The Royal Society of Chemistry 2025 |