
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLC-MS/MS-guided separation of guaiacolane-type sesquiterpenes with anti-inflammatory activities from Chrysanthemum indicum†
Xinyue Li a,
Yanfen Caia,
Yunshuang Hua,
Limei Miua,
Yanyu Zhangd,
Shiyun Huanga,
Min Weie,
Qing Mae,
Zhongqiu Liuab,
Hua Zhou*bc and
Peng Wu*ab
a,
Yanfen Caia,
Yunshuang Hua,
Limei Miua,
Yanyu Zhangd,
Shiyun Huanga,
Min Weie,
Qing Mae,
Zhongqiu Liuab,
Hua Zhou*bc and
Peng Wu*ab
aGuangdong Provincial Key Laboratory of Translational Cancer Research of Chinese Medicines, Joint International Research Laboratory of Translational Cancer Research of Chinese Medicines, International Institute for Translational Chinese Medicine, School of Pharmaceutical Sciences, Guangzhou University of Chinese Medicine, Guangzhou 510006, P. R. China. E-mail: wupeng@gzucm.edu.cn; Fax: +86-20-39358071; Tel: +86-20-39358651
bChinese Medicine Guangdong Laboratory (Hengqin Laboratory), Guangdong-Macao In-Depth Cooperation Zone in Hengqin, 519000, P. R. China. E-mail: gutcmzhs@hotmail.com
cState Key Laboratory of Traditional Chinese Medicine Syndrome, State Key Laboratory of Dampness Syndrome of Chinese Medicine, Guangdong Provincial Hospital of Chinese Medicine, Guangdong Provincial Academy of Chinese Medical Sciences, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, 510006, P. R. China. Fax: +86-20-81867705; Tel: +86-20-39318475
dHenan Key Laboratory of Traditional Chinese Medicine Prescription and Syndrome Signaling & Henan International Joint Laboratory of Traditional Chinese Medicine Prescription and Syndrome Signaling, Henan University of Chinese Medicine, Zhengzhou 450046, P. R. China
eChina Resources Sanjiu Medical & Pharmaceutical Co., Ltd, Shenzhen 518110, P. R. China
First published on 22nd July 2025
Abstract
Four previously undescribed compounds, including three guaiacane-type sesquiterpenoids (1–3) and one oleanane-type triterpenoid (4), along with seven known sesquiterpenoids, were isolated from the aerial parts of Chrysanthemum indicum using LC-MS/MS-guided fractionation. Structures were elucidated by IR, UV, HR-ESI-MS, and 1D/2D NMR analyses, and their absolute configurations were determined by ECD calculations. The anti-inflammatory activity was evaluated in LPS-stimulated RAW264.7 cells with NF-κB translocation by high-content imaging (HCI). Compound 2 reduced NF-κB translocation (IC50 = 9.70 μM) without cytotoxicity at 20 μM. Compounds 8 and 9 showed potent activity (IC50 = 2.04 and 1.21 μM, respectively) and no cytotoxicity at 6.25 μM.
1. Introduction
Chrysanthemum indicum (Asteraceae), a perennial herb widely utilized in traditional Chinese medicine, has been historically prescribed for treating inflammatory disorders, hypertension, and respiratory diseases.1,2 Modern pharmacological studies have validated its ethnomedicinal applications, revealing that C. indicum extracts possess multifaceted bioactivities, including anti-inflammatory,3 antioxidant,4 antimicrobial,5 and anticancer properties.1,2,5 Notably, its anti-inflammatory efficacy is attributed to the suppression of pro-inflammatory cytokines and modulation of the nuclear factor κB (NF-κB) signaling pathway.6 These pharmacological effects are predominantly mediated by bioactive constituents such as sesquiterpenes,7–9 flavonoids,10 and phenolic acids.11 Among these, guaiane-type sesquiterpenes have garnered significant attention due to their distinctive chemical scaffolds and potent anti-inflammatory efficacy.12–15As previous reported,15 LC-MS/MS is effective in isolating guaianolides from C. indicum extracts. In this study, the 95% ethanol extract of C. indicum was fractionated using a petroleum ether (PE)/ethyl acetate (EA) gradient, LC-MS analysis revealed varying distributions of guaiane-type sesquiterpenes, with higher abundance detected in the PE/EA (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1) fractions. Additionally, eleven purified compounds were isolated from the 95% ethanol extract through gradient PE/EA fractionation, including eight guaianolide-type sesquiterpenoids (1–3 being undescribed, and 5–9 being known), two caryolane-type sesquiterpenoids (10 and 11), and a previously undescribed triterpenoid (4) (Fig. 1). This contribution comprehensively details their isolation protocols, structural elucidation, and preliminary evaluation of NF-κB inhibitory activity, revealing promising anti-inflammatory potential that aligns with the plant's traditional uses.
:1 and 1:1) fractions. Additionally, eleven purified compounds were isolated from the 95% ethanol extract through gradient PE/EA fractionation, including eight guaianolide-type sesquiterpenoids (1–3 being undescribed, and 5–9 being known), two caryolane-type sesquiterpenoids (10 and 11), and a previously undescribed triterpenoid (4) (Fig. 1). This contribution comprehensively details their isolation protocols, structural elucidation, and preliminary evaluation of NF-κB inhibitory activity, revealing promising anti-inflammatory potential that aligns with the plant's traditional uses.
 | ||
| Fig. 1 Structures of compounds 1–11. | ||
2. Results and discussion
2.1. LC-MS guided isolation
After LC-MS analysis of the extracts using different gradients of PE/EA solvent systems, the extraction ion flow chromatogram (EIC, Fig. 2) with molecular weight of 229.12 (ref. 15) was extracted from their total ion flow chromatogram (TIC). Due to the molecular structure of exact mass of m/z 229.12 (Fig. 3) as a general guaiacolane-type sesquiterpene cleavage products, whose associated quasimolecular ion peaks (m/z 210.1039, 211.1117, 228.1145, 228.1323, 229.1223) were extracted in pure PE, PE/EA (2:1), PE/EA (1:1) as well as H2O phase. Obviously, the extracts of PE/EA (2:1) and PE/EA (1:1) showed higher abundance of relevant ions, while the pure PE and H2O phase extracted lower abundance of ions, and the relevant ion peaks of the pure EA site were not detected, which were presumed to contain lower content of guaiacolane-type sesquiterpenoids, thus provide guidance for the subsequent isolation of the guaiacolane-type sesquiterpenoids (Fig. 4).
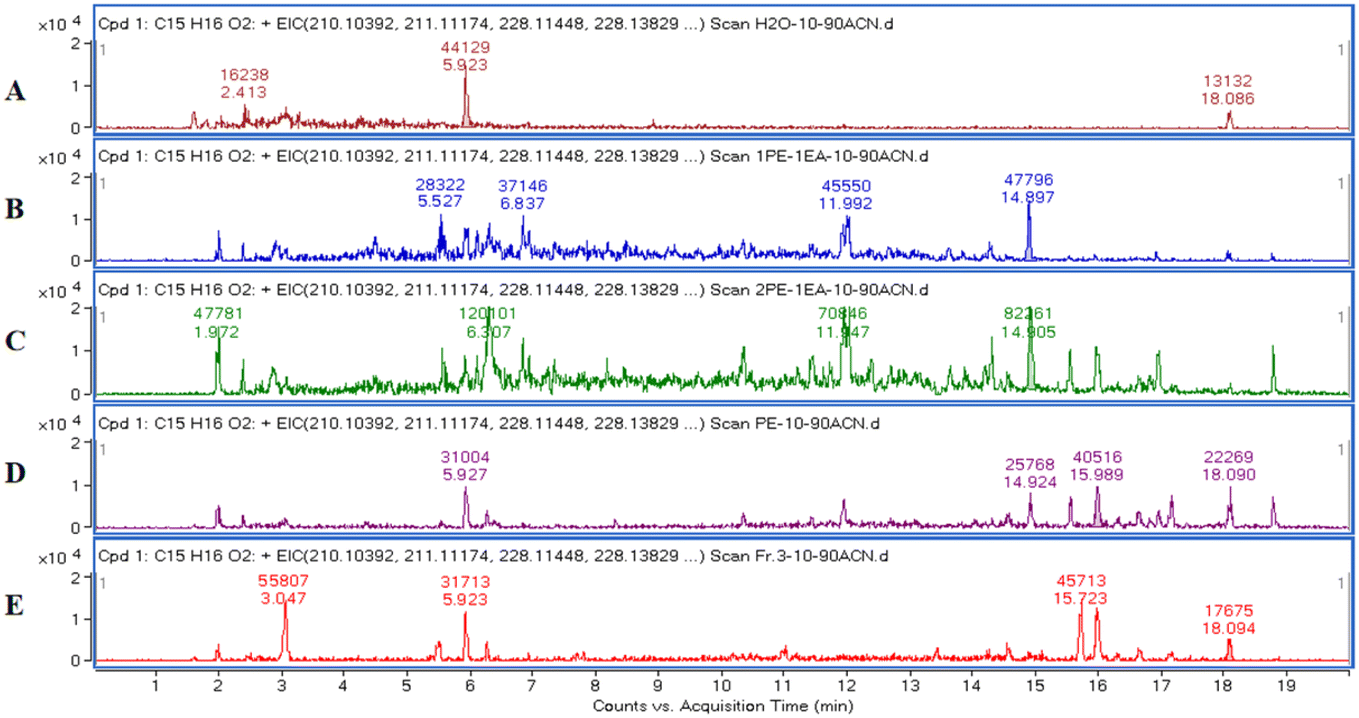 | ||
| Fig. 2 EIC of m/z 229.12, representing guaiacolane-type sesquiterpene cleavage products. Samples: H2O phase (A); PE/EA (1:1, v/v) extract (B); PE/EA (2:1, v/v) extract (C); pure PE extract (D); fraction 3 (in methanol) (E). | ||
 | ||
| Fig. 3 Histogram (right) and structure (left) of m/z 229.1223 in the EIC from the PE/EA (2:1) extract (A). Histogram (right) and structure (left) of m/z 347.1831 ([M + H]+, compound 9) in the EIC from fraction 3 (B). | ||
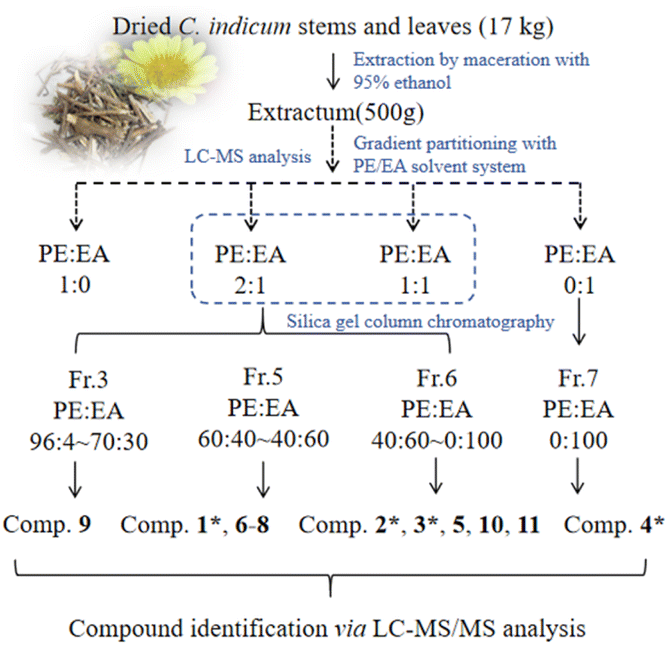 | ||
| Fig. 4 LC-MS-guided isolation workflow. | ||
2.2. Structural elucidation
8-Senecioylchrysanolide D (1), isolated as a colorless oil, displayed a sodium adduct ion at m/z 615.2923 [M + Na]+ (calcd for C35H44O8Na, 615.2928) in HR-ESI-MS analysis, confirming the molecular formula C35H44O8 (Ω = 14). The IR spectrum revealed the presence of hydroxyl (3516 cm−1), carbonyl (1759 cm−1), and olefinic (1653, 1446 cm−1) functionalities. The 1H-NMR and 13C NMR spectra (Table 1) exhibited 35 carbon signals, including four methyl singlets at δH 1.21 (3H, s, H-14), δH 1.29 (3H, s, H-14′), δH 1.45 (3H, s, H-15′) and δH 1.89 (3H, s H-15), three oxygenated methines (δC/H 69.2/5.51, 79.2/3.95 and 79.2/4.09), two oxygenated tertiary carbons (δC 73.0 and 73.6), an exocyclic double bond (δH 5.31, d, J = 3.6 Hz, 6.03, d, J = 3.6 Hz; δC 118.4, 141.4), two carbonyl carbons and (δC 170.4 and 178.2) and characteristic signals for a senecioyl moiety. The senecioyl moiety shows two methyl groups at δH 2.08 (3H, d, J = 1.2 Hz, CH3-4′′) and δH 1.88 (3H, s, CH3-5′′), along with an olefinic proton signal at δH 5.64 (1H, m, H-2′′), in conjunction with 13C-NMR signals at δC 165.4 (C-1′′), 115.2 (C-2′′), 161.7 (C-3′′), 20.6 (C-4′′), and 27.8 (C-5′′). The NMR data were nearly identical to those of chrysanolide D,16 except for an additional senecioyl group at C-8, as confirmed by the HMBC correlation (Fig. 5) between δH 5.51 (H-8) and δC 165.4 (C-1′′). The 1H–1H COSY spectrum revealed four isolated spin-coupling systems of H-2/H-3, H-5/H-6/H-7/H-8/H2-9, H-2′/H-3′ and H-5′/H-6′/H-7′/H2-8′/H2-9′. Furthermore, HMBC (H-15′/C-11; H-13a, H-13b/C-1′, C-4′, C-10′) correlations suggested that the two guaianolide units are connected through C-13 to C-1′ and C-11 to C-4′. These spectral features suggested that compound 1 is a guaianolide-type sesquiterpene dimer.| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a δ in ppm; J in Hz; in CDCl3. 1H-NMR data (δ) were measured at 400 MHz; 13C-NMR data (δ) were measured at 100 MHz. | ||||||
| 1 | 2.57 (1H, m) | 54.4 | 2.86 (1H, m) | 56.3 | 2.53 (1H, m) | 58.8 |
| 2 | 2.15 (1H, m); 2.04 (1H, m) | 33.4 | — | 207.5 | 4.26 (1H, d, 6.0) | 85.9 |
| 2-OCH | 3.32 (3H, s) | 56.2 | ||||
| 3 | 5.49 (1H, m) | 125.8 | 5.93 (1H, overlap) | 134.6 | 5.72 (1H, m) | 126.7 |
| 4 | — | 145.0 | — | 177.4 | — | 147.1 |
| 5 | 2.74 (1H, m) | 54.8 | 3.02 (1H, dd, 9.6, 6.4) | 54.8 | 3.07 (1H, dd, 9.6, 9.2) | 54.0 |
| 6 | 3.95 (1H, dd, 10.4, 10.0) | 79.2 | 3.81 (1H, dd, 10.0, 9.6) | 80.8 | 3.93 (1H, dd, 10.8, 9.6) | 81.5 |
| 7 | 3.43 (1H, dd, 10.0, 8.4) | 48.3 | 3.12 (1H, dd, 9.6, 9.6) | 47.5 | 3.80 (1H, m) | 47.5 |
| 8 | 5.51 (1H, m) | 69.2 | 5.41 (1H, m) | 70.3 | 5.22 (1H, m) | 72.1 |
| 9 | 2.24 (1H, m); 1.98 (1H, m) | 38.4 | 1.76 (2H, m) | 36.2 | 2.33 (1H, dd, 5.2, 16.0), 1.98 (1H, d, 4.4) | 43.4 |
| 10 | — | 73.6 | 2.46 (1H, m) | 26.9 | — | 73.1 |
| 11 | — | 58.6 | — | 59.8 | — | 137.8 |
| 12 | — | 178.2 | — | 177.7 | — | 169.5 |
| 13 | 2.37 (1H, d, 12.0), 1.48 (1H, m) | 37.3 | 2.34 (1H, m), 1.52 (1H, m) | 37.9 | 6.22 (1H, m), 5.58 (1H, d, 3.2) | 122.4 |
| 14 | 1.21 (3H, s) | 33.7 | 1.37 (3H, d, 7.2) | 21.5 | 1.30 (3H, s) | 31.8 |
| 15 | 1.89 (3H, s) | 18.5 | 2.27 (3H, s) | 20.7 | 1.95 (3H, s) | 18.1 |
| 1′ | — | 64.6 | — | 65.0 | — | 167.0 |
| 2′ | 6.22 (1H, d, 5.6) | 141.0 | 6.08 (1H, d, 5.2) | 139.6 | — | 127.2 |
| 3′ | 5.87 (1H, d, 5.6) | 133.6 | 5.94 (1H, m) | 131.6 | 6.20 (1H, m) | 140.3 |
| 4′ | — | 57.5 | — | 56.6 | 2.04 (1H, dd, 1.6, 7.2) | 16.1 |
| 5′ | 1.92 (1H, m) | 65.8 | 2.15 (1H, m) | 66.3 | 1.93 (1H, m) | 20.8 |
| 6′ | 4.09 (1H, dd, 10.0, 9.6) | 79.2 | 4.07 (1H, dd, 9.6, 9.6) | 79.4 | ||
| 7′ | 3.01 (1H, m) | 43.1 | 3.01 (1H, dd, 9.6, 6.4) | 43.2 | ||
| 8′ | 2.20 (1H, d, 4.4), 1.44 (1H, m) | 23.9 | 2.17 (1H, m), 1.44 (1H, m) | 23.7 | ||
| 9′ | 1.81 (2H, m) | 35.0 | 1.81 (2H, m) | 34.9 | ||
| 10′ | — | 73.0 | — | 72.7 | ||
| 11′ | — | 141.4 | — | 141.0 | ||
| 12′ | — | 170.4 | — | 170.3 | ||
| 13′ | 6.03 (1H, d, 3.6), 5.31 (1H, d, 3.6) | 118.4 | 6.05 (1H, d, 3.2), 5.33 (1H, d, 3.2) | 118.7 | ||
| 14′ | 1.29 (3H, s) | 29.9 | 1.29 (3H, s) | 29.9 | ||
| 15′ | 1.45 (3H, s) | 15.5 | 1.48 (3H, s) | 15.6 | ||
| 1′′ | — | 165.4 | — | 166.7 | ||
| 2′′ | 5.64 (1H, m) | 115.5 | — | 127.4 | ||
| 3′′ | — | 161.7 | 6.12 (1H, m) | 141.6 | ||
| 4′′ | 2.08 (1H, d, 1.2) | 20.6 | 1.93 (3H, dd, 1.2, 7.6) | 16.2 | ||
| 5′′ | 1.88 (1H, d, 1.2) | 27.8 | 1.85 (3H, s) | 20.6 | ||
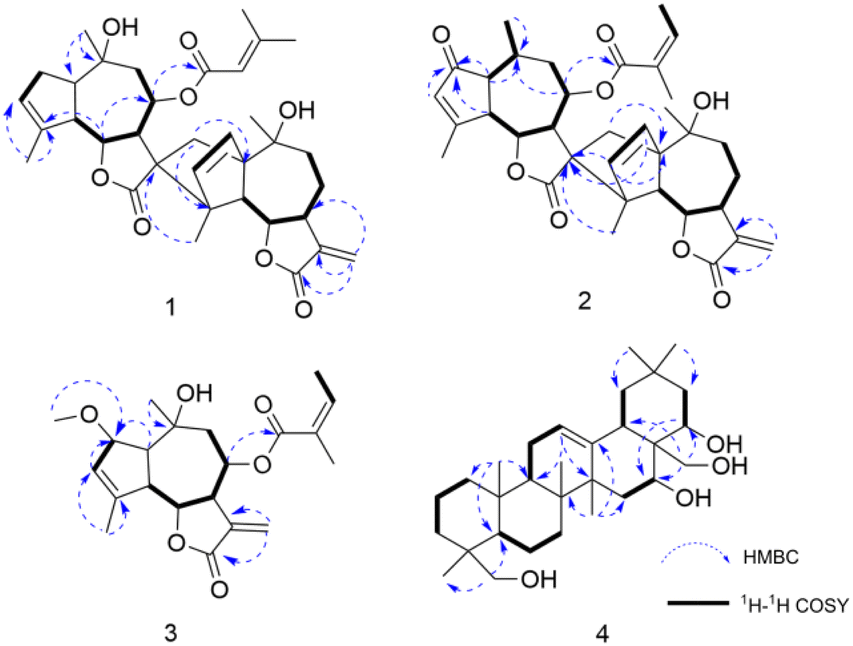 | ||
| Fig. 5 Selected HMBC (arrow) and 1H–1H COSY (bold) correlations of compounds 1–4. | ||
The relative configuration of 1 was established through a combination of NOESY correlations, biogenetic pathway and J-based configurational analysis. As guaianolide sesquiterpene lactones characteristically contain both cyclopentane and α-methylene-γ-butenolide moieties, their dimers are primarily formed via enzymatically catalyzed Diels–Alder reactions.17 Following the Geissman rule, the H-7 proton was determined to be α-oriented.12,18 NOESY (Fig. 6) experiment revealed key spatial relationships: H-7/H-5, H-5/H-1, H-5′/H-7′, H-7/H-5′, H-8/H-6, H-8/H2-13, H-6/H3-14′, H-6/H3-15′, H-2′/H-6′, H3-15′/H-6′, H-6′/H3-15, H3-14/H-9a and H-9a/H3-15′. The coupling constant (JH-6/H-7 = 10.0 Hz) indicated that these protons occupy an axial position on opposite sides of the molecule; thus, H-6 were deduced to have a β-orientation. The observable NOESY correlation of H-7/H-5, H-5/H-1, H-5′/H-7′ and H-7/H-5′, supported their cofacial spatial arrangement, confirming their α-orientation. In contrast, the NOESY cross-peaks of H-8/H-6, H-8/H2-13, H-6/H3-14′, H-6/H3-15′, H-2′/H-6′, H-15′/H-6′, H-6′/H3-15, H3-14/H-9a and H-9a/H3-15′ established the β-orientation of H-6, H-8, H-6′, CH2-13, CH3-14, CH3-14′, CH3-15 and CH3-15′. The β-orientation of the CH2-13, providing key evidence for determining the absolute configuration of the spiro center at C-11. Similarly, the β-orientation of H-2′ was used to assign the absolute configuration of C-1′.
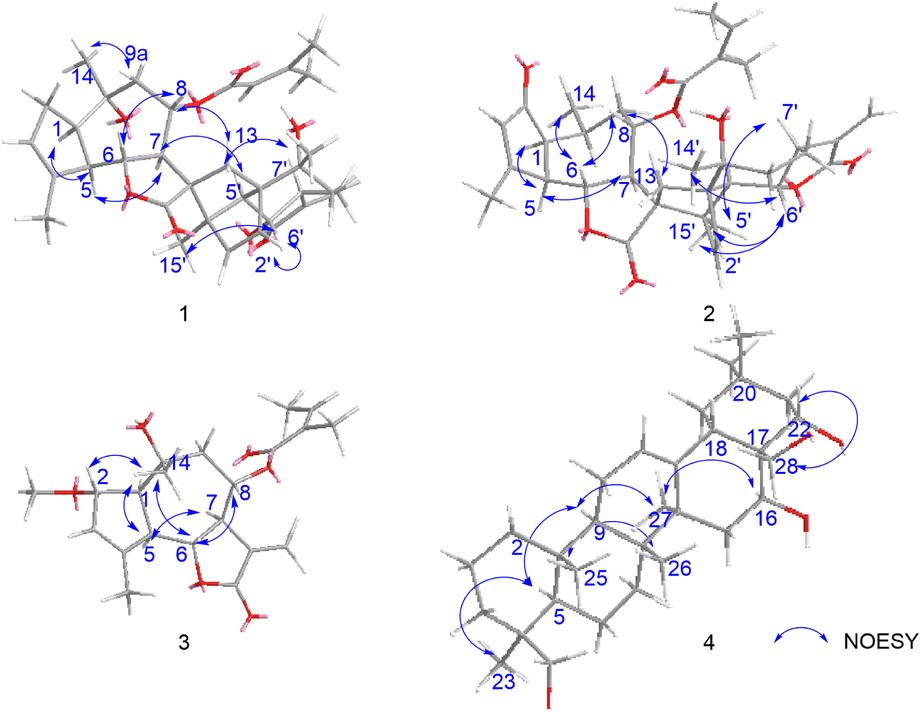 | ||
| Fig. 6 Selected NOESY (arrow) correlations of compounds 1–4. | ||
The absolute configuration of compound 1 was determined through comparison of its experimental electronic circular dichroism (ECD) spectrum (Fig. 7) with the theoretically calculated ECD spectrum as (1R,5R,6R,7R,8S,10R,11R,1′R,4′R,5′S,6′S,7′S,10′R). Accordingly, the structure of compound 1 was unequivocally established and named 8-senecioylchrysanolide D.
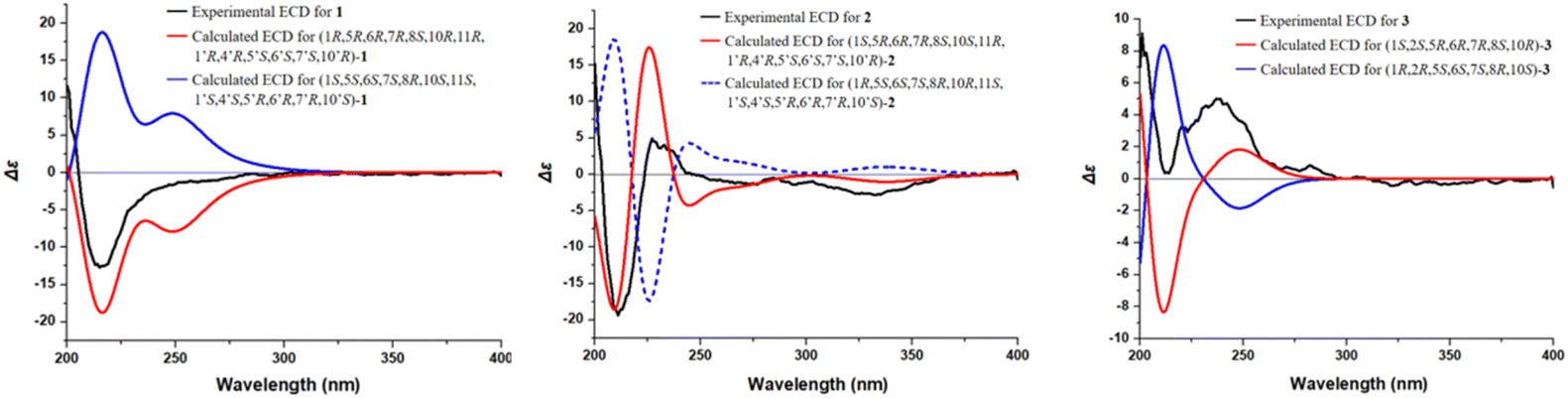 | ||
| Fig. 7 ECD spectra of compounds 1–3. | ||
Chrysanolide J (2), isolated as a colorless oil, its molecular formula is C35H42O8 (Ω = 15) according to HR-ESI-MS (m/z 613.2824 [M + Na]+, calcd for C35H42O8Na, 613.2822). The IR spectrum revealed the presence of hydroxyl (3488 cm−1), carbonyl (1749 cm−1), and olefinic (1454 cm−1) functional groups. Comparative analysis with the known compound Chrysanolide C17 revealed similar NMR data, with the primary differences being the presence of a carbonyl signal at δC 207.5 (C-2) and the absence of a hydroxyl group at C-10 in compound 2. The 1H-NMR spectrum (Table 1) exhibited signals of an angeloyl moiety for two methyl groups at δH 1.93 (3H, dd, J = 1.2, 7.6 Hz, CH3-4′′) and δH 1.85 (3H, s, CH3-5′′), along with an olefinic proton signal at δH 6.11 (1H, m, H-3′′). The 1H–1H COSY spectrum revealed five isolated spin-coupling systems of H-5/H-6/H-7/H-8/H2-9, H-1/H-10/H3-14, H-2′/H-3′, H-5′/H-6′/H-7′/H2-8′/H2-9′ and H-3′′/H-4′′. HMBC (H-1, H-3, H-5/C-2) correlations (Fig. 5) confirmed the presence of a carbonyl group at C-2. NOESY (Fig. 6) experiments revealed key spatial relationships: H-5/H-1, H-5/H-7, H-6/H-8, H-6/H3-14, H-8/H2-13, H2-13/H3-14′, H-2′/H-6′, H3-15′/H-6′, H3-14′/H3-15′, H-7/H-5′, and H-5′/H-7′. Based on an accepted principle that H-7 is generally α-oriented in natural guaianolides, and three diagnostic coupling constants (J = 9.6 Hz for H-6/H-7, H-5/H-6, and H-6′/H-7′) revealed trans-diaxial for these three pairs of protons. Collectively, these data established the α-orientation of H-1, H-5, H-7, H-5′, and H-7′, while H-6, H-8, CH2-13, CH3-14, H-2′, H-6′, CH3-14′, and CH3-15′ were determined to be β-oriented. The absolute configuration of compound 2 was determined through comparison of its experimental ECD spectrum (Fig. 7) with the theoretically calculated ECD spectrum as (1S,5R,6R,7R,8S,10S,11R,1′R,4′R,5′S,6′S,7′S,10′R).
8-Angeloyl-2-methoxy-10-hydroxy-3,11(13)-guaiadien-12,6-olide (3), isolated as a colorless oil, its molecular formula is C21H28O6 (Ω = 8) according to HR-ESI-MS (m/z 399.1764 [M + Na]+, calcd for C21H28O6Na, 399.1770). IR spectroscopy indicated the presence of hydroxyl (3510 cm−1), carbonyl (1741 cm−1), and double bond (1454 cm−1) functional groups. Comparison with the known compound 8-acetoxy-2-methoxy-10-hydroxy-3,11(13)-guaiadien-12,6-olide,19 showed similar spectroscopic data, with the main difference being the substituent at C-8. The 1H–1H COSY spectrum revealed three isolated spin-coupling systems of H-2/H-3 H-5/H-6/H-7/H-8/H2-9 and H-3′′/H-4′′. HMBC spectra (Fig. 5) showed that δH 5.22 (H-8) was remotely correlated with δC 167.0 (C-1′), suggesting that an angelica acyl group was attached at the C-8. NOESY correlations (Fig. 6) (H-5/H-1, H-5/H-7, H-6/H-8, H-14/H-2, H-14/H-6) suggested that H-1, H-5, and H-7 are α-oriented, while H-2, H-6, H-8, and H-14 are β-oriented. The absolute configuration of compound 3 was determined to be (1S,2S,5R,6R,7R,8S,10R) (Fig. 7).
16,22,23,28-Tetrahydroxyolean-12-ene (4) was isolated as a white powder, HR-ESI-MS analysis showed a molecular ion peak at m/z 475.3864 [M]+ (calcd for C30H50O4, 475.3857), thus the molecular formula was deduced to be C30H50O4 (Ω = 6). IR spectroscopy indicated the presence of hydroxyl (3425 cm−1) and double bond (1689 cm−1, 1460 cm−1) functional groups. The 1H-NMR spectrum (Table 2) revealed six methyl proton signals(δH 0.93, 0.95, 0.98, 1.03, 1.16, and 1.43), two oxygenated methylene signals (δH 3.66, H-23a and δH 3.44, H-23b, J = 11.2 Hz; δH 3.61, H-28a and δH 3.32, H-28b, J = 10.8 Hz), two oxygenated methine proton signals (δH 4.03, 4.67), while a double bond proton signal was observed at δH 5.29 (1H, brs) in the downfield region. In the 13C-NMR spectrum (Table 2), four oxygenated carbon signals were observed at δC 67.2, 68.3, 76.7, and 73.2, while a pair of double bond carbon signals appeared at δC 122.8 and 142.7. These data suggested that compound 4 is a Δ12-oleanene-type triterpenoid. Compound 4 showed similar spectroscopic data to the known compound gymnemanol,20 with the main difference being the absence of a hydroxyl group at C-3. In the 1H–1H COSY spectrum, the following correlations (Fig. 5) were observed: δH 5.29 (H-12) with δH 1.94 (H-11); δH 2.04/1.45 (H-15) with δH 4.67 (H-16); and δH 1.77/1.58 (H-21) with δH 4.03 (H-22). HMBC (H-23/C-24, H-23/C-5, H-28/C-16, H-28/C-18, H-28/C-22, H-22/C-16) correlations (Fig. 5) indicated hydroxyl substitutions at C-16 and C-22, and the formation of –CH2OH structures at C-24 and C-28 due to hydroxylation of the angular methyl groups. NOESY correlations (Fig. 6) (H-5/H-9, H-5/H3-23, H-9/H3-27, H-16/H3-27) suggested that H-5, H-9, H-16, CH3-23, and CH3-27 are α-oriented, while correlations between H-28/H-22 and H3-25/H3-26 indicated that H-22, CH3-25, and CH3-26 are β-oriented.
| No. | δH | δC |
|---|---|---|
| a δ in ppm; J in Hz; in CDCl3. 1H-NMR data (δ) were measured at 400 MHz; 13C-NMR data (δ) were measured at 100 MHz. | ||
| 1 | 1.94 (1H, m), 1.40 (1H, m) | 39.1 |
| 2 | 2.63 (1H, m), 2.29 (1H, m) | 35.4 |
| 3 | 1.25 (2H, m) | 29.8 |
| 4 | — | 52.6 |
| 5 | 1.64 (1H, m) | 49.3 |
| 6 | 1.45 (1H, m), 1.55 (1H, m) | 19.3 |
| 7 | 1.67 (1H, m), 1.42 (1H, m) | 32.3 |
| 8 | — | 39.9 |
| 9 | 1.77 (1H, m) | 45.9 |
| 10 | — | 36.6 |
| 11 | 1.94 (2H, m) | 23.7 |
| 12 | 5.29, brs | 122.8 |
| 13 | — | 142.7 |
| 14 | — | 41.9 |
| 15 | 2.04(1H, m), 1.45(1H, m) | 33.8 |
| 16 | 4.67, brs | 68.3 |
| 17 | — | 44.0 |
| 18 | 1.94 (1H, m) | 42.5 |
| 19 | 2.27 (1H, m), 1.08 (1H, m) | 47.4 |
| 20 | — | 31.5 |
| 21 | 1.77 (1H, m),1.58 (1H, m) | 45.9 |
| 22 | 4.03, dd, 12.4, 6.0 | 76.7 |
| 23 | 3.66(1H, d, 11.2), 3.44(1H, d, 11.2) | 67.2 |
| 24 | 1.03(3H, s) | 17 |
| 25 | 1.16 (3H, s) | 15.6 |
| 26 | 0.98 (3H, s) | 17.0 |
| 27 | 1.43 (3H, s) | 27.1 |
| 28 | 3.61 (1H, d, 10.8), 3.32 (1H, d, 10.8) | 73.2 |
| 29 | 0.93 (3H, s) | 33.2 |
| 30 | 0.95 (3H, s) | 24.9 |
Apart from four previously undescribed compounds, the other known compounds were isolated and identified as handelin (5),21 8-tigloylchrysanolide D (6),16 chrysanolide C (7),17 cumambrin A (8),21 angeloylcumambrin B (9),22 caryolane-1β,9β-diol (10),23 (–)-clovane-2,9-diol (11) (Fig. 1).24
2.3. LC-MS/MS validation of isolated compounds
To verify the presence of both previously undescribed compounds and known compounds in the original plant material and their enrichment in fractions, LC-MS/MS analysis was conducted. Multiple structural isomers with the molecular formula C35H44O8 (m/z 590.30) were identified in C. indicum extracts, notably, EIC (Fig. 8) analysis of C35H44O8 revealed consistent co-elution of compounds 1 and 6 in both crude extract and fraction 5.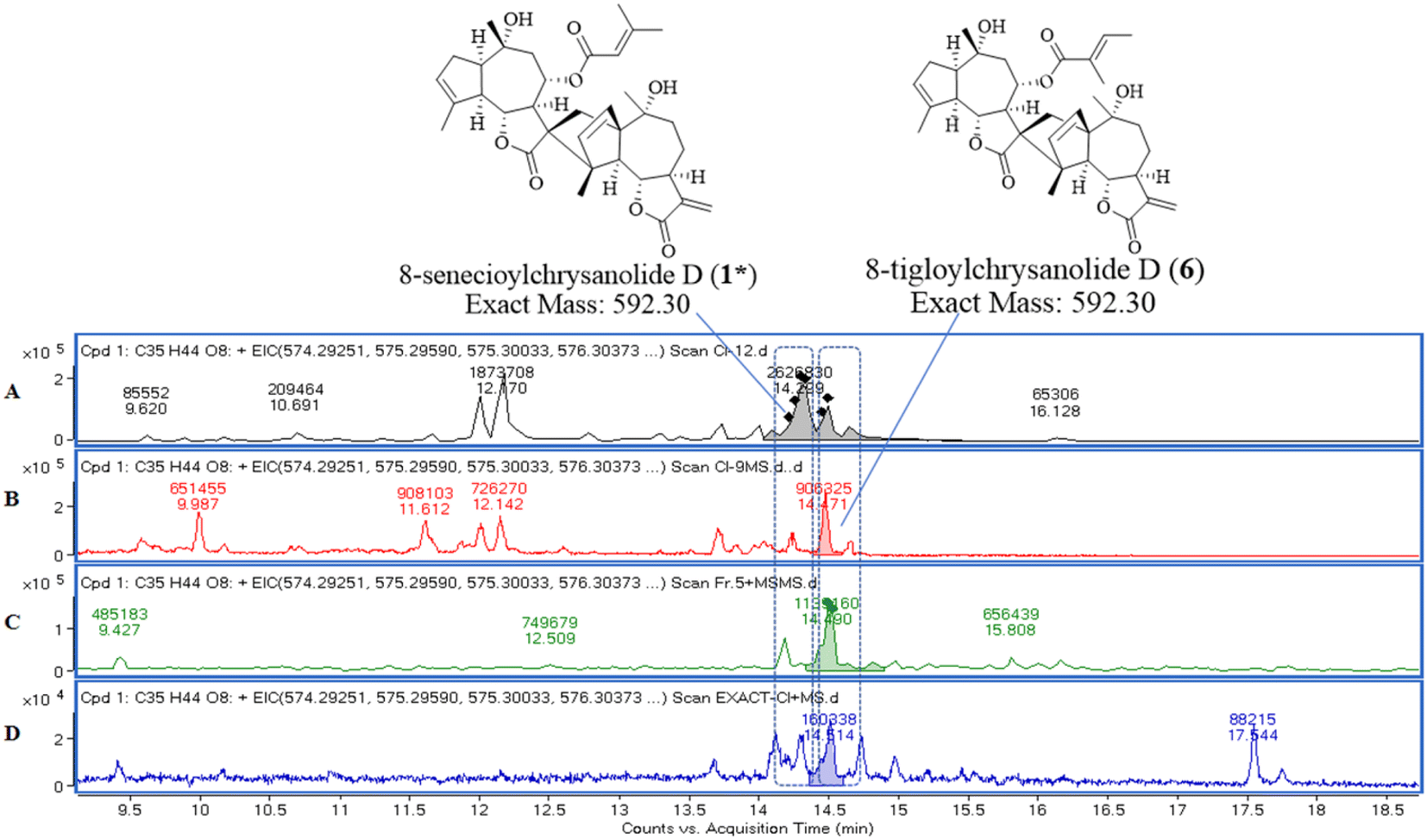 | ||
| Fig. 8 EIC about molecular formula of C35H44O8 (exact mass of m/z 590.30). Samples: compound 1 (A); compound 6 (B); fraction 5 (C); C. indicum crude extract (D). | ||
2.4. Biological activity
 | ||
| Fig. 9 Inhibitory effect of compounds 1–11 from C. indicum on NF-κB nuclear translocation in LPS-induced RAW264.7 cells. RAW264.7 cells were seeded at 10000 cells per well in 96-well plate overnight, and then treated with 10 μM of 11 compounds for 40 minutes followed by 200 ng mL−1 LPS stimulation for 40 minutes. The NF-κB nuclear translocation (green) was detected by HCI. The screening result of compound 1–11 on NF-κB nuclear translocation in LPS-induced RAW264.7 cells was detected by HCI (A). The NF-κB translocation was normalized to the LPS group data (100%) and control data (0%). The results are presented as means ± S.E, n = 3. The presentative HCI images of LPS-induced RAW264.7 cells treated without or with compound 2, 8 and 9 (B). Scale bar: 50 μm. ^^P < 0.01 vs. control alone, **P < 0.01 vs. LPS alone. | ||
 | ||
| Fig. 10 The cell cytotoxicity of compounds 2, 8 and 9 in with/without LPS-stimulated RAW264.7 cells by MTT and HCI. RAW264.7 cells were seeded at 8000 cells per well in 96-well plates overnight, and then treated with different concentrations of compounds 2, 8 and 9 for 1 h followed with or without 200 ng mL−1 LPS for 18 h. Cell viability of compounds in with/without LPS-stimulated RAW264.7 cells was analyzed by MTT (A–C) and HCI (D–F). The cell viability of control group was set to 100% and the results were presented as means ± S.E, n = 3. (G–I) The presentative cell cytotoxicity HCI images of compounds 2, 8 and 9. Scale bar: 100 μm. **P < 0.01, ***P < 0.001 vs. LPS alone. | ||
 | ||
| Fig. 11 The IC50 values of compounds 2, 8 and 9 on NF-κB nuclear translocation in LPS-stimulated RAW264.7 cells (A–C). RAW264.7 cells were seeded at 8000 cells per well in 96-well plates overnight, and then treated with or without compounds 2, 8 and 9 for 1 h followed with or without 200 ng mL−1 LPS for 40 minutes. NF-κB nuclear translocation was analysed by HCI. The NF-κB nuclear translocation of LPS group was set to 100% and control data was set 0%. The results are presented as means ± S.E, n = 3. | ||
3. Experimental
3.1. Plant material
The aerial parts of C. indicum were collected in Yangxin Chrysanthemum indicum L. Cultivation Base, Hubei, China in 2019. The samples (Batch no. 20190719) were identified by Min Wei, an engineer of China Resources Sanjiu Pharmaceutical Company Limited, and preserved in the International Institute of Translational Medicine of Traditional Chinese Medicine, Guangzhou University of Traditional Chinese Medicine, Guangzhou, China.3.2. LC-MS analysis
:0, 2:1, 1:1, 0:1) in order. After recycling the solvent, the products were then dosed to 10 mL with methanol. And all of them were filtered with 0.22 μm filter membrane as the samples for LC-MS analysis.3.3. General experimental procedures of isolation and structure elucidation
UV spectra were measured using a JASCO V-730 UV-vis spectrophotometer (Jasco, Tokyo, Japan), IR spectra were obtained using a JASCO FT/IR-4600 spectrophotometer (Jasco, Tokyo, Japan), and HR-ESI-MS data were acquired using an Agilent 6540 UPLC-Q-TOF-MS (Agilent, California, USA). NMR data were acquired using a Bruker AV-400 MHz NMR spectrophotometer (Bruker, Faellanden, Switzerland) in CDCl3 (Sigma-Aldrich, USA), with tetramethylsilane as the internal standard. ECD spectra were available on a Chirascan circular dichroism (Applied Photophysics, UK). Semi-preparative HPLC separations were carried out on an HPLC (Shimadzu, Japan) on an Inertsil PREP-ODS (10 μm, 20 × 250 mm) column (Gl Sciences Inc., Eindhoven, the Netherlands), and analysed and purified by HPLC on a COSMOSIL (5 μm, 4.6 × 150 mm) column (Nacalai Tesque, Japan). Silica gel (100–200, 200–300, 300–400 mesh, Qingdao Ocean Chemical Factory, China), Sephadex LH-20gel (25–100 μm, Fluka, Sweden), MCI (Mitsubishi Chemical Corporation, Japan), and ODS (YMC Corporation, Kyoto, Japan) were used for the column chromatography (CC) analysis. Silica gel thin-layer plates (GF254, Qingdao Ocean Chemical Factory, Qingdao, China), vanillin (Tianjin Komeo Company, Tianjin, China), and concentrated sulfuric acid (AR, Shanghai energy Chemical Co., Ltd, Shanghai, China) were used for thin-layer chromatography (TLC) analysis. Solvents used for analysis and preparation on HPLC were of HPLC grade (Merck, USA), and the others used for CC were of AR grade (Shanghai energy chemical Co.)3.4. Extraction and isolation
After crushing the dried C. indicum stems and leaves (17 kg), the crude powder (40–60 mesh) obtained was extracted by maceration with 95% ethanol (50 L in total) at room temperature (3 days × 3 times). The combined extracts were concentrated under reduced pressure until they were flavorless without alcohol to obtain a crude extract (500 g). The crude extract was eluted with a gradient of PE/EA (100:0–0:100) on a column chromatography of silica gel (100–200 mesh) to obtain seven fractions (1–7).
Fraction 3 (70 g, PE/EA, 96:4–7:3) was loaded on a silica gel (200–300 mesh) column eluting with a n-hexane–ethyl acetate gradient (100:0–1:1) to obtain eleven flow fractions (3A–3K), and Fr.3H (5 g, n-Hex/EA, 10:1–5:1) continued to be purified on a silica gel (300–400 mesh) column with elution of PE/EA (100:0–1:1),resulting in four subfractions (3H-1–3H-4), and Fr.3H-3 (PE/EA, 15:1–10:1) was again purified over a silica gel (300–400 mesh) column to obtain Fr.3H-3A–Fr.3H-3D, the oil on the bottle wall of Fr.3H–3C was taken out as YJ-3H–3C2 alone, and was prepared by analytical HPLC to obtain compound 9 (5 mg, ACN/H2O, 60:40, tR = 22.8 min, 1 mL min−1).
Fr.5 (24.31 g, PE/EA, 6:4–4:6), eluting its upsampled silica gel (200–300 mesh) column by a gradient of n-Hex/EA (9:1–1:1) yielded nine fractions (5A–5I), the Fr.5E (5.5 g, n-Hex/EA, 8:2) was given to 7 subfractions (5E-11–5E-7) by a silica gel (200–300 mesh) column eluting its up-loading through a PE/EA (7:1–0:10) gradient.Fr.5E-5 (1.6 g, PE/EA, 3:1) was converted to an n-Hex/EA (10:1–0:10) solvent system on a silica gel (300–400 mesh) column to purify another 7 fractions (5E-5A–5E-5G). Among them were identified by thin-layer chromatography and vanillin-concentrated sulfuric acid chromatography, all of which contained blue spots combined as Fr.5E-5F (n-Hex/EA, 4:1–3:1). Fr.5E-5F was purified on an ODS column with a gradient of MeOH/H2O (10:90–100:0) to obtain 12 fractions (5E-5F-1–5E-5F-12). Finally, Fr.5E-5F-4 (MEOH/H2O 45:55–50:50) was used to obtain compound 8 (30.5 mg, ACN/H2O, 40:60, tR = 26.5 min, 1 mL min−1) by analytical HPLC. Fr.5G (6.5 g, n-Hex/EA, 3:1), over a silica gel (300–400 mesh) column eluted with PE/EA (6:1–0:10) yielded 10 subfractions (5G-1–5G-10), and Fr.5G-8 (PE/EA, 3:1) was purified on an ODS column with MeOH/H2O (30:70–100:0) gradient elution, and the 21st sample bottle obtained through a Sephadex LH-20 column under an isocratic eluent of DCM/MeOH (1:1) and was given to four subfractions (5G-8-21-A–5G-8-21-D), of which Fr.5G-8-21-A was used on analytical HPLC, and the sample was separated on an ACN/H2O (60:40) gradient to obtain compounds 6 (6 mg, tR = 27.0 min, 1 mL min−1), 1 (1 mg, tR = 27.5 min, 1 mL min−1), and 7 (7.7 mg, tR = 31.0 min, 1 mL min−1) sequentially.
Fr.6 (25 g, PE/EA, 4:6–0:10) eluted with DCM/Me (100:1–1:1) gradient silica gel (200–300 mesh) column yielded 9 fractions (6A–6I), Fr.6D (1 g, DCM/ME, 100:1) eluted with PE/EA (100:1–2:1) gradient silica gel (300–400 mesh) column, leading to the separation of 9 subfractions (6D-1–6D-9), Fr.6D-6 (PE/EA, 4:1) was eluted with DCM/ME (1:1) isocratic elution of its up-sampling of Sephadex LH-20 column, the purified Fr.6D-6A was obtained, and the next analytical column was used to obtain by analytical HPLC the compound 5 (10 mg, ACN/H2O, 45:55, tR = 21.9 min, 1 mL min−1). Fr.6E (DCM/ME, 100:1–50:1) over PE/EA (10:0–0:10) gradient eluted silica gel (300–400 mesh) columns to obtain 7 subfractions (6E-1–6E-7), of which Fr.6E-3 (PE/EA, 3:1) over MeOH/H2O (50:50–100:0) gradient elution of the ODS column led to Fr.6E-3A–Fr.6E-3H, of which the sample of vial 7 and 8 was Fr.6E-3B (MeOH/H2O, 90:10), Fr.6E-3B was used to obtain compound 3 by analytical HPLC (4.3 mg, ACN/H2O, 40:60, tR = 33.8 min, 1 mL min−1); Fr.6 × 10−5 (PE/EA, 3:1–2:1) over MeOH/H2O (30:70–100:0) gradient elution of the ODS column to obtain Fr.6E-5A–Fr.6E-5J, Fr.6E-5H (MeOH/H2O, 80:20) was purified by analytical HPLC to obtain compound 2 (1 mg, ACN/H2O, 55:45, tR = 30.0 min, 1 mL min−1). Fr.6F (DCM/ME, 50:1–20:1) was eluted with MeOH/H2O (30:70–60:40) on an ODS column under a gradient to obtain Fr.6F-1–Fr.6F-7, of which Fr.6F-6 and Fr.6F-7 eluted from 60% methanol were recognized by thin-layer chromatography as uniform purple spots. Both fractions were further purified by isocratic elution with DCM/Me (1:1) on a Sephadex LH-20 column, and the resulting Fr.6F–6B and Fr.6F-7A were prepared by analytical HPLC to obtain the compounds 10 (25 mg, ACN/H2O, 30:70, tR = 34.8 min, 1 mL min−1) and 11 (5.5 mg, ACN/H2O, 35:65, tR = 22.8 min, 1 mL min−1), respectively.
Fr.7 (19.73 g, pure EA) was subjected to a silica gel (200–300 mesh) column eluted by a gradient of DCM/Me (99:1–0:100) to give 7 fractions (7A–7G), and Fr.7C (2.73 g, DCM/ME 98:2–97:3) was eluted on an MCI column with MeOH/H2O (20:80–100:0) gradient elution to obtain four subfractions (7C-1–7C-4), Fr.7C-4 (MeOH/H2O, 70:30–100:0) was isocratically eluted with DCM/Me (1:1) on its up-loading Sephadex LH-20 column to obtain Fr.7C-4A–Fr.7C–4F, Fr.7C–4C was purified by semipreparative HPLC to give compound 4 (23 mg, ACN/H2O, 75:25, tR = 35.0 min, 3 mL min−1).
8-Senecioylchrysanolide D (1): colorless oil; [α]25.1D = –4.2 (c = 0.1, MeOH); IR (KBr) υmax: 3516, 2931, 1759, 1653, 1446 cm−1; UV (MeOH) λmax: 198 nm; ECD (MeOH) λmax (Δε): 200 (+13.0), 221 (−12.8) nm; NMR spectroscopic data (CDCl3, 400/100 MHz), see Table 1; HR-ESI-MS m/z 615.2923 [M + Na]+ (calcd for C35H44O8Na, 615.2928).
Chrysanolide J (2): colorless oil; [α]25.1D = −4.0 (c = 0.1, MeOH); IR (KBr) υmax: 3489, 2927, 1749, 1454, 1373 cm−1; UV (MeOH) λmax: 200 nm; ECD(MeOH) λmax (Δε): 200 (+15.0), 208 (−19.8), 228 (+5.0) nm; NMR spectroscopic data (CDCl3, 400/100 MHz), see Table 1; HR-ESI-MS m/z 613.2824 [M + Na]+ (calcd for C35H42O8Na, 613.2822).
8-Angeloyl-2-methoxy-10-hydroxy-3,11(13)-guaiadien-12,6-olide (3): colorless oil; [α]25.1D = +13.0 (c = 0.1, MeOH); IR (KBr) υmax: 3510, 2945, 2866, 1741, 1454 cm−1; UV (MeOH) λmax: 198 nm; ECD (MeOH) λmax (Δε): 200 (+9.0), 212 (+0.1), 241 (+5.0) nm; NMR spectroscopic data (CDCl3, 400/100 MHz), see Table 1; HR-ESI-MS m/z 399.1764 [M + Na]+ (calcd for C21H28O6Na, 399.1770).
16,22,23,28-Tetrahydroxyolean-12-ene (4): white powder; [α]25.1D = +3.6 (c = 0.1, MeOH); IR (KBr) υmax: 3425, 3925, 1689, 1460, 1065 cm−1; UV (MeOH) λmax: 199 nm; NMR spectroscopic data (CDCl3, 400/100 MHz), see Table 2; HR-ESI-MS m/z 475.3864 [M]+ (calcd for C30H50O4, 475.3857).
3.5. ECD spectral computation
The initial conformations of two possible configurations for compounds 1–3 were constructed based on its NOESY correlations. Conformational searches were performed using the MMFF94s force field in SYBYL 8.1, with low-energy conformations within a 10 kcal mol−1 energy window collected to form an initial conformational ensemble. These preliminary conformations were subsequently optimized at the DFT (B3LYP)/6-31+G(d) level of theory in Gaussian 09. Further refinement selected conformations within a 3 kcal mol−1 energy cutoff to generate a stable conformational ensemble. Finally, ECD spectra for all stable conformers were calculated using time-dependent DFT (TDDFT) with the following parameters: TD (singlet, nstate = 50) scrf (solvent = methanol). Theoretical ECD spectra were generated by Boltzmann-weighted averaging based on thermodynamic statistics, employing the CPCM solvation model (methanol as solvent).3.6. Cells and treatments
The murine macrophages RAW264.7 cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Grand Island, New York, USA) supplemented with 10% heat-inactivated FBS (Gibco BRL Co, Grand Island, NY, USA), penicillin G (100 units per ml), streptomycin (100 μg mL−1), and L-glutamine (2 mM) (Gibco BRL Co, Grand Island, NY, USA). The cells were grown at 37 °C in a humidified atmosphere with 5% CO2. Compounds 1–11 were dissolved in DMSO and the final concentration is 20 mM. Pretreated the cells with the tested compounds and then stimulated with LPS. At the end of incubation, the anti-inflammatory activity was evaluated by assessing NF-κB translocation and cell viability. The cells stimulated by LPS without any intervention were used as model control. The cells incubated with DMEM medium were used as normal control.3.7. Measurement of NF-κB nuclear translocation
The NF-κB nuclear translocation was detected using a previously described HCI method.25 The cells were seeded in 96-well plates at a density of 1.0 × 104 cells per well for 18 h. The cells were then respectively pretreated with compounds (10 μM) and BAY-117085 (1 μM) for 1 h. Afterward, the cells were stimulated with or without LPS (200 ng mL−1) for 40 minutes. The cells were fixed with 4% paraformaldehyde solution for 15 minutes, permeabilized with 0.2% Trition-100X for 15 minutes, blocked with 3% BSA for 30 minutes, stained with a 1:500 dilution of p65-FITC antibody (Santa Cruz, CA, USA) for 3 h, and with DAPI solution (Thermo Scientific, NC, USA) for 5 minutes. Finally, the plates were imaged, and the nuclear translocation of NF-κB (green) was analysed using by HCI technology with the In Cell Analyzer 6000 imaging system (General Electric Company, Boston, USA).
3.8. Cell cytotoxicity assay
The RAW 264.7 cells were seeded at a density of 8000 cells per well in 96-well plates for 24 hours. The cells were then pretreated with the candidate compounds (2, 8 and 9) and DMSO (0.1%). After that, the cells were stimulated with or without LPS (200 ng mL−1) for 18 hours. After the treatment, different experiments were used to measure the cell viability.MTT method was used to assess the cells cytotoxicity of compounds. After the treatment, 100 μL of MTT solution per well was added for 4 hours, which followed by 10% SDS–HCl solution for 18 hours. Then thoroughly shake the 96-well plates for 5 min. Lastly, the OD value at 570 nm and 650 nm was measured, and the OD value of control cells was set as 100%.
HCI was used to assess the cells cytotoxicity of compounds. After the treatment, the cells were stained by CalceinAM (green) and propidium iodide (red) (Beyotime Biotechnology, shanghai, China) for 30 min at 37 °C. Then the cells were washed twice by PBS. Lastly, the lived cells (green) and dead cells (red) in 96-well plates were imaged by HCI. And the cell viability of control group was set to 100%.
3.9. Statistical analysis
The NucCyto difference was used to analyze NF-κB translocation via Nuclear Translocation Analysis on an HCS imager. The log (agonist) vs. normalized response was plotted using GraphPad Prism 7.0 software (GraphPad Software Inc, La Jolla, CA, USA). The data was expressed as the mean ± S. M. E. of two or three independent experiments. Statistical significance comparisons between the different compound-treated groups and the model control group were analysed by one-way ANOVA followed by post hoc analyses using the SNK method in GraphPad Prism 7.0 software. Compounds with 50% inhibition of NF-κB nuclear translocation were considered as potential candidates. P values less than 0.05 were considered statistically significant.4. Conclusion
In conclusion, three previously undescribed guaiacane-type sesquiterpenes (1–3) and one triterpene compound (4) were found in the aerial parts of C. indicum, along with seven known sesquiterpenoids. The structures of these terpenoids were elucidated by comprehensive spectral analysis. Then, in the LPS-stimulated RAW264.7 macrophages, compounds 2, 8 and 9 at the dosage of 10 μM had shown significant inhibitory effect on the NF-κB translocation. In addition, they exhibited no significant cytotoxicity toward LPS-induced RAW264.7 cells at therapeutic doses. Based on these results, compounds 2, 8 and 9 may have anti-inflammatory value for in-depth research.Data availability
The data supporting this study are included within the manuscript and its ESI† files.Author contributions
Xinyue Li: investigation, data curation, visualization, writing-original draft, writing review & editing. Yanfen Cai: data curation, writing review & editing. Yunshuang Hu: data curation. Limei Miu: investigation & conceptualization. Yanyu Zhang: data curation, visualization, writing review & editing. Shiyun Huang: data curation & visualization. Min Wei: resources. Qing Ma: resources. Zhongqiu Liu: resources, conceptualization, supervision & funding acquisition. Hua Zhou: resources, conceptualization, supervision & funding acquisition. Peng Wu: resources, conceptualization, supervision, funding acquisition, writing review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research received funding from National Key Research and Development Program of China (2023YFC3502800), Joint Funds of the National Natural Science Foundation of China (No. U22A20368), Natural Science Foundation of Guangdong Province (No. 2023A1515011761), Key Laboratory of Guangdong Drug Administration (No. 2021ZDB03), Talent Support Project of Guangdong (No. 2021JC050230), the Open Research Project of State Key Laboratory of Dampness Syndrome of Chinese Medicine (No. SZ2022KF09), Guangxi Science and Technology Major Program Grant (No. GUIKEAA23023035), and Guangzhou University of Chinese Medicine College Students' Innovation and Entrepreneurship Training Program Project (No. 202410572291).References
- G. Song, M. Choi, W. Y. Park, S. H. Kim, W. Jiao, J. Y. Park, K. S. Ahn, H. J. Kwak and J. Y. Um, Front. Pharmacol., 2024, 15, 1455805 CrossRef CAS PubMed
.
- Y. J. Wang, J. Su, J. J. Yu, M. Q. Yan, M. L. Shi, Q. D. Huang, B. Li, W. Y. Wu, R. S. Xia, S. F. Li, S. H. Chen and G. Y. Lv, Front. Pharmacol., 2021, 12, 755140 CrossRef CAS PubMed
- S. H. Yu, X. Sun, M. K. Kim, M. Akther, J. H. Han, T. Y. Kim, J. Jiang, T. B. Kang and K. H. Lee, J. Ethnopharmacol., 2019, 239, 111917 CrossRef CAS PubMed
- J. H. Yun, E. S. Hwang and G. H. Kim, Korean J. Food Sci. Technol., 2012, 44(1), 82–88 CrossRef
- S. Jiang, M. Y. Wang, Z. C. Jiang, S. Zafar, Q. Xie, Y. P. Yang, Y. Liu, H. W. Yuan, Y. Q. Jian and W. Wang, Molecules, 2021, 26(10), 3038 CrossRef CAS PubMed
- M. S. Cheon, T. Yoon, D. Y. Lee, G. Choi, B. C. Moon, A. Y. Lee, B. K. Choo and H. K. Kim, J. Ethnopharmacol., 2009, 122(3), 473–477 CrossRef PubMed
- H. J. Zhang, B. H. Wang, X. Wang, C. P. Huang, S. M. Xu, J. L. Wang, T. E. Huang, W. L. Xiao, X. L. Tian, X. Q. Lan, Q. Q. Wang and Y. Xiang, J. Cachexia Sarcopeni., 2023, 15(1), 173–188 CrossRef PubMed
- B. K. Ghimire, S. H. Kim, C. Y. Yu and I. M. Chung, Plants, 2022, 11(11), 1440 CrossRef CAS PubMed
- L. L. Liu, T. K. Q. Ha, W. Ha, W. K. Oh, J. L. Yang and Y. P. Shi, J. Nat. Prod., 2017, 80(2), 298–307 CrossRef CAS PubMed
- X. Y. Chen, J. Li, W. M. Cheng, H. Jiang, X. F. Xie and R. Hu, Am. J. Chin. Med., 2008, 36(04), 695–704 CrossRef CAS PubMed
- T. Zhang, D. R. Wan, Y. Y. Li, S. S. Wang, X. T. Zhou, F. Sefidkon and X. Z. Yang, Molecules, 2023, 28(5), 2022 CrossRef CAS PubMed
- G. M. Xue, X. Q. Li, C. Chen, K. Chen, X. B. Wang, Y. C. Gu, J. G. Luo and L. Y. Kong, J. Nat. Prod., 2018, 81(2), 378–386 CrossRef CAS PubMed
- J. G. Kim, J. W. Lee, T. P. L. Le, J. S. Han, Y. B. Cho, H. Kwon, D. Lee, M. K. Lee and B. Y. Hwang, J. Nat. Prod., 2021, 84(3), 562–569 CrossRef CAS PubMed
- A. Reddy, J. Lee, J. Seo, B. H. Kim, E. Chung, S. Ryu, Y. Kim, C. Lee, K. Min and Y. Kim, Arch. Pharm. Res., 2006, 29(7), 591–597 CrossRef CAS PubMed
- Z. G. Shao, L. Z. Li, Y. Z. Zheng, Q. Gong, C. Q. Ke, S. Yao, H. Y. Zhang, C. P. Tang and Y. Ye, Fitoterapia, 2022, 159, 105199 CrossRef CAS PubMed
- P. Luo, Y. F. Cheng, Z. Y. Yin, C. J. Li, J. Xu and Q. Gu, J. Nat. Prod., 2019, 82(2), 349–357 CrossRef CAS PubMed
- Q. Gu, Y. Y. Chen, H. Cui, D. Huang, J. W. Zhou, T. Z. Wu, Y. P. Chen, L. N. Shi and J. Xv, RSC Adv., 2013, 3(26), 10168 RSC
- W. Stöcklin, T. G. Waddell and T. A. Geissman, Tetrahedron, 1970, 26(10), 2397–2409 CrossRef
- J. Lee, M. Yang, J. Lee, S. Hwang, Y. Kho and K. H. Park, Planta Med., 2003, 69(9), 880–882 CrossRef CAS PubMed
- N. P. Sahu, S. B. Mahato, S. K. Sarkar and G. Poddar, Phytochemistry, 1996, 41(4), 1181–1185 CrossRef CAS PubMed
- N. D. Abdullaev, M. R. Yagudaev, V. A. Tarasov, Sh. Z. Kasymov and G. P. Sidyakin, Chem. Nat. Compd., 1979, 15(3), 285–289 CrossRef
- M. Haruna, M. Kato, K. Ito, T. Nikai, H. Sugihara and H. Muratat, Phytochemistry, 1981, 20(11), 2583–2584 CrossRef CAS
- H. Heymann, Y. Tezuka, T. Kikuchi and S. Supriyatna, Chem. Pharm. Bull., 1994, 42(1), 138–146 CrossRef CAS
- W. Y. Tsui and G. D. Brown, J. Nat. Prod., 1996, 59(11), 1084–1086 CrossRef CAS
- Y. Y. Zhang, Y. D. Yao, Q. Q. Cheng, Y. F. Huang and H. Zhou, Curr. Drug Metab., 2022, 23(5), 394–414 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03586d |
| This journal is © The Royal Society of Chemistry 2025 |