
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtozeolite seed-assisted construction of hierarchical nano-ZSM-5 zeolites and their catalytic application in Meinwald rearrangement†
Ming-Lei Gou *,
Xuan Ru,
Yongli Yang,
Zhen Liu and
Shuge Peng
*,
Xuan Ru,
Yongli Yang,
Zhen Liu and
Shuge Peng
School of Chemistry and Chemical Engineering, Henan University of Science and Technology, Luoyang, Henan 471023, PR China. E-mail: mingleigou@haust.edu.cn; Fax: +86 37964231914; Tel: +86 37964231914
First published on 2nd June 2025
Abstract
A facile strategy affording hierarchical nano-ZSM-5 zeolites with high yields (approaching 100%) and crystallinity using a seed-assisted dry-gel conversion (SA-DGC) method is developed. Only a small amount of tetrapropylammonium hydroxide (TPAOH) was used for the synthesis of protozeolite seeds without involving the use of any structure-directing agents (i.e., SiO2/TPA+ = 30). A very small amount of water (i.e., 1.0 mL) was required for crystallization conditions. Protozeolite seeds with a size not exceeding 20 nm can provide a high-activity nucleus for zeolite crystallization, and the precursor is not in direct contact with water and freely migrates, resulting in the formation of nano-zeolites (100–200 nm) around the microregion of the seeds during steaming treatment. Owing to the high-quality nanosized crystals, the as-prepared hierarchical nano-ZSM-5 zeolites exhibit outstanding performance in the rearrangement of styrene oxide and its derivatives compared with conventional ZSM-5 zeolites.
1. Introduction
Zeolites are a class of crystalline microporous aluminosilicates with a large surface area, uniform channels (typically 0.25–1.5 nm), tunable surface properties and excellent hydrothermal stability, which have been widely used in the fields of heterogeneous catalysts and adsorbents.1,2 In particular, MFI structure ZSM-5 zeolites are among the most industrially important zeolites in the catalytic field and can tailor reaction pathways to afford desirable products via reactants, intermediates or transition states.3,4 However, with an increasing need for faster diffusion rates and higher conversion rates of bulky molecules, the sole presence of micropores imposes diffusion limitations, and rapid deactivation limits the performance of industrial zeolitic catalysts.5 To overcome this limitation, approaches to decrease zeolite crystal sizes to the nanometer scale or introduce supplementary pores (meso-/macropores) into zeolite crystals have been extensively adopted.6–8Nano-zeolites with a large external surface area, high accessibility and abundant silicon hydroxyl groups offer numerous unique advantages in the chemical industry as heterogeneous catalysts.9,10 However, nano-zeolites are difficult to control and separate in solvent via the traditional hydrothermal method.11,12 Recently, using a series of growth modifiers13–18 or adopting a kinetic-modulated crystallization strategy,19–21 some zeolite mesocrystals composed of uniform nano-crystal domains were constructed via crystal–crystal oriented attachment in the hydrothermal synthesis process. However, hydrothermal synthesis often requires large amounts of solvents (e.g., water or alcohols). With incomplete crystallization, the solvent inevitably leads to some of the nutrients, such as alkalis, silicates, and aluminates, dissolving in waste solution, which increases the cost of separation and results in environmental pollution.22 In addition, organic templates used as structure-directing agents (SDAs) play an important role in the zeolite synthesis process.23,24 The heavy use of organic templates increases the cost of zeolite production and causes environmental pollution owing to the removal of organic templates via calcination.25 For instance, costly tetrapropylammonium (TPA+) cations are typically used as SDAs for the synthesis of ZSM-5 zeolites with a relatively low ratio of SiO2/TPA+ ranging from 1.7 to 4.0, but they usually suffer from the aforementioned drawbacks.26 Significant efforts have been undertaken using less amounts or no SDAs to prepare zeolites,27–32 but many difficulties, such as low yield, a low rate of crystallization, and uncontrollable size of crystals, have not been resolved.
In this study, a facile strategy affording hierarchical nano-ZSM-5 zeolites with high yields and crystallinity via a seed-assisted dry-gel conversion (SA-DGC) method was proposed. Only a small amount of TPA+ was used for the synthesis of the protozeolitic seeds without involving any SDAs in the subsequent processes, and a very small amount of solvent was required. Importantly, the as-prepared nano-ZSM-5 zeolites exhibit outstanding performance in the rearrangement of styrene oxide and its derivatives compared to conventional ZSM-5 zeolites.
2. Experimental
2.1. Reagents
Tetraethylorthosilicate (TEOS, Shanghai McLean Biochemical Technology Co., Ltd, >99%), sodium aluminate (NaAlO2, Shanghai McLean Biochemical Technology Co., Ltd, reagent grade), silica sol (TCI Development Co., Ltd, 30 wt% SiO2), tetrapropylammonium hydroxide (TPAOH, Shanghai McLean Biochemical Technology Co., Ltd, 1.0 M in H2O) and sodium (NaOH, Sinopharm Chemical Reagent Company, 98%) were supplied by commercial suppliers and used without further purification. Reagents for the catalytic tests were styrene oxide (98 wt%, TCI Development Co., Ltd), 4-methylstyrene oxide (95 wt%, WuXi AppTec), 4-fluorostyrene oxide (95 wt%, WuXi AppTec), 4-chlorinestyrene oxide (95 wt%, WuXi AppTec) and 4-nitrostyrene oxide (95 wt%, WuXi AppTec). All the reagents were used as purchased without further purification. The water used in this study was ultrapure deionized water self-made by the laboratory.2.2. Catalyst preparation
First, protozeolite seeds were hydrothermally synthesized with a molar ratio of 1SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.35TPAOH:5H2O. Typically, TEOS and deionized water were mixed at room temperature until complete hydrolysis was achieved, followed by the addition of TPAOH under fast stirring. Then, the gel was transferred into a Teflon-lined stainless steel autoclave and heated at 90 °C for 24 h under static conditions. The as-prepared solid sample was centrifuged, washed with deionized water, vacuum dried at 80 °C for 12 h, and then used as the seeds for the SA-DGC method.
:0.35TPAOH:5H2O. Typically, TEOS and deionized water were mixed at room temperature until complete hydrolysis was achieved, followed by the addition of TPAOH under fast stirring. Then, the gel was transferred into a Teflon-lined stainless steel autoclave and heated at 90 °C for 24 h under static conditions. The as-prepared solid sample was centrifuged, washed with deionized water, vacuum dried at 80 °C for 12 h, and then used as the seeds for the SA-DGC method.
Then, nano-ZSM-5 zeolites were synthesized by applying the SA-DGC method with a molar ratio of 1SiO2:0.01Al2O3:0.25NaOH:25H2O. Typically, silica sol (30 wt% SiO2) and deionized water were mixed at room temperature, followed by the addition of NaAlO2 and NaOH under stirring until a homogenous mixture was obtained. Then, different amounts of protozeolite seeds were added into the above mixture and stirred until homogeneity. Subsequently, the mixture was vacuum dried at 80 °C for 12 h and then placed in a Teflon cup, which was put into another larger autoclave, and different amounts of water were added outside the cup to create steam for the crystallization conditions. The autoclave was placed in an oven maintained at 150 °C for continuous crystallization. After cooling to room temperature, the solids were collected, then dried at 110 °C for 4 h, and calcined at 500 °C for 2 h. The resulting samples were denoted as SA-DGC-x-y-z, where x represents the ratio of SiO2/TPA+ calculated according to the amount of seeds added, y represents the volume (mL) of water added into the autoclave, and z represents the crystallization time (h).
For comparison, conventional ZSM-5 zeolites were prepared by applying the dry-gel conversion (DGC) method according to the above procedures by replacing the seeds with TPAOH. The resulting samples were named +DGC-x-y-z, where x, y, and z have the same meanings as above.
Before the catalytic test, all the samples were subjected to ion exchange with 1 M NH4NO3 solution at room temperature for 24 h, followed by drying at 110 °C for 4 h and calcining at 500 °C for 2 h.
2.3. Catalyst characterization
The structure and crystallinity of the samples were investigated via power X-ray diffraction (XRD) using a Bruker D8 Advance diffractometer with Cu kα radiation (λ = 0.1541 nm) at a scanning rate of 5° min−1 from 5° to 50°, and the relative crystallinity (RC) of the samples was calculated from the areas of the main peaks in the range of 2θ = 8–10°. The framework vibration bands of the samples were measured through Fourier Transform Infrared Spectroscopy (FT-IR) using a Nicolet 380 spectrometer by applying the KBr pellet technique. The morphologies of the samples were observed using a scanning electron microscope (SEM, FlexSEM-1000, Hitachi), field emission scanning electron microscope (FESEM, SU8010, Hitachi) and transmission electron microscopy (TEM, JEM-2100F, JEOL). X-ray fluorescence (XRF) was used to measure the SiO2/Al2O3 ratios of the samples using a Bruker S4 Pioneer X-ray fluorescence spectrometer. N2 adsorption–desorption isotherms were obtained at −196 °C using a Quantachrome Autosorb-1 instrument. The total surface area (Stotal) and pore volume (Vtotal) were calculated by applying the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) models, respectively. The micropore surface area (Smicro) and volume (Vmicro) were calculated by applying the t model. The external surface area (Sexter) and mesoporous volume (Vmeso) were obtained by subtracting Smicro and Vmicro from Stotal and Vtotal, respectively. 27Al magic-angle spinning nuclear magnetic resonance (MAS-NMR) spectra were recorded using a Varian Infinity-Plus 300 MHz spectrometer with a resonance frequency of 78.13 MHz. Al(NO3)3 was used as the reference for the 27Al chemical shift.The acidity of the samples was investigated by temperature-programmed desorption of ammonia (NH3-TPD) and FTIR spectra of adsorbed pyridine (Py-FTIR). NH3-TPD was performed using a quartz U-tube reactor equipped with a thermal conductivity detector (TCD). At a N2 flow of 50 mL min−1, the sample (0.1 g, 20–30 mush) was pretreated at 500 °C for 2 h. After cooling to 50 °C, NH3 was supplied to the sample until saturation adsorption. Then, the reactor was maintained at 150 °C for 1 h to remove physically adsorbed NH3. Finally, the sample was heated from 150 to 550 °C at a rate of 15 °C min−1.
Py-FTIR was analyzed using a Nicolet 380 spectrometer. All samples were pressed into self-supporting wafers (diameter: 13 mm, weight: 15 mg) and then pre-treated at 500 °C for 4 h under vacuum (10−3 Pa). The adsorption of pyridine was preceded in situ at room temperature and then evacuated at 200 °C for 2 h. Afterwards, the difference spectrum was obtained by subtracting the spectrum of the sample before the probe adsorption.
2.4. Catalytic tests
All reactions were carried out in a 50 mL batch glass reactor under a nitrogen atmosphere. The solvent and catalyst were initially placed in the flask with vigorous agitation (500–550 rpm) to overcome external mass-transfer limitations. A certain amount of styrene oxide or its derivatives was added directly to the mixture after reaching the reaction temperature. Samples of about 0.1 mL were extracted at various time intervals using a syringe equipped with 0.2 μm filters and analyzed by GC-MS (Agilent 6890/5973, MSD) and GC (Agilent 6820, FID) provided with a VF-5ms capillary column (5% phenyl–95% dimethylpolysiloxane, 30 m × 0.25 mm × 0.25 μm).When the reaction was completed, the catalyst was separated by centrifugation, washed three times with solvent and reused for another run.
3. Results and discussion
3.1. Crystallization and morphology
Protozeolite seeds were synthesized at a low temperature (90 °C), which favors nucleation over growth for the zeolite crystals.33 As shown in Fig. 1(a), the protozeolite synthesized at 90 °C for 24 h was still non-crystalline without any characteristic peaks of zeolites. However, the stretching or bending vibrations of SiO4 tetrahedral (1100, 793, and 453 cm−1) and five-membered ring (1222 and 545 cm−1) indicated that the protozeolite already had partial characters of the MFI structure, as shown in Fig. 1(b). The surface Si–OH groups (971 cm−1) were formed in increasing numbers, which can provide a high-activity nucleus for initiating zeolite crystallization. SEM (Fig. 1(c)) and TEM (Fig. 1(d)) images revealed that the protozeolite was irregular nanoparticles with a size not exceeding 20 nm. | ||
| Fig. 1 (a) XRD pattern of protozeolite seeds synthesized at 90 °C for 24 h. (b) FT-IR spectra of protozeolite seeds synthesized at 90 °C and at different times. (c) SEM and (d) TEM images of protozeolite seeds synthesized at 90 °C for 24 h. | ||
The XRD patterns of the samples synthesized by applying the SA-DGC and DGC methods are shown in Fig. 2. As shown in Fig. 2(a), without any seeds (i.e., SiO2/TPA+ = ∞) in the SA-DGC method, no crystal structure peaks were observed. By increasing the addition of the seeds, the SiO2/TPA+ ratios gradually reduced, and an increasing typical MFI topology and no other phases of the samples were found.34 When the SiO2/TPA+ ratios were reduced to the range of 20–30, the RC of the obtained samples increased to 96–100%. However, with the same SiO2/TPA+ ratio (SiO2/TPA+ = 30) in the DGC method, by replacing only the seeds with TPA+, the RC of the sample (DGC-30-1.0-72) was only 5%. It is demonstrated that ZSM-5 zeolite can be synthesized using a smaller amount of SDAs in the SA-DGC method. When the SiO2/TPA+ ratio was reduced to 3, the sample (DGC-3-1.0-72) had stronger and narrower diffraction peaks in the range of 2θ = 22–25° but slightly weaker diffraction peaks in the range of 2θ = 8–10° compared with SA-DGC-30-1.0-72, indicating that the nano-sized zeolites were obtained in the SA-DGC method and the traditional micro-sized zeolites were obtained in the DGC method, which can also be verified in SEM images, as shown in Fig. 4.
 | ||
| Fig. 2 XRD patterns of samples synthesized using the SA-DGC and DGC methods under different amounts of seeds (a) and water (b). | ||
The amount of water added outside the cup to create steam for crystallization conditions is another key element in the SA-DGC method. As shown in Fig. 2(b), the X-ray diffraction peak intensity became stronger as the amount of water increased. Compared with the conventional hydrothermal process, the precursor was not in direct contact with water and migrated freely using the SA-DGC method. The dry gel could be covered with a thin layer of water owing to capillary condensation of water and transformed into zeolite crystals around the micro region of seeds during the steaming treatment.35 The more amount of water added outside the cup, the more layers of water owing to capillary condensation formed, which led to the faster crystallization rate and higher crystallinity of samples in the water addition range of 0.2–1.0 mL. When the addition of water increased to 2.0 mL, the RC of the sample (i.e., SA-DGC-30-2.0-72) slightly decreased to 98% and the width of the peaks in the range of 2θ = 22–25° trended to narrow, which may be because the precursor could migrate within a larger scale region and grow into larger zeolite crystals. This can also be observed in the SEM images, as shown in Fig. 4.
As shown in Fig. 3, the main FT-IR bands of the samples at 453, 545, 793, 1100 and 1222 cm−1 are the characteristics of the ZSM-5 framework.36,37 The bands at 453, 793 and 1100 cm−1 are generally attributed to the stretching or bending vibration of the T–O tetrahedral (T = Si and Al), and the bands at 545 and 1222 cm−1 correspond to the vibration of the five-membered ring in the MFI structure. With the SiO2/TPA+ ratio decreased in Fig. 3(a) and water addition increased in Fig. 3(b), the characteristic bands of ZSM-5 samples synthesized using the SA-DGC method at 545 and 1222 cm−1 gradually increased. SA-DGC-30-1.0-72 showed obviously stronger intensity of bands than DGC-30-1.0-72, indicating that the SA-DGC method has a faster crystallization rate than the DGC method, which agrees well with the XRD results.
 | ||
| Fig. 3 FT-IR spectra of samples synthesized using the SA-DGC and DGC methods under different amounts of seeds (a) and water (b). | ||
The SEM images of the samples are presented in Fig. 4. With no seed addition, the sample (SA-DGC-∞-1.0-72) appeared to have a typical amorphous morphology without any crystal particles. With the SiO2/TPA+ ratios gradually reduced from 120 to 30, more and more crystal particles were formed on the surface of the amorphous precursor. The more the seeds added, the smaller the crystal particles formed, which was because the precursor was unable to freely migrate in the SA-DGC method and could only be crystallized around the micro region of seeds. For instance, as shown in Fig. 4(d), the particle diameter of SA-DGC-30-1.0-72 was about 100–200 nm. However, insufficient water added outside the cup in the SA-DGC method can cause incomplete crystallization, as shown in Fig. 4(e), and too much water can cause nanoparticles to grow into larger particles, as shown in Fig. 4(f). For comparison, with the same SiO2/TPA+ ratio of 30 in the DGC method, the sample (DGC-30-1.0-72) appeared to have an amorphous morphology. When the SiO2/TPA+ ratio was reduced to 3, a traditional micro-sized zeolite (DGC-3-1.0-72) was obtained, as shown in Fig. 4(h). It can be concluded that the nano-sized zeolites are obtained in the SA-DGC method because the precursor can only be crystallized around the micro region of seeds, but by replacing seeds with TPAOH in the DGC method, the traditional micro-sized zeolites are obtained. Using any method, the yields of all the above samples approached 100%.
 | ||
| Fig. 4 SEM images of samples synthesized using the SA-DGC and DGC methods: (a) SA-DGC-∞-1.0-72, (b) SA-DGC-120-1.0-72, (c) SA-DGC-60-1.0-72, (d) SA-DGC-30-1.0-72, (e) SA-DGC-30-0.2-72, (f) SA-DGC-30-2.0-72, (g) DGC-30-1.0-72, and (h) DGC-3-1.0-72. | ||
3.2. Textural and acidic properties
The nano-sized zeolite (SA-DGC-30-1.0-72) and micro-sized zeolite (DGC-3-1.0-72) are selected for further study. Fig. 5(a) shows the N2 adsorption–desorption isotherms and BJH mesopore size distribution of the samples, and their texture properties are summarized in Table 1. DGC-3-1.0-72 exhibited a type I isotherm with a limited external surface area (15.3 m2 g−1) and mesoporosity volume (0.012 cm3 g−1), which is typical of microporous materials. SA-DGC-1.0-72 showed a mixture of type I and IV isotherms with a hysteresis loop near p/p0 = 0.5–0.8, indicating that the sample contains a certain amount of mesopores with a size of 15–20 nm, as shown in the inset in Fig. 5(a). Owing to the nano-sized particles, SA-DGC-1.0-72 possessed more external surface area (98.4 m2 g−1) and mesoporosity volume (0.107 cm3 g−1), as shown in Table 1. | ||
| Fig. 5 (a) N2 adsorption–desorption isotherms and BJH mesopore size distribution, (b) 27Al MAS NMR spectra, (c) NH3-TPD curves and (d) FT-IR spectra of adsorbed pyridine of SA-DGC-30-1.0-72 and DGC-3-1.0-72. | ||
| Sample | SiO2/Al2O3 | Stotal (m2 g−1) | Smicro (m2 g−1) | Sexter (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) |
|---|---|---|---|---|---|---|---|
| DGC-3.0-1.0-72 | 47 | 295.4 | 280.1 | 15.3 | 0.145 | 0.133 | 0.012 |
| SA-DGC-30-1.0-72 | 45 | 355.2 | 256.8 | 98.4 | 0.229 | 0.122 | 0.107 |
27Al MAS NMR has been conducted to monitor the chemical states of Al species. As illustrated in Fig. 5(b), each sample showed a strong sharp peak at 53 ppm corresponding to the tetrahedral framework aluminum species,38 indicating that most of the aluminum species appeared dimensional a tetrahedral framework. Furthermore, SA-DGC-30-1.0-72 showed a relatively smaller peak at 0 ppm, which is attributed to octahedral extra-framework aluminum species.39 According to the above XRD and SEM results, the precursor can only be crystallized around the micro region of seeds in the SA-DGC method, resulting in a small number of aluminum atoms becoming octahedral extra-framework aluminum species in the area away from the seeds. The sum of the two Al species in both samples was nearly the same according to the similar SiO2/Al2O3 ratios, as shown in Table 1.
The acidity of the samples was investigated by NH3-TPD and FT-IR spectra of adsorbed pyridine. As shown in Fig. 5(c), each sample has three NH3 desorption peaks at <200, 240–300, and 350–450 °C, which were assigned to physisorbed NH3, weak acid sites, and strong acid sites, respectively.40 The strong acid sites are typically identified as Brønsted acid sites generated from tetrahedral framework aluminum species, while the weak acid sites are usually Lewis acid sites related to the octahedral extra-framework aluminum species.41 According to the 27Al MAS NMR results, most aluminum species appeared in the dimensional tetrahedral framework on DGC-3-1.0-72; hence, it contained stronger acid sites (421 °C) and less weak acid sites (254 °C), which corresponded to the Brønsted acid sites at 1545 cm−1 and Lewis acid sites at 1443 cm−1, as depicted in Fig. 5(d).42 A small number of aluminum atoms became octahedral extra-framework aluminum species on SA-DGC-30-1.0-72, leading to an increase in weak acid sites (285 °C) and Lewis acid sites (1443 cm−1). With the reduction of aluminum in the skeleton of SA-DGC-30-1.0-72, the strength and concentration of strong acid sites (395 °C) were significantly lower than those (421 °C) of DGC-3-1.0-72.
3.3. Catalytic performance
Epoxides are versatile and highly useful synthetic intermediates in organic synthesis. For example, under acidic conditions, epoxides can be isomerized to carbonyl compounds, and their halogenated derivatives are well-known as a reaction of the Meinwald rearrangement.43 Both Brønsted and Lewis acid sites can catalyze this reaction.44 However, it usually suffers from low yields due to side reactions, which produce allylic alcohols or polymers because of steric or diffusional limitations.45Initially, the rearrangement of 4-substituted styrene oxides (Scheme 1) over SA-DGC-30-1.0-72 and DGC-3-1.0-72 was investigated. Phenylacetaldehyde and its derivatives (i.e., 4-substituted phenylacetaldehydes) are all highly reactive substances, which then undergo aldol condensation or polymerization, forming large amounts of by-products in the subsequent separation process. Therefore, the rearrangement of 4-substituted styrene oxides in this work was carried out in large amounts of solvent (i.e., 1,2-dichloroethane), and reactants of about 0.1 mL were extracted at various time intervals and immediately analyzed by GC-MS and GC to verify the correctness of the reactions and the purity of the products. The mass spectra of the main products produced by GC-MS are illustrated in Fig. 6. The main products in the rearrangement of 4-substituted styrene oxides are fully identified as the corresponding 4-substituted phenylacetaldehydes, and other by-products are not further investigated in this work because our focus is the characterization of the Meinwald rearrangement.
 | ||
| Scheme 1 Rearrangement of 4-substituted styrene oxides (1a–e). | ||
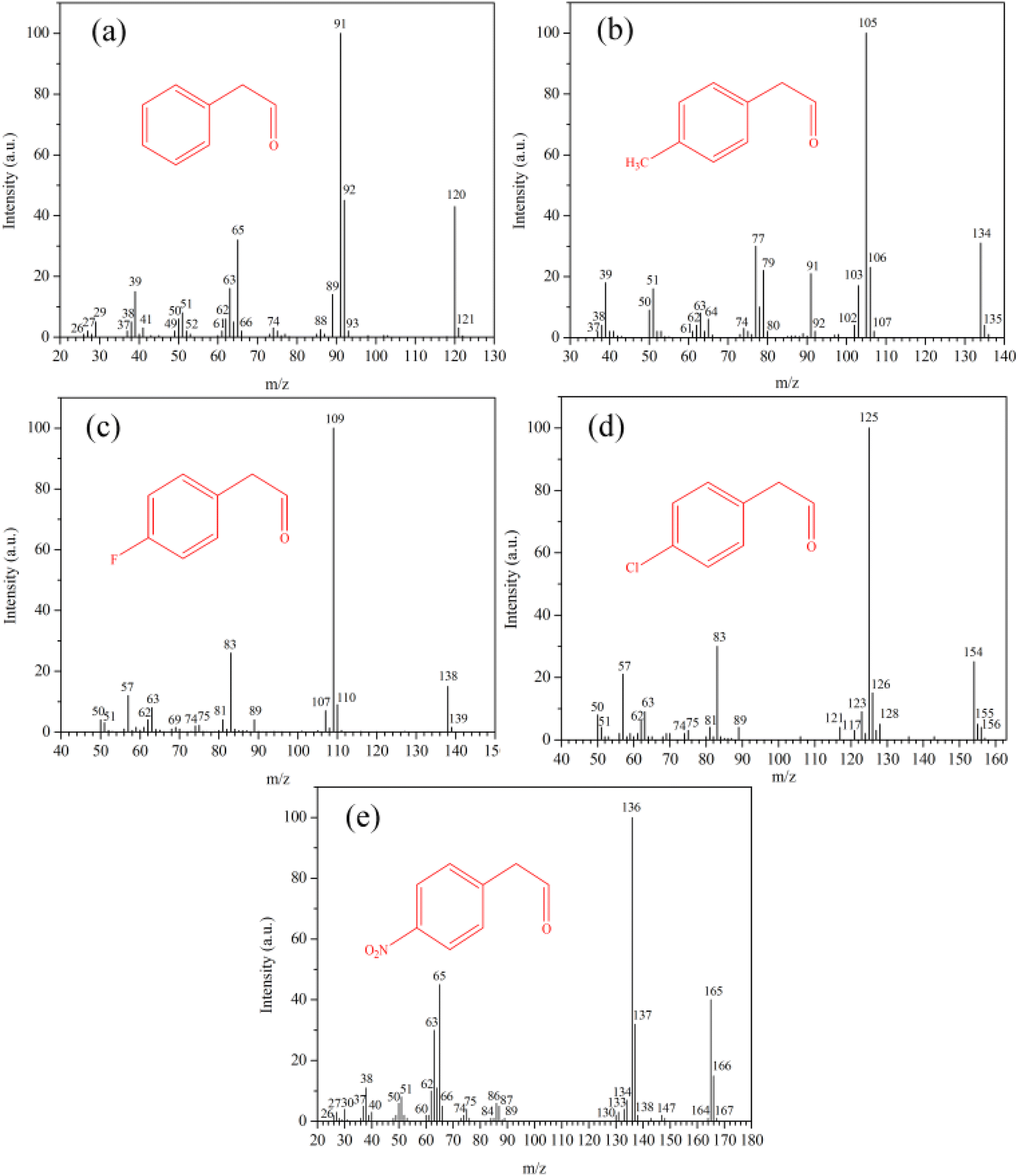 | ||
| Fig. 6 Mass spectra of phenylacetaldehyde (a), (4-methylphenyl)acetaldehyde (b), (4-fluorophenyl)acetaldehyde (c), (4-chlorophenyl)acetaldehyde (d) and (4-nitrophenyl)acetaldehyde (e) in the rearrangement of 4-substituted styrene oxides. | ||
The conversions of 4-substituted styrene oxides and yields of 4-substituted phenylacetaldehydes according to the results obtained by GC are presented in Table 2. DGC-3-1.0-72 had higher activity in the rearrangement of 1a owing to its higher strength and concentration of Brønsted acid sites according to the NH3-TPD results. However, somewhat low yields of 2a were obtained over DGC-3-1.0-72 because stronger acid sites favor the formation of condensation products and subsequent fast deactivation of the catalyst by coke.46 With the extension of reaction time, the yields of 2a decreased gradually owing to its side reactions under acidic conditions. Other 4-substituted styrene oxides with electron-donating (1b) or electron-withdrawing (1c–e) groups can also react completely over the catalysts in one hour, but slightly higher yields of 2c–e were obtained with electron-withdrawing groups under the same conditions. The rearrangement of 4-substituted styrene oxides under acidic conditions is likely to occur using a carbocation mechanism.47 The neighboring aryl and electron-withdrawing groups favor the formation of the carbocations by stabilizing the positive charge, which results in the high regioselectivity of 4-substituted phenylacetaldehydes. For instance, the yields of (4-fluorophenyl)acetaldehyde (94%), (4-chlorophenyl)acetaldehyde (93%) and (4-nitrophenyl)acetaldehyde (94%) were apparently higher than (4-methylphenyl)acetaldehyde (86%).
| Entry | X group | Catalyst | Reaction time/min | Conversiond/% | Yieldd/% |
|---|---|---|---|---|---|
| a Reactions were carried at 50 °C in 20 mL 1,2-dichloroethane using 1.0 g catalyst and 10 mmol 4-substituted styrene oxides in a nitrogen atmosphere.b After each run, the catalyst was separated via centrifugation, washed with 1,2-dichloroethane and reused in run 5 and run 6.c After each run, the catalyst was separated via centrifugation, washed with 1,2-dichloroethane and reused in run 2 and run 3.d All the conversions and yields were determined via GC-MS and GC analysis. | |||||
| 1 | 1a, H | SA-DGC-30-1.0-72 | 15 | 68 | 66 |
| 2 | 1a, H | SA-DGC-30-1.0-72 | 30 | 87 | 83 |
| 3 | 1a, H | SA-DGC-30-1.0-72 | 60 | 100 | 92 |
| 4 | 1a, H | SA-DGC-30-1.0-72 | 120 | 100 | 89 |
| 5 | 1b, Me | SA-DGC-30-1.0-72 | 60 | 100 | 86 |
| 6 | 1c, F | SA-DGC-30-1.0-72 | 60 | 100 | 94 |
| 7 | 1d, Cl | SA-DGC-30-1.0-72 | 60 | 100 | 93 |
| 8 | 1e, NO2 | SA-DGC-30-1.0-72 | 60 | 100 | 94 |
| 9b | 1a, H | SA-DGC-30-1.0-72 | 60 | 100 | 93 |
| 10b | 1a, H | SA-DGC-30-1.0-72 | 60 | 98 | 90 |
| 11 | 1a, H | DGC-3.0-1.0-72 | 15 | 77 | 62 |
| 12 | 1a, H | DGC-3.0-1.0-72 | 30 | 95 | 76 |
| 13 | 1a, H | DGC-3.0-1.0-72 | 60 | 100 | 80 |
| 14 | 1a, H | DGC-3.0-1.0-72 | 120 | 100 | 78 |
| 15 | 1b, Me | DGC-3.0-1.0-72 | 60 | 100 | 75 |
| 16 | 1c, F | DGC-3.0-1.0-72 | 60 | 100 | 81 |
| 17 | 1d, Cl | DGC-3.0-1.0-72 | 60 | 100 | 80 |
| 18 | 1e, NO2 | DGC-3.0-1.0-72 | 60 | 100 | 82 |
| 19c | 1a, H | DGC-3.0-1.0-72 | 60 | 91 | 77 |
| 20c | 1a, H | DGC-3.0-1.0-72 | 60 | 78 | 66 |
Furthermore, several experiments were conducted under the same conditions in the presence of reused catalysts, as shown in Table 2 (entries 9, 10, 19, and 20). After each run, the catalyst was separated by centrifugation, washed and reused without calcination for the rearrangement of 1a. SA-DGC-30-1.0-72 could be recycled at least 5 times without losing its activity. However, the activity of DGC-3-1.0-72 began to decline in runs 2 and 3; for example, the conversions of 1a decreased to 91% and 78%, respectively. SA-DGC-30-1.0-72 exhibited higher yields than DGC-3-1.0-72 in all the reactions. Owing to the sensitivity of phenylacetaldehyde to acidic reaction conditions, which leads to competing aldol and polymerisation reactions, catalysts displaying high acidity and diffusional problems are easily deactivated.48 On one hand, SA-DGC-30-1.0-72 had mild strong acid sites (395 °C), as shown in NH3-TPD; on the other hand, nano-sized SA-DGC-30-1.0-72 can alleviate diffusion limitations via a shorter diffusion distance of reactants, reducing the probabilities of side reactions and coke deposition.
4. Conclusions
Hierarchical nano-ZSM-5 zeolites with high yields (approaching 100%) and crystallinity are synthesized using a seed-assisted dry-gel conversion (SA-DGC) method. Compared with the traditional method, only a small amount of TPA+ (SiO2/TPA+ = 20–30) is used for the synthesis of the protozeolite seeds without involving any SDAs in the subsequent processes, and a very small amount of water (0.2–1.0 mL) is required to create the steam for the crystallization conditions. The precursor can be covered within a thin layer of water owing to capillary condensation of water and transformed into nano-crystals around the micro region of seeds. Owing to the high-quality nanosized crystals (100–200 nm), mesopores (15–20 nm) and mild strong acid sites (395 °C), the as-prepared nano-ZSM-5 catalyst (i.e., SA-DGC-30-1.0-72) can alleviate diffusion limitations via a shorter diffusion distance of reactants, exhibiting higher conversions, yields and lifetime in the rearrangement of styrene oxide and its derivatives than the conventional microporous ZSM-5 catalyst (i.e., DGC-3-1.0-72).Data availability
Data for this article are available at https://www.scidb.cn/en/s/VbUnUb.Author contributions
Ming-Lei Gou: formulation of overarching research goals and aims, writing original draft preparation, reviewing and editing. Xuan Ru: experiments and other research outputs. Yongli Yang: development of methodology and analysis of study data. Zhen Liu: investigation, methodology and formal analysis. Shuge Peng: review, editing, supervision, and resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 22175057) and Experimental Technology Development Fund of Henan University of Science and Technology (SY2425045).References
- E. Perez-Botella, S. Valencia and F. Rey, Chem. Rev., 2022, 122, 17647–17695 CrossRef CAS.
- S. Lee, Y. Park and M. Choi, ACS Catal., 2024, 14, 2031–2048 CrossRef CAS.
- S. Xu, P. Dugkhuntod, S. Ding, Y. Zhang, P. Gosalvitr, S. Chen, J. Huang, S. Klinyod, S. Chansai, C. Hardacre, C. Wattanakit and X. Fan, Appl. Catal. B Environ. Energy, 2024, 348, 123826 CrossRef CAS.
- A. C. P. Guimaraes, L. R. F. Coelho, A. A. A. Silva, Y. Xing, R. C. Colman, L. V. Mattos and C. A. Henriques, Catal. Today, 2025, 447, 115159 CrossRef CAS.
- G. Song, D. Xue, J. Xue and F. Li, Microporous Mesoporous Mater., 2017, 248, 192–203 CrossRef CAS.
- K. Fu, G. Li, F. Xu, T. Dai, W. Su, H. Wang, T. Li, Y. Wang and J. Wang, Nanomaterials, 2023, 13, 1923 CrossRef CAS PubMed.
- M. Hartmann, A. Machoke and W. Schwieger, Chem. Soc. Rev., 2016, 45, 3313–3330 RSC.
- M. Pan, X. Jiang, Y. Pang, X. Pan, Q. Zhang, R. Yang and J. Zheng, Appl. Catal., A, 2023, 656, 119132 CrossRef CAS.
- S. Mintova, M. Jaber and V. Valchev, Chem. Soc. Rev., 2015, 44, 7207–7233 RSC.
- A. Javdani, J. Ahmadpour and F. Yaripour, Microporous Mesoporous Mater., 2019, 284, 443–458 CrossRef CAS.
- B. Gao, P. Tian, M. Li, M. Yang, Y. Qiao, L. Wang, S. Xu and Z. Liu, J. Mater. Chem. A, 2015, 3, 7741–7749 RSC.
- P. Feng, C. Kang, X. Yue, Z. Zhou, Z. Liu, Y. Gai, J. Shi and B. Zong, RSC Adv., 2024, 14, 19264–19270 RSC.
- Y. Zhao, L. Wang, Z. Ye, H. Zhang, S. Xie, Y. Zhang and Y. Tang, Cryst. Growth Des., 2019, 19, 6192–6198 CrossRef CAS.
- Y. Zhao, Z. Ye, L. Wang, H. Zhang, F. Xue, S. Xie, X. M. Cao, Y. Zhang and Y. Tang, Cryst. Growth Des., 2018, 18, 1101–1108 CrossRef CAS.
- Y. Zhao, H. Zhang, P. Wang, F. Xue, Z. Ye, Y. Zhang and Y. Tang, Chem. Mater., 2017, 29, 3387–3396 CrossRef CAS.
- H. Zhang, H. Zhang, Y. Zhao, Z. Shi, Y. Zhang and Y. Tang, Chem. Mater., 2017, 29, 9247–9255 CrossRef CAS.
- D. Jin, G. Ye, J. Zheng, W. Yang, K. Zhu, M.-O. Coppens and X. Zhou, ACS Catal., 2017, 7, 5887–5902 CrossRef CAS.
- Y. Jia, J. Wang, K. Zhang, W. Feng, S. Liu, C. Ding and P. Liu, Microporous Mesoporous Mater., 2017, 247, 103–115 CrossRef CAS.
- Q. Zhang, A. Mayoral, O. Terasaki, Q. Zhang, B. Ma, C. Zhao, G. Yang and J. Yu, J. Am. Chem. Soc., 2019, 141, 3772–3776 CrossRef CAS PubMed.
- Y. Shao, Y. Wang, X. Liu, T. Li, P. R. Haydel, T. Tatsumi and J. Wang, ChemCatChem, 2020, 12, 2702–2707 CrossRef CAS.
- D. Fan, Y. Diao, K. Cao, L. Sun, S. Xu, P. Tian and Z. Liu, Microporous Mesoporous Mater., 2020, 300, 110156 CrossRef CAS.
- Q. Wu, X. Meng, X. Gao and F. S. Xiao, Acc. Chem. Res., 2018, 51, 1396–1403 CrossRef CAS.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef CAS.
- X. Y. Yang, L. H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B. L. Su, Chem. Soc. Rev., 2017, 46, 481–558 RSC.
- L. Bu, Y. Wang, W. Liu, K. Chu, N. Guo, Y. Huang, L. Qu, X. Su, X. Zhang and Y. Li, Appl. Catal., A, 2023, 665, 119393 CrossRef CAS.
- Y. Jia, J. Wang, K. Zhang, G. Chen, Y. Yang, S. Liu, C. Ding, Y. Meng and P. Liu, Powder Technol., 2018, 328, 415–429 CrossRef CAS.
- K. M. Leung, P. Edwards, E. Jones and A. Sartbaeva, RSC Adv., 2015, 5, 35580–35585 RSC.
- B. Xie, H. Zhang, C. Yang, S. Liu, L. Ren, L. Zhang, X. Meng, B. Yilmaz, U. Muller and F. S. Xiao, Chem. Commun., 2011, 47, 3945–3947 RSC.
- S. Inagaki, S. Shinoda, Y. Kaneko, K. Takechi, R. Komatsu, Y. Tsuboi, H. Yamazaki, J. N. Kondo and Y. Kubota, ACS Catal., 2013, 3, 74–78 CrossRef CAS.
- H. Zhang, L. Wang, D. Zhang, X. Meng and F. S. Xiao, Microporous Mesoporous Mater., 2016, 233, 133–139 CrossRef CAS.
- H. Zhang, C. Wu, M. Song, T. Lu, W. Wang, Z. Wang, W. Yan, P. Cheng and Z. Zhao, Microporous Mesoporous Mater., 2021, 310, 110633 CrossRef CAS.
- S. Prodinger, I. C. Berdiell, T. Cordero-Lanzac, O. R. Bygdnes, B. G. Solemsli, K. Kvande, B. Arstad, P. Beato, U. Olsbye and S. Svelle, J. Mater. Chem. A, 2023, 11, 21884–21894 RSC.
- Q. Zhang, J. Li, X. Wang, G. He, L. Li, J. Xu, D. Mei, O. Terasaki and J. Yu, J. Am. Chem. Soc., 2023, 145, 21231–21241 CrossRef CAS PubMed.
- Y. Guo, R. Wang, L. Zhong, T. Fu and Z. Li, Appl. Catal., A, 2025, 689, 120008 CrossRef CAS.
- T. Ge, Z. Hua, X. He, J. Lv, H. Chen, L. Zhang, H. Yao, Z. Liu, C. Lin and J. Shi, Chem.–Eur. J., 2016, 22, 7895–7905 CrossRef CAS PubMed.
- X. Chen, R. Jiang, Y. Gao, Z. Zhou and X. Wang, CrystEngComm, 2021, 23, 2793–2800 RSC.
- Z. Chen, Z. Li, Y. Zhang, D. Chevella, G. Li, Y. Chen, X. Guo, J. Liu and J. Yu, Chem. Eng. J., 2020, 388, 124322 CrossRef.
- Z. Buniazet, A. Cabiac, S. Maury, D. Bianchi and S. Loridant, Appl. Catal. B Environ. Energy, 2019, 243, 594–603 CrossRef CAS.
- L. Meng, B. Mezari, M. G. Goesten, W. Wannapakdee, R. Pestman, L. Gao, J. Wiesfeld and E. J. M. Hensen, Catal. Sci. Technol., 2017, 7, 4520–4533 RSC.
- L. Tao, L. Chen, S. F. Yin, S. L. Luo, Y. Q. Ren, W. S. Li, X. P. Zhou and C. T. Au, Appl. Catal., A, 2009, 367, 99–107 CrossRef CAS.
- Y. Jia, Q. Shi, J. Wang, C. Ding and K. Zhang, RSC Adv., 2020, 10, 29618 RSC.
- J. Cao, G. Qi, B. Yao, Q. He, R. J. Lewis, X. Li, F. Deng, J. Xu and G. J. Hutchings, ACS Catal., 2024, 14, 1797–1807 CrossRef CAS.
- J. L. Jat and G. Kumar, Adv. Synth. Catal., 2019, 361, 4426–4441 CrossRef CAS.
- Y. Zhang, B. Hu, Y. Chen and Z. Wang, Chem.–Eur. J., 2024, 30, e202402469 CrossRef CAS PubMed.
- L. A. Gallego-Villada, J. Cueto, M. D. Alonso-Doncel, M. A. Paivi, E. A. Alarcon, D. P. Serrano and D. Y. Murzin, Green Chem., 2024, 26, 10512–10528 RSC.
- M. L. Gou, J. Cai, W. Song, Z. Liu, Y. L. Ren and Q. Niu, RSC Adv., 2017, 7, 44247–44253 RSC.
- M. L. Gou, J. Cai, W. Song, Y. Duan, Z. Liu and Q. Niu, Microporous Mesoporous Mater., 2020, 297, 110037 CrossRef.
- T. E. Davies, S. A. Kondrat, E. Nowicka, J. L. Kean, C. M. Harris, J. M. Socci, D. C. Apperley, S. H. Taylor and A. E. Graham, Appl. Catal., A, 2015, 493, 17–24 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03351a |
| This journal is © The Royal Society of Chemistry 2025 |