
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of surface functionalization on the electrosynthesis of molecularly imprinted polymers (MIPs) and the detection of per- and polyfluoroalkyl substances (PFAS)†
Daniel A. Bellido-Aguilar,
McKenna Dunmyer,
Cameron S. Malloy,
Matthew J. Danley,
Vasiliki Karanikola and
Suchol Savagatrup*
and
Suchol Savagatrup*
Department of Chemical and Environmental Engineering, University of Arizona, 1133 E. James E. Rogers Way, Tucson, Arizona 85721, USA. E-mail: suchol@arizona.edu
First published on 16th June 2025
Abstract
Recently, the US Environmental Protection Agency (EPA) has established stringent maximum contaminant levels (MCLs) for multiple per- and polyfluoroalkyl substances (PFAS) in drinking water. These compounds pose substantial environmental and health risks due to their bioaccumulative properties. While low concentrations can be detected quantitatively and selectively by liquid chromatography with tandem mass spectrometry (LC-MS/MS), this technique is cost-prohibitive, time-consuming, and not suitable for rapid and on-site measurements. Electrochemical sensors have the potential to provide a fast and portable alternative with sufficient selectivity and sensitivity for early screening of potential contaminated sources. These sensors rely on a layer of molecularly imprinted polymers (MIPs) that are synthesized through electrochemical oxidation of monomers (e.g., o-phenylenediamine, o-PD) in the presence of targeted molecules (e.g., perfluorooctane sulfonic acid, PFOS) as the template for selective binding sites. In this study, we test the hypothesis that the physicochemical properties of the electrode surface dictate the electropolymerization of MIPs and the resulting physical morphology and sensing properties. Specifically, MIP-based sensors prepared on hydrophobic surfaces exhibit improved sensing performance toward PFOS than the ones prepared on hydrophilic surfaces. We attribute the increased sensitivity to the stronger attraction of the hydrophobic surfaces to PFOS during the electropolymerization, which leads to enhanced imprinting of the MIPs and more selective binding sites. Our results, with PFOS as a model compound, demonstrate the importance of surface functionalization to the formation, physical morphologies, and sensing properties of a promising class of materials for environmental monitoring.
1. Introduction
Per- and polyfluoroalkyl substances (PFAS) are synthetic chemicals that have brought environmental concerns due to their toxicity and non-biodegradability.1 Specifically, toxicological studies have revealed that they are endocrine disruptors and potential human carcinogens.2,3 Due to their widespread applications, PFAS have been detected in water sources and soil, posing a serious threat to the ecosystem.4 One of the most studied PFAS is perfluorooctane sulfonic acid (PFOS).5 The World Health Organization (WHO) recommends a maximum concentration of PFOS in drinking water of 100 ppt (or 100 ng L−1).6 Additionally, the US Environmental Protection Agency (EPA) has recently proposed a regulatory limit of 4 ppt (4 ng L−1).7 While the current gold-standard technologies—such as liquid chromatography with tandem mass spectrometry (LC-MS/MS)—are capable of detecting such concentrations, they are often impractical due to high cost, lengthy procedures, and lack of portability for on-site detections.8–11 Therefore, there are demands for new and alternative sensing platforms that provide cost-effective and rapid detection to complement traditional analytical instruments.12An emerging area of alternative sensors for PFAS combines electrochemical platforms with molecularly imprinted polymers (MIPs).13,14 MIPs are thin polymeric coatings that are polymerized in the presence of the targeted analytes as the templating molecules.15,16 These templating molecules can then be removed by either a physical or chemical treatment to create selective molecular cavities.17 In ideal situations, the selectivity of MIPs toward the templating molecules mimics the lock-and-key mechanisms found in antigen–antibody interactions.18 For example, MIPs have been incorporated into various sensing platforms for PFAS, including electrochemical,19–23 photoelectrochemical,24–27 and photoluminescent.28–30 These reported sensors have demonstrated their capability to selectively detect PFOS with sufficiently low limits of detection (∼1 ng L−1). Furthermore, MIP-based sensing platforms are transportable and convenient, with fast analytical time (∼minutes).
An important principle in the development of MIP-based sensors has been the rational selection of the monomers and the templating molecules, based on their physicochemical properties. That is, a functional MIP can be achieved when the monomers and the templating molecules form favorable interactions, to generate selective molecular cavities within the polymer matrix. For example, several intermolecular interactions are possible between the commonly used monomer, ortho-phenylenediamine (o-PD), and PFOS. PFOS can form hydrogen bonds and electrostatic interactions when ionized through its polar sulfonic group, and hydrophobic interactions through its fluorinated alkyl chain.31 Therefore, the selected monomer for the preparation of MIPs must be able to form hydrogen bonds or hydrophobic interactions with PFOS.32
However, the monomer–template interaction is not the only interaction to consider if MIP sensors are electrochemically synthesized on an electrode. The surface energy of the electrode will also impact the electropolymerization of MIPs and the formation of selective binding sites.33,34 For example, Nishizawa et al. evaluated the effect of hydrophobic and hydrophilic surfaces on the electropolymerization of pyrrole for electronic applications.33 Their results showed that lateral growth of polypyrrole occurred only on hydrophobic surfaces but not on hydrophilic surfaces. Similarly, Gerasimov et al. studied the effect of surface modification on the electropolymerization of a thiophene-based monomer in the preparation of transistors.34 They functionalized the surface with hydrophobic molecules and with molecules bearing positive or negative charges. Their results demonstrated that the polymerization varied according to the surface nature. While these reports have focused only on the interaction between the monomer and the modified electrodes, the development of electrosynthesized MIP-based sensors for PFOS requires the understanding of the simultaneous interactions of three components: the monomers, the electrode surface, and the template molecules (PFOS). Therefore, the monomer–template, monomer–electrode, and template–electrode interactions must be considered for the electrochemical preparation of MIP.
Here, we investigated the effects of surface properties of gold electrodes on both the electropolymerization of MIP and its sensing properties. Specifically, we modified the gold electrodes by surface functionalization with polar and non-polar molecules and evaluated their interactions with the monomers (i.e., o-PD) and the templating molecules (i.e., PFOS). Since PFOS and o-PD can form polar and non-polar intermolecular bonds, they can have different interfacial behaviors depending on the surface energy of the electrodes during polymerization. Therefore, we aim to understand the relationship between the electrode surface properties and the structures of MIPs, to enhance the sensing performance by using PFOS as a model compound. Considering only the template–electrode interactions, we hypothesized that the strong affinity of PFOS to hydrophobic surfaces, due to their long-fluorinated alkyl chain, would enhance the formation of selective cavities and increase sensitivity.35,36 In relation to the monomer–template interactions, our results showed that different polymer coatings are formed depending on the electrode surface chemistry. Overall, our results demonstrated that the surface properties of the electrode significantly impact the formation and the sensing properties of MIPs.
2. Results and discussion
2.1 Functionalization and characterization of electrodes
We began by preparing hydrophobic and hydrophilic electrodes to study the interaction of PFOS and o-PD with the electrode surface during electrosynthesis, using methods adapted from previous reports (Fig. 1).37,38 Briefly, we functionalized UV-cleaned gold electrodes by soaking them in a solution containing either 1-octanethiol (OT) or L-cysteine (Cys) for 24 hours, followed by thorough rinsing and drying steps. OT and Cys were chosen because they can form a hydrophobic and a hydrophilic surface, respectively. | ||
Fig. 1 Schematic of the preparation of gold electrodes prior to electrosynthesis of molecularly imprinting polymers (MIPs). Hydrophilic and hydrophobic gold electrodes were prepared by surface functionalization of L-cysteine (Cys) and 1-octanethiol (OT), respectively. The polymerization of o-PD in the presence of PFOS was then carried out on the functionalized gold electrodes. The template removal was carried out by solvent extraction using a mixture of water and methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). :1, v/v). | ||
We characterized the functionalized surfaces by first measuring the water contact angles (Fig. 2a). Prior to functionalization, UV-cleaning treatment produced a bare gold electrode (Au) with hydrophilic properties (20.55° ± 2.12°). Similarly, the water contact angle of L-cysteine functionalized gold electrode (Au–Cys) was 15.85° ± 1.31°, indicating a hydrophilic coating as expected. The hydrophilicity of Cys arises from the presence of the amino and carboxyl groups. On the other hand, gold electrode with 1-octanethiol (Au–OT) exhibited hydrophobic properties with a water contact angle of 103.40° ± 0.59°, which was expected for a surface functionalized with long alkyl chains.39 To confirm the functionalization, we then used X-ray photoelectron spectroscopy (XPS) to characterize the chemical properties of the different electrode surfaces. XPS results of the bare Au showed the presence of carbon and oxygen, which we attributed to the natural contamination of the surface (Table S1†). In the case of Au–Cys, the XPS results showed the additional presence of nitrogen and sulfur. Moreover, its XPS S 2p spectrum showed a characteristic doublet, indicative of the presence of thiol groups arising from the presence of Cys on the gold electrode (Fig. 2b). Similarly, the XPS spectrum of Au–OT also detected a sulfur peak with a characteristic doublet. We attributed these results to the presence of both S–Au and S–H with the binding energies of 162.5 and 164 eV, respectively.
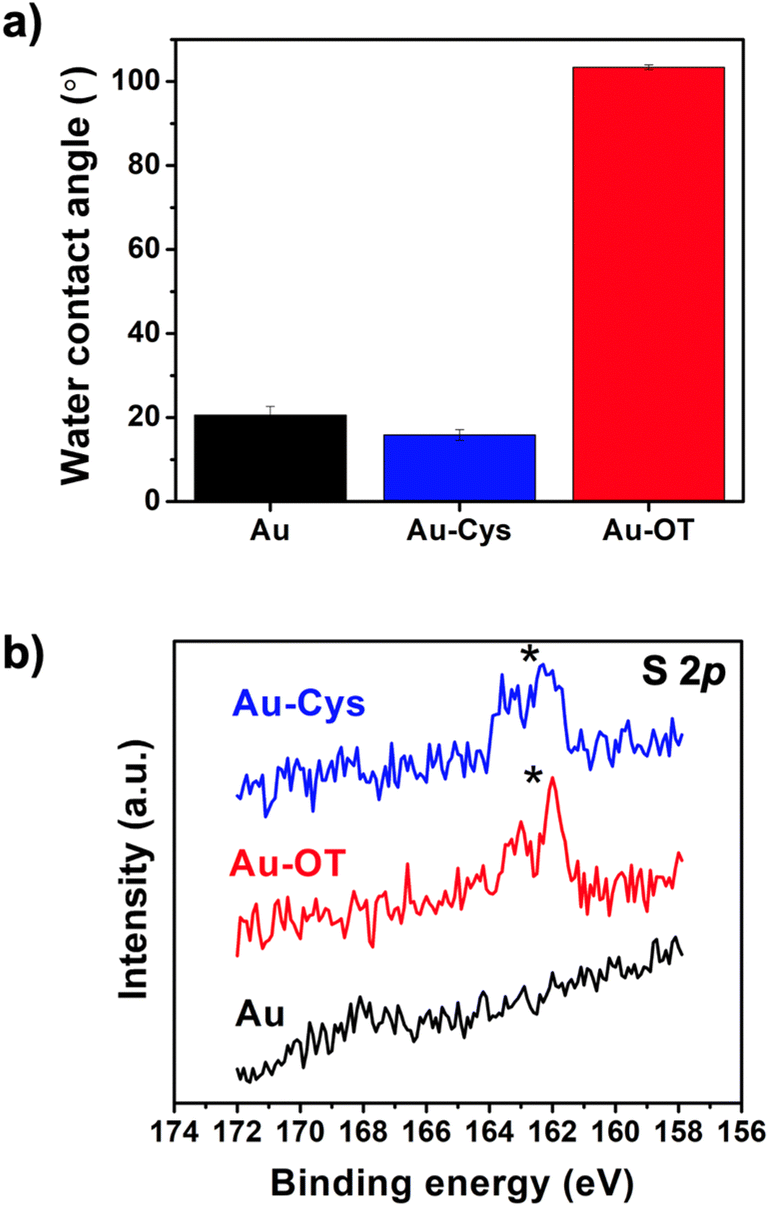 | ||
| Fig. 2 (a) Water contact angles of the bare Au and the Cys and OT functionalized Au electrodes, (b) XPS S 2p spectrum of Au, Au–Cys, and Au–OT. The appearance of doublets indicates the presence of thiol groups. | ||
2.2 Preparation and characterization of MIP films
After functionalization of the electrodes, we sought to evaluate their effects on both the physical and sensing properties of the MIP films. Thus, we fabricated poly(o-phenylenediamine) (PoPD) MIP films from a solution of o-PD with PFOS as the templating molecules. We selected o-PD for this study because it has been previously shown to produce functional MIP-based sensors for legacy PFAS, such as PFOS.20,21,40 We adapted the synthesis parameters of the anodic electrochemical polymerization from previous reports.20,21,40 Briefly, we prepared a solution containing 10 mM of o-PD and 0.05 mM of PFOS in a 2:1 (v/v) mixture of acetate buffer (pH = 5.8) and methanol solution. We reduced the relative concentration of PFOS, in comparison to previous reports, to minimize the formation of defects and nonspecific binding sites that could lead to lower sensitivity and signal drifts.41 The polymerization was carried out via cyclic voltammetry (CV) by scanning 25 cycles in the potential range of 0 to 1.0 V (vs. Ag/AgCl) at 50 mV s−1 to polymerize MIP films onto the gold electrodes. We then removed the templating PFOS by soaking the coated electrodes in a mixture of methanol and water (1:1, v/v) for 20 minutes. Full details of the synthesis parameters are provided in the ESI.† We hypothesized that the surface chemistry of the electrode dictates the interfacial behaviors and the localized concentration of PFOS.35,36 More importantly, we expected that the changes in surface energy will also affect the electropolymerization of o-PD and imprinting process of PFOS into the PoPD MIP films.33,34 For each functionalized surface, we also fabricated non-imprinted polymer films (NIPs) as the controls by electropolymerizing o-PD using the same synthetic parameters without the presence of PFOS.
We first evaluated the molecular imprinting of PFOS within the MIP films fabricated on each surface via XPS. Specifically, we observed the presence of fluorine in MIP-Au, MIP-Cys, and MIP-OT before the template removal step and the absence of F 1s peak after the template removal (Table S1†). While these results only suggested that PFOS imprinting may have occurred and the templating molecules were mostly removed from the MIP surfaces, we will further evaluate the formation of selective binding sites via electrochemical methods in the next section. We then measured the film thickness and surface roughness using a stylus profilometer and atomic force microscopy (AFM) in tapping mode, respectively. Due to the thinness of most of the samples, we focused primarily on surface characterizations instead of the bulk characteristic. Similar to previous reports by our group, we observed that the thickness of an MIP film after undergoing a template removal (TR) process via solvent extraction was significantly reduced compared to the freshly synthesized film (no template removal, NTR) (Fig. 3a).41 This reduction in thickness is associated with the removal of weakly adhered polymer on the surface.42,43 We also observed a clear deviation in film thickness for MIP films fabricated on electrodes functionalized with OT (MIP-OT). Specifically, the hydrophobic OT surface resulted in 200 nm MIP films, compared to <10 nm films on bare Au (MIP-Au) and Au–Cys (MIP-Cys). Interestingly, the hydrophobic surface of OT did not lead to a significant increase in thickness for the control NIP film (NIP-OT). This result suggested that the polymerization and imprinting process of MIP films were affected by the strong interactions of PFOS toward the hydrophobic surfaces.44,45 Despite the large difference in thickness for MIP-OT, the surface roughness after TR (both average and RMS roughness, Ra and Rq) for all MIP and NIP films were relatively smooth (<3 nm) (Fig. 3b) with minor differences in the grain size of the surface morphology (Fig. S1, ESI†).
 | ||
| Fig. 3 (a) Thickness values of the polymer coatings. The abbreviations NTR and TR stand for no template removal and template removal steps. (b) Roughness values expressed as Rq (RMS roughness) and Ra (average roughness) of the polymer coatings after template removal step obtained from 1 × 1 μm2 AFM images. | ||
To uncover the underlying causes of the different physical properties of MIP films fabricated on OT, we started by analyzing the cyclic voltammograms (CV) of the functionalized electrodes during the electropolymerization (Fig. 4). Two oxidation peaks were observed for the polymerization of o-PD on bare gold (MIP-Au, 0.65 V and 0.85 V) and Cys (MIP-Cys, 0.65 V and 0.93 V), which were consistent with previous reports.20,21,40 These peaks are related to the chain propagation of o-PD polymerization by oxidative coupling and intramolecular oxidations.46–48 We observed that the peak at 0.93 V of MIP-Cys exhibited a higher current than that of MIP-Au, which we suspected to be the result of additional oxidation from Cys. On the other hand, the CV of MIP-OT showed only one distinctive oxidation peak at 0.61 V, followed by a shoulder during the first and second scans. This result suggested that OT had a distinct effect on the electropolymerization of o-PD when compared to Cys. While these results indicated that surface functionalization affected MIP synthesis, additional control experiments were conducted to explore the underlying mechanisms.
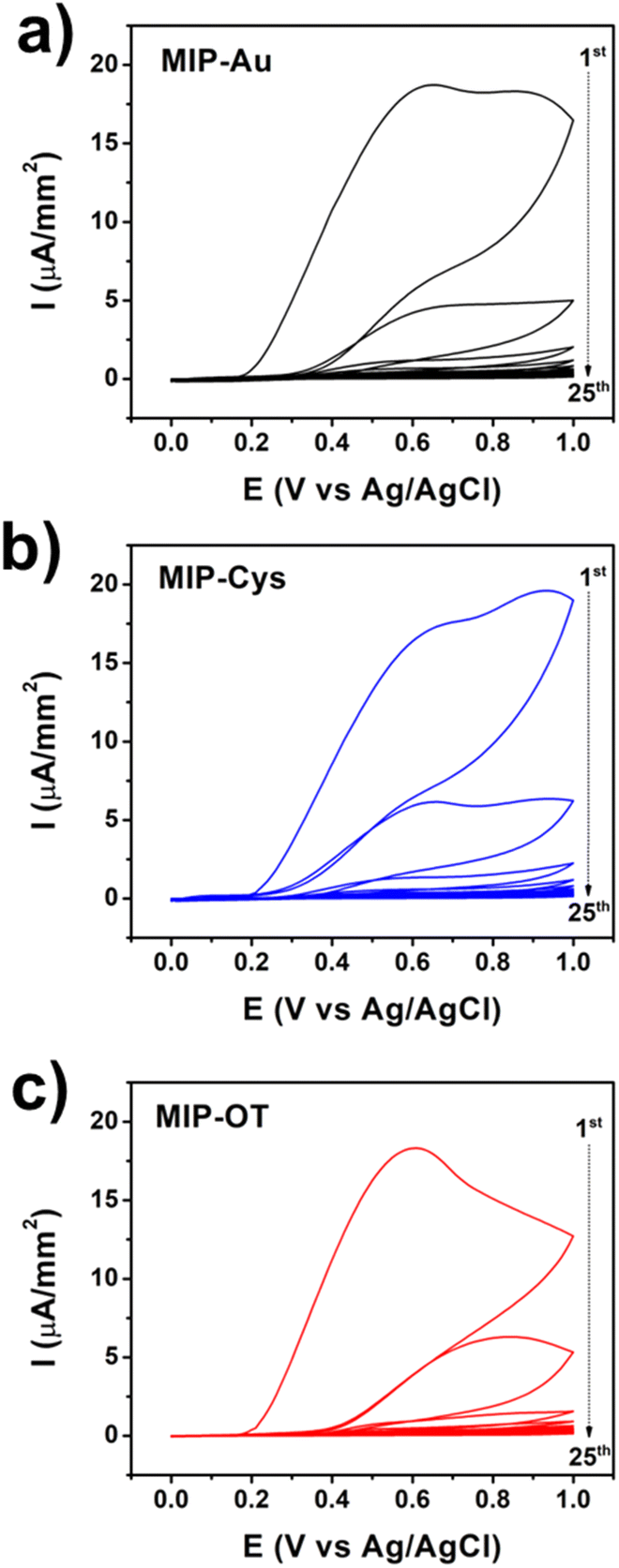 | ||
| Fig. 4 Representative CV curves (25 scans) of the synthesis of MIP samples for (a) MIP-Au, (b) MIP-Cys, and (c) MIP-OT. The arrows indicate the sequence of CV scans. | ||
As a first control, we collected the electrochemical responses of the functionalized surfaces without the presence of o-PD monomers to isolate the responses of the surfaces with and without PFOS. For samples without PFOS, we observed two oxidation peaks in the first cycles for Au–Cys (0.7 V and 1.0 V) and a single peak for Au–OT (1.0 V), which were absent on bare Au electrodes (ESI, Fig. S2a, c and e†). These processes are indicative of the oxidation of thiol groups in Cys and OT, respectively.38,49 Specifically, they indicated the oxidative desorption of both Cys and OT, which was confirmed by the drastic reduction of the S 2p peaks in XPS (Fig. S3†). Furthermore, these results showed that almost all Cys molecules were quickly oxidized and desorbed in the first cycle. However, we observed that the oxidative desorption of OT occurred at a much slower rate and took multiple cycles to complete. We attributed this observation to the different oxidative potentials of Cys and OT, where Cys oxidizes at a lower potential than that of OT. In fact, the oxidative desorption of OT occurs at a potential higher than the potential window used in this study (ESI, Fig. S2g†). This difference in the oxidative potentials of Cys and OT may have also contributed to the distinctive behavior of the electropolymerization of MIP-OT. We proposed that the lack of oxidative desorption of OT from the surface at lower potential resulted in the attenuation of the second peak, in comparison to those of MIP-Au and MIP-Cys (Fig. 4). Interestingly, in the presence of PFOS, we observed that the oxidation of OT was further delayed as indicated by the higher number of cycles before achieving the characteristic cyclic voltammetry curve of the buffer (ESI, Fig. S2f†). In contrast, PFOS did not attenuate the oxidation of Cys. A plausible explanation of the attenuation effect by PFOS is its strong interaction with OT, which may arise from the surfactant properties of PFOS, specifically at the liquid–solid interface.44,45
In the second set of control experiments, we fabricated non-imprinted polymer (NIP) films to evaluate the interactions of o-PD monomers with the functionalized surfaces in the absence of PFOS (ESI, Fig. S4†). We observed that the attenuation in synthesis current over multiple cycles was much slower for the OT functionalized surface, compared to Cys and bare gold surfaces. We attributed this observation to the physical resistance provided by OT to prevent o-PD monomers from reaching the surface and polymerizing. This transport limitation was expected because OT is larger than Cys. We also observed similar behavior when the surface is functionalized with thiols with an even longer alkyl group (ESI, Fig. S5†). In contrast, polymerization of o-PD on bare gold and Cys functionalized surfaces occurred rapidly, as observed by the rapid attenuation in CV current over just two cycles. Additionally, XPS results of NIP-Cys after template removal showed complete desorption of Cys, while OT persisted in the sample of NIP-OT (ESI, Fig. S6†). This observation indicated that the polymerization of o-PD prevents the complete oxidative desorption of OT. Moreover, we conducted XPS to evaluate if incomplete oxidative desorption of OT occurred during the synthesis of MIP-OT as it happened in NIP-OT. Due to the thickness of the MIP-OT film (∼200 nm), XPS results of the fully fabricated films showed no presence of OT. However, XPS results (ESI, Fig. S7†) of thin MIP-OT prepared with one and two synthesis scans suggested that some amount of OT remained on the surface of gold during the polymerization. Moreover, we have attributed the higher thickness of MIP-OT (∼200 nm) to the favorable conditions for o-PD polymerization to grow thicker when PFOS and OT are present.
2.3 Sensing performance of MIP-based sensors
We then evaluated the PFOS sensing performance of the MIP coatings by measuring the oxidative currents of the redox probe (ferrocene carboxylic acid, FcCOOH) at different PFOS concentrations. Since PFOS is not electrochemically active, the oxidative current from the redox probe decreases with increasing concentration of PFOS due to the transport competition between PFOS and the redox probe.20,21,23,41 Specifically, we measured the change in current density (ΔJ = J0 − Ji), where J0 and Ji are the current densities at zero concentration of PFOS and at a specific concentration of PFOS, respectively (Fig. 5a). We characterized the sensing performance in terms of sensitivity (the absolute values of the fitting slope of the calibration curves, Fig. 5b) and the change in current at the lowest tested concentration of 1 nM of PFOS (Fig. 5c). For the tested concentration range (from 0 to 1000 nM), we observed a single linear relationship between ΔJ and PFOS concentration, which is consistent with our previous results.41 However, previously reported calibration curves comprising a larger concentration range have exhibited two linear regions.20,21 The observed slope values indicated that the PFOS sensitivity decreased as MIP-OT > MIP-Cys > MIP-Au. Additionally, the values of ΔJ at 1 nM of PFOS showed a similar trend of MIP-OT > MIP-Cys ∼ MIP-Au. We note here that 1 nM of PFOS was the lowest non-zero concentration tested for this study. No further optimization of the sensors was performed to isolate the variable of surface functionalization and to produce a fair comparison. From these two metrics of performance, we observed that the hydrophobic MIP-OT outperformed both MIP-Cys and MIP-Au, which were hydrophilic surfaces. This result suggested the importance of understanding the effects of surface functionalization in the optimization of electrochemical fabrication of MIPs, in addition to other parameters traditionally explored (e.g., scan rate, potential window, molar ratio, pH).20,41 A plausible explanation for the positive effect of hydrophobic functionalization is the favorable molecular imprinting condition. A key requirement to obtain imprinting cavities is that PFOS must be attracted to the electrode surface during electropolymerization. PFOS had a stronger attraction to Au–OT than Au–Cys (ESI, Fig. S2d and f†). To demonstrate the preference of PFOS on hydrophobic surfaces, we obtained the absorption profiles of PFOS on hydrophobic and hydrophilic gold surfaces using a quartz crystal microbalance with dissipation (QCM-D) (ESI, Fig. S9†). The results showed a mass increase (frequency decay) in the Au–OT surface but almost no mass change was detected in Au and Au–Cys. These results demonstrated that PFOS was deposited on Au–OT but not on the hydrophilic surfaces (Au and Au–Cys), which agreed very well with the electrochemical results described in previous sections and our previous results.35,36 Therefore, our results suggest that better imprinting conditions are obtained in hydrophobic surfaces, and therefore, the MIP sensors exhibited enhanced sensing performances. | ||
| Fig. 5 (a) Calibration curves of MIP samples. Straight lines are the linear fittings. (b) Absolute slope values of the linear fittings. (c) Absolute ΔJ values at 1 nM PFOS. Statistical analysis was performed using on-way ANOVA; differences were considered significant when P ≤ 0.05 (“*”). | ||
A potential limitation to MIP-based sensors is the confounding signals arising from non-specific binding due to defects and non-uniformity of the polymeric films. To this end, we observed that our non-imprinted control (NIP) samples also showed responses to PFOS (ESI, Fig. S8†). We attributed this non-specific response to the presence of defects in the NIP samples, which were consistent with our previous work.41 We have previously shown that film defects increase with faster synthesis scan rates, which control the rate of electropolymerization and quality of molecular imprinting. This observation also implied that these NIP samples exhibited wildly different morphologies than the MIP samples and ultimately were not a true control. Therefore, we attributed these apparent sensing properties of NIP samples to the non-specific adsorption of PFOS onto defective sites of the NIP surface. A similar result was found in our recent report on the relationship mechanical and sensing properties of MIP sensors.41 Thus, further research is needed to decouple the PFOS absorption signals originating from specific and non-specific interactions on the polymeric sensors and evaluate their implications on the sensing mechanism. Additionally, complexities arising from interfering effects from real-world samples must be evaluated and mitigated to enhance the robustness of such sensing technology.
3. Conclusions
We tested the hypothesis that the physicochemical properties of the electrode will affect the PFOS sensing properties of the electropolymerized MIPs. The PFOS sensing performance of MIPs was studied on coatings prepared on bare and functionalized Au electrodes with a hydrophobic OT and a hydrophilic Cys. Electrochemical and QCM-D results showed that PFOS had a strong attraction with OT while weak interaction with Cys. Moreover, the MIP prepared on hydrophobic surfaces (MIP-OT) exhibited higher PFOS sensitivity than samples prepared on hydrophilic surfaces (MIP-Cys, MIP-Au). This result highlights the importance of the electrode's surface chemistry when designing MIP sensors.4. Experimental details
4.1 Materials
Perfluorooctane sulfonic acid (PFOS) solution (40% in H2O), L-cysteine (97%), 1-octanethiol (98.5%), and ferrocene carboxylic acid (FcCOOH, 97%) were obtained from Millipore Sigma. Ortho-Phenylenediamine (o-PD, 98%) was purchased from Thermo Scientific Chemicals. All solvents were obtained from Millipore Sigma. All chemicals were used as received without further purification.4.2 Preparation of electrodes
Gold electrodes were prepared on glass substrates via electron-beam physical vapor deposition with a thickness of 100 nm with 10 nm of chromium as the adhesive layer. The gold electrodes were cleaned by bath sonication in a detergent solution for 30 min, followed by rinsing with DI water, drying with a stream of compressed air, and 10 min of UV/ozone treatment. To functionalize the gold electrode, L-cysteine (Cys) and 1-octanethiol (OT) were used to create hydrophilic and hydrophobic surfaces, respectively. The clean electrodes were soaked in either a 10 mM solution of Cys in DI water or a 10 mM solution of OT in pure ethanol for 24 h. Afterward, the electrodes were then gently rinsed with DI water and sonicated in either DI water or ethanol for 10 min to remove excess Cys or OT. We chose to perform this study on gold electrodes because of the facile processes of functionalization with Cys and OT to generate hydrophilic and hydrophobic surfaces. Gold surfaces have also been extensively used for the preparation of MIP-based sensors for PFAS.20,21,40 Additionally, our group had reported the adhesion behaviors of PFAS on modified gold surfaces.354.3 Electrosynthesis of MIP
The electropolymerization of o-PD was performed on the pristine and functionalized gold electrodes, following previously reported methods with minor modifications.20,21,40,41 Specifically, we performed the synthesis in a three-electrode electrochemical cell with the gold electrode, a platinum sheet electrode, and a Ag/AgCl electrode as the working, counter, and reference electrodes, respectively. For all MIP samples, the concentrations of o-PD (the monomer) and PFOS (the templating molecule) were 10 mM of o-PD and 0.05 mM PFOS in the solution of acetate buffer (pH = 5.8) and methanol (2:1 v/v). The solution was mixed for 1 h prior to synthesis. The synthesis was carried out by cyclic voltammetry of 25 total scans from 0.0 to 1.0 V vs. Ag/AgCl at the scan rate of 50 mV s−1. Subsequently, the templating molecules were removed via a solvent treatment, which consisted of soaking in methanol for 15 s, followed by soaking in a 1:1 mixture of methanol and water for 20 min under mild stirring, and a final soaking in methanol for 30 s. As controls, the synthesis and treatment of NIP samples were identical to that of the MIP samples without PFOS.
4.4 Characterization
Elemental analysis was carried out by X-ray photoelectron spectroscopy (XPS) using a Kratos Axis Ultra X-ray photoelectron spectrometer with a pass energy of 20 eV. The X-ray source was Al Kα, operated at 15 kV and 20 mA. Water contact angle measurements were performed via a goniometer (Kruss Scientific DSA25E) with 5 μL DI water droplets. Thickness measurement of MIP and NIP layers were measured using a profilometer (KLA-Tencor 15) with at least seven measurements for each sample. Atomic force microscopy (Park NX20) under non-contact mode was used to obtain 0.5 × 0.5 μm2 and 1 × 1 μm2 topographic images and roughness values from five locations. The sensing properties of MIP towards PFOS were evaluated electrochemically by assessing the change in the measured current of the redox reaction of FcCOOH. Briefly, each tested concentration of PFOS solution comprised 0.5 mM of FcCOOH in 0.01 M ammonia buffer (pH = 8.4). The MIP-modified electrodes were submerged in the PFOS and FcCOOH solutions for 15 min prior to each measurement. We used cyclic voltammetry of 20 scans from 0.0 to 0.5 V vs. Ag/AgCl reference electrode at the scan rate of 50 mV s−1. The average of 20 scans was taken for each concentration. Quartz-crystal microbalance with dissipation (QCM-D) experiments were carried out by using a Q-Sense Explorer (Biolin Scientific) on pristine gold and functionalized gold sensors. We cleaned and functionalized the gold sensors in the same manner as the preparation for MIP-modified electrodes. For QCM-D measurement, an aqueous solution of PFOS at 0.05 mM was fed into the cells at a constant flow rate of 300 μL min−1 until steady state was reached. DI water was used to establish a baseline before the injection of PFOS solution.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Regents Research Grant awarded by the Arizona Board of Regents (ABOR). Additionally, this work was also supported by startup funds from the University of Arizona through the Department of Chemical and Environmental Engineering, The BIO5 Institute, and Research, Innovation, and Impact (RII). This work was performed in part at the Micro/Nano Fabrication Center (MNFC), the Keck Center for Nanoscale Imaging and the Laboratory for Electron Spectroscopy (LESSA) at the University of Arizona. Additional support was provided by the National Science Foundation Graduate Research Fellowship (NSF GRFP) under Grant No. DGE-2137419, awarded to C. S. M. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.References
- A. Florentin, T. Deblonde, N. Diguio, A. Hautemaniere and P. Hartemann, Impacts of Two Perfluorinated Compounds (PFOS and PFOA) on Human Hepatoma Cells: Cytotoxicity but No Genotoxicity?, Int. J. Hyg. Environ. Health, 2011, 214(6), 493–499, DOI:10.1016/j.ijheh.2011.05.010
.
- N. Singh and C. Y. J. Hsieh, Exploring Potential Carcinogenic Activity of Per- and Polyfluorinated Alkyl Substances Utilizing High-Throughput Toxicity Screening Data, Int. J. Toxicol., 2021, 40(4), 355–366, DOI:10.1177/10915818211010490
- D. Patch, N. O'Connor, T. Vereecken, D. Murphy, G. Munoz, I. Ross, C. Glover, J. Scott, I. Koch and S. Sauvé, et al., Advancing PFAS Characterization: Enhancing the Total Oxidizable Precursor Assay with Improved Sample Processing and UV Activation, Sci. Total Environ., 2024, 909, 168145, DOI:10.1016/j.scitotenv.2023.168145
- B. A. Schumacher, J. H. Zimmerman, A. C. Williams, C. C. Lutes, C. W. Holton, E. Escobar, H. Hayes and R. Warrier, Distribution of Select Per- and Polyfluoroalkyl Substances at a Chemical Manufacturing Plant, J. Hazard. Mater., 2024, 464(210), 133025, DOI:10.1016/j.jhazmat.2023.133025
- N. Hamid, M. Junaid, R. Manzoor, M. Sultan, O. M. Chuan and J. Wang, An Integrated Assessment of Ecological and Human Health Risks of Per- and Polyfluoroalkyl Substances through Toxicity Prediction Approaches, Sci. Total Environ., 2023, 905, 167213, DOI:10.1016/j.scitotenv.2023.167213
- E. Southerland and L. S. Birnbaum, What Limits Will the World Health Organization Recommend for PFOA and PFOS in Drinking Water?, Environ. Sci. Technol., 2023, 57(18), 7103–7105, DOI:10.1021/acs.est.3c02260
- US EPA, Per- and Polyfluoroalkyl Substances (PFAS) Final PFAS National Primary Drinking Water Regulation, https://www.epa.gov/sdwa/and-polyfluoroalkyl-substances-pfas.
- M. Trojanowicz, K. Bobrowski, B. Szostek, A. Bojanowska-Czajka, T. Szreder, I. Bartoszewicz and K. Kulisa, A Survey of Analytical Methods Employed for Monitoring of Advanced Oxidation/Reduction Processes for Decomposition of Selected Perfluorinated Environmental Pollutants, Talanta, 2018, 177, 122–141, DOI:10.1016/j.talanta.2017.09.002
- L. J. Winchell, M. J. M. Wells, J. J. Ross, X. Fonoll, J. W. Norton, S. Kuplicki, M. Khan and K. Y. Bell, Analyses of Per- and Polyfluoroalkyl Substances (PFAS) through the Urban Water Cycle: Toward Achieving an Integrated Analytical Workflow across Aqueous, Solid, and Gaseous Matrices in Water and Wastewater Treatment, Sci. Total Environ., 2021, 774, 145257, DOI:10.1016/j.scitotenv.2021.145257
- M. K. Gupta, A. Ghuge, M. Parab, Y. Al-Refaei, A. Khandare, N. Dand and N. Waghmare, A Comparative Review on High-Performance Liquid Chromatography (HPLC), Ultra Performance Liquid Chromatography (UPLC) & High-Performance Thin Layer Chromatography (HPTLC) with Current Updates, Curr. Issues Pharm. Med. Sci., 2022, 35(4), 224–228, DOI:10.2478/cipms-2022-0039
- A. U. Rehman, M. Crimi and S. Andreescu, Current and Emerging Analytical Techniques for the Determination of PFAS in Environmental Samples, Trends Environ. Anal. Chem., 2023, 37, e00198, DOI:10.1016/j.teac.2023.e00198
- D. Thompson, N. Zolfigol, Z. Xia and Y. Lei, Recent Progress in Per- and Polyfluoroalkyl Substances (PFAS) Sensing: A Critical Mini-Review, Sens. Actuators Rep., 2024, 7, 100189, DOI:10.1016/j.snr.2024.100189
- A. Tasfaout, F. Ibrahim, A. Morrin, H. Brisset, I. Sorrentino, C. Nanteuil, G. Laffite, I. A. Nicholls, F. Regan and C. Branger, Molecularly Imprinted Polymers for Per- and Polyfluoroalkyl Substances Enrichment and Detection, Talanta, 2023, 258, 124434, DOI:10.1016/j.talanta.2023.124434
- R. F. Menger, E. Funk, C. S. Henry and T. Borch, Sensors for Detecting Per- and Polyfluoroalkyl Substances (PFAS): A Critical Review of Development Challenges, Current Sensors, and Commercialization Obstacles, Chem. Eng. J., 2021, 417, 129133, DOI:10.1016/j.cej.2021.129133
- Y. Li, L. Luo, Y. Kong, Y. Li, Q. Wang, M. Wang, Y. Li, A. Davenport and B. Li, Recent Advances in Molecularly Imprinted Polymer-Based Electrochemical Sensors, Biosens. Bioelectron., 2024, 249, 116018, DOI:10.1016/j.bios.2024.116018
- A.-M. Gavrilă, E.-B. Stoica, T.-V. Iordache and A. Sârbu, Modern and Dedicated Methods for Producing Molecularly Imprinted Polymer Layers in Sensing Applications, Appl. Sci., 2022, 12(6), 3080, DOI:10.3390/app12063080
- M. Garg and N. Pamme, Strategies to Remove Templates from Molecularly Imprinted Polymer (MIP) for Biosensors, TrAC, Trends Anal. Chem., 2024, 170, 117437, DOI:10.1016/j.trac.2023.117437
- J. J. Belbruno, Molecularly Imprinted Polymers, Chem. Rev., 2019, 119(1), 94–119, DOI:10.1021/acs.chemrev.8b00171
- C. Fang, Z. Chen, M. Megharaj and R. Naidu, Potentiometric Detection of AFFFs Based on MIP, Environ. Technol. Innovation, 2016, 5, 52–59, DOI:10.1016/j.eti.2015.12.003
- N. Karimian, A. M. Stortini, L. M. Moretto, C. Costantino, S. Bogialli and P. Ugo, Electrochemosensor for Trace Analysis of Perfluorooctanesulfonate in Water Based on a Molecularly Imprinted Poly(o-Phenylenediamine) Polymer, ACS Sens., 2018, 3(7), 1291–1298, DOI:10.1021/acssensors.8b00154
- R. Kazemi, E. I. Potts and J. E. Dick, Quantifying Interferent Effects on Molecularly Imprinted Polymer Sensors for Per-A Nd Polyfluoroalkyl Substances (PFAS), Anal. Chem., 2020, 92(15), 10597–10605, DOI:10.1021/acs.analchem.0c01565
- D. Lu, D. Z. Zhu, H. Gan, Z. Yao, J. Luo, S. Yu and P. Kurup, An Ultra-Sensitive Molecularly Imprinted Polymer (MIP) and Gold Nanostars (AuNS) Modified Voltammetric Sensor for Facile Detection of Perfluorooctance Sulfonate (PFOS) in Drinking Water, Sens. Actuators, B, 2022, 352(P1), 131055, DOI:10.1016/j.snb.2021.131055
- R. B. Clark, D. C. Wagner, D. T. Holden, J. J. P. Roberts, E. Zumbro, L. Goodnight, K. T. Huynh, R. B. Green, J. A. Grove and J. E. Dick, PFAS Electroanalysis in Low-Oxygen River Water Using Electrogenerated Dioxygen, Environ. Sci. Technol., 2023, 57(51), 21815–21822, DOI:10.1021/acs.est.3c03967
- T. T. Tran, J. Li, H. Feng, J. Cai, L. Yuan, N. Wang and Q. Cai, Molecularly Imprinted Polymer Modified TiO2 Nanotube Arrays for Photoelectrochemical Determination of Perfluorooctane Sulfonate (PFOS), Sens. Actuators, B, 2014, 190, 745–751, DOI:10.1016/j.snb.2013.09.048
- J. Gong, T. Fang, D. Peng, A. Li and L. Zhang, A Highly Sensitive Photoelectrochemical Detection of Perfluorooctanic Acid with Molecularly Imprined Polymer-Functionalized Nanoarchitectured Hybrid of AgI–BiOI Composite, Biosens. Bioelectron., 2015, 73, 256–263, DOI:10.1016/j.bios.2015.06.008
- X. Li, X. Wang, T. Fang, L. Zhang and J. Gong, Disposable Photoelectrochemical Sensing Strip for Highly Sensitive Determination of Perfluorooctane Sulfonyl Fluoride on Functionalized Screen-Printed Carbon Electrode, Talanta, 2018, 181, 147–153, DOI:10.1016/j.talanta.2018.01.005
- S. Chen, A. Li, L. Zhang and J. Gong, Molecularly Imprinted Ultrathin Graphitic Carbon Nitride Nanosheets–Based Electrochemiluminescence Sensing Probe for Sensitive Detection of Perfluorooctanoic Acid, Anal. Chim. Acta, 2015, 896, 68–77, DOI:10.1016/j.aca.2015.09.022
- H. Feng, N. Wang, T. T. Tran, L. Yuan, J. Li and Q. Cai, Surface Molecular Imprinting on Dye–(NH2)–SiO2 NPs for Specific Recognition and Direct Fluorescent Quantification of Perfluorooctane Sulfonate, Sens. Actuators, B, 2014, 195, 266–273, DOI:10.1016/j.snb.2014.01.036
- Z. Jiao, J. Li, L. Mo, J. Liang and H. Fan, A Molecularly Imprinted Chitosan Doped with Carbon Quantum Dots for Fluorometric Determination of Perfluorooctane Sulfonate, Microchim. Acta, 2018, 185(10), 473, DOI:10.1007/s00604-018-2996-y
- L. Zheng, Y. Zheng, Y. Liu, S. Long, L. Du, J. Liang, C. Huang, M. T. Swihart and K. Tan, Core-Shell Quantum Dots Coated with Molecularly Imprinted Polymer for Selective Photoluminescence Sensing of Perfluorooctanoic Acid, Talanta, 2019, 194, 1–6, DOI:10.1016/j.talanta.2018.09.106
- Z. Abbasian Chaleshtari and R. Foudazi, A Review on Per- and Polyfluoroalkyl Substances (PFAS) Remediation: Separation Mechanisms and Molecular Interactions, ACS ES&T Water, 2022, 2(12), 2258–2272, DOI:10.1021/acsestwater.2c00271
- T. Sajini and B. Mathew, A Brief Overview of Molecularly Imprinted Polymers: Highlighting Computational Design, Nano and Photo-Responsive Imprinting, Talanta Open, 2021, 4, 100072, DOI:10.1016/j.talo.2021.100072
- M. Nishizawa, Y. Miwa, T. Matsue and I. Uchida, Surface Pretreatment for Electrochemical Fabrication of Ultrathin Patterned Conducting Polymers, J. Electrochem. Soc., 1993, 140(6), 1650–1655, DOI:10.1149/1.2221617
- J. Y. Gerasimov, A. Halder, A. H. Mousa, S. Ghosh, P. C. Harikesh, T. Abrahamsson, D. Bliman, J. Strandberg, M. Massetti and I. Zozoulenko, et al., Rational Materials Design for In Operando Electropolymerization of Evolvable Organic Electrochemical Transistors, Adv. Funct. Mater., 2022, 32(32), 2202292, DOI:10.1002/adfm.202202292
- J. Welchert, M. Dunmyer, L. Carroll, I. Martinez, T. J. Lane, D. A. Bellido-Aguilar, S. Savagatrup and V. Karanikola, Investigation into the Adhesion Properties of PFAS on Model Surfaces, RSC Appl. Interfaces, 2024, 1(6), 1265–1275, 10.1039/D4LF00228H
- M. Dunmyer, J. Welchert, D. A. Bellido-Aguilar, M. Brusseau, S. Savagatrup and V. Karanikola, Molecular Scale Adsorption Behavior of Per- and Poly-Fluoroalkyl Substances (PFAS) on Model Surfaces, Chem. Eng. J., 2024, 497, 154286, DOI:10.1016/j.cej.2024.154286
- A. Tewari, K. Björkström, A. M. Ghafari, E. Macchia, L. Torsi and R. Österbacka, Stability of Thiol-Based Self-Assembled Monolayer Functionalized Electrodes in EG-OFET-Based Applications, FlatChem, 2023, 42, 100553, DOI:10.1016/j.flatc.2023.100553
- F. Liu, L. Huang, X. Duan, Y. y. Li, J. q. Hu, B. h. Li and J. Lu, A Facile Method to Prepare Noble Metal Nanoparticles Modified Self-Assembly (SAM) Electrode, J. Exp. Nanosci., 2018, 13(1), 1–10, DOI:10.1080/17458080.2017.1373202
- Z. Li, S.-C. Chang and R. S. Williams, Self-Assembly of Alkanethiol Molecules onto Platinum and Platinum Oxide Surfaces, Langmuir, 2003, 19(17), 6744–6749, DOI:10.1021/la034245b
- M. W. Glasscott, K. J. Vannoy, R. Kazemi, M. D. Verber and J. E. Dick, μ-MIP: Molecularly Imprinted Polymer-Modified Microelectrodes for the Ultrasensitive Quantification of GenX (HFPO-DA) in River Water, Environ. Sci. Technol. Lett., 2020, 7(7), 489–495, DOI:10.1021/acs.estlett.0c00341
- C. S. Malloy, M. J. Danley, D. A. Bellido-Aguilar, L. Partida, R. Castrejón-Miranda and S. Savagatrup, Effects of Fabrication Parameters on the Mechanical and Sensing Properties of Molecularly Imprinted Polymers (MIPs) for the Detection of Per- and Polyfluoroalkyl Substances (PFAS), ACS Appl. Polym. Mater., 2024, 6(16), 9914–9921, DOI:10.1021/acsapm.4c01818
- M. Cieplak, R. Węgłowski, Z. Iskierko, D. Węgłowska, P. S. Sharma, K. R. Noworyta, F. D'souza and W. Kutner, Protein Determination with Molecularly Imprinted Polymer Recognition Combined with Birefringence Liquid Crystal Detection, Sensors, 2020, 20(17), 1–12, DOI:10.3390/s20174692
- C. E. Buensuceso, B. D. B. Tiu, L. P. Lee, P. M. G. Sabido, G. M. Nuesca, E. B. Caldona, F. R. del Mundo and R. C. Advincula, Electropolymerized-Molecularly Imprinted Polymers (E-MIPS) as Sensing Elements for the Detection of Dengue Infection, Anal. Bioanal. Chem., 2022, 414(3), 1347–1357, DOI:10.1007/s00216-021-03757-y
- P. Meng, S. Deng, Z. Du, B. Wang, J. Huang, Y. Wang, G. Yu and B. Xing, Effect of Hydro-Oleophobic Perfluorocarbon Chain on Interfacial Behavior and Mechanism of Perfluorooctane Sulfonate in Oil-Water Mixture, Sci. Rep., 2017, 7(1), 44694, DOI:10.1038/srep44694
- Y. He, X. Cheng, S. J. Gunjal and C. Zhang, Advancing PFAS Sorbent Design: Mechanisms, Challenges, and Perspectives, ACS Mater. Au, 2024, 4(2), 108–114, DOI:10.1021/acsmaterialsau.3c00066
- I. Losito, F. Palmisano and P. G. Zambonin, O -Phenylenediamine Electropolymerization by Cyclic Voltammetry Combined with Electrospray Ionization-Ion Trap Mass Spectrometry, Anal. Chem., 2003, 75(19), 4988–4995, DOI:10.1021/ac0342424
- G. Camurri, P. Ferrarini, R. Giovanardi, R. Benassi and C. Fontanesi, Modelling of the Initial Stages of the Electropolymerization Mechanism of o-Phenylenediamine, J. Electroanal. Chem., 2005, 585(2), 181–190, DOI:10.1016/j.jelechem.2005.08.016
- F. Zivari-Moshfegh, D. Nematollahi, M. M. Khoram and A. Rahimi, Electrochemical Oxidation of o-Phenylenediamine and 1,3 Dihydrospiro[Benzo[d]Imidazole-2,1′-Cyclohexane]. A Comprehensive Study and Introducing a Novel Case of CE Mechanism, Electrochim. Acta, 2020, 354, 136700, DOI:10.1016/j.electacta.2020.136700
- M. Pazalja, E. Kahrović, A. Zahirović and E. Turkušić, Electrochemical Sensor for Determination of L-Cysteine Based on Carbon Electrodes Modified with Ru(III) Schiff Base Complex, Carbon Nanotubes and Nafion, Int. J. Electrochem. Sci., 2016, 11(12), 10939–10952, DOI:10.20964/2016.12.86
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03253a |
| This journal is © The Royal Society of Chemistry 2025 |