
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable thermosensitive hydrogel co-loading with ATRi and doxorubicin for the treatment of triple-negative breast cancer†
Lan Wei‡
a,
Jiaru Zhu‡a,
Qi Wanga,
Yuanfang Hea,
Haili Yanb,
Long Gao*c,
Chenyang Zhangd and
Jiangfeng Du *bef
*bef
aSchool of Pharmacy, Medicinal Basic Research Innovation Center of Chronic Kidney Disease, Ministry of Education, Shanxi Provincial Key Laboratory of Drug Synthesis and Novel Pharmaceutical Preparation Technology, Shanxi Medical University, Taiyuan 030001, Shanxi Province, China
bDepartment of Medical Imaging, Shanxi Key Laboratory of Intelligent Imaging and Nanomedicine, First Hospital of Shanxi Medical University, Taiyuan, 030001, Shanxi Province, China. E-mail: dujf@sxmu.edu.cn
cDepartment of Oncological and Vascular Intervention, First Hospital of Shanxi Medical University, Taiyuan 030001, Shanxi Province, China. E-mail: gaolong@sxmu.edu.cn
dDepartment of Biotherapy, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing 100730, China
eKey Laboratory of Cellular Physiology at Shanxi Medical University, Ministry of Education, Taiyuan 030001, Shanxi Province, China
fCollaborative Innovation Center for Molecular Imaging of Precision Medicine, Shanxi Medical University, Taiyuan 030001, Shanxi Province, China
First published on 16th June 2025
Abstract
Chemotherapy has been the first-line treatment option for cancer. However, acquired chemo-resistance led by DNA damage repair (DDR) of cancer cells and serious side-effects of chemotherapeutic agents are huge hurdles to effectively suppress metastatic tumors. Herein, we developed an injectable thermosensitive hydrogel for localized co-delivery of ATRi-BAY-1895344 (BAY) and doxorubicin (DOX), serving as a localized drug depot to minimize systemic toxicity while ensuring sustained tumor-specific drug release exceeding 4 days. The in vitro cumulative drug release rate of DOX and BAY reached up to 73.9% and 63.3% under pH 6.5 conditions. This study pioneers the synergistic combination of a DNA-damaging agent and Ataxia telangiectasia and RAD3-related (ATR) kinase inhibitor ATRi to disrupt the DDR pathway. The ATRi-mediated inhibition of ATR kinase effectively disrupts the replication stress response by impairing the repair of DOX-induced DNA lesions. This dual mechanism significantly enhances tumor cell vulnerability to chemotherapy, ultimately achieving an 8-fold increase in chemosensitivity compared to monotherapy regimens. In triple-negative breast cancer models, the hydrogel-based DOX + BAY@Gel formulation achieved a tumor inhibition rate of 79.4%, significantly surpassing the 58% observed with free DOX monotherapy. This dual-action strategy overcomes chemo-resistance by disabling DDR compensatory mechanisms and prolongs tumor suppression through controlled drug release. The hydrogel platform represents a functional innovation in localized combination therapy, integrating stimuli-responsive drug delivery with DDR pathway disruption for synergistic efficacy.
1 Introduction
Representing 15–20% of breast cancer subtypes, triple-negative breast cancer (TNBC) demonstrates aggressive biological behavior characterized by early metastatic propensity and dismal prognosis in patients with advanced or recurrent disease. Contemporary therapeutic strategies therefore primarily focus on delaying disease progression and extending progression-free survival through optimized clinical management. In the current therapeutic paradigm, cytotoxic chemotherapy continues to serve as the cornerstone intervention in the clinical management of TNBC.1 Doxorubicin, cisplatin, and gemcitabine, exert anti-cancer effects by inducing DNA damage.2 Unfortunately, cancer cells have a DNA damage repair (DDR) network that repairs all spontaneously occurring and exogenous factors-induced DNA damage.3 An increased DNA repair capacity is a major factor contributing to acquired chemo-resistance, which is the main obstacle to effective breast cancer management.4 DDR promotes cell survival by detecting DNA damage and repairing it to maintain genomic integrity. Therefore, DNA damage repair inhibitors may be useful to enhance the sensitivity of cancer cells to chemotherapeutics.5Therapeutic strategies combining DNA damage repair inhibitors have demonstrated remarkable antitumor efficacy in preclinical and clinical settings. Multiple DDR-targeting agents have advanced into clinical development pipelines, with some achieving FDA approval.6 Notably, the Chk1 inhibitor prexasertib has exhibited favorable safety profiles in squamous cell carcinoma clinical trials,7 while the PARP inhibitor veliparib enhances chemosensitivity in cervical cancer xenograft models.8 Furthermore, synergistic effects were observed when combining the ATR inhibitor VE-822 with radiotherapy, significantly augmenting local tumor control efficacy.9 These advances collectively establish a robust preclinical foundation for addressing therapeutic challenges in triple-negative breast cancer management.
Emerging as a pivotal therapeutic paradigm in oncology research, DNA damage repair – targeted strategies are gaining substantial momentum in anticancer drug development.10 Ataxia telangiectasia and Rad3-related protein (ATR) with ataxia telangiectasia mutated (ATM) and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), these proteins form the core of the so-called DNA damage repair.11 ATR is a serine/threonine protein kinase involved in coordinating cell-cycle checkpoints and DNA damage repair caused by DNA replication-associated stress and sending it to S and G2/M checkpoints to facilitate DNA damage repair.12 Preclinical investigations have validated multiple ATR inhibitors (ATRis) with promising antitumor profiles,13 where pharmacological ATR blockade potentiates chemotherapeutic agents like doxorubicin by compromising tumor cell damage repair capacity.14 Among clinically developed ATRi, the novel oral agent BAY-1895344 (BAY) demonstrates exceptional selectivity,15 with ongoing clinical trials evaluating its therapeutic potential across malignancies.16 Particularly, its combination with cisplatin exhibits synergistic antitumor activity in advanced solid tumors, notably urothelial carcinoma,15 while coadministration with the PI3K inhibitor Copanlisib shows enhanced efficacy in lymphoma models.17 These findings collectively lay the preclinical foundation for optimizing ATR-based combination therapies.
Persistent systemic toxicity remains a principal constraint in conventional chemotherapy regimens.18 Anthracycline agents exemplified by doxorubicin, while demonstrating potent antitumor efficacy, are significantly limited in clinical applicability due to dose-limiting cardiotoxicity such as myocardial fibrosis stemming from mitochondrial dysfunction in cardiomyocytes.19 Compared to conventional intravenous administration that induces peak-trough plasma concentration fluctuations, hydrogel-based drug delivery systems enable sustained release through their three-dimensional polymeric networks.20
Hydrogels are hydrophilic cross-linked polymer networks capable of absorbing substantial amounts of water or biological fluids, and they have been extensively employed as carrier materials for controlled drug delivery.21 Thermosensitive hydrogels are a class of smart hydrogels that respond to thermal stimuli and undergo sol–gel phase transitions within specific temperature ranges. Due to their excellent biocompatibility and tunable physical properties, thermosensitive hydrogels have also been extensively investigated for biomedical applications.22 Chitosan-based hydrogels have garnered significant attention due to their exceptional biocompatibility and biodegradability.23 Chenite et al. developed novel injectable chitosan/polyol salt-based hydrogels exhibiting in situ biodegradability for in vivo delivery of bioactive growth factors24 Kolawole demonstrated that the chitosan/β-glycerophosphate thermo-gelling system exhibits exceptional gelation properties enabling sustained intravesical delivery of mitomycin-C.25 Building upon these advancements, this study developed an injectable thermoresponsive hydrogel co-encapsulating chemotherapeutic agents with ATR inhibitors. And the unique sustained-release properties of thermosensitive hydrogels not only extend the therapeutic window within tumor microenvironments, but more critically, significantly reduce peak drug concentrations (Cmax), thereby effectively mitigating off-target drug accumulation in vital organs. This spatiotemporal drug release platform represents a translatable therapeutic paradigm that synchronizes drug exposure kinetics to potentiate synergistic antitumor effects.
This study presents an injectable thermoresponsive hydrogel engineered for co-encapsulation of a DNA-damaging chemotherapeutic agent and a highly selective ATR inhibitor. The latter, as a novel potent therapeutic agent, has demonstrated remarkable antitumor activity in preclinical models with compromised DNA damage repair pathways.26 To address conventional therapeutic limitations including frequent dosing regimens and systemic toxicity, we formulated a chitosan/β-glycerophosphate-based thermoresponsive hydrogel platform. This biomaterial enables controlled intratumoral co-delivery of DOX and BAY through temperature-triggered gelation, achieving precise spatiotemporal synchronization of drug release kinetics. Experimental validation confirmed that this integrated delivery system potentiated synergistic therapeutic effects between the two agents (Scheme 1). Our findings not only validate the efficacy of thermoresponsive hydrogels as advanced drug carriers but also establish an innovative treatment paradigm for combinatorial regimens involving DDR inhibitors and genotoxic agents.
 | ||
| Scheme 1 Schematic illustration of DOX + BAY@Gel localized and extended release of DOX and BAY in the treatment of tumor. DOX + BAY@Gel is prepared by cross-linking CS and β-GP before performing thermosensitive gelation in vivo. Then the localized injection of DOX + BAY@Gel reduces the size of the tumor by increasing the concentration of DOX and BAY in the tumor. DOX can trigger DSBs and BAY induces more severe DNA damage by inhibiting the DNA damage repair (DDR). | ||
2 Results and discussion
2.1. Characterization of hydrogel
To perform the thermosensitive gelation of blank-gel, CS (2%, dissolved in 0.1 mol per L acetic acid, w/w) was cross-linked with β-GP (60%, dissolved in DI water, w/w) at different ratios. Thermosensitive gelation was only observed at CS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β-GP = 7:3 and 6:4 (v/v) after incubating at 37 °C for 3 min. Thermosensitive gelation was not observed at the CS:β-GP ratios of 8:2, 4:6, 3:7, and 2:8 (Fig. 1a). Unlike the SEM image of the CS/β-GP solution in Fig. 1b, both blank-gel (Fig. 1c) and DOX + BAY@Gel (Fig. 1d) had a typical porous network structure and the surface of DOX + BAY@Gel has become rougher and more textured noticed that DOX and BAY have been encapsulated in hydrogel.
:β-GP = 7:3 and 6:4 (v/v) after incubating at 37 °C for 3 min. Thermosensitive gelation was not observed at the CS:β-GP ratios of 8:2, 4:6, 3:7, and 2:8 (Fig. 1a). Unlike the SEM image of the CS/β-GP solution in Fig. 1b, both blank-gel (Fig. 1c) and DOX + BAY@Gel (Fig. 1d) had a typical porous network structure and the surface of DOX + BAY@Gel has become rougher and more textured noticed that DOX and BAY have been encapsulated in hydrogel.
 | ||
| Fig. 1 Characterization of hydrogel. (a) The blank gel was prepared using different CS to β-GP ratios at 25 °C before undergoing thermosensitive gelation at 37 °C. (b) SEM images of a mixture of CS and β-glycerol phosphate disodium salt pentahydrate. (c) SEM images of blank hydrogel. (d) SEM images of DOX + BAY@Gel. Scale bar = 100 μm. (e) FT-IR spectra of a hydrogel. (f) Rheological analysis of blank hydrogel at 37 °C. (g) Rheological analysis of blank hydrogel at different temperatures (G′ is the storage modulus, G′′ is the loss modulus). (h) The release curve of DOX in DOX + BAY@Gel at pH = 7.4, 6.5 and 5.5. (i) The release curve of BAY in DOX + BAY@Gel at pH = 7.4, 6.5 and 5.5. | ||
Furthermore, structural characterization of the thermosensitive hydrogel was performed using FT-IR on lyophilized powder samples to prove that there have a crosslinking between CS and β-GP components at physiological temperature (37 °C). As shown in Fig. 1e, the stretching vibration bands of N–H in CS and O–H in β-GP exhibited a downward shift from 3183 cm−1 to 3127 cm−1, indicative of intermolecular hydrogen bonding formation between these functional groups. Concomitantly, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations (1636 cm−1 → 1604 cm−1) and N–H bending vibrations (1596 cm−1 → 1569 cm−1) of CS demonstrated pronounced red-shifting phenomena, and the attenuation of distinct peaks at 1047 cm−1 and 944 cm−1 in the hydrogel further corroborated the intermolecular interactions between amine groups on CS chains and both –OH and PO42− in β-GP, involving both hydrogen bonding and coordination complex formation. These results provide conclusive evidence of temperature-dependent crosslinking between CS and β-GP components at physiological temperature (37 °C), resulting in the formation of an injectable thermoresponsive hydrogel matrix.
O stretching vibrations (1636 cm−1 → 1604 cm−1) and N–H bending vibrations (1596 cm−1 → 1569 cm−1) of CS demonstrated pronounced red-shifting phenomena, and the attenuation of distinct peaks at 1047 cm−1 and 944 cm−1 in the hydrogel further corroborated the intermolecular interactions between amine groups on CS chains and both –OH and PO42− in β-GP, involving both hydrogen bonding and coordination complex formation. These results provide conclusive evidence of temperature-dependent crosslinking between CS and β-GP components at physiological temperature (37 °C), resulting in the formation of an injectable thermoresponsive hydrogel matrix.
Finally, in order to determine the gelation temperature of the chitosan gel-forming solution, an MCR92 rheometer (Anton Paar) was used to measure the storage (elastic) modulus (G′), and loss (viscous) modulus (G′′). Under the condition of 37 °C, with the increase of time, G′ has been larger than G′′, and the value of G′′/G′ is close to 0 (Fig. 1f), which indicates that the hydrogel can be rapidly gelatinized at physiological temperature and can form a stable gel system at 37 °C. With increasing temperature from 10 °C to 60 °C, all samples had a rapid increase of the storage modulus (G′), showing the phase transition of the CS/β-GP solution to the CS/β-GP gel around 37 °C (Fig. 1g).
2.2. In vitro anti-tumor efficacy
Successful synthesis of thermosensitive hydrogels has been achieved. Building upon their favorable temperature-dependent phase transition properties and long-term stability, we subsequently explored the in vitro release rate of DOX and BAY from the DOX + BAY@Gel, which was subsequently used in the in vitro release experiments performed in PBS with different pH (pH = 5.5, 6.5, 7.4). As expected, all the DOX + BAY@Gel formulations in PBS with different pH achieved a slow and sustained release of DOX and BAY. Drug release remained minimal during the initial four hours (<20%). A progressive release acceleration commenced at the 4-hour mark, reaching a plateau phase beyond 48 hours, the cumulative release of DOX was 53.6%, 73.9%, and 81.5 for the 3 pH, respectively (Fig. 1h). The cumulative release of BAY was 57.3%, 63.3%, and 74.0% for the three pH, respectively (Fig. 1i). This phenomenon suggests the potential emergence of a dose–response plateau, and also showed that DOX + BAY@Gel can release more drugs in a low pH, which indicated that DOX + BAY@Gel can have a better release performance in the tumor microenvironment.Given the favorable physicochemical properties and controlled drug release profile of the material established in preceding sections, we subsequently evaluated in vitro antitumor efficacy of DOX + BAY. Fig. 2a shown that as the dosage increases, the cytotoxicity of DOX to normal cells gradually intensifies. Nevertheless, at the same concentration of DOX, it exhibits relatively lower cytotoxicity toward tumor cells, which indicates that DOX lacks specificity for tumor cells and is excessively toxic to normal cells. Fig. 2b shows, that as the drug concentration increases, the anti-tumor effect of BAY becomes more significant, and it also exhibits lower cytotoxicity to normal cells at the same concentration of BAY, which is attributed to the that numerous DNA damage repair pathways in tumor cells are flawed compared to normal cells and so tumor cells are more sensitive to ATR inhibitor than normal cells. Next, we aim to investigate whether different drug administration sequences will impact their curative potency on 4T1 cells. The findings are presented in Fig. 2c, the group treated with DOX in combination with BAY demonstrates optimal therapeutic efficacy, outperforming the other two groups. Notably, as depicted in Fig. 2d, in contrast to the sole application of DOX or BAY, the combination of DOX and BAY can markedly impede the proliferation of tumor cells, Fig. S1† can also prove it. We also investigated whether BAY and DOX could act synergistically in 4T1 cells. Next, we tested whether there was a synergistic effect between DOX and BAY on 4T1 cells. The combination index (CI) of BAY and DOX was less than 1.0 in 4T1 cells (Fig. 2e), suggesting a synergistic effect in vitro. Meanwhile, flow cytometry was employed to detect cell apoptosis status. Comparisons at different time points (co-incubation for 24 h, 48 h, and 72 h) are shown in Fig. 2f, g, S2 and S3.† The apoptosis rate of 4T1 cells in the DOX + BAY group after 48 h of co-incubation showed a substantial increase compared to the 24 h group, while the difference between 72 h and 48 h was not significant which indicates the potential existence of a dose–response plateau under sustained release conditions. At the 48 h time point, the apoptosis rates in the DOX + BAY group were 26.09% higher than those in the DOX group and 42.38% higher than those in the control group. This outcome strongly suggests that the synergistic interplay between the chemotherapeutic agent (DOX) and the ATR inhibitor (BAY) culminates in the most severe cytotoxic impact. Such a combined effect effectively triggers and enhances the process of tumor-cell apoptosis, highlighting the potential of this dual-agent approach in anti-tumor treatment.
 | ||
| Fig. 2 Therapeutic efficacy of DOX + BAY in vitro. (a) CCK-8 assay of co-incubate MCF-10A and 4T1 cells with 0, 0.125, 0.25, 0.5, 0.75, 1 μmol per L DOX for 48 h. (b) CCK-8 assay of co-incubate MCF-10A and 4T1 cells with 0, 0.1, 0.2, 0.4, 0.6, 0.8, 1 μmol per L BAY for 48 h. (c) Different administration of DOX and BAY on 4T1 cells. (d) CCK-8 assay of co-incubate 4T1 cells with 0.5 μmol per L DOX plus 0, 0.1, 0.2, 0.4, 0.6, 0.8, 1 μmol per L BAY for 48 h. (e) Combination index (CI) in 4T1 cells upon treatment with BAY and DOX. The CI was calculated using cell viability data presented in (a, b and d). (f) The statistic of cell apoptosis at 48 h. (g) The analysis of cell apoptosis by flow cytometric at 48 h. Statistical analysis: group DOX + BAY was compared with other groups. Data are presented as mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
In pre-clinical studies, a wide variety of inhibitors targeting the DNA damage repair pathway have been thoroughly probed into. Specifically, recent DDR-targeting compounds have been crafted to exploit the marked difference in the prevalence of defective G1/S checkpoints between cancer cells and normal tissues. This strategic approach has spurred the development of G2/M checkpoint-targeting agents, such as ATR inhibitors.27 Furthermore, it is hypothesized that the peak clinical efficacy of these agents is most likely attainable when they are combined with exogenous sources of DNA damage, potentially paving the way for more effective cancer treatment modalities.28 In our proposed approach, the combination of BAY and DOX can effectively potentiate chemotherapy by impeding DNA repair through the regulation of the S and G2/M checkpoints. First, we evaluated the level of DNA damage by the phosphorylated histone H2AX (γ-H2AX) detection. In Fig. 3a, DOX + BAY can induce the most severe DNA damage. The number of γ-H2AX foci in the group treated with DOX + BAY is 1.8 times that in the group treated with DOX (Fig. 3b), which demonstrates that BAY can enhance the DNA breaks induced by DOX. Additionally, the protein level of γ-H2AX in the group treated with DOX + BAY is much higher than that in the other groups (Fig. 3c and S4†). Based on the above results, BAY can inhibit the ATR-related DNA repair pathway, thereby enhancing the DNA damage induced by DOX and ultimately promoting DNA damage. Therefore, we examined the key proteins within this pathway. As depicted in Fig. 3d and S4,† when comparing the group treated with the combination of DOX and BAY to the group treated with DOX, there was a decline in the expression level of pChk1, an increase in the expression level of phosphorylated Cdc25C (pCdc25C), and a marked decrease in the expression level of phosphorylated CDK1 (pCDK1). These findings strongly suggest that BAY holds the potential to inhibit the ATR-related DNA-repair pathway. Moreover, the disparities in the cell cycle following different treatments further confirm the inhibitory effect of BAY on DNA repair (Fig. 3e and 4f). Specifically, the group that received DOX treatment exhibited an arrest at the G2/M phase. However, the group treated with the combination of DOX and BAY exhibited the alleviation of G2/M phase arrest, which indicated a pronounced interference effect on the cell cycle. Notably, the inhibition of ATR reversed the G2/M phase arrest induced by DOX. All the results demonstrate that DOX can cause severe DNA damage, and the presence of BAY can further amplify this damage.
 | ||
| Fig. 3 The mechanism of DSBs. (a) Images of DSBs tested by γ-H2AX foci. Scale bar = 20 μm. (b) The corresponding number of γ-H2AX foci in cells. (c) The protein expression level of γ-H2AX. (d) The corresponding protein expression level of the G2/M pathway. (e) Cell cycle analysis by flow cytometric. (f) The statistic of the cell cycle. Statistical analysis: group control was compared with other groups. Data are presented as mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
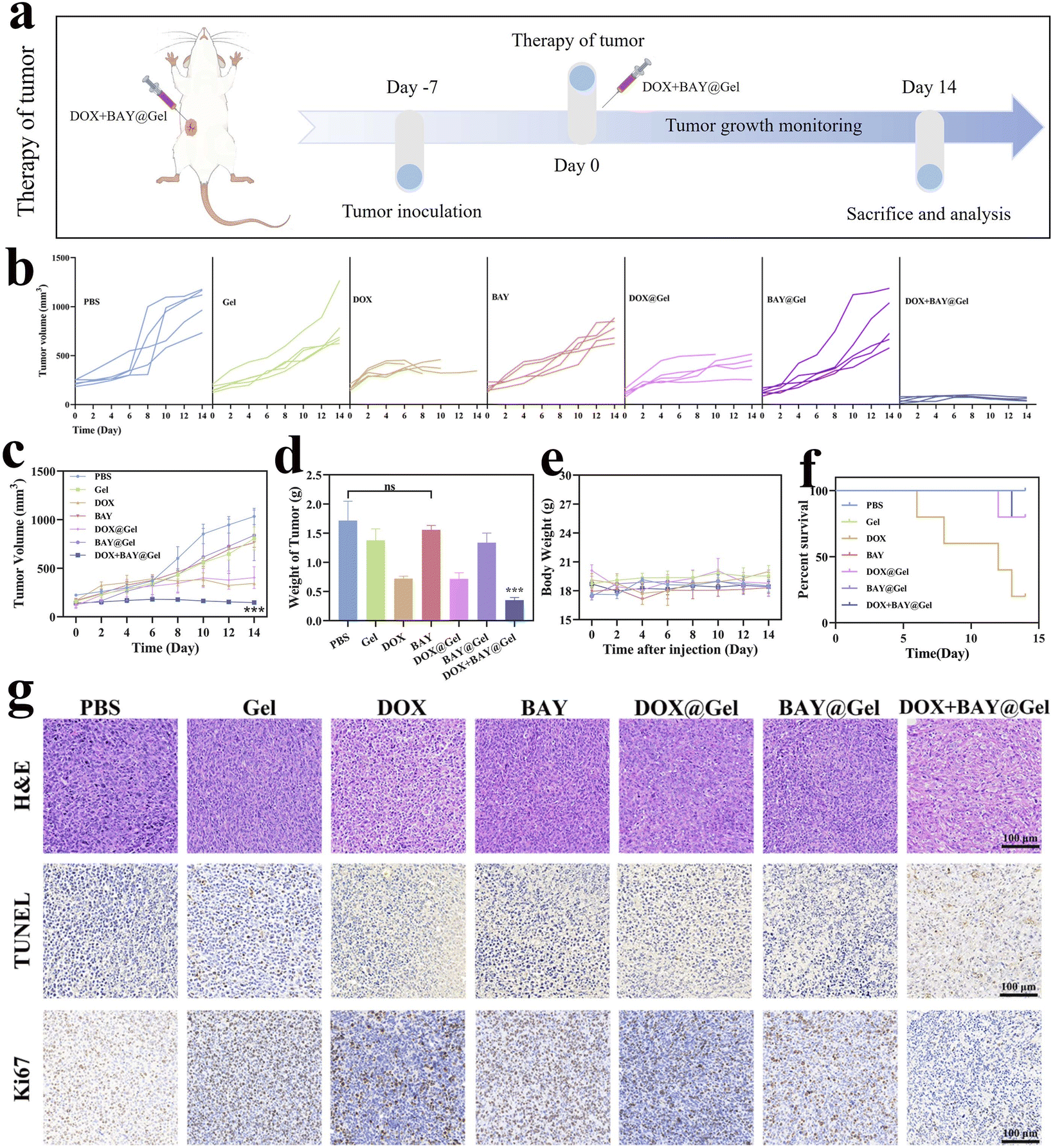 | ||
| Fig. 4 The antitumor efficacy of DOX + BAY@Gel in vivo. (a) Schematic illustration therapeutic design (b) The volume of tumors in each group. (c) Tumor volume growth curves of mice. (d) The average weight of the tumor from each group. Statistical analysis: group DOX + BAY@Gel was compared with other groups. Data are presented as mean ± SD (***p < 0.001). (e) Body weight curves of mice. (f) The survival curves of mice. (g) Images of H&E, TUNEL, and ki67 staining of tumor slices. Scale bar = 100 μm. | ||
2.3. Antitumor efficacy assay of DOX + BAY@Gel in vivo
Inspired by the in vitro anti-tumor efficacy of DOX + BAY, our subsequent investigation aimed to determine whether DOX + BAY@Gel could effectively suppress the growth of 4T1 tumors in vivo (Fig. 4a). To this end, we administered treatments to mice bearing 4T1 tumors, including free DOX, free BAY, DOX@Gel, BAY@Gel, and DOX + BAY@Gel. While all treatment groups demonstrated efficacy in suppressing tumor progression, the combination group exhibited the most pronounced antitumor effect, achieving a 79.4% inhibition rate compared to 58.0% observed in the free DOX group (Fig. 4b–d) During the experiment, the body weights of mice in each group were continuously monitored. The results showed that there were no significant variation in the body weight of mice for all cases (18.5 ± 3 g) throughout the 14-day treatment period (Fig. 4e). However, the treatment effects varied significantly among different groups. After 14 days of treatment, mice in all groups except the DOX group showed relatively stable survival status. In the group DOX, a severe mortality event occurred. Among the original five mice in this group, only one survived at the end of the treatment (Fig. 4f). Subsequently, the tumor tissues were harvested for pathological examination, which involved H&E staining and immunohistochemical analysis. As illustrated in Fig. 4g, the micrographs of tumor slices stained with Hematoxylin and Eosin (H&E), TdT-mediated dUTP nick end labeling (TUNEL), and Ki67 vividly demonstrated that DOX + B@BAYGel inflicted more severe damage on tumor cells compared to the other treatment groups.Taken together, the robust antitumor efficacy and reduced mortality observed in the DOX + BAY@Gel group established the therapeutic advantage of our hydrogel delivery system. However, given the known organ toxicity associated with chemotherapeutic agents – particularly evidenced by the severe mortality in free DOX group – it became imperative to systematically evaluate whether the CS/β-GP hydrogel carrier itself contributes to biosafety risks.
2.4. The biosafety of CS/β-GP gel
We therefore proceeded to validate the hydrogel's biosafety in therapeutic contexts where it functions as a drug carrier. To assess the biocompatibility of the CS/β-GP gel, we conducted an in vivo degradation experiment of the hydrogel. As Fig. 5a and b show that the muscle tissues surrounding the gel in the mice showed no abnormalities, and the hydrogel gradually diminished in size, demonstrating that the CS/β-GP Gel has no obvious toxicity in vivo. Next, we employed the Cell Counting Kit-8 (CCK-8) assay. As depicted in Fig. 5c, the extracts of the CS/β-GP gel at different time points exerted no significant adverse effects on cell proliferation, providing compelling evidence that the CS/β-GP gel exhibits no obvious toxicity in vitro. | ||
| Fig. 5 The safety and biocompatibility of DOX + BAY@Gel in vivo. (a) Images of in vivo degradation of hydrogels (n = 3 mice/group). (b) The statistic of (a). Group 21 was compared with group 0. Data are presented as mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001). (c) Cell viability of HUVEC cells after indirect treatment with CS/β-GP Gel. (d) The blood routine analysis of BALB/c mice (n = 5 mice/group) after 14 days of different treatment. (e) The pivotal blood biochemical indicators of BALB/c mice (n = 5 mice/group) after 14 days of different treatment. Data are presented as mean ± SD (nsp > 0.05, *p < 0.05, **p < 0.01). (f) H&E staining of the major organs. Scale bar = 100 μm. | ||
This subsequent phase focused specifically on its capacity to mitigate toxicity during antitumor treatment when delivering drugs. This assessment encompassed hematological examinations and histological analyses. The results of the blood routine test are shown in Fig. 5d, only in the groups treated with DOX@Gel and DOX + BAY@Gel, the counts of white blood cells (WBC) and lymphocytes (Lymph) are within the normal range. The relevant indicators of the other groups exceed the normal range. As vividly presented in Fig. 5e, a notable pattern emerged, with the sole exception of the DOX group, there were no statistically significant disparities in blood biochemical parameters across the groups that underwent different treatments. This observation strongly implies that the hydrogel serves as an effective buffer against the toxic side effects of DOX. H&E staining was used to evaluate the side effects on major organs, including the heart, liver, spleen, lung, and kidney. As shown in Fig. 5f, no obvious differences were observed among the tissue sections of each group, except for the DOX group.
Collectively, these multi-modal analyses validate that the CS/β-GP gel not only exhibits inherent biocompatibility but also effectively mitigates drug-induced systemic toxicity.
3 Experimental section
3.1. Reagents and materials
Reagents and materials: chitosan (CS) was purchased from Shanghai Yuanye Bio-technology Co., Ltd (China). Sodium β-glycerophosphate pentahydrate (97%, β-GP) was purchased from Thermo Fisher Scientific (USA). Acetic acid (99%) was purchased from Beichen Fangzheng (China). Doxorubicin hydrochloride and BAY-1895344 hydrochloride were purchased from MedChemExpress (MCE, USA). Antibodies against γ-H2AX (S139), pChk1 were purchased from Cell Signaling Technology (CST, USA). Antibodies against CDK1, CyclinB, and Chk1 were purchased from Abcam (USA). Antibodies against pCdc25C and pCDK1 were purchased from Immunoway (China). Antibody against Cdc25C was purchased from HUABIO (China). Annexin V-FITC Apoptosis Detection Kit and Cell Cycle and Apoptosis Analysis Kit were purchased from Beyotime Biotechnology Co., Ltd (China).3.2. Preparation of CS/β-GP gel
A 2% (w/v) chitosan solution was prepared by stirring powdered chitosan in 0.1 mol per L aqueous acetic acid at room temperature overnight. The insoluble particles in the chitosan solution were removed by filtration. A 60% (w/v) β-GP solution was prepared in distilled water and stored at 4 °C. At the end, 3 mL sterilized, ice-cold β-GP solution was added drop by drop to the 7 mL CS solution under stirring conditions in an ice bath, stirring for 15 min.24 To determine the gelation time, the CS/β-GP solution was prepared. The 1 mL of each sample was tested in 5 mL glass vials. Gelation time was measured as a function of time at constant temperatures of 37 °C in a water bath. The test tube inverting method was used to determine the gelation time.29 The samples were incubated in a water bath at the predetermined temperatures, and time measurements were initiated. The flowability of the samples was observed every 30 s by tilting the tubes. The time at which the flowing of the samples stopped was taken as the gelation time and the values were recorded.3.3. Preparation of DOX + BAY@Gel
The preparation of DOX + BAY@Gel was done by adding DOX (3 mg) and BAY (1 mg) into 2% CS solution, with constant stirring at an ice bath for 30 min. Finally, they were mixed with the β-GP solution as described above.3.4. Characterization of the CS/β-GP gel
The shapes and surface morphology of CS/β-GP solution, CS/β-GP gel, and DOX + BAY@Gel were observed by field emission scanning electron microscope (ThermoFisher, ApreoC). The CS/β-GP gels were prepared at 37 °C for 2 min. Next, samples were lyophilized overnight (SCIENTZ, SCIENTZ-10ND/A). The samples were fixed with conductive tape on a metal stub, sputtered with gold under vacuum, and examined at a 20 kV accelerating voltage with a working distance of 17 mm. To characterize the chemical structure of the CS/β-GP solution and CS/β-GP gel, FT-IR spectra were obtained using a Nicolet6700 FT-IR spectrometer. All spectra were recorded by transmittance mode (32 times scanning, 400–4000 cm−1). Rheological measurement30 was performed with an MCR92 rheometer (Anton Paar) equipped with PP25 flat plates. All the hydrogel samples required about 3 mL solution sample and then placed between parallel plates with a gap of 1 mm. Dynamic frequency sweeps were measured at a constant strain of 1% along the constant frequency of 1 Hz at 37 °C. The dynamic temperature sweep was carried out between 10 and 60 °C at a heating rate of 1 °C min−1 and fixed parameters of 1% strain and 1 Hz.3.5. In vitro drug release test
DOX and BAY released from the CS/β-GP gel were quantified over time by an in vitro release assay. 1 mL of DOX + BAY@Gel solution, DOX@Gel, and BAY@Gel were pipetted to 50 mL centrifuge tubes and allowed to completely become a gel at 37 °C for 30 min. 10 mL of PBS with different pH (pH = 5.5, 6.5, 7.4) was pipetted into each tube. At designated time points (1, 2, 4, 8, 12, 24, 48, 72, and 96 hours), 100 μL aliquots of the release medium were sampled and the same amount of fresh PBS with different pH was added to the centrifuge tubes. In the collected fractions, the cumulative amount of DOX and BAY released as a function of time was determined by a Microplate Reader (BioTek Epoch) at 488 and 396 nm. Finally, the drug release was calculated by Dual-wavelength Ultraviolet Spectrophotometry.313.6. Cell culture
The 4T1 cell and MCF 10A cell lines were obtained from the Beijing Seven (Beijing, China) and Procell (Wuhan, China), respectively. 4T1 cells were cultured in RPMI-1640 medium with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin. MCF-10A cells were cultured in an MCF-10A cell-specialized culture medium, which was obtained from Procell (Wuhan, China). The cells were cultured in an incubator supplied with 5% CO2 at 37 °C.3.7. In vitro cytotoxicity of DOX and BAY
4T1 and MCF 10A cells were seeded in 96 well plates at a density of 2 × 104 cells per well. After 24 h of adherence, the cells were treated with culture medium, DOX, BAY, and DOX + BAY, respectively. After 24, 48, and 72 h of coincubation, the cells were washed with PBS, then CCK-8 (10 μL per well) was added to each well, and the cells were further incubated for about 20 min. The absorbance was measured at 450 nm using a microplate reader (iMark, Bio-Rad, MA, USA). To determine the synergistic effect of BAY and DOX on 4T1 cells, the combination index (CI) value was determined using CompuSyn software 1.0 (ComboSyn Inc. Paramus, NJ). Synergy was defined as a CI value less than 1.0.3.8. Cell apoptosis analysis
Cellular apoptosis was conducted using the Annexin V-FITC apoptosis detection kit. Briefly, the cells were seeded in a 6 cm cell culture dish at a density of 1 × 106 cells per dish and incubated with culture medium, 0.5 μmol per L DOX, 0.1 μmol per L BAY, and DOX + BAY for 48 h. Thereafter, the cells were processed according to the kit's description. Finalized experimental data obtained through flow cytometry were processed and analyzed using FlowJo analytical software (BD Biosciences).3.9. Cell cycle analysis
The cell cycle analysis was conducted using the cell cycle and apoptosis analysis kit. Briefly, the cells were seeded in 6-well plates at a density of 5 × 105 cells per well and incubated with culture medium, 0.5 μmol per L DOX, 0.1 μmol per L BAY, and DOX + BAY for 48 h. Thereafter, the cells were processed according to the kit's description. Finalized experimental data obtained through flow cytometry were processed and analyzed using FlowJo analytical software (BD Biosciences).3.10. DNA double-strand breaks detection
The DNA double-strand breaks (DSBs) levels were detected by a DNA damage assay kit by γ-H2AX immunofluorescence. Cells were inoculated on coverslips, and incubated with culture medium, 0.5 μmol per L DOX, 0.1 μmol per L BAY, and DOX + BAY for 48 h. Thereafter, the cells were processed according to the kit's description. The confocal images were captured by a Nikon Ti-A1 laser confocal microscope. Representative images are shown.3.11. Orthotopic breast cancer model
The female BALB/c mice (6–8 weeks) were provided by Beijing HFK Bioscience Co., Ltd All experiments were carried out strictly under the ethical regulations permitted by the Animal Experiments Ethics Committee of Shanxi Medical University (SYXK-JIN2019-0007). The mice were randomly assigned and kept in standard conditions.Exponentially growing 4T1 cells were enzymatically detached, washed twice with PBS (pH = 7.4) through centrifugation, and resuspended in ice-cold sterile PBS at 5 × 106 cells per mL. Following isoflurane-induced anesthesia, a dermal incision was surgically created along the fourth mammary gland pair using aseptic techniques. The mammary fat pad was carefully dissected through blunt dissection. 100 μL cell suspension was injected into the fat pad. Post-injection wound closure was achieved using tissue adhesive. Animals were monitored postoperatively and maintained under standard vivarium conditions.
3.12. Antitumor efficacy assay in vivo
4T1 cells were orthotopic injected into the mammary fat pad of mice, 5 × 105 cells per mouse. When the tumors grew to about 100 mm3, the mice were divided into different groups (n = 5), including the group treated with PBS (1), the group treated with only Gel (2), the group treated with free DOX (3), the group treated with free BAY (4), the group treated with DOX@Gel (5), the group treated with only BAY@Gel (6) as well as the group treated with DOX + BAY@Gel (7). The mice were injected intratumorally with 100 μL Gel or DOX@Gel (3 mg mL−1) or BAY@Gel (1 mg mL−1) or DOX + BAY@Gel or PBS, respectively. The tumor volume (volume = 0.5 × width2 × length) was measured once every two days. On day 14, the tumors were isolated and weighed. At the same time, the isolated tumors were obtained for the next experiments.3.13. In vitro biosafety of gel
Cytotoxicity of CS/β-GP gel was studied by using HUVEC cells. Firstly, we prepared the hydrogel extract, put 1 mL of hydrogel into a 6-well plate, and put it into an incubator for 30 minutes to completely become a gel. Then we added 5 mL of culture medium according to the volume ratio of hydrogel to a complete culture medium of 1:5 and put it in the incubator for 24, 48, and 72 hours to obtain the complete hydrogel extract. Finally, dilute with a complete culture medium to obtain a hydrogel extract solution with different percentage content. Cells were seeded at 1 × 104 cells per well.
3.14. In vivo biosafety
The biosafety evaluation system is established through multi-dimensional indicators: (1) dynamic body weight monitoring: beginning on day 0 of the experiment, body weight changes in mice are systematically recorded at 48-hour intervals. (2) Hematological analysis: as previously mentioned, retro-orbital venous blood samples are collected before euthanasia. Whole blood specimens are EDTA-anticoagulated for complete blood count analysis, while separated serum is analyzed for biochemical indicators such as liver function (ALT/AST) and renal function (UREA/CREA). (3) Histopathological evaluation: during tumor tissue collection, organs (heart, liver, spleen, lung, kidney) are excised, thoroughly rinsed with PBS, and fixed in 4% paraformaldehyde. Tissue sections are then prepared via standard paraffin embedding and sectioning, followed by H&E staining for microscopic pathological observation.3.15. In vivo degradation test of the hydrogel
Female Balb/c mice (6–8 weeks old) were used to evaluate the hydrogel's in vivo degradation. A 100 μL blank hydrogel was subcutaneously injected into the right dorsal region of each mouse. At each time point (days 0, 3, 7, 14, 21), three mice were euthanized, and the injection site skin was dissected to assess hydrogel degradation and surrounding tissue status, with photographic documentation.3.16. Statistical analysis
Data were shown as mean ± standard deviation (SD). The statistical analyses and graphing were performed by GraphPad Prism. Student's t-test and one-way analysis of variance (ANOVA) were used for data comparison (*p < 0.05, **p < 0.01, ***p < 0.001 was considered statistically significant).4 Conclusions
This study successfully developed an innovative injectable thermo-responsive hydrogel drug delivery system (DOX + BAY@Gel), aiming to achieve efficient breast cancer treatment through a synergistic mechanism. The core strategy of this system lies in the co-delivery of a DNA-damaging agent (DOX) and an inhibitor (BAY) targeting a key DNA damage repair pathway (ATR). This approach simultaneously induces DNA damage in cancer cells and blocks their repair mechanisms, thereby triggering more severe DNA damage accumulation and cell death, forming a “synthetic lethality” effect.The hydrogel's unique thermo-responsive physical characteristics constitute a key advantage. It exists in a fluid state at room temperature, facilitating injection, and rapidly transforms into a stable gel state upon reaching the tumor site triggered by body temperature. This in situ phase transition effectively confines the co-encapsulated DOX and BAY within the local tumor tissue, significantly enhancing drug enrichment at the target site, laying the foundation for precise therapy. Concurrently, the gel matrix endows it with excellent sustained-release properties, ensuring both drugs are released in a sustained, stable, and controllable manner, avoiding the potential drug burst release associated with conventional administration methods. This controlled release profile not only maintains long-term effective therapeutic concentrations but, more importantly, significantly reduces the risk of systemic toxic side effects induced by chemotherapeutic agents like DOX, offering patients a safer therapeutic option.
Comprehensive results from both in vitro and in vivo experiments demonstrate the potent anti-tumor efficacy of this drug delivery system (DOX + BAY@Gel). The significant tumor reduction observed in vivo experiments robustly validates the effectiveness of its targeted delivery and synergistic therapeutic strategy.
In conclusion, the strategy adopted in this study, utilizing an injectable thermo-responsive hydrogel for the co-delivery of a DNA-damaging agent and a DNA repair inhibitor, provides a highly promising novel approach for the local treatment of breast cancer. Its advantages of precise positioning, sustained release, and reduced systemic toxicity hold the potential to offer novel alternatives for improving treatment outcomes in breast cancer patients.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. W., J.-R. Z.: writing – original draft and investigation. Q. W.: investigation. Y.-F. H., H-L. Y., C.-Y. Z.: data curation. L. G., J.-F. D.: project administration, supervision, and writing review and editing.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the Four “Batches” Innovation Project of Invigorating Medical through Science and Technology of Shanxi Province (2023XM036); National Natural Science Foundation of China (32201160, 52402348); Shanxi Applied Basic Research Project (202203021222374). We are grateful to the Shared Instrument Platform of the School of Pharmacy, Shanxi Medical University.References
- P. Yuan and B. H. Xu, Chin. J. Oncol., 2024, 46, 471–480 Search PubMed.
- Z. Zhang, Y. Wang, Q. Ma, S. Zhang, H. Liu, B. Zhao, R. Liu, W. Wang, B. Du, Y. Zhong and D. Kong, Chem. Eng. J., 2021, 406, 126801 CrossRef CAS.
- R. Huang and P. K. Zhou, Signal Transduct. Targeted Ther., 2021, 6, 254 CrossRef CAS PubMed.
- A. I. Gomaa and I. Waked, World J. Hepatol., 2015, 7, 673–687 CrossRef PubMed.
- J. L. Weintraub and R. Salem, J. Vasc. Intervent. Radiol., 2013, 24, 1123–1134 CrossRef PubMed.
- M. L. Abel, N. Takahashi, C. Peer, C. E. Redon, S. Nichols, R. Vilimas, M.-J. Lee, S. Lee, M. Shelat, R. Kattappuram, L. Sciuto, D. Pinkiert, C. Graham, D. Butcher, B. Karim, A. K. Sharma, J. Malin, R. Kumar, C. W. Schultz, S. Goyal, J. Del Rivero, M. Krishnamurthy, D. Upadhyay, B. Schroeder, T. Sissung, M. Tyagi, J. Kim, Y. Pommier, M. Aladjem, M. Raffeld, W. D. Figg, J. Trepel, L. Xi, P. Desai and A. Thomas, Clin. Cancer Res., 2023, 29, 3603–3611 CrossRef CAS PubMed.
- L. H. Cossar, A. G. Schache, J. M. Risk, J. J. Sacco, N. J. Jones and R. Lord, Clin. Oncol., 2017, 29, 626–634 CrossRef CAS PubMed.
- L. R. Kelland and K. S. Tonkin, Radiother. Oncol., 1989, 15, 363–369 CrossRef CAS PubMed.
- R. Liu, C. Zhang, X. Wu, C. Wang, M. Zhao, C. Ji, X. Dong, R. Wang, H. Ma, X. Wang, Y. Tan, J. Du and Z. Gu, Chem. Eng. J., 2023, 461, 142085 CrossRef CAS.
- E. Lecona and O. Fernandez-Capetillo, Nat. Rev. Cancer, 2018, 18, 586–595 CrossRef CAS PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- J. V. Forment and M. J. O'Connor, Pharmacol. Ther., 2018, 188, 155–167 CrossRef CAS PubMed.
- Z. Wilson, R. Odedra, Y. Wallez, P. W. G. Wijnhoven, A. M. Hughes, J. Gerrard, G. N. Jones, H. Bargh-Dawson, E. Brown, L. A. Young, M. J. O'Connor and A. Lau, Cancer Res., 2022, 82, 1140–1152 CrossRef CAS PubMed.
- J. Cui, D. Dean, F. J. Hornicek, R. E. Pollock, R. M. Hoffman and Z. Duan, Am. J. Cancer Res., 2022, 12, 1577–1592 CAS.
- R. Leibrandt, P. H. Frankel, J. H. Beumer, N. Takebe, M. Yin, H. Emamekhoo, P. L. N. Lara, S. Gore and M. Parikh, J. Clin. Oncol., 40, TPS3174 CrossRef.
- F. F. Pusch, H. Dorado García, R. Xu, D. Gürgen, Y. Bei, L. Brückner, C. Röefzaad, J. Von Stebut, V. Bardinet, R. Chamorro Gonzalez, A. Eggert, J. H. Schulte, P. Hundsdörfer, G. Seifert, K. Haase, B. W. Schäfer, M. Wachtel, A. A. Kühl, M. V. Ortiz, A. M. Wengner, M. Scheer and A. G. Henssen, Mol. Cancer Ther., 2024, 23, 507–519 CrossRef CAS PubMed.
- G. Sartori, C. Tarantelli, F. Spriano, E. Gaudio, L. Cascione, M. Mascia, M. Barreca, A. J. Arribas, L. Licenziato, G. Golino, A. Ferragamo, S. Pileri, G. Damia, E. Zucca, A. Stathis, O. Politz, A. M. Wengner and F. Bertoni, Br. J. Haematol., 2024, 204, 191–205 CrossRef CAS PubMed.
- B. Mapes, O. Charif, S. Al-Sawwaf and E. Dolan, Clin. Cancer Res., 2017, 23, 4010–4019 CrossRef PubMed.
- K. T. Sawicki, V. Sala, L. Prever, E. Hirsch, H. Ardehali and A. Ghigo, Annu. Rev. Pharmacol. Toxicol., 2021, 61, 309–332 CrossRef CAS PubMed.
- J. Zhao, L. Cui, X. R. Wang and C. Y. Deng, Colloid Polym. Sci., 2022, 300, 1075–1086 CrossRef CAS.
- S. Yan, T. Wang, X. Li, Y. Jian, K. Zhang, G. Li and J. Yin, RSC Adv., 2017, 7, 17005–17019 RSC.
- S. Shi, Y. Chang, K. Fu, N. Fu, X. Hu, B. Zhao, B. Chen, X. Yun and E. Shi, RSC Adv., 2024, 14, 19134–19146 RSC.
- R. A. A. Muzzarelli, Carbohydr. Polym., 2009, 76, 167–182 CrossRef CAS.
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. D. Buschmann, C. D. Hoemann, J. C. Leroux, B. L. Atkinson, F. Binette and A. Selmani, Biomaterials, 2000, 21, 2155–2161 CrossRef CAS PubMed.
- O. M. Kolawole, W. M. Lau and V. V. Khutoryanskiy, Int. J. Pharm.: X, 2019, 1, 100007 CAS.
- T. A. Yap, D. S. P. Tan, A. Terbuch, R. Caldwell, C. Guo, B. C. Goh, V. Heong, N. R. M. Haris, S. Bashir, Y. Drew, D. S. Hong, F. Meric-Bernstam, G. Wilkinson, J. Hreiki, A. M. Wengner, F. Bladt, A. Schlicker, M. Ludwig, Y. Zhou, L. Liu, S. Bordia, R. Plummer, E. Lagkadinou and J. S. de Bono, Cancer Discov., 2021, 11, 80–91 CrossRef CAS PubMed.
- A. M. Wengner, G. Siemeister, U. Lücking, J. Lefranc, L. Wortmann, P. Lienau, B. Bader, U. Bömer, D. Moosmayer, U. Eberspächer, S. Golfier, C. A. Schatz, S. J. Baumgart, B. Haendler, P. Lejeune, A. Schlicker, F. von Nussbaum, M. Brands, K. Ziegelbauer and D. Mumberg, Mol. Cancer Ther., 2020, 19, 26–38 CrossRef CAS PubMed.
- H. Hafsi, M. T. Dillon, H. E. Barker, J. N. Kyula, U. Schick, J. T. Paget, H. G. Smith, M. Pedersen, M. McLaughlin and K. J. Harrington, Front. Oncol., 2018, 8, 245 CrossRef PubMed.
- H. Chen, Z. Zhang, J. Qi, C. Cao, M. Lin, L. Lyu and D. Xu, ACS Omega, 2025, 10, 7964–7972 CrossRef CAS PubMed.
- N. Hirun, P. Kraisit and S. Santhan, Pharmaceutics, 2024, 16(8), 1055 CrossRef CAS PubMed.
- Y. Ma, Y. Tang, J. Fan, T. Sun, X. Qiu, L. Wei and X. Zhang, RSC Adv., 2024, 14, 38353–38363 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03120f |
| ‡ Lan Wei and Jiaru Zhu contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2025 |