
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally-driven interface engineering of PMo12/BiOBr heterojunctions for enhanced artificial photosynthesis of CO2 in water vapor†
Yan Shia,
Shiqin Liua,
Hui Chena,
Zaihui Fu b,
Youji Lia and
Senpei Tang*a
b,
Youji Lia and
Senpei Tang*a
aHunan Province Key Laboratory of Mineral Cleaner Production and Green Functional Materials, College of Chemistry and Chemical Engineering, Jishou University, Jishou, 416000, Hunan, PR China. E-mail: ChemTangJSU@163.com
bCollege of Chemistry and Chemical Engineering, Hunan Normal University, 410081, Hunan, PR China
First published on 30th May 2025
Abstract
In this study, a heterojunction material composed of Keggin-type H3PMo12O40 and BiOBr (PM/BOB) was synthesized by a hydrothermal-calcination method, and its photocatalytic CO2 reduction performance and mechanism were investigated. Structural characterization through XRD, SEM, XPS, and UV-vis DRS revealed that calcination at 200 °C facilitated tight interfacial bonding between PM and BiOBr. The BiOBr sheets fragmented into nano-sized particles that uniformly integrated with PM, while partial reduction of PM generated active species containing mixed Mo5+/Mo6+ valence states. Under optimal conditions, the t200-PM/BOB0.5 composite demonstrated exceptional CO2 reduction performance without sacrificial agents, achieving a CO production rate of 18.82 μmol g−1 h−1, representing 10.06-fold and 7.13-fold enhancements over pristine BiOBr and PM, respectively. Mechanistic studies unveiled a Z-scheme electron transfer pathway where the reduced-state intervalence charge transfer (IVCT) excited species in PM act as electron mediators to drive CO2 reduction, while the BiOBr valence band holes participate in water oxidation, achieving spatial separation of redox sites. This work provides a novel strategy for designing efficient Keggin-type molybdenum-based photocatalysts and advances the development of solar-driven CO2 utilization technologies.
Introduction
The accelerated industrialization process has led to excessive consumption of fossil fuels, resulting in a sharp rise in atmospheric CO2 concentration, which profoundly disrupts the global carbon balance and exacerbates the climate crisis.1 Under this context, developing CO2 utilization technologies has become a crucial pathway to achieve the “dual carbon” goals.2 Among existing conversion strategies, photocatalytic reduction technology has attracted significant attention due to its unique advantage of directly utilizing solar energy to convert CO2 into fuel molecules (CO, CH4, CH3OH, etc.).3 This process triggers redox reactions through photogenerated electron–hole pairs, theoretically enabling a fully cyclic carbon-neutral process.4 However, the inherent chemical inertness of CO2 molecules (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation energy reaching 750 kJ mol−1) and multiple kinetic bottlenecks in photocatalytic systems (including insufficient light absorption, charge separation, and surface reaction synergy) severely restrict practical applications.5
O bond dissociation energy reaching 750 kJ mol−1) and multiple kinetic bottlenecks in photocatalytic systems (including insufficient light absorption, charge separation, and surface reaction synergy) severely restrict practical applications.5
To overcome these limitations, researchers have focused on developing efficient photocatalytic systems, with the core strategy being the optimization of charge carrier dynamics and reaction microenvironments. Heterojunction engineering has emerged as an effective approach to enhance photocatalytic performance by modulating band structures to promote interfacial charge separation.6 Most of the traditional heterostructures have been semiconductor–semiconductor composites (e.g. BiOBr/TiO2, g-C3N4/CdS), while polyoxometalate (POM)-based heterostructures remain relatively underexplored.7 POMs are nanoscale clusters composed of high-valence transition metals (Mo6+, W6+, V5+, etc.) coordinated with oxygen atoms, exhibiting semiconductor-like characteristics that include:6d,7a,8 (i) well-defined HOMO–LUMO energy gaps (2–4 eV), enabling broad-spectrum light absorption; (ii) multi-electron storage capacity (e.g., Keggin-type structures can accommodate 6–12 electrons), which is particularly suitable for the multi-step reduction of CO2; and (iii) reversible redox properties that help maintain catalytic cycle stability. These unique attributes make POMs ideal building blocks for designing novel photocatalysts for CO2 reduction.8b,9
A representative case was demonstrated by Li et al.,10 who achieved spatially confined encapsulation of CoNi bimetallic active sites within hollow dodecahedral K3PW12O40 frameworks through a hydrothermal-annealing synergistic strategy. This unique architecture effectively suppressed metal cluster aggregation via steric hindrance effects, yielding a CO production rate of 15.1 μmol g−1 h−1 after 6 hours of illumination in a [Ru(bpy)3]2+ photosensitizer-containing liquid-phase system. In another approach, Wang's group9a developed a diffusion-driven self-assembly method to precisely incorporate silicomolybdic acid into the mesoporous channels of sulfonic acid-functionalized MIL-101-Cr, constructing a hierarchical SiMo12O40@SO3H-MIL-101-Cr composite. This design maintained the parent material's high specific surface area and chemical stability while significantly improving active site accessibility, achieving 93.2% CO selectivity with a production rate of 82.95 μmol g−1 h−1 in triethanolamine/acetonitrile mixed solution. Such MOF pore-confined POM encapsulation strategies systematically enhance photocatalytic CO2 reduction performance by creating highly dispersed atomic-level contact interfaces, enabling efficient electron transport channels and multicomponent coupling effects.11 However, these systems represent the complex solid–liquid phase reaction mode. Although dominant in photocatalytic CO2 reduction research, such three-phase systems (comprising photocatalysts, solvents, and photosensitizers/sacrificial agents) face inherent limitations:9d,10,12 (i) poor dispersion stability of catalyst particles in liquid phase, leading to aggregation-induced deactivation; (ii) severely limited mass transfer efficiency due to low CO2 solubility (e.g., ∼33 mM in water) and weak adsorption capacity in conventional solvents; (iii) difficult product separation, particularly for liquid organic compounds, which substantially increases purification costs.
In contrast, the gas–solid reaction mode demonstrates distinctive advantages:13 (i) direct utilization of CO2/H2O vapor as reactants not only provides proton sources but also suppresses the competitive hydrogen evolution reaction, while circumventing solvent-induced mass transfer limitations; (ii) precise control of reaction kinetics through adjustable CO2 concentration; (iii) dominant gaseous products (CO, CH4) enabling real-time monitoring and facile collection. Representative studies include Zhang et al. on constructing Lewis basic group-functionalized BiOBr heterojunctions through NH2-UiO-66 modification. The optimized BiOBr/NH2-UiO-66 exhibited a CO production rate of 9.19 μmol g−1 h−1, representing a 4-fold enhancement over pristine BiOBr.14 In another approach, Liu et al. developed Au@NENU-10 composite photocatalysts utilizing the dual proton-electron storage capability of Keggin-type [PTi2W10O40]7− clusters, achieving 12.8 μmol per g per h CO yield under visible light irradiation.15 Yang et al. engineered 1D/2D BiOBr/CdS heterostructures that delivered 13.6 μmol per g per h CO output through band structure optimization.16 Nevertheless, the currently reported CO2 photoreduction efficiencies in gas–solid systems remain substantially below industrial requirements. This performance gap highlights the urgent need for fundamental breakthroughs in interfacial charge transfer efficiency, CO2 adsorption-activation synergy, and long-term operational stability to address existing technical bottlenecks.13
Notably, IVCT effects in reduced polyoxometalates provide critical advantages for CO2 photoreduction.17 As Neumann demonstrated,18 these mixed-valence POMs exhibit visible-light-responsive IVCT transitions (λmax ≈ 725 nm) through W5+/W6+ electron hopping, enabling efficient directional electron transfer to catalytic sites. Haviv's work19 confirmed this mechanism, showing a 3.8-fold CO yield enhancement in POM-Re hybrids via IVCT-mediated pathways. Crucially, IVCT effects simultaneously broaden light absorption to near-infrared and synchronize multi-electron transfer kinetics, effectively addressing Z-scheme recombination.
Herein, we developed a t200-PM/BOB0.5 heterojunction composite through a hydrothermal-calcination approach. Comprehensive characterization revealed that thermal treatment facilitated intimate interfacial contact between PM and BOB, while simultaneously inducing partial reduction of PM to form mixed-valence active centers (Mo5+/Mo6+). This structural optimization resulted in significantly enhanced visible-light absorption and charge separation efficiency. Remarkably, the t200-PM/BOB0.5 system exhibits significantly enhanced properties in CO2 photoreduction performance under sacrificial agent-free and photosensitizer-free gas–solid conditions, with CO as the predominant product (selectivity ∼100%). This findings provide fundamental insights for developing sustainable solar fuel generation systems with industrial viability.
Results and discussion
Microstructure and structure characterization
SEM characterization of the material reveals distinct morphological features (Fig. 1). BOB exhibits well-defined square nanosheets with smooth surfaces and uniform thickness at micro–nano scales (Fig. 1a). The PM film deposited on glass substrates displays pronounced surface undulations with extensive crack networks (Fig. 1b), which surface cracks was conducive to increasing the specific surface area and enhancing the gas adsorption capacity. The uncalcined t25-PM/BOB0.5 composite material shows that BOB and PM form a physical mixed system through mechanical grinding (Fig. 1c and d). Although a small amount of irregular fragments were generated, the main body still maintains the flake shape of BOB, indicating that the interface interaction between the components was limited when only grinding was performed. Notably, calcination at 200 °C induces significant structural reorganization in t200-PM/BOB0.5 (Fig. 1e and f), where BOB nanosheets fragment into nanoscale irregular debris while forming intimate heterointerfaces with PM, a critical microstructural foundation for multicomponent synergistic catalytic effects. EDS elemental mapping confirms homogeneous dispersion of Bi, O, Br, P, and Mo across the t200-PM/BOB0.5 surface (Fig. 1g–k). | ||
| Fig. 1 SEM images of catalyst (a) BOB, (b) PM, (c and d) t25-PM/BOB0.5 and (e and f) t200-PM/BOB0.5; (g–k) elements mapping of Bi, O, Br, P, and Mo in the corresponding region. | ||
To elucidate the surface functional groups, FT-IR spectroscopy was employed to characterize BOB, PM, and the t200-PM/BOB0.5 composite (Fig. 2a). The BOB spectrum exhibits a prominent absorption band at 514 cm−1, attributed to the Bi–O stretching vibration. The PM spectrum displays four characteristic bands at 1064, 966, 867, and 719 cm−1, corresponding to P–Oa (oxygen atoms in PO4 tetrahedra), MoOd (terminal oxygen atoms), Mo–Ob–Mo (corner-sharing bridging oxygen), and Mo–Oc–Mo (edge-sharing bridging oxygen), respectively, confirming the intact Keggin structure of PM.20 Notably, FT-IR spectra (Fig. 2a) demonstrate that the characteristic vibration peaks of PM in the composite of t200-PM/BOB0.5 remain largely consistent with pure PM, confirming the intact preservation of its fundamental structure during compounding. The significant shift of the Bi–O vibrational characteristic peak in the t200-PM/BOB0.5 (501 cm−1) compared to pure BOB,providing direct spectroscopic evidence for effective interfacial coupling between the two components.
 | ||
| Fig. 2 (a) FT-IR spectra of BOB, PM, and t200-PM/BOB0.5; (b) the XRD pattern of BOB, t25-PM/BOB0.5, and t200-PM/BOB0.5. | ||
The crystal structure of the sample was analyzed by XRD. The PM sample exhibits characteristic diffraction peaks matching the standard card (JCPDS No. 01-0032), though with reduced intensity and minor impurity peaks, indicative of its low crystallinity (Fig. S3†), which is consistent with the characteristics of this material reported in the literature.21 In contrast, BOB and its composites (t25-PM/BOB0.5 and t200-PM/BOB0.5) display sharp, high-intensity diffraction peaks in Fig. 2b, showing excellent agreement with the BiOBr standard card (JCPDS No. 78-0348) and confirming their high crystallinity and well-ordered atomic arrangement. The position and intensity of BOB's diffraction peak did not change significantly either by mechanical grinding or calcination at 200 °C, indicating that its crystal structure remained stable without phase change or structural damage. However, due to the material fragmentation caused by calcination process, the relative intensity of (001) crystal plane diffraction peaks decreases, while the relative intensity of (102) and (110) crystal plane diffraction peaks increases, which should be a slight change in the crystal orientation ratio due to the increase in cross section. Additionally, although PM exhibits low crystallinity and is highly dispersed within the composite, leading to weak diffraction signals, PM diffraction peaks could still be identified in the XRD pattern of t200-PM/BOB0.5.
The surface chemical states of the samples were characterized by XPS as shown in Fig. 4. The XPS survey spectrum (Fig. 3a) confirmed the coexistence of five elements (Bi, O, Br, P, and Mo) in the t200-PM/BOB0.5 composite, with all high-resolution spectra calibrated using the C 1s reference peak at 284.8 eV. The high-resolution Bi 4f spectrum (Fig. 3b) of pristine BOB exhibited characteristic doublet peaks at 159.6 eV (Bi 4f7/2) and 164.9 eV (Bi 4f5/2), corresponding to Bi3+ species.22 Notably, the composite material showed significant positive shifts of approximately 0.4 eV in both Bi 4f7/2 (160.0 eV) and Bi 4f5/2 (165.3 eV) peaks, suggesting potential electron interaction between PM and BOB components. Similar chemical shifts were observed in the Br 3d spectrum (Fig. 3c), where the characteristic Br− peaks at 68.6 eV (Br 3d5/2) and 69.6 eV (Br 3d3/2) for BOB23 shifted to 69.0 eV and 69.9 eV in the composite, respectively, indicating interfacial electronic redistribution. The high-resolution P 2p spectrum (Fig. 3d) revealed stable phosphorus chemical states in both PM and composite materials, showing characteristic doublet peaks at 134.2 eV (P 2p3/2) and 135.1 eV (P 2p1/2) corresponding to P5+ in phosphate groups.24 For molybdenum species analysis (Fig. 3e), the PM component exhibited Mo 3d peaks at 233.4 eV (Mo 3d5/2) and 236.6 eV (Mo 3d3/2), characteristic of Mo6+ species.25 In the t200-PM/BOB0.5, these peaks shift slightly to lower binding energies (233.3 eV and 236.5 eV), accompanied by additional peaks at 232.2 eV (3d5/2) and 235.2 eV (3d3/2) assigned to Mo5+, indicating partial reduction of Mo6+ at the interface. The Mo5+ content accounts for 4.7% of total Mo species, as quantified by peak area integration.26 In the O 1s high-resolution spectrum analysis (Fig. 3f), the peaks at 530.6 eV and 531.5 eV for BOB correspond to lattice oxygen in Bi–O bonds and surface hydroxyl groups (–OH) or adsorbed water molecules, respectively.27 For the PM, the peaks at 531.3 eV and 532.4 eV are attributed to bridging oxygen (Mo–O–Mo) and terminal oxygen (MoO).28 Because PM covers the surface of BOB, XPS detects more of the oxygen signals of the surface PM. Therefore, in the t200-PM/BOB0.5, the O 1s peaks shifted to 531.2 eV and 532.3 eV, closely aligning with PM's spectral features and exhibiting minor deviations (−0.1 eV). These XPS characterization results strongly indicate that there is an electron transfer between BOB and PM during the calcination process, which may enhance the catalytic activity by generating oxygen vacancies or exposing the active sites of Mo5+.
 | ||
| Fig. 3 (a) XPS survey spectrum and high-resolution XPS of (b) Bi 4f, (c) Br 3d, (d) P 2p, (e) Mo 3d and (f) O 1s in catalyst BOB, PM and t200-PM/BOB0.5. | ||
Optical absorption and photoelectrochemical properties
The visible light absorption characteristics and the separation efficiency of photogenerated electron hole pairs of catalysts are the key factors determining their photocatalytic activity. UV-vis DRS analysis revealed distinct absorption edges for pristine BOB (450 nm) and PM (550 nm), corresponding to electron transitions in Bi–O/Bi–Br and Mo–O bonds, respectively (Fig. 4a). Remarkably, the t200-PM/BOB0.5 composite exhibited an extended absorption edge (∼525 nm) with a broadened new absorption band centered at 680 nm, attributable to the IVCT effect induced by reduced Mo5+ species in PM. Kubelka–Munk transformation analysis yielded bandgap values of 2.79 eV (BOB), 2.28 eV (PM), and 2.34 eV (t200-PM/BOB0.5), confirming their visible-light-responsive characteristics (insert of Fig. 4a). | ||
| Fig. 4 (a) UV-vis DRS spectra of catalysts BOB, PM and t200-PM/BOB0.5, with the band gap diagram of the corresponding catalyst illustrated; (b) solid PL spectra of BOB, PM and t200-PM/BOB0.5, with amplified fluorescence spectra of PM and t200-PM/BOB0.5 shown in the embedded spectrogram; (c) photocurrent response and (d) EIS of BOB, PM, t25-PM/BOB0.5, and t200-PM/BOB0.5. | ||
PL spectroscopy under 320 nm excitation provided critical verification of separation efficiency of electron–hole pairs (Fig. 4b). PM exhibited a weak emission peak at 480 nm, while BOB showed relatively intense fluorescence at 440 nm, indicative of rapid electron–hole recombination. The t200-PM/BOB0.5 demonstrated the lowest PL intensity, verify that the interaction between BOB and PM can improve the separation efficiency of electron–hole pairs (insert of Fig. 4b). The interfacial charge transfer properties of catalysts were investigated through electrochemical characterization. As shown in Fig. 4c, the photocurrent response of t200-PM/BOB0.5 (0.46 μA cm−2) significantly exceeded that of t25-PM/BOB0.5 (0.41 μA cm−2), pristine BOB (0.08 μA cm−2), and PM (0.03 μA cm−2), indicating that the photoelectric conversion efficiency of the composite material is improved after calcination treatment. EIS analysis further revealed the charge transfer resistance through Nyquist plot comparison (Fig. 4d). The t200-PM/BOB0.5 composite exhibited the smallest arc radius, indicating the lowest interfacial charge transfer resistance among the tested materials. The quantitative correlation between the reduced semi-circular size and the improved charge separation efficiency confirms that, compared with individual components, the composite material structure significantly enhances photoinduced charge separation and interface carrier transport, which is highly consistent with the observed photocatalytic performance enhancement.
Photocatalytic CO2 reduction activity
The photocatalytic CO2 reduction (PCR) performance of the catalyst was investigated at gas–solid interface with water as electron sacrificial agent under simulated sunlight. As shown in Fig. 5a and b, pristine BOB and PM exhibited CO evolution rates of 1.87 and 2.64 μmol g−1 h−1, respectively. The mechanically mixed t25-PM/BOB0.5 showed marginal improvement (3.55 μmol g−1 h−1), whereas the calcined t200-PM/BOB0.5 achieved a remarkable 18.82 μmol g−1 h−1, representing 10.06 and 7.13-fold enhancement over individual components. Control experiments demonstrate that individual components (uncompounded PM or BOB, denoted as t200-PM and t200-BOB) calcined at 200 °C for identical duration exhibited photocatalytic CO2 reduction only 6.29 and 3.55 μmol g−1 h−1 respectively, showing little improvement compared with before calcination. The significant changes in catalytic performance under these different conditions highlight the key role of calcination induction interface engineering.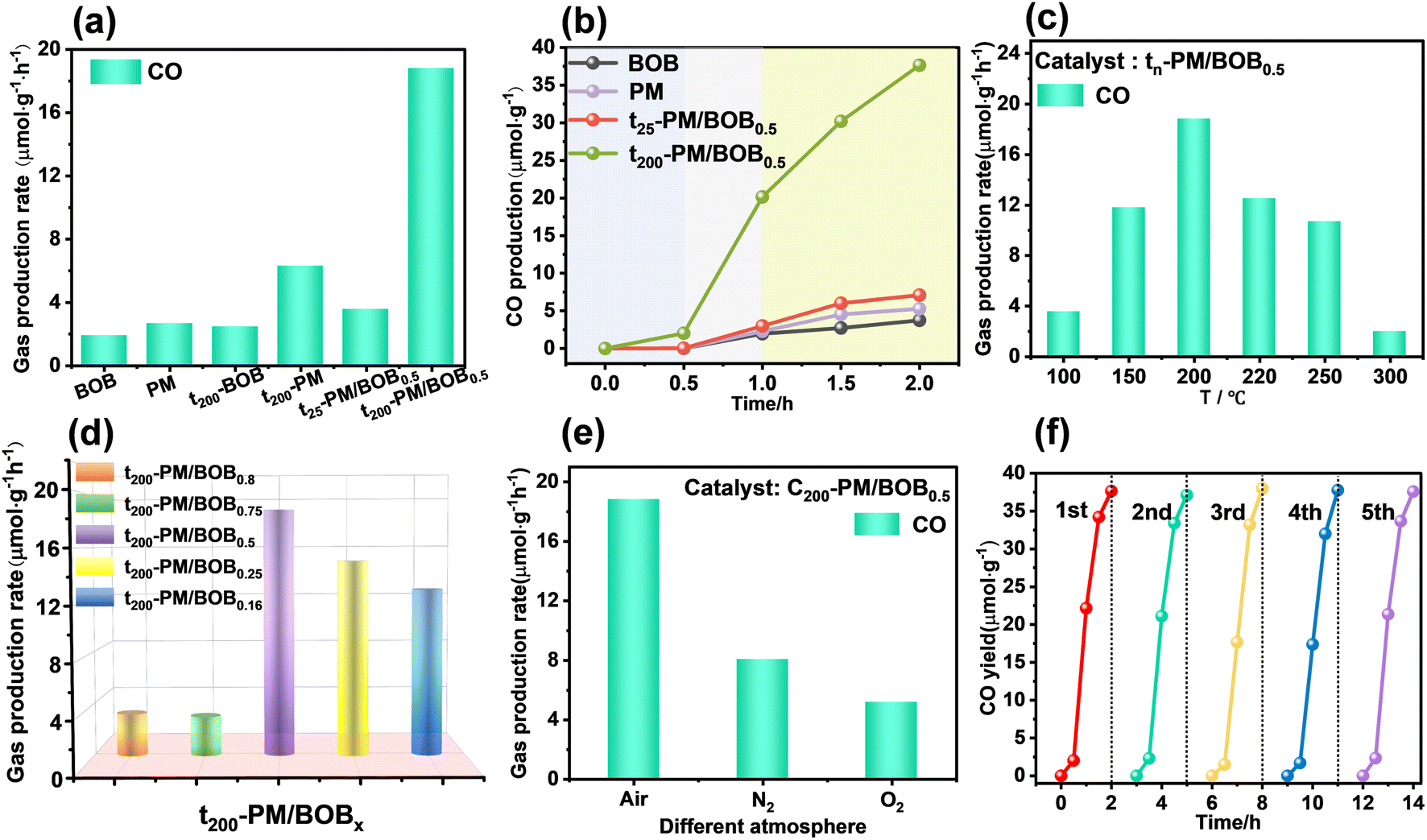 | ||
| Fig. 5 (a) The PCR performance diagram of BOB, PM, t25-PM/BOB0.5, t200-PM/BOB0.5, t200-BOB, t200-PM and (b) their corresponding CO yields over time; (c) PCR performance diagram of tn-PM/BOB0.5 synthesized at different temperatures; (d) PCR performance of t200-PM/BOBx with different ratios of PM/BOB; (e) The PCR performance diagram of t200-PM/BOB0.5 synthesized under different calcination atmosphere; (f) the PCR cycle test performance of t200-PM/BOB0.5. | ||
The calcination temperature optimization study (Fig. 5c) revealed 200 °C as the optimal thermal treatment condition. Below this threshold, insufficient heterointerface formation limited charge separation efficiency. When the temperature exceeds 200 °C, the thermal instability of PM components leads to structural decomposition, resulting in a downward trend in catalytic activity. The synchronized physical photos show that with the increase of calcination temperature (25–300 °C), the color of the material changes from light yellow to dark green (Fig. S4†), which is consistent with the characteristic color development of the reduced Mo5+ state of phosphomolybdenum heteropolyacid. The UV-vis DRS (Fig. S5†) further confirmed that the characteristic absorption peak intensity of the material at 600–800 nm was significantly enhanced with the increase of the treatment temperature, indicating that the concentration of Mo5+ reduced species was positively correlated with the calcination temperature.
Compositional optimization studies at 200 °C identified a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PM/BOB mass ratio as the optimal configuration (Fig. 5d). PM-rich composites (t200-PM/BOB0.8 and t200-PM/BOB0.75) exhibited significantly reduced activity (3.21–4.52 μmol g−1 h−1), while BOB-dominant formulations (t200-PM/BOB0.25 and t200-PM/BOB0.16) maintained moderate performance (13.5–15.5 μmol g−1 h−1). At the same time, different atmosphere during calcination will also affect the PCR efficiency of t200-PM/BOB0.5. As shown in Fig. 5e, when the composite material was calcined in N2, air, and O2 atmosphere, the CO conversion rates are 8.06, 18.82, and 5.16 μmol g−1 h−1, respectively. This atmosphere-dependent behavior originates from dual modulation effects: (i) oxygen partial pressure regulates Mo5+ enrichment in PM, enhancing reducibility; (ii) calcination atmosphere tunes oxygen vacancy concentration in BOB, which serves as oxidative active sites and promotes the formation of heterojunction interface through coordination with phosphomolybdenum heteropoly acid.
:1 PM/BOB mass ratio as the optimal configuration (Fig. 5d). PM-rich composites (t200-PM/BOB0.8 and t200-PM/BOB0.75) exhibited significantly reduced activity (3.21–4.52 μmol g−1 h−1), while BOB-dominant formulations (t200-PM/BOB0.25 and t200-PM/BOB0.16) maintained moderate performance (13.5–15.5 μmol g−1 h−1). At the same time, different atmosphere during calcination will also affect the PCR efficiency of t200-PM/BOB0.5. As shown in Fig. 5e, when the composite material was calcined in N2, air, and O2 atmosphere, the CO conversion rates are 8.06, 18.82, and 5.16 μmol g−1 h−1, respectively. This atmosphere-dependent behavior originates from dual modulation effects: (i) oxygen partial pressure regulates Mo5+ enrichment in PM, enhancing reducibility; (ii) calcination atmosphere tunes oxygen vacancy concentration in BOB, which serves as oxidative active sites and promotes the formation of heterojunction interface through coordination with phosphomolybdenum heteropoly acid.
The photocatalytic performance of t200-PM/BOB0.5 under varying CO2 concentrations to evaluate the practical applicability of the catalyst, as illustrated in Fig. S7.† The CO production rate increased significantly from 1.33 to 18.83 μmol g−1 h−1 as the CO2 concentration rose from 2 to 18 vol%. This concentration-dependent behavior demonstrates the material's direct applicability potential for medium-to-low concentration CO2 mitigation scenarios. Finally, durability tests confirmed exceptional stability of t200-PM/BOB0.5, maintaining 18.8 ± 0.24 μmol g−1 h−1, CO production over 5 cycles with <3% activity decay (Fig. 5f). In addition, XRD and FT-IR spectra were used to conduct comparative analyses before and after the photocatalytic reaction of the t200-PM/BOB0.5. The characteristic diffraction peaks in the XRD pattern and the functional group vibrations in the FT-IR pattern all maintained their positions and intensities, and there was no obvious structural change (Fig. S6†). These robust performance stems from the engineered heterointerface's structural integrity and inherent anti-photocorrosion properties. Finally, a comparative analysis of the photocatalytic CO2 reduction performance at the gas–solid interface with the recently reported POM/BiOBr-based analogues indicates that our material outperforms many of the reported similar materials, demonstrating its superior activity.
Photocatalytic mechanism
Based on the aforementioned experimental results, it can be speculated that the enhanced photocatalytic CO2 reduction mechanism primarily originates from the construction of high-quality heterojunction structures. To validate this hypothesis, investigations into the band alignment and heterojunction type of the catalysts were conducted. As shown in Fig. 6a, the positive slope of the MS plot for BOB confirms its n-type semiconductor characteristics. The flat-band potential (Efb) of BOB was determined to be −0.57 V. For n-type semiconductors, the Efb is approximately equivalent to the Fermi level (EF) under most conditions.29 Based on the Boltzmann distribution relationship between EF and conduction band potential (ECB), along with the intrinsic properties of n-type semiconductors, the ECB of BOB was calculated to be −0.77 V. Furthermore, the UV-vis DRS results revealed a Eg of 2.79 eV for BOB, representing the energy difference between its valence band maximum (EVB) and ECB. Using the equation Eg = EVB − ECB, the EVB of BOB was subsequently determined as 2.02 V. | ||
| Fig. 6 (a) The M–S curve of BOB, (b) the CV curve of PM; and (c) the mechanism diagram of photocatalytic CO2 reduction reaction based on t200-PM/BOB0.5. | ||
To investigate the energy band structure of PM, CV curves was employed to estimate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels. As shown in Fig. 6b, the onset potential of the first reduction peak in the CV curve for oxidized PM (PMox) was measured at 0.71 V, corresponding to its LUMO potential (ELUMO). Combined with the Eg of PM (2.28 eV) determined from UV-vis DRS, the HOMO potential (EHOMO) was calculated as 2.79 V. Notably, the LUMO level of PMox aligns with the HOMO level of reduced PM (PMred).30 UV-vis DRS analysis of the composite (Fig. S7†) revealed an IVCT absorption edge at 823 nm for the PMred component in t200-PM/BOB0.5. Using the equation ΔEgap = 1239.8/λedge, the IVCT transition bandgap was calculated as 1.51 eV. This indicates that photoexcitation induces charge transfer between Mo5+ and Mo6+ in PMred, generating an excited state with an IVCT band (EIVCT) electron reduction potential of −0.80 V.31 Both the conduction band potential of BOB (ECB = −0.77 V) and the EIVCT of PMred are significantly more negative than the CO2/CO reduction potential (−0.53 V vs. NHE),32 confirming their thermodynamic capability to drive CO2 reduction to CO. Incidentally, macroscopic color changes before and after calcination of the catalyst and IVCT characteristic absorption peaks (600–900 nm) in the UV-vis DRS support the formation of reduced PMred (Fig. S4 and S5†), indicating partial reduction of PM in t200-PM/BOB0.5 and formation of mixed valence active centers.
Based on the aforementioned experimental results, the photocatalytic CO2 reduction mechanism of the t200-PM/BOB0.5 composite can be summarized as follows (Fig. 6c). Under light excitation, BOB semiconductor generates photogenerated electron–hole pairs, with electrons transitioning from the valence band (VB) to the conduction band (CB). Due to the significant energy difference between the LUMO level of PM (0.71 V) and the conduction band of BOB (−0.77 V), electrons spontaneously transfer from the CB of BOB to the LUMO of PM, forming reduced PMred containing mixed Mo5+/Mo6+ valence states. Studies reveal that the IVCT excited state (EIVCT = −0.8 V) in PMred exhibits semiconductor-like impurity level characteristics, serving as an electron transfer mediator. Its reduction potential is significantly more negative than the standard CO2/CO potential (−0.53 V), thereby efficiently driving CO2 reduction. Simultaneously, the VB holes in BOB are consumed through water oxidation (H2O + h+ → O2 + H+), achieving charge balance and supplying protons for CO2 reduction. This Z-scheme electron transfer mechanism not only effectively suppresses carrier recombination but also significantly enhances photocatalytic efficiency through spatial separation of redox sites.
Conclusion
In general, the t200-PM/BOB0.5 composite with heterojunction structure was synthesized via hydrothermal-calcination method. Systematic characterization revealed that calcination at 200 °C facilitated intimate interfacial contact between PM and BOB, inducing partial reduction of PM to form mixed-valence active centers (Mo5+/Mo6+). The composite exhibited significantly enhanced visible-light absorption and charge separation efficiency. Optimization studies confirmed that air-calcination at 200 °C and a 1:1 PM/BOB mass ratio delivered optimal performance, achieving a CO production rate of 18.82 μmol g−1 h−1 under gas–solid with simulated sunlight and water vapor, which was 7.1–10.6 times higher than individual components. Furthermore, the material demonstrated exceptional stability with <3% activity decay over five consecutive cycles. Mechanistic investigations attributed the superior performance to synergistic Z-scheme electron transfer and spatially separated redox sites, where the IVCT excited state in reduced PMred acted as an electron relay to suppress carrier recombination and accelerate CO2 reduction kinetics. This work establishes novel design reference for developing high-performance Keggin-type molybdenum-based photocatalysts.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yan Shi: investigation, validation, formal analysis, writing – original draft. Shiqin Liu: investigation, validation. Hui Chen: investigation. Zaihui Fu: conceptualization. Youji Li: supervision. Senpei Tang: conceptualization, resources, project administration, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the financial support for this work by the National Natural Science Foundation of China (Grant No. 22469009, 22478102), the Research Foundation of Education Bureau of Hunan Province, China (24A0371), the Natural Science Fund of Changsha City (kq2208161), and Program for Science and Technology Innovative Research Team in Higher Educational Institutions of Hunan Province.References
- (a) J. Martínez, F. de la Cruz-Martínez, M. A. Gaona, E. Pinilla-Peñalver, J. Fernández-Baeza, A. M. Rodríguez, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, Inorg. Chem., 2019, 58, 3396–3408 CrossRef PubMed; (b) W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- (a) Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef; (b) Y. Ma, X. Yi, S. Wang, T. Li, B. Tan, C. Chen, T. Majima, E. R. Waclawik, H. Zhu and J. Wang, Nat. Commun., 2022, 13, 1400 CrossRef CAS PubMed; (c) S. R. Lingampalli, M. M. Ayyub and C. N. R. Rao, ACS Omega, 2017, 2, 2740–2748 CrossRef CAS PubMed; (d) Y.-S. Xia, M. Tang, L. Zhang, J. Liu, C. Jiang, G.-K. Gao, L.-Z. Dong, L.-G. Xie and Y.-Q. Lan, Nat. Commun., 2022, 13, 2964 CrossRef CAS PubMed.
- V. Humphrey, J. Zscheischler, P. Ciais, L. Gudmundsson, S. Sitch and S. I. Seneviratne, Nature, 2018, 560, 628–631 CrossRef CAS PubMed.
- (a) B. J. Soden, W. D. Collins and D. R. Feldman, Science, 2018, 361, 326–327 CrossRef CAS PubMed; (b) Q. Xu, Z. Xia, J. Zhang, Z. Wei, Q. Guo, H. Jin, H. Tang, S. Li, X. Pan, Z. Su and S. Wang, Carbon Energy, 2023, 5, e205 CrossRef CAS.
- (a) Y. Yu, X. Kuang, X. Jin, F. Chen, J. Min, H. Duan, J. Li, Z. Wu and B. Cao, Appl. Surf. Sci., 2024, 649, 159169 CrossRef CAS; (b) M. A. Mohd Adnan, N. Muhd Julkapli, M. N. I. Amir and A. Maamor, Int. J. Environ. Sci. Technol., 2019, 16, 547–566 CrossRef CAS; (c) X. Li, J. Xiong, Y. Xu, Z. Feng and J. Huang, Chin. J. Catal., 2019, 40, 424–433 CrossRef CAS; (d) A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239–272 CrossRef PubMed; (e) Y. Liu, Y. Dong, J. Yang, Y. Feng, M. Zhao, Y. Qin and H. Lv, Chem. Commun., 2025, 61, 5810–5813 RSC.
- (a) Y. Guo and C. Hu, J. Mol. Catal. A: Chem., 2007, 262, 136–148 CrossRef CAS; (b) Y.-H. Guo and C.-W. Hu, J. Cluster Sci., 2003, 14, 505–526 CrossRef CAS.
- (a) M. Stuckart and K. Y. Monakhov, J. Mater. Chem. A, 2018, 6, 17849–17853 RSC; (b) K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809–10825 CrossRef CAS; (c) J.-J. Ye and C.-D. Wu, Dalton Trans., 2016, 45, 10101–10112 RSC.
- (a) Y. Wang, Y. Xie, M. Deng, T. Liu and H. Yang, Eur. J. Inorg. Chem., 2021, 2021, 681–687 CrossRef CAS; (b) J. Lan, Y. Wang, B. Huang, Z. Xiao and P. Wu, Nanoscale Adv., 2021, 3, 4646–4658 RSC; (c) E. Papaconstantinou, D. Dimotikali and A. Politou, Inorg. Chim. Acta, 1980, 43, 155–158 CrossRef CAS; (d) J. Gu, W. Chen, G. G. Shan, G. Li, C. Sun, X. L. Wang and Z. Su, Mater. Today Energy, 2021, 21, 100760 CrossRef CAS; (e) J. Lan, Q. Zhu, X. Gu, M. Ma, D. Li, K. Huang, X. Fu, Y. Zhu and Y. Zhang, Sep. Purif. Technol., 2023, 321, 124228 CrossRef CAS.
- B. Li, M. Chen, Q. Hu, J. Zhu, X. Yang, Z. Li, C. Hu, Y. Li, P. Ni and Y. Ding, Appl. Catal., B, 2024, 346, 123733 CrossRef CAS.
- (a) A. Solé-Daura, Y. Benseghir, M.-H. Ha-Thi, M. Fontecave, P. Mialane, A. Dolbecq and C. Mellot-Draznieks, ACS Catal., 2022, 12, 9244–9255 CrossRef; (b) J. Du, Y.-Y. Ma, W.-J. Cui, S.-M. Zhang, Z.-G. Han, R.-H. Li, X.-Q. Han, W. Guan, Y.-H. Wang, Y.-Q. Li, Y. Liu, F.-Y. Yu, K.-Q. Wei, H.-Q. Tan, Z.-H. Kang and Y.-G. Li, Appl. Catal., B, 2022, 318, 121812 CrossRef CAS; (c) H. Li, M. Chi, X. Xin, R. Wang, T. Liu, H. Lv and G.-Y. Yang, Chin. J. Catal., 2023, 50, 343–351 CrossRef CAS.
- D. Zang and H. Wang, Polyoxometalates, 2022, 1, 9140006 CrossRef.
- H.-N. Wang, Y.-H. Zou, H.-X. Sun, Y. Chen, S.-L. Li and Y.-Q. Lan, Coord. Chem. Rev., 2021, 438, 213906 CrossRef CAS.
- J. Liu, W. Qin, Y. Wang, Q. Xu, Y. Xie, Y. Chen, Y. Dai and W. Zhang, Sep. Purif. Technol., 2024, 344, 127289 CrossRef CAS.
- S.-M. Liu, Z. Zhang, X. Li, H. Jia, M. Ren and S. Liu, Adv. Mater. Interfaces, 2018, 5, 1801062 CrossRef.
- Q. Yang, W. Qin, Y. Xie, K. Zong, Y. Guo, Z. Song, G. Luo, W. Raza, A. Hussain, Y. Ling, J. Luo, W. Zhang, H. Ye and J. Zhao, Sep. Purif. Technol., 2022, 298, 121603 CrossRef CAS.
- Y. Cao, Q. Chen, C. Shen and L. He, Molecules, 2019, 24, 2069 CrossRef CAS PubMed.
- H. Yu, E. Haviv and R. Neumann, Angew. Chem., Int. Ed., 2020, 59, 6219–6223 CrossRef CAS PubMed.
- E. Haviv, L. J. W. Shimon and R. Neumann, Chem.–Eur. J., 2017, 23, 92–95 CrossRef CAS PubMed.
- S. Tang, J. She, Z. Fu, S. Zhang, Z. Tang, C. Zhang, Y. Liu, D. Yin and J. Li, Appl. Catal., B, 2017, 214, 89–99 CrossRef CAS.
- J. She, Z. Fu, J. Li, B. Zeng, S. Tang, W. Wu, H. Zhao, D. Yin and S. R. Kirk, Appl. Catal., B, 2016, 182, 392–404 CrossRef CAS.
- T. Jia, J. Li, F. Long, F. Fu, J. Zhao, Z. Deng, X. Wang and Y. Zhang, Crystals, 2017, 7, 266 CrossRef.
- Y. Peng, J. Xu, T. Liu and Y.-g. Mao, CrystEngComm, 2017, 19, 6473–6480 RSC.
- (a) S. Damyanova, J. Fierro, I. Sobrados and J. Sanz, Langmuir, 1999, 15, 469–476 CrossRef CAS; (b) D. Barats-Damatov, L. J. Shimon, Y. Feldman, T. Bendikov and R. Neumann, Inorg. Chem., 2015, 54, 628–634 CrossRef CAS PubMed.
- L. Wang, P. Yin, J. Zhang, J. Hao, C. Lv, F. Xiao and Y. Wei, Chem.–Eur. J., 2011, 17, 4796–4801 CrossRef CAS PubMed.
- L. Wang, P. Yin, J. Zhang, J. Hao, C. Lv, F. Xiao and Y. Wei, Chem.–Eur. J., 2011, 17, 4796–4801 CrossRef CAS PubMed.
- J. Wang, J. Yang, S. Zeng, H. Liu, F. Li, H. Xie, S. Tang, Z. Fu and Y. Li, J. Catal., 2025, 443, 115996 CrossRef CAS.
- J. Xiang, P. Xu, J. Wang, Y. Shi, S. Yang, D. Tang, S. Tang and Y. Li, J. Alloys Compd., 2024, 1002, 175476 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- H. Yu, E. Haviv and R. Neumann, Angew. Chem., 2020, 132, 6278–6282 CrossRef.
- M. Dan, S. Yu, W. Lin, M. Abdellah, Z. Guo, Z. Q. Liu, T. Pullerits, K. Zheng and Y. Zhou, Adv. Mater., 2025, 37, 2415138 CrossRef CAS PubMed.
- E. Haviv, L. J. Shimon and R. Neumann, Chem.–Eur. J., 2017, 23, 92–95 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02980e |
| This journal is © The Royal Society of Chemistry 2025 |