
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegration of dual drugs into a collagen scaffold by a combination of apatite coating and impregnation with apatite particles for periodontal regeneration
Kaushita Banerjee a,
Ayako Oyane*a,
Maki Nakamuraa,
Tomoya Inosea,
Erika Nishidab,
Kanako Shitomib and
Hirofumi Miyajib
a,
Ayako Oyane*a,
Maki Nakamuraa,
Tomoya Inosea,
Erika Nishidab,
Kanako Shitomib and
Hirofumi Miyajib
aResearch Institute of Core Technology for Materials Innovation, National Institute of Advanced Industrial Science and Technology (AIST), AIST Tsukuba Central 5, 1-1-1 Higashi, Tsukuba 305-8565, Japan. E-mail: a-oyane@aist.go.jp
bGeneral Dentistry, Department of Oral Health Science, Faculty of Dental Medicine, Hokkaido University, N13W7, Kita-ku, Sapporo 060-8586, Japan
First published on 9th June 2025
Abstract
Bioresorbable porous scaffolds capable of promoting osteoregeneration while preventing bacterial infection are needed for regenerative periodontal therapy. Previously, a porous collagen sponge coated with low-crystalline apatite has been shown to possess superior bioresorption and osteogenic properties compared to the uncoated sponge. In this study, we integrated osteogenic and antibacterial dual drugs into the sponge utilizing two types of apatite matrices to achieve further functionalization. First, the collagen sponge was coated with apatite loaded with an osteogenic drug, L-ascorbic acid 2-phosphate (AS), using a metastable supersaturated calcium phosphate (CaP) solution supplemented with AS. Second, the coated sponge was impregnated with apatite particles loaded with an antibacterial drug, ciprofloxacin (CF), which were fabricated using a labile supersaturated CaP solution supplemented with CF. The resulting dual drug-immobilized sponge demonstrated biological activities arising from both AS and CF; it enhanced proliferation of osteoblastic MC3T3-E1 cells and exhibited antibacterial activity against the oral bacterium Actinomyces naeslundii. The proposed technique to fabricate multifunctional scaffolds would offer a solution to provide more effective, patient-tailored regenerative periodontal therapy.
1. Introduction
Periodontal disease, an infectious disease caused by oral bacteria, often results in the destruction of alveolar bone, which is essential to the tooth-supporting system.1 In the regenerative therapy of periodontal disease, three-dimensional (3D) porous scaffolds are used as a structural framework that supports cell growth and tissue regeneration. Conventional scaffolds are constructed from osteoconductive bioceramics, such as hydroxyapatite: Ca10(PO4)6(OH)2, carbonate apatite, and β-tricalcium phosphate, and/or bioresorbable polymers such as collagen and gelatin.2–6 However, these materials are inferior to autologous bones in osteogenic activity. Because autologous bones are difficult to obtain, scaffolds with improved osteogenic activity have been in great demand. On the other hand, since bone defects caused by periodontal disease are located close to the oral cavity, scaffolds are prone to bacterial infection. To reduce the risk of infection, scaffolds should be resorbed and replaced rapidly with the regenerated bone tissue, while being strong enough to retain their porous structure in the initial stage.7 Thus, ideal scaffolds for periodontal regeneration are composed of a 3D porous framework that has sufficient initial strength, is bioresorbable, and can promote bone cell growth and osteoregeneration while preventing bacterial infection. Because systemically administered drugs hardly reach the affected site owing to the destructed vascular network, various osteogenic and antibacterial drugs have been incorporated together or separately into a scaffold.5,8,9 However, even with the use of multiple drugs, the management of both large bone loss and bacterial infection remains a major challenge owing to the difficulty in controlling local concentrations of multiple drugs and the trade-off between the antibacterial and osteogenic activities. In this study, we propose a new approach for the development of a multifunctional scaffold immobilizing osteogenic and antibacterial drugs based on the technique of fabricating drug-immobilized apatite coatings and particles using supersaturated calcium phosphate (CaP) solutions.10,11 A collagen sponge was used as a 3D porous framework, and it was functionalized with osteogenic and antibacterial drugs utilizing low-crystalline apatite as a matrix for drug immobilization. As osteogenic and antibacterial drugs, we selected L-ascorbic acid 2-phosphate (AS) and ciprofloxacin (CF), respectively. AS, a stable derivative of L-ascorbic acid (vitamin C), is a nutrient that can enhance collagen synthesis and osteoblastic proliferation and differentiation, and help in the repair and maintenance of healthy bones and gingiva.12,13 CF is a broad-spectrum fluoroquinolone antibiotic that has been used clinically to treat bacterial infection.14,15 Both AS and CF show high affinity with CaPs and can coprecipitate with CaP in supersaturated CaP solutions.16,17The purpose of this study was to integrate AS and CF into a collagen sponge utilizing two types of apatite matrices through apatite coating, followed by impregnation with apatite particles, and to demonstrate the potential of the resulting sponge as a multifunctional scaffold. First, the collagen sponge was coated with AS-immobilized apatite using a metastable supersaturated CaP solution supplemented with AS based on our coating technique.18,19 Second, the coated sponge was impregnated with CF-immobilized apatite particles17 which were fabricated using a labile supersaturated CaP solution supplemented with CF. The two supersaturated CaP solutions are different in stability, the metastable solution stays transparent without inducing homogeneous CaP precipitation during coating, whereas the labile solution, with a higher degree of supersaturation, induces homogeneous CaP precipitation soon after preparation. We hypothesized that the resulting dual drug-immobilized sponge should exhibit both osteogenic and antibacterial activities. As a preliminary evaluation, a cell proliferation assay using osteoblastic MC3T3-E1 cells and an antibacterial assay using Actinomyces naeslundii (A. naeslundii), an oral bacterium associated with periodontal infections, were performed.
2. Materials and methods
2.1 Preparation of collagen sponges
Sterile sponge sheets (3 mm-thick) made from calf dermal-derived type I and III atelocollagen (Terudermis; GC corporation, Tokyo, Japan) were used. These sponge sheets feature a 3D porous structure with a longer axis of pores of 178 ± 54 μm.20 The sheet was cut into 5 mm-diameter circular discs. The resulting collagen sponges, referred to as Col, were stored at room temperature until required for the experiments. In the sample preparation for cell proliferation (Section 2.5) and antibacterial (Section 2.6) assays, the sponges were treated under aseptic conditions until the final step, i.e., lyophilization in Sections 2.2 and 2.3.2.2 Coating with apatite and AS-immobilized apatite
Prior to the coating process, an initial stock of a metastable supersaturated CaP solution was prepared following the previously published protocol.18 Briefly, NaCl (142 mM), K2HPO4·3H2O (1.5 mM), HCl (40 mM), and CaCl2 (3.75 mM) (all from Nacalai Tesque, Inc., Kyoto, Japan) were dissolved in ultrapure water one by one, and the solution was buffered to pH 7.40 at 25 °C with 50 mM of tris(hydroxymethyl)aminomethane and HCl. The prepared solution was stored at 4 °C and used within a month of preparation. Just before the coating process, the supersaturated CaP solution was supplemented with 60 μg/mL L-ascorbic acid phosphate magnesium salt n-hydrate (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan, Lot. WTF5181; n = 4.4, water content = 21.5 mass%) as an AS source. The solutions with and without AS were then filtered using a 0.22 μm-pore-sized filter.The collagen sponges (Col) were precoated with CaP by the plasma and alternate immersion treatments.18,19 The plasma treatment was performed for 2 min in an O2 atmosphere under a pressure of 30 Pa and power density of 0.1 W/cm2 using a compact ion etcher (FA-1, SAMCO Inc., Kyoto, Japan) operating at 13.56 MHz. In the subsequent alternate immersion treatment, each sponge was immersed in 10 mL of a 200 mM CaCl2 aqueous solution for 20 min under reduced pressure using a 6-well plate and washed with 10 mL of ultrapure water. The sponge was then immersed in 10 mL of a 200 mM K2HPO4·3H2O aqueous solution for 20 min under reduced pressure. The resulting CaP-precoated sponge was immersed in 10 mL of the supersaturated CaP solution with and without AS supplementation after prewashing with the same solution, and it was then incubated at 25 °C for 24 h with orbital shaking at 150 rpm in a thermostatic shaker (M-BR-104P, Taitec Corporation, Koshigaya, Japan). As a control, the AS-supplemented supersaturated CaP solution without any sponge was also incubated under the same conditions. After incubation, the sponges were thoroughly washed with ultrapure water and frozen at −80 °C followed by lyophilization. The lyophilized sponges, prepared using the supersaturated CaP solutions with and without AS, are referred to as ApAS and Ap (Fig. 1), respectively.
 | ||
| Fig. 1 Fabrication scheme of Ap, ApAS, Ap-CF, and ApAS-CF. | ||
The AS-supplemented supersaturated CaP solutions after incubation with (test solution) and without sponge (control solution) were analyzed for residual AS quantification by measuring the absorbance at 260 nm using a UV-vis spectrometer (UV-2450, Shimadzu Corporation, Kyoto, Japan). The AS content immobilized in ApAS was estimated by subtracting the amount of AS in the test solution from that in the control solution. Three independent batches were used to obtain the average and standard deviation (SD).
2.3 Impregnation with CF-immobilized apatite particles
CF-immobilized apatite particles were prepared following the previously published protocol17 with slight modifications. First, three source solutions, solutions A, B, and C, were prepared in separate 50 mL centrifuge tubes. Solution A was prepared by mixing a 500 mM calcium ion solution (Calcium Chloride Corrective Injection 1 mEq/mL, Otsuka Pharmaceutical Co., Ltd, Tokyo, Japan) and ultrapure water in a volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :21. Solution B was prepared by mixing a 500 mM phosphate ion solution (Dibasic Potassium Phosphate Injection 20 mEq Kit, Terumo Corporation, Tokyo, Japan), a 500 mM sodium carbonate solution (prepared by dissolving Na2CO3 in ultrapure water), and ultrapure water in a volume ratio of 4:4:17. Solution C, 2 mg/mL CF solution, was prepared by dissolving CF (Fluorochem Ltd, Hadfield, UK) in 7.5 mM HCl solution. Second, a labile supersaturated CaP solution supplemented with CF was prepared by sequential addition of 1 mL of solution B, 1 mL of solution C, and 2 mL of solution A in a 15 mL centrifuge tube maintained at 25 °C. The final 4 mL of the labile supersaturated CaP solution was vortexed for 60 s, followed by incubation for 24 h at 25 °C with shaking at 150 rpm in the thermostatic shaker (M-BR-104P). Incubation for 24 h leads to the formation of the CF-immobilized apatite particles. The particles were collected via centrifugation for 5 min at 6000 rpm and washed with ultrapure water twice and then with aqueous solution of 50 vol% ethanol.
:21. Solution B was prepared by mixing a 500 mM phosphate ion solution (Dibasic Potassium Phosphate Injection 20 mEq Kit, Terumo Corporation, Tokyo, Japan), a 500 mM sodium carbonate solution (prepared by dissolving Na2CO3 in ultrapure water), and ultrapure water in a volume ratio of 4:4:17. Solution C, 2 mg/mL CF solution, was prepared by dissolving CF (Fluorochem Ltd, Hadfield, UK) in 7.5 mM HCl solution. Second, a labile supersaturated CaP solution supplemented with CF was prepared by sequential addition of 1 mL of solution B, 1 mL of solution C, and 2 mL of solution A in a 15 mL centrifuge tube maintained at 25 °C. The final 4 mL of the labile supersaturated CaP solution was vortexed for 60 s, followed by incubation for 24 h at 25 °C with shaking at 150 rpm in the thermostatic shaker (M-BR-104P). Incubation for 24 h leads to the formation of the CF-immobilized apatite particles. The particles were collected via centrifugation for 5 min at 6000 rpm and washed with ultrapure water twice and then with aqueous solution of 50 vol% ethanol.
The final CF-immobilized apatite particles were dispersed in 200 μL of an aqueous solution of 50 vol% ethanol through sonication for 3 min. This particle dispersion was diluted with aqueous solution of 50 vol% ethanol with a dilution factor of 2 or 3. Then, a 50 μL aliquot of the particle dispersion with and without dilution was drop-casted onto the sponge of ApAS. Immediately after drop-casting, the whole sponge image (side-view image) was captured using a digital camera (Tough TG-6, Olympus Corporation, Tokyo, Japan) to assess the sponge's impregnating ability of the particle dispersion. The particle dispersion (50 μL), diluted by the selected factor of 3, was drop-casted onto the sponges of Ap and ApAS, which were subsequently frozen at −80 °C, followed by lyophilization to prepare Ap-CF and ApAS-CF (Fig. 1), respectively.
The CF content in Ap-CF and ApAS-CF was estimated as follows. The particle dispersion after 3-fold dilution was further diluted with 0.1 M HCl aqueous solution with a dilution factor of 50 for complete dissolution of the particles. The CF concentration of the resulting solution was determined by measuring the absorbance at 277.5 nm using the UV-vis spectrometer. From the measured CF concentration, we calculated the CF content in 50 μL of the particle dispersion used for drop-casting (corresponding to the CF content in Ap-CF and ApAS-CF). Two independent batches were used to obtain the average and SD.
2.4 Material characterizations
The as-prepared CF-immobilized apatite particles were analyzed using a field emission scanning electron microscope (FE-SEM; SU-8020, Hitachi High-Tech Corporation, Tokyo, Japan) and an analytical transmission electron microscope (TEM; Tecnai Osiris, FEI, Hillsboro, OR, USA) operated at 200 kV, in conjunction with an energy dispersive X-ray spectrometer (EDX; Super-X system, FEI) and a high-angle annular dark-field scanning TEM (HAADF-STEM) system with a probe diameter smaller than 1 nm. Before FE-SEM observation, the particle dispersion was drop-casted onto a silicon substrate, dried under reduced pressure, and sputter-coated with Au using a sputtering system (SC-701MkII, Sanyu Electron Co., Ltd, Akishima, Japan). Before TEM analysis, the particle dispersion was drop-casted onto a Cu grid covered with holey carbon film. Four distinct regions were analyzed by STEM–EDX to estimate Ca/P atomic ratio of the particles as average ± SD.The prepared sponges (Ap, ApAS, Ap-CF, and ApAS-CF) and untreated Col were characterized using a tabletop scanning electron microscope (SEM; TM4000Plus II, Hitachi High-Tech Corporation) equipped with an EDX (AZtecOne, Oxford Instruments plc, Abingdon, UK), and an X-ray diffractometer (XRD; MiniFlex600-C, Rigaku Corporation, Tokyo, Japan). EDX analysis was performed without any conductive coating, whereas SEM observation was performed after sputter-coating with Au. In the XRD measurements, CuKα radiation was used at 40 kV and 15 mA. The measurements were conducted with a 2θ step width of 0.01°, and a scan speed of 4° per minute.
The precipitates on selected sponges (Ap and ApAS) were further analyzed using the analytical TEM in conjunction with the EDX and HAADF-STEM system. Before TEM analysis, the precipitates were carefully collected from the sponge surface and transferred onto a Cu grid covered with holey carbon film. Crystalline structure of the precipitates was examined by selected area electron diffraction (SAED) analysis. In the STEM–EDX analysis, three distinct regions (regions non-overlapping with the grid) were analyzed to estimate C/Ca, O/Ca, and Ca/P atomic ratios of the precipitates as average ± SD.
2.5 Cell proliferation assay
Mouse osteoblastic MC3T3-E1 cells (RIKEN BioResource Center, Tsukuba, Japan) were cultured in a minimum essential medium (MEM α-GlutaMAX-I, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Qualified FBS, BioWest, Nuaillé, France) and 1% penicillin–streptomycin antibiotic cocktail (10000 U/mL, Thermo Fisher Scientific Inc.) under humidified atmospheric conditions of 5% CO2 at 37 °C.
Just before cell seeding, each sponge was put in 0.2 mL of the culture medium in each well of a 96-well plate and subjected to vacuum degassing for 30 min to remove air bubbles and ensure complete immersion of the sponge in the medium. The cells (1 × 104 cells/0.2 mL per well) were seeded on each sponge and cultured for 3 d. After culture, the cells were treated with a water-soluble tetrazolium salt to quantify the relative number of viable cells in each well using a cell counting kit (CCK-8, Dojindo Laboratories, Mashiki, Japan). Absorbance at 450 nm, proportional to the number of viable cells, was recorded using a microplate reader (Multiskan™ FC, Thermo Fisher Scientific Inc.). The absorbance data were normalized to the absorbance of Ap.
2.6 Antibacterial assay
Freeze-dried pellet stock of the anaerobic bacterium A. naeslundii was obtained from American Type Culture Collection (ATCC27039, Manassas, VA, USA). The frozen bacterial stock was subjected to thawing and anaerobically cultured at 37 °C in a brain heart infusion medium (Pearl Core®, Eiken Chemical Co., Ltd, Tokyo, Japan). The culture was then stored at −80 °C until further experiments.Each sponge was placed in 0.2 mL of the culture medium in each well of a 96-well plate and subjected to vacuum degassing for 30 min to remove air bubbles. The bacteria (2.0 × 105 colony-forming units/0.2 mL per well) were inoculated on each sponge and incubated anaerobically at 37 °C. After 24 h of incubation, 180 μL of the medium was collected, and 10 μL of adjusted chromogenic reagents (WST solution and electron mediator reagent from Microbial Viability Assay Kit-WST, Dojindo Laboratories) were added. Following incubation for 2 h, absorbance at 450 nm, proportional to the number of bacteria, was measured using the microplate reader. The absorbance data were normalized to the absorbance of Ap.
2.7 Statistical analysis
For cell proliferation and antibacterial assays, 6 and 5 sponges were used, respectively, for each sample group to obtain average and SD. The statistical differences were analyzed using a two-tailed one-way ANOVA with the post hoc Turkey HSD test (for comparisons among the sample groups) using the SPSS software package (version 11.0; IBM Corporation, Armonk, NY, USA). Differences with p < 0.05 were taken to be statistically significant.3. Results and discussion
3.1 Coating with apatite and AS-immobilized apatite
In the present coating process, the surface of the collagen sponge (Col) was coated with apatite in the metastable supersaturated CaP solutions with (for ApAS) and without (for Ap) AS supplementation. As shown in the SEM images (Fig. 2), Col had a smooth surface, whereas Ap and ApAS had micron-sized particulate layers on their surfaces. These particulate layers on Ap and ApAS were composed of CaP from their EDX spectra (Fig. 3a). The Na and Cl peaks detected for all sponges were attributed to NaCl contained in the pristine collagen sponge. On the XRD patterns, diffraction peaks ascribed to NaCl were clearly detected for Col (Fig. 3b). These NaCl peaks decreased in intensity, whereas broad peaks ascribed to apatite (more specifically, hydroxyapatite: JCPDS 09-0432) appeared on both Ap and ApAS. This result indicates that the main CaP phase in the particulate layers on Ap and ApAS was low-crystalline apatite. This conclusion was further supported by the results of SAED analysis. The CaP precipitates (Fig. 4a) collected from Ap and ApAS both provided Debye rings attributed to apatite (Fig. 4b). | ||
| Fig. 2 SEM images of the surfaces of Col, Ap, ApAS, Ap-CF, and ApAS-CF. | ||
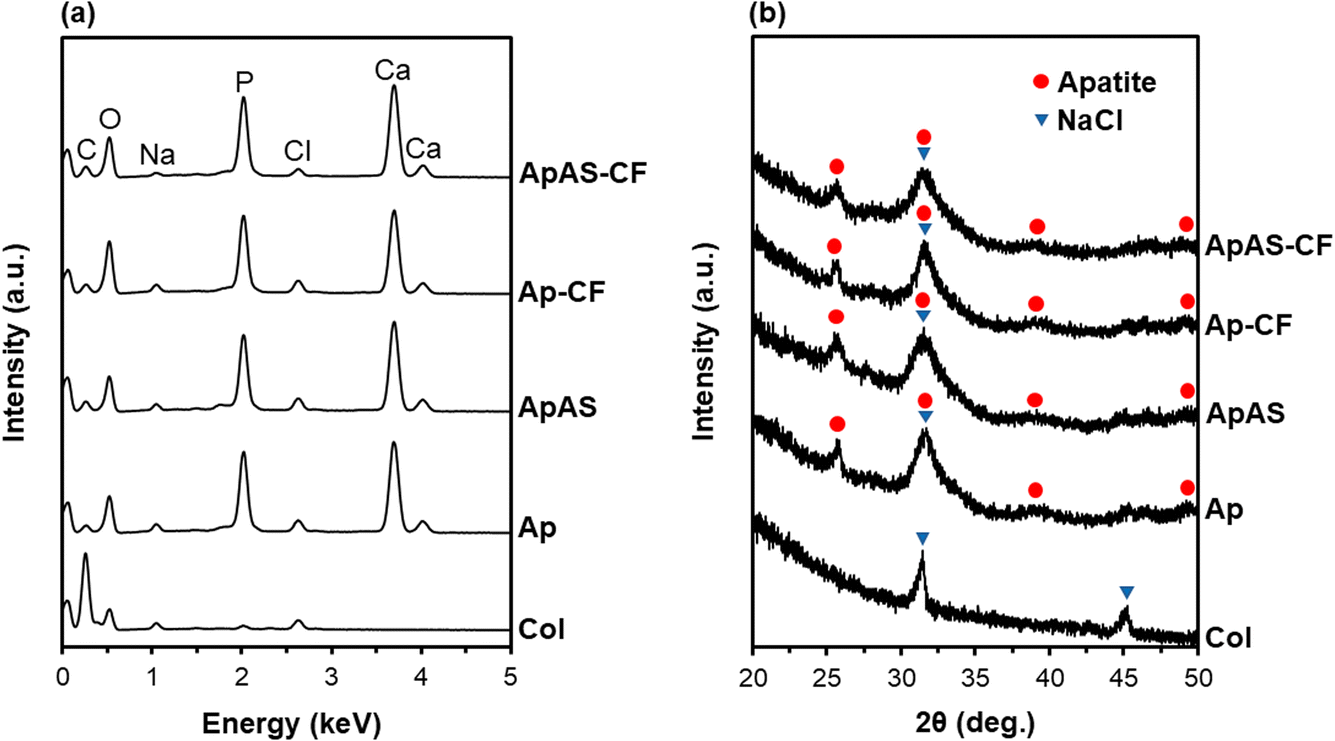 | ||
| Fig. 3 (a) SEM–EDX and (b) XRD patterns of the surfaces of Col, Ap, ApAS, Ap-CF, and ApAS-CF. | ||
 | ||
| Fig. 4 (a) TEM images, (b) SAED patterns obtained from the circled regions in (a), and (c) STEM–EDX spectra of the precipitates collected from Ap and ApAS. The Cu peak in (c) is derived from the TEM grid. | ||
Supplementation of AS to the supersaturated CaP solution led to the immobilization of AS within the apatite coating; the coating on ApAS was AS-immobilized apatite. According to the chemical analysis using UV-vis spectroscopy, the amount of AS in the metastable supersaturated CaP solution decreased after the coating process (incubation of the CaP-precoated sponge for 24 h). From the difference in residual AS between this CaP solution and the control solution (CaP solution incubated without any sponge), the AS content in ApAS was estimated at 201 ± 9 μg (per sponge). That is, the loading efficiency of AS was approximately 48%; about half of AS supplemented to the supersaturated CaP solution was immobilized within the apatite coating on ApAS. The STEM–EDX analysis revealed the difference in the coating composition between Ap and ApAS. In the STEM–EDX spectra (Fig. 4c), the precipitates collected from ApAS showed stronger peaks of C (at 0.28 keV) and O (at 0.53 keV) with respect to the Ca peak (at 3.69 keV) compared to those collected from Ap. This observation was confirmed by the quantitative analysis using three different precipitates; precipitates collected from ApAS showed higher C/Ca and O/Ca atomic ratios compared to those from Ap as summarized in Table 1. Considering the chemical formula of AS, C6H9O9P, the compositional difference described above should be due to the presence of AS in the apatite coating on ApAS.
| C/Ca | O/Ca | Ca/P | |
|---|---|---|---|
| Ap | 1.39 ± 0.34 | 2.03 ± 0.31 | 1.51 ± 0.01 |
| ApAS | 3.56 ± 0.81 | 2.83 ± 0.25 | 1.43 ± 0.01 |
The presence of AS might account for the lower Ca/P atomic ratio of the apatite coating on ApAS than that on Ap (Table 1). Even without AS, the apatite coating on Ap had a lower Ca/P atomic ratio (∼1.5) than that of stoichiometric hydroxyapatite (1.67). The coating on Ap might contain Ca-deficient apatite together with its precursor phases; amorphous CaP and octacalcium phosphate, although these precursor phases were not clearly detected either by XRD or SAED analysis. A Mg peak (at 1.25 keV) was hardly detected in the STEM–EDX spectrum of ApAS (Fig. 4c), because magnesium in the AS source is ionized and dissociate from AS in the supersaturated CaP solution.
The supplementation of AS to the supersaturated CaP solution affected not only the coating composition but also the coating morphology. As can be seen in Fig. 2, the particulate size in the AS-immobilized apatite coating on ApAS was smaller than that in the apatite coating on Ap. A possible cause of this phenomenon is the inhibitory effect of AS and magnesium ions (derived from the AS source) on crystal growth of apatite in the supersaturated CaP solution. Certain molecules21,22 and magnesium ions23,24 are known to inhibit growth of apatite crystals by adsorbing onto their growth sites, thereby influencing the morphology and maturation of the grown apatite. Such an inhibitory effect might be another reason (besides the presence of P-containing AS) for the lower Ca/P atomic ratio of the apatite coating on ApAS.
3.2 Impregnation with CF-immobilized apatite particles
The CF-immobilized apatite particles were prepared as reported previously.17 The prepared particles were a few tens of nanometers in size from their FE-SEM image (Fig. 5a). From the STEM–EDX analysis (Fig. 5b) and our previous results,17 these particles were composed of CF-immobilized, Ca-deficient apatite with a Ca/P atomic ratio of 1.53 ± 0.02 (n = 4). The small F peak in the STEM–EDX spectrum (Fig. 5c) substantiates the presence of CF (chemical formula of C17H18FN3O3) immobilized in the particles. According to our previous results,17 a single batch, i.e., 4 mL of the labile supersaturated CaP solution in one tube, yields 15.8 mg of apatite particles immobilizing 1.35 ± 0.02 mg of CF, with the CF loading efficiency of ∼68%. This means that approximately 68% of CF supplemented to the labile supersaturated CaP solution is immobilized in the final particles. | ||
| Fig. 5 (a) FE-SEM image and (b and c) STEM–EDX spectra, in (b) wider and (c) narrower ranges, of the CF-immobilized apatite particles. The Cu peak in (b) and (c) is derived from the TEM grid. | ||
The CF-immobilized apatite particles were incorporated into the mineralized sponges (Ap and ApAS) via drop-casting of the particle dispersion after dilution with an adequate dilution factor. Without dilution (1-fold), the particle dispersion was very dense and hardly infiltrated into the sponge of ApAS as shown in the left image of Fig. 6. With 2-fold dilution, the particle dispersion penetrated the sponge (Fig. 6, middle); however, a small amount of the dispersion remained outside the sponge most likely due to the pore clogging. With 3-fold dilution, the entire particle dispersion penetrated throughout the sponge (Fig. 6, right). In the following experiments, the particle dispersion (50 μL) with a fixed dilution factor of 3-fold was drop-casted onto the sponges of Ap and ApAS. The CF content in the resulting sponges (Ap-CF and ApAS-CF, respectively) was estimated to be 21.3 ± 0.2 μg (per sponge) from the chemical analysis of the particle dispersion using UV-vis spectroscopy.
 | ||
| Fig. 6 Side view images of the ApAS sponge after drop-casting of the differently diluted (dilution factor: 1-fold, 2-fold, and 3-fold) dispersions (50 μL) of the CF-immobilized apatite particles. | ||
Impregnation with the CF-immobilized apatite particles delivered no apparent changes in the surface morphology, composition, and crystalline structure of the mineralized sponges under the tested analytical conditions. Under SEM observation, Ap-CF and ApAS-CF proved similar surface morphologies to those of Ap and ApAS, respectively (Fig. 2). The CF-immobilized apatite particles on Ap-CF or ApAS-CF were unidentifiable by FE-SEM due to their small size. There were no noticeable differences in either SEM–EDX spectra (Fig. 3a) or XRD patterns (Fig. 3b) between the sponges before (Ap and ApAS) and after (Ap-CF and ApAS-CF, respectively) particle impregnation. These results are not surprising because the particles were similar in composition and crystalline structure to the mineralized coatings on these sponges. From these results, the CF-immobilized apatite particles should have only limited effects on the physicochemical properties of the sponge, such as porosity, flexibility, and mechanical strength, although they brought about an antibacterial effect as will be described later.
3.3 Cell proliferation assay
AS immobilized in the scaffold as a form of the AS-immobilized apatite coating enhanced proliferation of osteoblastic MC3T3-E1 cells. After incubation for 3 d, the relative numbers of viable cells were higher for ApAS and ApAS-CF than for Ap and Ap-CF, respectively (Fig. 7). AS released from the apatite coating and its hydrolyzed product (L-ascorbic acid, vitamin C) should be responsible for the enhanced cell proliferation on the AS-immobilized sponges as reported in previous reports.12,16,25 AS has been known to hydrolyze into L-ascorbic acid in the presence of serum and accelerate proliferation of osteoblast-like cells (MG-63) at a concentration range from 0.25 to 1.0 mM in a culture medium.12 Assuming that only 7% of AS immobilized in ApAS is released into the medium (0.2 mL), the nominal AS concentration in the medium reaches 0.27 mM, that falls within the effective concentration range. Note that the relative number of viable cells was higher for ApAS-CF than for ApAS, although there was no significant difference between Ap and Ap-CF (Fig. 7). This might be owing to the synergistic effect of AS and CF, although details remain uncertain.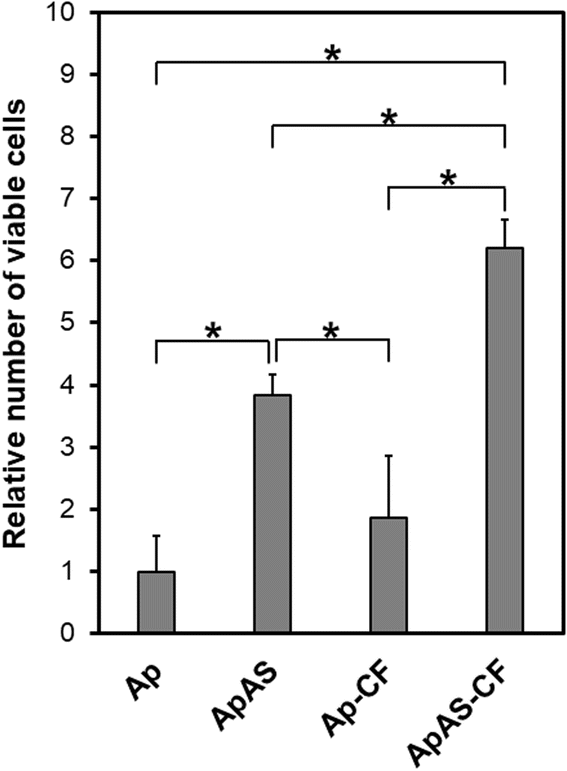 | ||
| Fig. 7 Relative number of viable MC3T3-E1 cells (absorbance at 450 nm normalized to that of Ap) after incubation for 3 d on Ap, ApAS, Ap-CF, and ApAS-CF (average + SD, n = 6, * p < 0.05). | ||
3.4 Antibacterial assay
CF immobilized in the scaffold as a form of the CF-immobilized particles exhibited antibacterial activity against A. naeslundii. The relative numbers of bacteria after 24 h of incubation were significantly smaller for Ap-CF and ApAS-CF than for Ap and ApAS (Fig. 8). This should be owing to the bacteriostatic and bactericidal activities of CF specific to bacterial DNA.14,15,26 The differences between Ap-CF and ApAS-CF and between Ap and ApAS were not significant, indicating that AS showed no antibacterial effect. | ||
| Fig. 8 Relative number of viable A. naeslundii (absorbance at 450 nm normalized to that of Ap) after incubation for 24 h on Ap, ApAS, Ap-CF, and ApAS-CF (average + SD, n = 5, * p < 0.05). | ||
Considering the estimated CF content in one sponge (∼21 μg, described in Section 3.2) and the minimal inhibitory concentration (MIC) of CF for A. naeslundii (3.9 μg/mL),27 at least 3.7% of CF immobilized in Ap-CF and ApAS-CF should be released into the medium (0.2 mL) during incubation (24 h). It has been reported that similarly prepared CF-immobilized apatite particles (“CF-CaP24h” in ref. 17) release approximately 19% of immobilized CF in a physiological salt solution through a dialysis membrane after incubation for 24 h. Therefore, the estimated minimum release fraction of 3.7% seems to be a reasonably achievable level, although the release conditions are different. It should be emphasized that the CF-immobilized apatite particles have been shown to exhibit not only antibacterial activities against other oral bacteria (Streptococcus mutans and Porphyromonas gingivalis) but also an acid-neutralizing effect.17 It is known that the implantation of a scaffold is liable to acidify the surrounding environment by postoperative inflammation reactions.28 Proliferation of osteoclasts29 and certain bacteria30 also acidify the surrounding environment. An acidified environment is unfavorable for biomineralization, because solubilities of apatite and its precursor phases increase by a decrease in environmental pH from the physiological pH (∼7.4).31 Therefore, the acid-neutralizing effect of the CF-immobilized apatite particles should be a side benefit for periodontal regeneration.
3.5 Potential as a scaffold for periodontal regeneration
The mineralized, dual-drug-immobilized collagen sponge (ApAS-CF) has potential as a multifunctional scaffold for periodontal regeneration. The base framework of this scaffold is a 3D porous collagen sponge coated with low-crystalline apatite (Ap), which mimics the natural bone tissue comprised of collagen and low-crystalline apatite. It has been known that low-crystalline apatite can be replaced with regenerated bone tissue through bioresorption different from sintered hydroxyapatite.32 Thus, from the low-crystalline apatite coating, the collagen sponge gains osteogenic activity while retaining its bioresorbability. Our previous in vivo study confirmed substantial resorption of a similarly prepared apatite-coated collagen sponge (corresponding to Ap) and its superior osteogenic activity compared to the untreated sponge (corresponding to Col).7,33 The integration of osteogenic and antibacterial drugs (AS and CF, respectively) proposed in this study should further improve the functionality of Ap as a scaffold for periodontal regeneration. According to the manufacturer's information, the AS source, when dissolved in water, retains 85% activity for 1 week at 37 °C, whereas CF has been reported to be stable for 2 days at 37 °C.34 Therefore, AS immobilized in the apatite coating in ApAS-CF should exert its osteogenic activity; it should promote osteoblastic cell proliferation and differentiation, and enhance the regeneration of bones and gingiva.12,13,16 In the meantime, CF immobilized in ApAS-CF as a form of the CF-immobilized apatite particles should exert its antibacterial activity; it should prevent bacterial growth and biofilm formation around the sponge,17 thereby decreasing the risk of infection. Our preliminary results demonstrated that ApAS-CF is endowed with biological activities arising from both AS and CF; it showed antibacterial activity without deteriorating the sponge's good cytocompatibility (Fig. 7 and 8). Further in vitro and in vivo studies are needed to fully clarify the functionality of this scaffold and its clinical potential in regenerative periodontal therapy.In conventional regenerative therapy for periodontal disease, a scaffold is implanted into a periodontal bone defect, and then antibiotics are administered orally to prevent postoperative infection. However, infection control with oral antibiotics is extremely difficult because the defect site lacks functional vascular networks, teems with bacteria, and is located close to the oral cavity. Localized delivery of antibiotics from the scaffold should ensure the reduction of infection risk, although it is not easy to bring out sufficient antibacterial activity without deteriorating the biocompatible and osteogenic properties of the scaffold. In this study, we integrated osteogenic and antibacterial drugs within a scaffold utilizing low-crystalline apatite as a matrix for drug immobilization. Low-crystalline apatite, a major component of human biominerals, has several advantages over other biodegradable materials; it shows good biocompatibility and osteogenic activity,7,32,33 dissolves into calcium and phosphate ions, and exhibits acid-neutralizing effects,17 all of which are beneficial for periodontal regeneration. In addition, a mineralized coating can increase the compressive strength of a collagen sponge,35 that would help retain its porous structure, without the complete loss of its flexible and compressible nature. In fact, a similarly prepared apatite-coated collagen sponge (corresponding to Ap) could be compressed to fulfill a deep (5–6 mm) furcation defect upon implantation.7 Such mechanical properties of the mineralized sponge profitable as a scaffold should be retained even after particle impregnation, as it caused no apparent physicochemical difference on the mineralized sponge (Fig. 2 and 3). Moreover, low-crystalline apatite can immobilize various drugs and allows their sustained release.10,11 Drug release from a low-crystalline apatite matrix is finely tunable by controlling parameters of the coprecipitation process, such as drug concentration of the solution36,37 and supplementation of certain ions to the solution.19 In this study, we combined two processes for drug loading, AS-immobilized apatite coating and impregnation with CF-immobilized apatite particles, so that the release of each drug can be controlled independently.
The present methodology potentially allows integration of a variety of drug combinations into a scaffold. Besides AS and CF, various types of drugs, such as lactoferrin,38 tetracycline,39 heparin,38 and basic fibroblast growth factor,19,36 have been immobilized within apatite coatings and particles via coprecipitation in supersaturated CaP solutions.10,11 By choosing an appropriate combination of drugs, the function of the resulting scaffold could be customized to meet the needs of each patient, which may lead to a new platform to fabricate a multifunctional, customized scaffold for periodontal regeneration.
4. Conclusion
An osteogenic drug and an antibacterial drug, AS and CF, respectively, were integrated into the porous collagen sponge utilizing two types of apatite matrices through apatite coating followed by impregnation with apatite particles. The resulting dual-drug-immobilized sponge enhanced proliferation of osteoblastic cells and exhibited antibacterial activity against A. naeslundii and thus demonstrated its potential as a multifunctional scaffold for periodontal regeneration.Data availability
The data are available upon reasonable request.Author contributions
Kaushita Banerjee: methodology, investigation, formal analysis, data curation, funding acquisition, writing – original draft. Ayako Oyane: conceptualization, methodology, formal analysis, data curation, funding acquisition, supervision, project administration, writing – original draft. Maki Nakamura: methodology, supervision, visualization, writing – review & editing. Tomoya Inose: investigation, data curation, writing – review & editing. Erika Nishida: investigation, formal analysis, data curation, funding acquisition, writing – review & editing. Kanako Shitomi: investigation, formal analysis, data curation, writing – review & editing. Hirofumi Miyaji: conceptualization, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Mr Noriyuki Saito (AIST) for TEM analyses, and Ms Miyabi Makino, Ms Hiroko Araki, and Ms Ikuko Sakamaki (AIST) for their technical support. This study was supported by JSPS KAKENHI Grant Numbers JP23KF0095, JP22H05148, and JP22K10012.References
- E. Könönen, M. Gursoy and U. K. Gursoy, J. Clin. Med., 2019, 8, 1135 CrossRef PubMed.
- K. Aoki, H. Ideta, Y. Komatsu, A. Tanaka, M. Kito, M. Okamoto, J. Takahashi, S. Suzuki and N. Saito, Bioengineering, 2024, 11, 180 CrossRef CAS PubMed.
- R. Rohanizadeh, M. V. Swain and R. S. Mason, J. Mater. Sci.: Mater. Med., 2008, 19, 1173–1182 CrossRef CAS.
- M. Mallick, R. P. Are and A. R. Babu, Materialia, 2022, 22, 101391 CrossRef CAS.
- S. Jiang, M. Wang and J. He, Bioeng. Transl. Med., 2021, 6, e10206 CrossRef PubMed.
- H. Chen, G. Song, T. Xu, C. Meng, Y. Zhang, T. Xin, T. Yu, Y. Lin and B. Han, J. Funct. Biomater., 2024, 15, 233 CrossRef CAS PubMed.
- Y. Kanemoto, H. Miyaji, E. Nishida, S. Miyata, K. Mayumi, Y. Yoshino, A. Kato, T. Sugaya, T. Akasaka, A. J. Nathanael, S. Santhakumar and A. Oyane, J. Periodontal Res., 2022, 57, 205–218 CrossRef CAS PubMed.
- C. Zhao, W. Liu, M. Zhu, C. Wu and Y. Zhu, Bioact. Mater., 2022, 18, 383–398 CAS.
- Y. Cui, H. Liu, Y. Tian, Y. Fan, S. Li, G. Wang, Y. Wang, C. Peng and D. Wu, Mater. Today Bio, 2022, 16, 100409 CrossRef CAS PubMed.
- X. Wang, A. Oyane and A. Ito, in Advances in Calcium Phosphate Biomaterials, ed. B. Ben-Nissan, Springer Berlin, Heidelberg Germany, 2014, pp. 171–197 Search PubMed.
- X. Wang, A. Ito, X. Li, Y. Sogo and A. Oyane, Biofabrication, 2011, 3, 022001 CrossRef PubMed.
- S. Takamizawa, Y. Maehata, K. Imai, H. Senoo, S. Sato and R. I. Hata, Cell Biol. Int., 2004, 28, 255–265 CrossRef CAS PubMed.
- K. Tsutsumi, H. Fujikawa, T. Kajikawa, M. Takedachi, T. Yamamoto and S. Murakami, J. Periodontal Res., 2012, 47, 263–271 CrossRef CAS PubMed.
- B. A. A. Almutairi, A. A. Almotawa, M. A. M. Almutairi, R. S. Baabbad, A. A. Abdullah S, H. S. K. Alagedi, E. A. H. Aljaizani, M. A. Alahmar, F. A. A. Shabi, S. A. Alharbi, I. S. A. Alaboudi, A. A. Kaabi and A. A. A. Saleh, J. Ecohumanism, 2024, 3, 9328–9336 Search PubMed.
- A. Shariati, M. Arshadi, M. A. Khosrojerdi, M. Abedinzadeh, M. Ganjalishahi, A. Maleki, M. Heidary and S. Khoshnood, Front. Public Health, 2022, 10, 1025633 CrossRef.
- X. Wang, A. Ito, Y. Sogo, X. Li, H. Tsurushima and A. Oyane, Acta Biomater., 2009, 5, 2647–2656 CrossRef CAS PubMed.
- A. Pal, A. Oyane, T. Inose, M. Nakamura, E. Nishida and H. Miyaji, Materials, 2024, 17, 2035 CrossRef CAS.
- J. A. Nathanael, A. Oyane, M. Nakamura, I. Sakamaki, E. Nishida, Y. Kanemoto and H. Miyaji, ACS Appl. Mater. Interfaces, 2017, 9, 22185–22194 CrossRef PubMed.
- A. Pal, A. Oyane, M. Nakamura, K. Koga, E. Nishida and H. Miyaji, Int. J. Mol. Sci., 2024, 25, 1495 CrossRef CAS PubMed.
- K. Hori, A. Osada, T. Isago and H. Sakurai, Burns, 2017, 43, 846–851 CrossRef.
- R. W. Romberg, P. G. Werness, B. L. Riggs and K. G. Mann, Biochemistry, 1986, 25, 1176–1180 CrossRef CAS PubMed.
- A. Zieba, G. Sethuraman, F. Perez, G. H. Nancollas and D. Cameron, Langmuir, 1996, 12, 2853–2858 CrossRef CAS.
- N. Kanzaki, K. Onuma, G. Treboux, S. Tsutsumi and A. Ito, J. Phys. Chem. B, 2000, 104, 4189–4194 CrossRef CAS.
- T. A. Fuierer, M. LoRe, S. A. Puckett and G. H. Nancollas, Langmuir, 1994, 10, 4721–4725 CrossRef CAS.
- M. C. Ronzière, S. Roche, J. Gouttenoire, O. Démarteau, D. Herbage and A. M. Freyria, Biomaterials, 2003, 24, 851–861 CrossRef.
- F. Silva, O. Lourenço, J. A. Queiroz and F. C. Domingues, J. Antibiot., 2011, 64, 321–325 CrossRef CAS.
- M. Elshikh, I. Moya-Ramírez, H. Moens, S. Roelants, W. Soetaert, R. Marchant and I. M. Banat, J. Appl. Microbiol., 2017, 123, 1111–1123 CrossRef CAS.
- L. Borkowski, M. Kiernicka, A. Belcarz, K. Pałka, M. Hajnos and G. Ginalska, J. Biomed. Mater. Res., Part B, 2017, 105B, 1178–1190 Search PubMed.
- F. Barrère, C. A. van Blitterswijk and K. de Groot, Int. J. Nanomed., 2006, 1, 317–332 Search PubMed.
- N. Takahashi, Int. Congr. Ser., 2005, 1284, 103–112 CrossRef CAS.
- Q. Liu, S. Huang, J. P. Matinlinna, Z. Chen and H. Pan, BioMed Res. Int., 2013, 2013, 929748 Search PubMed.
- S. Y. Park, S. M. Yi, S. W. On, S. A. Che, J. Y. Lee and B. E. Yang, J. Dentistry, 2025, 154, 105597 CrossRef CAS PubMed.
- S. Santhakumar, A. Oyane, M. Nakamura, Y. Yoshino, M. K. Alruwaili and H. Miyaji, Materials, 2021, 14, 5860 CrossRef CAS PubMed.
- M. P. Kane, G. R. Bailie, D. G. Moon and I. Siu, Am. J. Health Syst. Pharm., 1994, 51, 373–377 CrossRef CAS.
- S. Murakami, H. Miyaji, E. Nishida, K. Kawamoto, S. Miyata, H. Takita, T. Akasaka, B. Fugetsu, T. Iwanaga, H. Hongo, N. Amizuka, T. Sugaya and M. Kawanami, Dent. Mater. J., 2017, 36, 573–583 CrossRef CAS PubMed.
- H. Tsurushima, A. Marushima, K. Suzuki, A. Oyane, Y. Sogo, K. Nakamura, A. Matsumura and A. Ito, Acta Biomater., 2010, 6, 2751–2759 CrossRef CAS PubMed.
- X. Zhu, H. Zhang, X. Zhang, C. Ning and Y. Wang, Dent. Mater. J., 2017, 36, 677–685 CrossRef CAS PubMed.
- M. Nakamura, A. Oyane, T. Inose, Y. Kanemoto and H. Miyaji, Int. J. Mol. Sci., 2025, 26, 852 Search PubMed.
- A. Oyane, Y. Yokoyama, M. Uchida and A. Ito, Biomaterials, 2006, 27, 3295–3303 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |