
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of amino and methyl groups on the loading capacity of UiO-66 for 8-hydroxyquinoline: an in-depth experimental and computational analysis
Jiao-Jiao Cao ab,
Jin-Long Geab,
Yan Gaoab and
Bo-Kai Liao*c
ab,
Jin-Long Geab,
Yan Gaoab and
Bo-Kai Liao*c
aSchool of Material and Chemical Engineering, Bengbu University, Bengbu, 233030, China
bEngineering Technology Research Center of Silicon-based Materials, Bengbu, 233030, China
cSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou, 510006, China
First published on 17th June 2025
Abstract
This research systematically investigates a library of UiO-66(Zr) dicarboxylate metal–organic frameworks (MOFs), functionalized with polar and nonpolar substituents (–H, –CH3, –NH2), to evaluate their potential as carriers for inhibitor encapsulation, using 8-hydroxyquinoline (8-HQ) as a model compound. The encapsulation process was conducted under vacuum, and 8-HQ uptake was quantified using UV-visible absorption spectroscopy, allowing for precise control over host–guest interactions. Quantitative assessment of 8-HQ loading within each functionalized UiO-66 variant revealed significant variations in uptake efficiency, ranging from 14.66% to 19.45% across the modified frameworks. Furthermore, a density functional theory-based quantitative structure–activity relationship (QSAR) approach was employed to identify key chemical and structural attributes of UiO-66 that significantly influence inhibitor uptake. Multivariate analysis identified two critical physicochemical parameters governing encapsulation efficiency: (1) the dipole moment of the functionalized linkers, which dictates framework-polarity-mediated adsorption, and (2) the hydrogen-bond-donating capacity, which modulates specific interactions with 8-HQ's phenolic oxygen. These descriptors exhibited strong linear correlations with experimental loading data, demonstrating that strategic enhancement of linker polarity and H-bonding capability can amplify inhibitor uptake by 10–20% compared to pristine UiO-66. The QSAR model deciphers MOF performance trends and predicts optimal functional groups via descriptor interpolation, streamlining MOF carrier design for corrosion and drug applications through targeted linker engineering.
1. Introduction
In oil and natural gas extraction, carbon dioxide corrosion of pipeline steel and associated equipment is a prevalent issue, potentially leading to severe safety incidents. Consequently, CO2 corrosion has attracted considerable international attention and investment, making the investigation of its mechanisms and protective strategies a critical focus within the global oil industry's corrosion prevention efforts. While complete prevention of corrosion remains elusive, its impact can be mitigated through various approaches, including the application of corrosion inhibitors and protective coatings.1–3 However, the performance of these physical barriers, particularly after exposure to water and ions, can deteriorate significantly, especially in the case of epoxy resin (EP) coatings. Although corrosion inhibitors are employed to combat corrosion, many are toxic and expensive. Encapsulating corrosion inhibitors within a matrix is therefore being explored as a promising strategy to prevent premature failure of protective coatings under corrosive conditions.4–6 Furthermore, for carriers impregnated with corrosion inhibitors, sustained corrosion protection can be achieved by extending the carrier film's lifespan, thereby enabling prolonged inhibitor release.7,8Recently, metal–organic frameworks (MOFs), intricate structures formed by metal ions linked with organic ligands, have garnered considerable interest as drug-delivery nanocarriers. This is primarily due to their high loading capacity, exceptional biocompatibility, and impressive thermal stability, making them promising candidates for advanced pharmaceutical applications.9–11 Among MOFs, zirconium-based MOFs, particularly UiO-66, are attractive for controlled inhibitor delivery due to their abundant active sites, outstanding chemical stability, and ultra-high thermal stability. For instance, Zhao et al.12 synthesized graphene oxide/UiO-66 nanocontainers encapsulating benzotriazole (BTA), which were successfully applied to protect copper and its alloys. Moreover, UiO-66 can be customized by incorporating functional metal nodes or groups with varying polarities, hydrophilicities, and acidities to achieve high loading efficiency.13,14 Chen et al.15 used epichlorohydrin to graft UiO-66 onto chitosan, creating a porous chitosan/Zr-MOF composite foam. Their results showed that the hierarchical porosity and abundant functional groups of the composite foam effectively facilitated the removal of ketoprofen from water, achieving a maximum adsorption capacity of 209.7 mg g−1 (at pH = 4), surpassing most reported adsorbents. Modifying organic linkers within the MOF structure is another common and effective approach, with various functional groups demonstrating efficacy.13 Sun et al.16 investigated the adsorption of two common anti-inflammatory drugs, ibuprofen (IBP) and naproxen (NAP), on UiO-66 and UiO-66-NH2. Their results indicated that the presence of additional binding site types and fewer adsorption sites on UiO-66-NH2, compared to UiO-66, led to weaker competitive adsorption of IBP and NAP. Gaudin et al.17 explored the impact of grafting various organic linkers onto the MOF structure for caffeine encapsulation, using QSAR models to demonstrate that grafting polar and H-donor groups onto the MOF's organic linkers increased drug uptake.
Although batch experiments are frequently employed to investigate the encapsulation or loading of guest molecules by various materials, discerning the underlying adsorption mechanisms can be complex. To overcome this limitation, researchers are increasingly integrating spectroscopic analysis with density functional theory (DFT) calculations to elucidate the molecular-level details of adsorption processes.18–20 DFT calculations offer valuable insights into molecular structures, binding sites, and binding energies associated with the adsorption of metals and organic chemicals onto diverse material.21,22 For instance, Jiang et al.23 investigated eco-friendly corrosion inhibitors (chitosan, ferulic acid, cysteine, methionine) derived from natural bio-based materials for steel protection in simulated concrete pore solutions. Quantum chemical (QC) calculations revealed electron-donating capacities and reactive sites, while molecular dynamics (MD) simulations visualized stable inhibitor adsorption on steel surfaces, corroborating experimental data. These inhibitors also enhanced mortar strength (up to an 11.18% compressive strength increase) without compromising workability, presenting a sustainable strategy for improving reinforced concrete durability. Similarly, Zhou et al.24 explored the corrosion inhibition effect of 5-methoxy-indazole (MIA) and methyl 1H-indazole-5-carboxylate (MIC) on copper in H2SO4 through theoretical calculations, employing DFT and MD simulations to further elucidate the corrosion inhibition mechanism of these imidazole derivatives and highlighting their potential application in acidic media. Moreover, Keshmiri et al.25 successfully grew a highly porous and stable covalent organic framework (COF) structure on a multi-walled carbon nanotube (CNT), using it as a nanocontainer for zinc as an inhibitor to produce a coating with superior active/passive anti-corrosion and thermo-mechanical properties.
Currently, 8-hydroxyquinoline (8-HQ) and its derivatives are recognized as effective corrosion inhibitors for steel, aluminum, copper, and magnesium alloys, demonstrating superior inhibition performance in acidic, saline, and other corrosive environments.26 To the best of my knowledge, systematic research examining the impact of carrier structure on the loading capacity of these inhibitors is limited. Therefore, in this study, UiO-66 was modified with 2-amino-terephthalic acid and 2-methyl terephthalic acid via solvothermal synthesis, resulting in UiO-66-NH2 and UiO-66-CH3, respectively. Subsequently, the adsorption of 8-HQ inhibitors by these modified MOFs was investigated. The UiO-66 derivatives and UiO-66-X@8-HQ composites were characterized using transmission electron microscopy (TEM), powder X-ray diffraction analysis (XRD), Fourier transform infrared spectroscopy (FT-IR), thermogravimetric analysis (TGA), and UV-vis spectroscopy. The corrosion inhibiting effectiveness of the UiO-66-X@8-HQ composites was evaluated for steel in CO2-saturated 0.5 mol per L NaCl solution using FE-SEM, XPS, and electrochemical methods. Finally, the adsorption behavior of the degradation products released from the UiO-66-X particles on the steel surface was investigated through theoretical calculations. Comprehensive spectroscopic characterization confirms the successful incorporation of 8-hydroxyquinoline (8-HQ) into UiO-66-X (X= –H, –NH2, –CH3) frameworks while preserving their crystalline integrity, as evidenced by XRD and FT-IR analyses. Thermogravimetric analysis (TGA) quantified distinct loading capacities of 16.01%, 14.66%, and 19.45% for UiO-66, UiO-66-NH2, and UiO-66-CH3, respectively, with methyl-functionalized variants demonstrating superior loading performance. UV-vis kinetic studies revealed time-dependent 8-HQ release profiles adhering to first-order kinetics (R2 > 0.98), achieving equilibrium after sustained release. The release rates followed the hierarchy: UiO-66 > UiO-66-CH3 > UiO-66-NH2, suggesting that amino groups enhance host–guest interactions to retard molecular diffusion. Systematic QSAR modeling decoupled structure–property relationships, identifying key molecular descriptors governing MOF performance. This integrated experimental-computational strategy establishes a robust framework for rational MOF carrier design, enabling precise linker engineering to advance corrosion-inhibitive and controlled drug delivery systems.
2. Experimental part
2.1 Materials and solution
A standard carbon steel grade N80 was employed for a suite of experiments. The cylindrical metal samples whose exposed surface area were 1.0 cm × cm serving as the working electrode, were coated with epoxy resin to seal all other surfaces. For weight loss tests, the specimens were fashioned to dimensions of 5.0 cm × 1.0 cm × 0.3 cm, with an naked surface area of 13.6 cm2 in contact with the solution. The samples were meticulously pre-treated, involving sequential polishing with silicon carbide sandpapers of 180, 800, and 2000 grit, followed by rinsing with deionized water, degreasing, and drying under a stream of nitrogen.The reactants, including terephthalic acid (H2BDC), 2-aminoterephthalic acid (H2BDC-NH2), 2-methylterephthalic acid (H2BDC-CH3), and 8-hydroxyquinoline (8-HQ), were procured from Aladdin. Zirconium chloride (ZrCl4) was sourced from Merck Chemicals Co. Solvents including N,N-dimethylformamide (DMF), methanol (CH3OH), and ethanol (C2H5OH) were sourced from Tianjin Guangfu Chemicals. The solvents utilized in our experiments were of analytical grade and employed without the need for additional purification. A CO2-saturated 0.5 mol per L NaCl solution served as the testing medium, and the solution was continuously sparged with CO2 to maintain saturation throughout the duration of the tests.
2.2 Preparation of UiO-66-X and UiO-66-X@8-HQ(X = –H, –CH3, –NH2)
UiO-66 solid powder was synthesized via the solvothermal method as described in the literature.27 In a concise procedure, ZnCl4 (0.4614 g) and H2BDC-NH2 (0.3289 g) were dissolved in 70 mL of DMF solvent and stirred at room temperature to achieve a clear and transparent solution, followed by the addition of 0.34 mL of acetic acid. The mixture was then subjected to ultrasonic treatment for 2 minutes to ensure homogeneity. The resulting solution was transferred into a Teflon-lined autoclave and heated to 120 °C for a duration of 24 hours. Upon cooling to room temperature, the white powder was collected via centrifugation and washed repeatedly with DMF and CH3OH, followed by drying under vacuum at 70 °C overnight. UiO-66-NH2 and UiO-66-CH3 were synthesized using the same method, employing H2BDC-NH2 and H2BDC-CH3 as the linkers, respectively (Fig. 1). | ||
| Fig. 1 Synthesis diagram of isostructural UiO-66, UiO-66-NH2 and UiO-66-CH3. | ||
To impregnate the 8-HQ inhibitor within UiO-66, 0.2 g of pre-dried UiO-66 (dry at 70 °C for 24 h) was introduced into 250 mL of an ethanolic solution containing 0.4 g of 8-HQ. The mixture was stirred continuously under vacuum for 24 hours to facilitate loading, followed by centrifugation and thorough washing with ethanol. The resulting UiO-66@8-HQ particles were then dried in an oven. Following the same procedure, UiO-66 was replaced with UiO-66-NH2 and UiO-66-CH3 to synthesize UiO-66-NH2@8-HQ and UiO-66-CH3@8-HQ, respectively.
2.3 Weight loss measurement
Dynamic weight loss tests were conducted in a CO2-saturated 0.5 mol per L NaCl solution (500 mL) at 30 °C for a duration of 12 hours. The test specimens were initially weighed with precision using an electronic balance accurate to 0.1 mg. Post-corrosion, the specimens were retrieved, and any corrosion byproducts were removed using a descaling solution. Subsequently, they were rinsed sequentially with distilled water and ethanol. Once dried with cool air, the specimens were re-weighed multiple times to ensure accuracy, and the average corrosion rates (vcorr) were determined using the following equation:
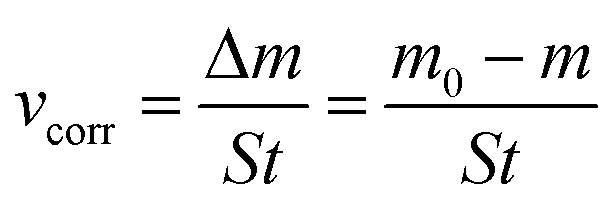 | (1) |
 | (2) |
2.4 Characterization
2.5 Inhibitors release studies
The concentration of UiO-66@8-HQ, UiO-66-NH2@8-HQ and UiO-66-CH3@8-HQ in the supernatant post-centrifugation were ascertained using a UV-vis spectrophotometer, which allowed for the determination of the inhibitor's absorption amount. The loading capacity was calculated as the percentage of the inhibitor in the UiO-66 derivatives relative to the total amount of inhibitors used in the preparation process. For the controlled release study, 100 mg of UiO-66@8-HQ, 100 mg of UiO-66-NH2@8-HQ, and 100 mg of UiO-66-CH3@8-HQ were each immersed in separate beakers containing 500 mL of 0.5 mol per L NaCl solution, with stirring frequently to facilitate slow release. At various time intervals, a 0.22 μM microporous cellulose membrane was utilized to filter and collect 2 mL of the supernatant, which was then replaced with an equivalent volume of fresh medium.282.6 Electrochemical measurements
Electrochemical measurements were conducted using the Wuhan Corrtest CS350 electrochemical workstation in a CO2-saturated 0.5 mol per L NaCl solution at 25 °C. And the electrochemical cell utilized a three-electrode configuration, featuring a saturated calomel electrode as the reference electrode and a platinum plate as the counter electrode. A sinusoidal excitation signal with an amplitude of 5 mV was applied for electrochemical impedance spectroscopy (EIS) measurements at the open circuit potential (OCP), covering a frequency range from 100 kHz to 0.01 Hz. The EIS data were subsequently analyzed using ZView software. Following a 2 hours immersion period, the potentiodynamic polarization scans were conducted separately, with a scanning range of 0 to −200 mV and 0 to 200 mV relative to the OCP, at a scanning rate of 1.0 mV s−1. The inhibition efficiency (η) of the inhibitor is calculated using the formula:
 | (3) |
2.7 Computational methodology
Density functional theory (DFT)-based calculations were employed to determine reactivity indices, shedding light on the development of quantitative structure–activity relationships. The geometries of UiO-66 derivatives and 8-HQ were optimized using the B3LYP functional and the 6-31d basis set, as implemented in the Gaussian 09 computational chemistry software. The resulting data encompassed the energy of the highest occupied molecular orbital (EHOMO), the energy of the lowest unoccupied molecular orbital (ELUMO), and the energy gap. Furthermore, the atomic–scale interactions between the 8-HQ inhibitor and UiO-66 derivatives were simulated using molecular dynamics (MD) simulation methodology, integrated with the COMPASS force field in the Materials Studio 8.0 package. This process necessitated the use of the Forcite module.MD simulations, can provide insights into the most probable inhibitor adsorption orientations on the target metal surface. Initially, an iron crystal was imported, cleaved along the Fe(110) plane, and truncated into three layers. A supercell (6 × 6) was then constructed with dimensions of (24.82 Å × 24.82 Å × 8.11 Å) to provide an ample surface for interaction with the tested inhibitor. Subsequently, a vacuum slab of zero thickness was created. An amorphous cell containing 500 H2O molecules, 5 Cl− ions, 5 H3O+ ions, and one molecule of the inhibitor of interest was assembled. The simulation was conducted at 298 K under the NVT ensemble with a time step of 1 fs and a simulation duration of 500 ps, utilizing the COMPASS force field. Ultimately, the interaction force between the corrosion inhibitor and the carbon steel is calculated using the subsequent equation:29
| Eint = Etot − (Esur+sol + Einh+sol) + Esol | (4) |
3. Results
3.1 Characterization of prepared UiO-66 derivatives and UiO-66-X@8-HQ
Scanning electron microscopy (SEM) images, as presented in Fig. 2, reveal a structural morphology characterized by cubic particles present on the surface of the MOFs. The incorporation of CH3 and NH2 functional groups into the MOF structure appears to induce particle agglomeration on the surface, as illustrated in Fig. 2a–c.30 However, a comparative analysis of UiO-66@8-HQ, UiO-66-CH3@8-HQ, and UiO-66-NH2@8-HQ, depicted in Fig. 2d–f, indicates only minimal morphological variation between these materials.31 | ||
| Fig. 2 SEM images of (a) UiO-66, (b) UiO-66-CH3, (c) UiO-66-NH2, (d) UiO-66@8-HQ, (e) UiO-66-CH3@8-HQ and (f) UiO-66-NH2@8-HQ. | ||
Fig. 3 presented TEM images and EDX dot mappings, detailing the elemental composition and distribution within the selected samples. As shown in Fig. 3(a–c), the pristine UiO-66 particles exhibited a consistent octahedral morphology with diameters exceeding 100 nm. The octahedral geometry was largely maintained following linker modification.32 Similarly, minimal morphological alterations are observed between UiO-66@8-HQ, UiO-66-CH3@8-HQ, and UiO-66-NH2@8-HQ. However, the elemental dot mappings for these modified species clearly demonstrate the uniform distribution of carbon, nitrogen, oxygen, and zirconium throughout the UiO-66-X framework, which suggested incomplete or non-ideal encapsulation of the 8-HQ inhibitor within UiO-66, UiO-66-NH2, and UiO-66-CH3.27
 | ||
| Fig. 3 TEM images of (a) UiO-66, (b) UiO-66-CH3, (c) UiO-66-NH2, (d) UiO-66@8-HQ, (e) UiO-66-CH3@8-HQ, (f) UiO-66-NH2@8-HQ and EDX dot mappings of (g) UiO-66@8-HQ, (h) UiO-66-CH3@8-HQ, (i) UiO-66-NH2@8-HQ. | ||
Fig. 4a presented the X-ray diffraction (XRD) patterns of the UiO-66 derivatives and the UiO-66-X@8-HQ composites. Diffraction peaks observed at 2θ values of 7.38° and 8.50°, corresponding to the (1 1 1) and (2 0 0) planes, respectively, align with previously reported results,27,33,34 confirming the successful synthesis of UiO-66. Furthermore, the near-identical XRD patterns of UiO-66 and its derivatives, UiO-66-X, indicate that these materials maintain the same three-dimensional porous crystalline lattice structure as the parent UiO-66, characterized by hexanuclear zirconium oxyhydroxide clusters and octahedral cages assembled with modified terephthalate linkers.13 Following the synthesis of UiO-66-X@8-HQ, the characteristic peaks of the UiO-66 derivatives remain, demonstrating that the composites retain their crystalline structures without significant loss of crystallographic integrity. However, a subtle shift of the peaks toward lower angles was observed in the UiO-66-X@8-HQ patterns. These variations in the XRD patterns are attributed to the successful incorporation of 8-HQ molecules within the UiO-66-X@8-HQ framework.
 | ||
| Fig. 4 Characterization of UiO-66 derivatives and UiO-66-X@8-HQ (X = –H/–CH3/–NH2) (a) XRD pattern, (b) FTIR spectra, (c) N2 adsorption isotherms, (d) pore volume, (e) high-resolution XPS spectra in N 1s region of UiO-66-X@8-HQ, (f) zeta potentials. | ||
Fig. 4b presented a comparative analysis of the FTIR spectra obtained for UiO-66 derivatives and UiO-66-X@8-HQ composites. In the UiO-66 spectrum, characteristic vibrations of Zr–O bonds were identified at 667 cm−1 and 558 cm−1.35,36 Peaks observed at 2920 cm−1, 1662 cm−1, 1581 cm−1, and 1396 cm−1 corresponded to the stretching vibrations of aromatic C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and CC bonds, respectively, originating from the organic linkers. The peak at 3450 cm−1 was attributed to hydroxyl groups and/or adsorbed water molecules present on the sample surface.37 For UiO-66 containing an amino group, the peak at 3328 cm−1 signified the characteristic absorption of the amino group (–NH2), the peak at 1439 cm−1 corresponded to the stretching vibration of the C–N bond, and the peak at 581 cm−1 indicates the Zr–O bond, collectively confirming the successful synthesis of UiO-66-NH2.38 The peak at 1379 cm−1 was identified as the characteristic absorption of the methyl group,39 while the peak at 590 cm−1 was attributed to the Zr–O bond, confirming the successful preparation of UiO-66-CH3. Moreover, in the spectra of UiO-66-NH2@8-HQ and UiO-66-CH3@8-HQ, peaks observed at 1326.00 cm−1, 1468.55 cm−1, and 1501.56 cm−1 were indicative of the C–C and C–N stretching vibrations within the aromatic ring structure of 8-HQ,40 thereby demonstrating the successful synthesis of the UiO-66-X@8-HQ hybrids.
O, and CC bonds, respectively, originating from the organic linkers. The peak at 3450 cm−1 was attributed to hydroxyl groups and/or adsorbed water molecules present on the sample surface.37 For UiO-66 containing an amino group, the peak at 3328 cm−1 signified the characteristic absorption of the amino group (–NH2), the peak at 1439 cm−1 corresponded to the stretching vibration of the C–N bond, and the peak at 581 cm−1 indicates the Zr–O bond, collectively confirming the successful synthesis of UiO-66-NH2.38 The peak at 1379 cm−1 was identified as the characteristic absorption of the methyl group,39 while the peak at 590 cm−1 was attributed to the Zr–O bond, confirming the successful preparation of UiO-66-CH3. Moreover, in the spectra of UiO-66-NH2@8-HQ and UiO-66-CH3@8-HQ, peaks observed at 1326.00 cm−1, 1468.55 cm−1, and 1501.56 cm−1 were indicative of the C–C and C–N stretching vibrations within the aromatic ring structure of 8-HQ,40 thereby demonstrating the successful synthesis of the UiO-66-X@8-HQ hybrids.
Nitrogen adsorption–desorption isotherms and pore size distribution curves for the UiO-66 derivatives and UiO-66-X@8-HQ composites were presented in Fig. 4c and d, respectively. As shown in Fig. 4c, all samples exhibited type I isotherms, consistent with IUPAC classification,41 indicative of microporous materials. Specific surface areas, determined using the Brunauer–Emmett–Teller (BET) method, were 1271.22 m2 g−1 for UiO-66, 771.10 m2 g−1 for UiO-66-CH3, and 846.42 m2 g−1 for UiO-66-NH2 (Table 1). Following the incorporation of 8-HQ, the specific surface areas decreased to 803.17 m2 g−1, 637.30 m2 g−1, and 284.06 m2 g−1 for UiO-66@8-HQ, UiO-66-CH3@8-HQ, and UiO-66-NH2@8-HQ, respectively. Non-local density functional theory (NLDFT) analysis revealed pore sizes primarily within the 0–3 nm range (Fig. 4d), further confirming the presence of micropores.42 The average pore diameters for UiO-66, UiO-66-CH3, and UiO-66-NH2 were 1.59 nm, 1.77 nm, and 1.62 nm, respectively. Correspondingly, the total pore volumes were 0.51 cm3 g−1 for UiO-66, 0.34 cm3 g−1 for UiO-66-CH3, and 0.45 cm3 g−1 for UiO-66-NH2. Notably, the pore diameters and volumes of the UiO-66-X@8-HQ composites were lower than those of the pristine UiO-66 and its derivatives, suggesting that the introduction of 8-HQ molecules into the pores of the UiO-66 derivatives leads to a partial pore blockage and consequently, a decrease in specific surface area.15,43,44
| Samples | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore width (nm) |
|---|---|---|---|
| UiO-66 | 1271.22 | 0.51 | 1.59 |
| UiO-66@8-HQ | 803.17 | 0.35 | 1.56 |
| UiO-66-CH3 | 771.10 | 0.45 | 1.77 |
| UiO-66-CH3@8-HQ | 637.30 | 0.27 | 10.25 |
| UiO-66-NH2 | 846.42 | 0.34 | 1.62 |
| UiO-66-NH2@8-HQ | 284.06 | 0.11 | 1.53 |
XPS analysis, as depicted in Fig. 4e, furnishes compelling evidence of the successful loading of 8-HQ onto functionalized UiO-66 carriers. The appearance of characteristic 8-HQ peaks in the XPS spectra confirms the effective incorporation of the inhibitor. Specifically, the observed chemical shift in the N 1s spectrum, for instance, from 399.8 eV to 400.5 eV, is attributable to the presence of amino groups, thereby unambiguously verifying 8-HQ loading on the UiO-66-NH2 variant. Concurrently, the C 1s spectrum reveals discernible alterations in bonding states, including the emergence of a new component at 285.2 eV corresponding to methyl groups, consistent with the structural characteristics of the UiO-66-CH3 carrier. These collective spectral shifts strongly suggest that hydrogen-bonding interactions between the functional groups (–NH2 and –CH3) and 8-HQ are the predominant mechanism governing the loading process, an observation that aligns favorably with our density functional theory (DFT) simulations.45,46 Fig. 4f displayed the zeta potential profiles of UiO-66-X and UiO-66-X@8-HQ (X = –H, –CH3, –NH2).28 Prior to loading, the pristine 8-HQ exhibits a negative zeta potential (−2.3 ± 0.5 mV), while the UiO-66-X series showed positive surface charges (+15.2 to +18.6 mV), consistent with their respective intrinsic physicochemical properties. Upon encapsulation, a significant reduction in zeta potential was observed for all UiO-66-X@8-HQ composites, with values decreasing to +12.1 ± 0.3 mV (UiO-66-NH2@8-HQ), +8.7 ± 0.4 mV (UiO-66-CH3@8-HQ), and +10.2 ± 0.2 mV (UiO-66@8-HQ). This systematic charge neutralization phenomenon provided compelling evidence for the successful incorporation of 8-HQ into the MOF carriers, likely mediated by electrostatic interactions between the negatively charged inhibitor and the positively functionalized UiO-66 frameworks.
3.2 Load capacity and release rate
Fig. 5 illustrated the thermogravimetric analysis (TGA) curves of the UiO-66 series, 8-HQ, and UiO-66-X@8-HQ composites under an air atmosphere. As depicted, the TGA profile of UiO-66@8-HQ exhibited three distinct weight loss stages: 30–110 °C, 110–494 °C, and 500–800 °C. The initial stage corresponded to the removal of residual solvent molecules. The second stage, accounting for approximately 16.01% weight loss, was attributed to the desorption of the loaded 8-HQ. This assignment was based on the thermal stability of UiO-66 up to 500 °C, suggesting that the observed weight loss in the 110–494 °C range was primarily due to 8-HQ. The final stage represented the decomposition of the UiO-66 framework, leaving behind approximately 47.82% zirconia residue.47 Similarly, UiO-66-CH3@8-HQ displayed three weight loss stages within the ranges of 30–110 °C, 110–468 °C, and 490–800 °C. The initial stage was again associated with solvent removal. The second stage, representing around 19.45% weight loss, was attributed to 8-HQ desorption, considering the thermal stability of UiO-66-CH3 up to 490 °C. The final stage corresponded to the framework degradation of UiO-66-CH3, resulting in a residual zirconia content of 47.8%.48 Finally, the TGA profile of UiO-66-NH2@8-HQ also revealed three weight loss stages: 30–110 °C, 110–316 °C, and 350–800 °C. The initial stage was attributed to solvent loss, while the second stage, accounting for approximately 14.66% weight loss, was associated with the desorption of loaded 8-HQ. Given that UiO-66-NH2 was thermally stable up to 500 °C, this weight loss was attributed to 8-HQ. The final stage corresponded to the decomposition of the UiO-66-NH2 framework, yielding approximately 49.2% zirconia residue.49 | ||
| Fig. 5 TGA of UiO-66-X@8-HQ (X = –H/–CH3/–NH2). | ||
To investigate the mechanism of UiO-66 and its derivatives loading the 8-HQ, kinetic models such as pseudo first-order was applied to analyze the adsorption data. Eqn (5) describes the pseudo-first-order:16
| Ct/Ce = 1 − Ae(−kt) | (5) |
Ct and Ce represent the concentrations of 8-HQ at time t and at equilibrium, respectively. A is a constant, t denotes the time of release, and k signifies the release rate constant. The release profile initially exhibited a rapid rate, which subsequently decelerated as the concentration increased over time. To investigate the 8-HQ release mechanism and the influence of the UiO-66 series framework, the experimental data were analyzed using the pseudo-first-order kinetic model (eqn (5)).45 This model suggests an initial phase of rapid release, followed by a gradual slowdown as the concentration approaches equilibrium. Fig. 6a presented the standard curve, while Fig. 6b illustrated the time-dependent release behavior of UiO-66-X@8-HQ. The observed rapid release within the first two hours, followed by a sustained, slower release until equilibrium, can be attributed to the materials' large specific surface area and ordered pore structure. Consequently, the release rates for the modified materials followed the order UiO-66@8-HQ < UiO-66-CH3@8-HQ < UiO-66-NH2@8-HQ, with UiO-66-NH2 exhibiting a higher equilibrium release than both UiO-66 and UiO-66-CH3. Table 2 summarized the kinetic parameters for 8-HQ adsorption by UiO-66-X@8-HQ. The high fitting determination coefficients (R2) of 0.97, 0.99, and 0.94 for UiO-66@8-HQ, UiO-66-NH2@8-HQ, and UiO-66-CH3@8-HQ, respectively, indicated that their release profiles are well-described by the pseudo-first-order kinetic model. The release rate constant (k) values for UiO-66 and UiO-66-CH3 were higher than that for UiO-66-NH2, suggesting faster release kinetics for the former two.50 This may be related to the enhanced transfer of hydrophilic 8-HQ to the carrier surface in solution. The higher k values observed for UiO-66-NH2 and UiO-66-CH3 compared to UiO-66 can potentially be attributed to the smaller BET surface areas and comparatively larger pore sizes of the modified UiO-66-NH2 and UiO-66-CH3 materials.
 | ||
| Fig. 6 (a) Standard curve of 8-HQ and (b) release concentrations of UiO-66-X@8-HQ of UiO-66-X@8-HQ (X = –H/–CH3/–NH2). | ||
| Samples | Ce (mg L−1) | A | k (h−1) | (R2) |
|---|---|---|---|---|
| UiO-66@8-HQ | 2.15 | −1.13 | 0.13 | 0.97 |
| UiO-66-CH3@8-HQ | 5.24 | −3.47 | 0.58 | 0.94 |
| UiO-66-NH2@8-HQ | 3.74 | −3.36 | 1.23 | 0.99 |
3.3 Inhibition efficiency measurements
Polarization curves for carbon steel immersed for one hour in a 0.5 mol per L NaCl solution saturated with CO2, both with and without the presence of 100 mg per L UiO-66-X@8-HQ (where X = –H, –CH3, –NH2), at 25 °C are presented in Fig. 7. The data indicated a positive shift in the corrosion potential and a substantial decrease in both anodic and cathodic current densities upon the introduction of UiO-66-X@8-HQ. Furthermore, the electrochemical parameters derived from fitting these polarization curves are summarized in Table 3. These parameters reveal that the corrosion potential (Ecorr) of carbon steel shifted towards more positive values, and the corrosion current density (icorr) was significantly reduced in the presence of UiO-66-X@8-HQ. The observed trend in icorr values follows the order: UiO-66@8-HQ > UiO-66-CH3@8-HQ > UiO-66-NH2@8-HQ. Consequently, UiO-66-NH2@8-HQ demonstrates the highest corrosion inhibition efficiency for carbon steel, while UiO-66@8-HQ exhibited the lowest. This variation in inhibition performance can be attributed to differences in the loading capacity and release rate of 8-HQ from UiO-66@8-HQ, UiO-66-NH2@8-HQ, and UiO-66-CH3@8-HQ.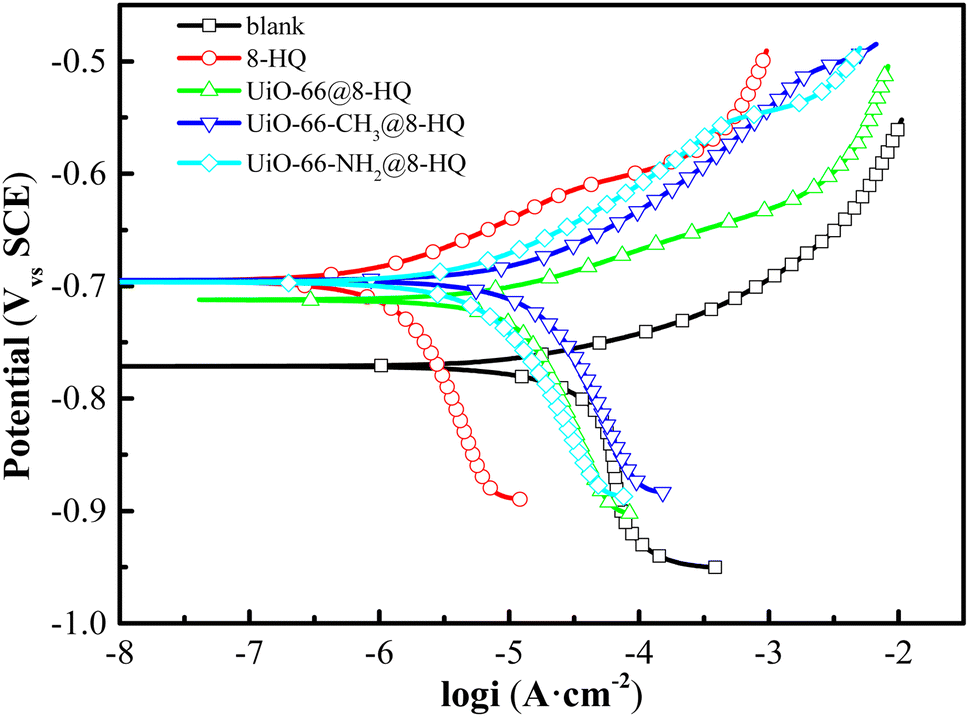 | ||
| Fig. 7 Polarization curves of carbon steel in CO2-saturated 0.5 mol per L NaCl solution with 100 mg per L UiO-66-X@8-HQ (X = –H/–CH3/–NH2) at 25 °C. | ||
| Samples | Ecorr (mV) | ba (mV dec−1) | bc (mV dec−1) | icorr (A cm−2) | IE (%) |
|---|---|---|---|---|---|
| Blank | −770 | 43 | −96 | 2.36 × 10−5 | |
| 8-HQ | −695 | 37 | −65 | 3.88 × 10−6 | 83.58 |
| UiO-66@8-HQ | −696 | 52 | −88 | 6.99 × 10−6 | 70.42 |
| UiO-66-CH3@8-HQ | −760 | 71 | −125 | 6.44 × 10−6 | 72.14 |
| UiO-66-NH2@8-HQ | −712 | 36 | −96 | 5.86 × 10−6 | 75.24 |
To elucidate the corrosion mechanism of carbon steel in solutions with and without various UiO-66-X@8-HQ additives, electrochemical impedance spectroscopy (EIS) was performed. Fig. 8 presented the impedance spectra obtained for carbon steel immersed in 0.5 mol per L NaCl solution saturated with CO2, both with and without the UiO-66-X@8-HQ compounds. A summary diagram of the AC impedance spectra of carbon steel in the presence of UiO-66-X@8-HQ was shown in Fig. 9. Equivalent circuits (Fig. 10) were employed to fit the corresponding EIS data, where n represents the dispersion effect index, Rs is the solution resistance, Rf is the resistance of the corrosion product film, Rct is the charge transfer resistance of the corrosion reaction, and W is the Warburg impedance, which reflects the diffusion process of electroactive species. CPEf and CPEdl are constant phase elements (CPEs) representing the capacitive response of the corrosion product film and the double-layer capacitance, respectively.51,52 Specifically, Fig. 8a displayed the fitted data for the blank solution and the solution containing UiO-66@8-HQ, while Fig. 8b showed the fitted data for the blank solution and the solutions containing 8-HQ, UiO-66-NH2@8-HQ, and UiO-66@8-HQ. The fitting parameters derived from these analyses are detailed in Table 4.
 | ||
| Fig. 8 (a) Nyquist and (b) Bode plots of carbon steel in CO2-saturated 0.5 mol per L NaCl solution containing 100 mg per L UiO-66-X@8-HQ (X = –H/–CH3/–NH2) at 25 °C. | ||
 | ||
| Fig. 9 Time dependence of Rct + Rf for carbon steel in CO2-saturated 0.5 mol per L NaCl solution containing 100 mg per L UiO-66-X@8-HQ (X = –H/–CH3/–NH2) at 25 °C. | ||
 | ||
| Fig. 10 Electrochemical equivalent circuits for fitting the EIS (a) blank (b) UiO-66-X@8-HQ (X = –H/–CH3/–NH2). | ||
| Rs (Ω cm2) | CPEdl-T (Sn1 Ω−1 cm−2) | n1 | Rct (Ω cm2) | CPEf-T (Sn2 Ω−1 cm−2) | n2 | Rf (Ω cm2) | |
|---|---|---|---|---|---|---|---|
| Blank | 3.8 | 2.51 × 10−5 | 0.92 | 41.5 | 1.01 × 10−4 | 0.60 | 269 |
| 8-HQ | 3.1 | 1.60 × 10−5 | 0.95 | 97.9 | 1.11 × 10−5 | 0.60 | 1841 |
| UiO-66@8-HQ | 6.9 | 2.92 × 10−5 | 0.87 | 67.1 | 1.46 × 10−4 | 0.65 | 861 |
| UiO-66-NH2@8-HQ | 6.2 | 1.96 × 10−5 | 0.89 | 89.0 | 5.71 × 10−5 | 0.57 | 1453 |
| UiO-66-CH3@8-HQ | 3.3 | 1.06 × 10−5 | 0.99 | 83.8 | 7.31 × 10−5 | 0.60 | 1058 |
EIS analysis of carbon steel in the blank solution, as depicted in the Nyquist and Bode plots, revealed distinct features attributable to different processes. The high-frequency region exhibited an arc corresponding to the capacitive behavior of the corrosion product film formed on the electrode surface. Conversely, the low-frequency region displayed an arc related to the charge transfer resistance. Furthermore, the observed tail in the response suggested a diffusion-controlled process, likely associated with the transport of corrosion products. As illustrated in Fig. 8a, the addition of MOF matrix composites, such as UiO-66-X@8-HQ, to the solution results in the disappearance of the Warburg impedance in the low-frequency region and a significant increase in the capacitive-reactance arc diameter, particularly evident with the addition of UiO-66-X@8-HQ and 8-HQ. The Bode plots in Fig. 8b demonstrated that the impedance modulus and phase angle values increase successively upon the introduction of UiO-66-X@8-HQ compared to the blank solution. This observation indicated that the presence of these MOF matrix composites impedes the corrosion reaction on the carbon steel surface, thus inhibiting its corrosion.53 Consistent with these findings, Table 4 showed a substantially higher impedance value for carbon steel in the solution containing UiO-66-X@8-HQ compared to the blank solution, suggesting that the incorporation of UiO-66-X@8-HQ effectively reduces the corrosion rate of carbon steel. The impedance values follow the order UiO-66-NH2@8-HQ > UiO-66-CH3@8-HQ > UiO-66@8-HQ, suggesting that the released 8-HQ contributed to the protective layer formed on the carbon steel surface.54 As observed in Table 4, the impedance value of carbon steel in the solution containing UiO-66-X@8-HQ was significantly higher compared to that in the blank solution. This suggested that the incorporation of UiO-66-X@8-HQ effectively diminished the corrosion rate of carbon steel.
To further elucidate the influence of 8-HQ released from the UiO-66 series on the corrosion behavior of carbon steel, EIS measurements were conducted on carbon steel at 25 °C after varying immersion times in solutions containing UiO-66-X@8-HQ. The EIS fitting results were presented in Fig. 9. As depicted, the impedance values of carbon steel increased progressively with extended immersion time, eventually reaching a plateau, indicating the maximal release of 8-HQ corrosion inhibitor from the UiO-66 series composites. The observed trend in impedance values (Rct + Rf) follows the order: UiO-66-NH2@8-HQ > UiO-66-CH3@8-HQ > UiO-66@8-HQ. These findings suggested that UiO-66-X@8-HQ and other MOF-based composites can be effectively utilized as corrosion inhibitors in corrosive media, providing long-term protection for the carbon steel substrate.
SEM images revealed the corrosive effects on carbon steel surfaces following a 24 hours immersion in a 0.5 mol per L NaCl solution, both with and without the addition of various UiO-66-X@8-HQ composites under saturated CO2 conditions. As depicted in Fig. 11a, the carbon steel surface in the blank solution exhibited a layer of loosely adhered corrosion products and a notably uneven texture. Conversely, the surface treated with UiO-66@8-HQ (Fig. 11b) displayed residual mechanical scratches from the abrasive pretreatment, accompanied by only a limited amount of corrosion products. Furthermore, the addition of UiO-66-CH3@8-HQ (Fig. 11c) resulted in a smoother carbon steel surface with a sparse distribution of corrosion products. Most notably, the presence of UiO-66-NH2@8-HQ (Fig. 11d) yielded a remarkably flat surface with a near absence of corrosion products. These observations suggest that the incorporation of UiO-66-X@8-HQ and related MOF matrix composites into the corrosive medium effectively mitigates corrosion and provides a protective effect for the carbon steel substrate.
 | ||
| Fig. 11 SEM images of carbon steel samples immersed in CO2-saturated 0.5 mol per L NaCl solution containing 100 mg per L UiO-66-X@8-HQ (X = –H/–CH3/–NH2) for 24 h at 25 °C (a) blank, (b) UiO-66@8-HQ, (c) UiO-66-CH3@8-HQ and (d) UiO-66-NH2@8-HQ. | ||
XPS spectra were acquired to elucidate the surface chemical composition of carbon steel samples following a 24 hours immersion in a 0.5 mol per L NaCl solution containing 100 mg per L UiO-66-X@8-HQ and saturated with CO2 (Fig. 12). To gain a deeper understanding of the elemental chemical environments and to attribute specific components to their corresponding chemical states, peak fitting analysis was performed on selected high-resolution spectra.
 | ||
| Fig. 12 Full-survey XPS spectra of carbon steel in CO2-saturated 0.5 mol per L NaCl solution containing 100 mg per L UiO-66-X@8-HQ (X = –H/–CH3/–NH2) for 24 h at 25 °C. | ||
Fig. 13 illustrates the high-resolution C 1s, N 1s, and Fe 2p spectra.53 The C 1s spectrum (Fig. 13a) obtained from the UiO-66@8-HQ treated carbon steel surface exhibits multiple bands at 289.22 eV, 285.92 eV, and 284.82 eV, corresponding to CN, C–N, and C–C/CH bonds, respectively. On the surface of carbon steel treated with UiO-66-NH2@8-HQ, peaks at 286.89 eV, 285.00 eV, and 283.50 eV were assigned to C–N and C–C/CH bonds, respectively. For the UiO-66-CH3@8-HQ treated carbon steel surface, peaks at 287.52 eV, 286.17 eV, and 284.77 eV were attributed to CN, C–N, and C–C/CH bonds, respectively.55 The N 1s spectrum (Fig. 13b) of the UiO-66@8-HQ treated carbon steel surface revealed peaks at 401.67 eV, 400.37 eV, and 398.97 eV, corresponding to Fe–N, C–N/CN, and N–N/NN bonds, respectively. The energy peak positions of the corresponding bonds on the UiO-66-NH2@8-HQ treated carbon steel surface were shifted to approximately 400.95 eV, 399.55 eV and 398.95 eV, respectively. Similarly, on the surface of the UiO-66-CH3@8-HQ treated carbon steel, these bonds were observed at 401.17 eV, 400.17 eV, and 398.92 eV, respectively.56 The O 1s spectrum (Fig. 13c) exhibited three components with peaks at 529.9 eV, 531.2 eV, and 532.4 eV, corresponding to Fe2O3, CO, and C–O groups, respectively. The Fe 2p3/2 spectrum (Fig. 13d) displayed fitted peaks at 709.7 eV, 710.8 eV, and 712.1 eV, which are associated with Fe–N, Fe2O3, and FeOOH states, respectively.57,58 The presence of Fe–N peaks indicated the formation of coordination bonds between 8-HQ and the carbon steel surface, suggesting a strong chemisorption of 8-HQ. This process involved the donation of lone pairs of electrons from nitrogen atoms and hydroxyl groups of 8-HQ to unoccupied molecular orbitals of the carbon steel, where the corrosion inhibitor inhibited corrosion ions on the carbon steel surface through chemisorption.59
 | ||
| Fig. 13 High-resolution XPS spectra of carbon steel in CO2-saturated 0.5 mol per L NaCl solution with UiO-66-X@8-HQ (X = –H/–CH3/–NH2) for 24 h at 25 °C (a), C 1s, (b) N 1s, (c) O 1s and (d) Fe 2p. | ||
3.4 Theoretical calculation
The interactions between 8-HQ and UiO-66 derivatives were computationally assessed using density functional theory with dispersion corrections (DFT-D). The initial and optimized geometries, illustrated in Fig. 14, were employed to evaluate these interactions. As presented in Table 5, the computational analysis depicted in Table 5 offers a comprehensive investigation of the quantum chemical parameters characterizing 8-HQ, UiO-66, its methylated (UiO-66-CH3), and aminated (UiO-66-NH2) derivatives, as well as their respective complexes with 8-HQ. Starting with the free molecules, 8-HQ exhibited an EHOMO value of −6.16 eV and an ELUMO of −1.68 eV, resulting in an energy gap (ΔE) of 4.48 eV and a notable electronegativity (χ) of −3.92 eV. UiO-66 itself showed lower frontier orbital energies (EHOMO = −7.41 eV, ELUMO = −2.12 eV, ΔE = 5.28 eV), indicating increased chemical hardness (σ = 0.38 eV) relative to 8-HQ. Substitution with a methyl group resulted in UiO-66-CH3 (EHOMO = −7.09 eV, ELUMO = −2.06 eV, ΔE = 5.03 eV), modestly reducing the gap and increasing polarity (μ = 3.02 D). In contrast, UiO-66-NH2 had notably higher HOMO energy (−5.89 eV), a decreased band gap (ΔE = 4.01 eV), and an elevated dipole moment (μ = 4.45 D), reflecting the electron-donating effect of the amino substituent. | ||
| Fig. 14 Initial and final DFT-optimized geometry for UiO-66-X@8-HQ (X = –H/–CH3/–NH2). | ||
| Sample | EHOMO (eV) | ELUMO (eV) | γ (eV) | χ (eV) | σ (eV) | ΔE (eV) | ΔN | μ (D) | Ebinding (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| 8-HQ | −6.16 | −1.68 | 2.24 | −3.92 | 0.45 | 4.48 | 1.88 | 2.99 | — |
| UiO-66 | −7.41 | −2.12 | 2.64 | −4.76 | 0.38 | 5.28 | 1.75 | 2.65 | — |
| UiO-66-CH3 | −7.09 | −2.06 | 2.51 | −4.58 | 0.40 | 5.03 | 1.80 | 3.02 | — |
| UiO-66-NH2 | −5.89 | −1.87 | 2.01 | −3.88 | 0.50 | 4.01 | 2.08 | 4.45 | — |
| UiO-66@8-HQ | −6.05 | −1.65 | 2.20 | −3.85 | 0.46 | 4.39 | 1.90 | 5.57 | −3.04 |
| UiO-66-CH3@8-HQ | −5.52 | −2.04 | 1.74 | −3.78 | 0.57 | 3.48 | 2.37 | 5.46 | −12.54 |
| UiO-66-NH2@8-HQ | −5.57 | −2.78 | 1.40 | −4.18 | 0.72 | 2.79 | 3.10 | 4.67 | −22.33 |
Upon complexation with 8-HQ, significant changes in electronic structure were observed. For UiO-66@8-HQ, EHOMO and ELUMO both increase, and the gap narrows (ΔE = 4.39 eV), while the dipole moment rises to 5.57 D, revealing enhanced charge separation in the assembly. Critically, the binding energy (Ebinding) was calculated as −3.04 kJ mol−1, evidencing a spontaneous but modestly strong interaction between 8-HQ and pristine UiO-66. For the methylated complex (UiO-66-CH3@8-HQ), the EHOMO further increases to −5.52 eV, the ΔE contracts to 3.48 eV, and dipole moment decreases to 5.46 D; notably, the binding energy becomes substantially more negative (−12.54 kJ mol−1), indicating increased thermodynamic favorability. This enhancement was likely driven by the methyl group modifying the local electron density and perhaps facilitating van der Waals or weak π–π interactions. The aminated derivative displayed the most dramatic effects: UiO-66-NH2@8-HQ attains a significantly elevated ELUMO (−2.78 eV), a considerably narrowed gap (ΔE = 2.79 eV), and a reduced dipole moment (μ = 4.67 D) relative to its unbound analogue. Its remarkably negative binding energy (−22.33 kJ mol−1) suggested the interaction is highly spontaneous, which is plausibly attributable to strong hydrogen bonding between the NH2 group and 8-HQ's heteroatoms, as further evidenced by the largest difference in chemical hardness (σ = 0.72 eV) and softness (γ = 1.40 eV).
Overall, these data delineated a clear trend: functionalization of UiO-66, particularly with electron-donating groups such as NH2, drastically enhances the host–guest interaction with 8-HQ, both by promoting electronic compatibility (reduced ΔE, increased charge transfer, and dipole moments) and providing sites for specific non-covalent interactions. The methyl group, while not as effective as NH2, still favorably shifts the binding energy compared to unmodified UiO-66, supporting the hypothesis that modulation of framework functionality offers a powerful lever for tuning guest adsorption properties in metal–organic frameworks. This insight not only deepens our understanding of host–guest chemistry in UiO-66 derivatives but also underscores the critical role of molecular design in optimizing sorbent materials for targeted applications.
Molecular dynamics (MD) simulations were conducted to analyze the adsorption behavior of 8-HQ on the surfaces of the UiO-66 series. Fig. 15 presented simulated side and top views of the 8-HQ inhibitor adsorbed on these surfaces, while Table 6 summarizes the corresponding theoretical calculations. As illustrated in Fig. 15a and d, 8-HQ molecules tended to adsorb onto the UiO-66 surface at an inclined angle. A similar inclined orientation is observed for 8-HQ adsorption on UiO-66-CH3 (Fig. 15b and e) and UiO-66-NH2 (Fig. 15c and f). These MD simulations demonstrated the feasibility of 8-HQ adsorption on all UiO-66 series surfaces. Furthermore, the adsorption energies listed in Table 6 for UiO-66@8-HQ, UiO-66-CH3@8-HQ, and UiO-66-NH2@8-HQ are −240.287 kJ mol−1, −465.807 kJ mol−1, and −617.032 kJ mol−1, respectively. A greater absolute adsorption energy indicates a stronger interaction between the carrier and the guest molecule. Therefore, 8-HQ exhibited a spontaneous binding affinity for the UiO-66 series surfaces, with the following order of binding energies: UiO-66-NH2@8-HQ > UiO-66-CH3@8-HQ > UiO-66@8-HQ.
 | ||
| Fig. 15 MD simulation diagrams for 8-HQ on the UiO-66 series: top view (a) UiO-66, (b) UiO-66-CH3 and (c) UiO-66-NH2, side view: (d) UiO-66, (e) UiO-66-CH3 and (f) UiO-66-NH2. | ||
| Samples | Etot | EUiO-66-X | Einh | Eint |
|---|---|---|---|---|
| UiO-66@8-HQ | −285.240 | −176.174 | 131.221 | −240.287 |
| UiO-66-CH3@8-HQ | −501.748 | −249.659 | 213.718 | −465.807 |
| UiO-66-NH2@8-HQ | −648.402 | −269.920 | 238.550 | −617.032 |
The differential loading of 8-HQ onto the UiO-66 series can be attributed to the influence of the functional groups. The presence of the amino group on UiO-66-NH2 likely promotes preferential interactions between the UiO-66-NH2 carrier and the ethanol solvent, thereby reducing the loading capacity for the 8-HQ inhibitor. Conversely, the methyl group on UiO-66-CH3 enhances hydrophobicity, leading to an improved loading capacity for 8-HQ. The observed differences in the 8-HQ release rates from the UiO-66 series are likely due to the formation of hydrogen bonds between the amino group on UiO-66-NH2 and 8-HQ, strengthening the binding interaction and resulting in the slowest release rate. The methyl group enhances van der Waals forces, leading to a stronger binding interaction between UiO-66-CH3 and 8-HQ compared to UiO-66@8-HQ.
Fig. 16 illustrated the electron density distribution of the optimized 8-HQ molecule. As depicted, electron density was largely delocalized across the conjugated bonds within the nitrogen-containing heterocyclic ring and adjacent hydroxyl groups. Furthermore, the electrostatic potential map revealed that regions of nucleophilic reactivity (red, negative charge) and electrophilic reactivity (green, positive charge) are primarily concentrated around the oxygen and nitrogen atoms. The calculated energy level of 8-HQ (4.48 eV) was below the Fermi level of iron (Fe = 4.5 eV), suggesting the likelihood of electron transfer from the corrosion inhibitor to the metal surface. These observations underscore the significant contribution of the N–N bond and the –OH group to the overall molecular activity of the 8-HQ inhibitor.
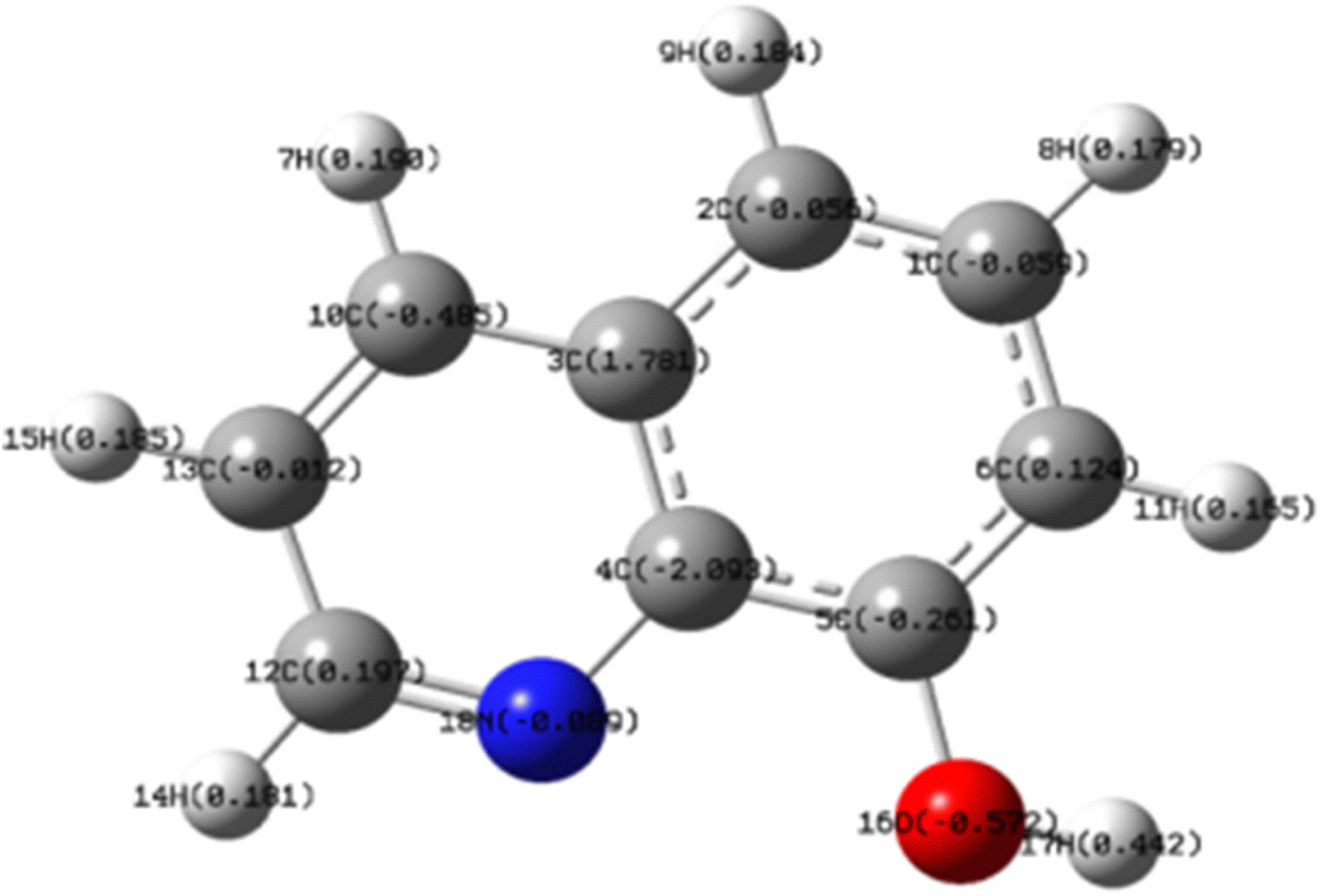 | ||
| Fig. 16 Molecular charge distribution of 8-HQ after DFT optimization (legend: grey represents carbon; white represents hydrogen, blue represents nitrogen, and magenta represents oxygen). | ||
MD simulations were employed to investigate the inhibition mechanism of 8-hydroxyquinoline (8-HQ) on carbon steel corrosion. A (6 × 6) supercell configuration of the Fe(110) surface was constructed, and the interactions between this surface and 8-HQ inhibitor molecules were simulated within a simulation box, utilizing periodic boundary conditions to replicate the corrosive environment. The simulated adsorption system incorporated Cl−, H+, CO2, and H2O molecules, alongside the 8-HQ inhibitor molecules, while other simulation parameters remained constant. As illustrated in Fig. 17, 8-HQ exhibited a predominantly parallel adsorption orientation on the carbon steel surface, as observed from both side and top views. Furthermore, the calculated adsorption energy of 8-HQ on the Fe(110) surface was −309.30 kJ mol−1, indicative of a strong interaction. This robust adsorption was attributed to the presence of electron-donating nitrogen atoms within the N–N group and the hydroxyl group in the 8-HQ molecule, which facilitated favorable interactions with the iron surface.
 | ||
| Fig. 17 Simulation of MD adsorption digrams of 8-HQ on carbon steel surface. | ||
4. Discussion
4.1 Mechanism underlying differential 8-HQ loading in the UiO-66 series
Comprehensive spectral analyses confirm that 8-HQ can be successfully loaded onto the UiO-66-X series without compromising their crystalline structure. TGA and UV-vis spectroscopy were employed to quantify the loading capacity and release kinetics of 8-HQ from these carriers. The observed loading capacities for UiO-66, UiO-66-NH2, and UiO-66-CH3 were 16.01%, 14.66%, and 19.45%, respectively. The release profiles of 8-HQ from the UiO-66-X materials followed first-order kinetics, characterized by an initial increase in release rate that gradually approached equilibrium. The observed release rates followed the sequence: UiO-66 > UiO-66-CH3 > UiO-66-NH2.The variations in 8-HQ loading across the UiO-66 series are attributed to the distinct polar functionalities present on each carrier. The amino groups in UiO-66-NH2, acting as hydrophilic and hydrogen bond-accepting moieties, can serve as preferential anchor points for solvent molecules. This preferential interaction with the solvent may hinder the effective adsorption of 8-HQ, resulting in a comparatively lower loading capacity. Conversely, increasing the hydrophobicity of the UiO-66 framework favors the adsorption of 8-HQ over ethanol. Consequently, both bare UiO-66 and the methyl-functionalized UiO-66-CH3 exhibit enhanced hydrophobicity, leading to higher 8-HQ loading. Furthermore, theoretical calculations indicate that the amino groups grafted onto UiO-66-NH2 can enhance the binding affinity between 8-HQ and the carrier through the formation of hydrogen bonds. UiO-66-CH3, on the other hand, strengthens its interaction with 8-HQ by increasing the van der Waals surface area. This enhanced binding affinity correlates with slower release rates, thereby explaining the observed differences in both loading capacity and release kinetics of 8-HQ across the UiO-66 series.4.2 Corrosion inhibition mechanism of 8-HQ
Electrochemical testing revealed that the released 8-HQ formed a protective layer on the carbon steel surface, effectively inhibiting corrosion. The observed differences in drug loading and release kinetics resulted in the following sequence of corrosion inhibition effectiveness: UiO-66@8-HQ < UiO-66-CH3@8-HQ < UiO-66-NH2@8-HQ. XPS analysis indicated that 8-HQ adsorbs onto the carbon steel surface via the formation of Fe–N bonds. Theoretical calculations demonstrated that the nitrogen atoms located outside the 8-HQ molecular ring exhibit a negative charge. Moreover, resonance within the molecular structure enhances the reactivity of the nitrogen atoms within the ring, rendering them as active adsorption sites. MD simulations further revealed that 8-HQ can spontaneously adsorb onto the carbon steel surface. Therefore, the corrosion inhibition mechanism of 8-HQ released from the UiO-66 series on carbon steel, as depicted in Fig. 18, involves the interaction between the π orbitals or lone pairs of electrons on the nitrogen heteroatoms of 8-HQ with the d orbitals of the carbon steel, thereby protecting the metal from corrosion. | ||
| Fig. 18 Schematic diagram of the corrosion inhibition of the released 8-HQ on carbon steel surface. | ||
5. Conclusions
This study investigated the use of UiO-66-X (X = –H, –NH2, –CH3) as host matrices for encapsulating the corrosion inhibitor 8-hydroxyquinoline (8-HQ), systematically examining the influence of functional group polarity on loading efficacy. The results confirm the successful encapsulation and pH-responsive release of 8-HQ from these MOF carriers, with the resulting MOF-inhibitor hybrids demonstrating significant corrosion suppression on metallic substrates. The key findings are summarized as follows:(1) UiO-66, UiO-66-NH2, and UiO-66-CH3 exhibited distinct 8-HQ loading capacities of 16.01%, 14.66%, and 19.45%, respectively, with corresponding release efficiencies of 98.89%, 97.70%, and 98.89%. Competitive solvation analysis indicated that the amino group in UiO-66-NH2 preferentially interacts with ethanol solvent molecules, consequently reducing 8-HQ uptake. Conversely, methyl-functionalized UiO-66-CH3 enhanced hydrophobic affinity for 8-HQ, leading to optimized loading performance. Molecular simulations further revealed that amino-functionalization strengthens 8-HQ binding energy through intensified host–guest interactions, while methyl substitution enhances van der Waals forces at the interface.
(2) The mechanism of corrosion inhibition involves the chemisorption of 8-HQ onto carbon steel surfaces via Fe–N coordination bonds, forming a protective molecular layer that effectively impedes electrolyte penetration. This adsorption-driven barrier effect significantly suppresses both anodic dissolution and cathodic oxygen reduction reactions, thereby achieving robust corrosion protection.
Data availability
The data are available from the first author on reasonable request.Author contributions
Jiao-Jiao Cao: conceptualization, writing – original draft, methodology. Jin-Long Ge: investigation, methodology. Yan Gao: investigation, methodology, formal analysis. Bo-Kai, Liao: methodology, data curation, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors express their gratitude for the financial support provided by the National Natural Science Foundation of China (Grant No. 2024AH040186), Bengbu University Applied Science Research Project (Grant No. 2024YYX26QD), National Natural Science Foundation of China (Grant No. 52001080), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2024A1515010905).References
- L.-C. Jing, W.-H. Geng, Z.-L. Bao, P.-F. Qian, R.-Y. Chang, T.-Y. Li, Y.-L. Guo, D. Zhang and H.-Z. Geng, Synthesis of polyaniline tannate-modified carbon nanotubes by oxidative copolymerization as highly efficient corrosion inhibitors for mild steel in HCl solution, J. Colloid Interface Sci., 2024, 680, 479–495 CrossRef PubMed.
- B.-K. Liao, R.-X. Quan, P.-X. Feng, H. Wang, W. Wang and L. Niu, Carbon steel anticorrosion performance and mechanism of sodium lignosulfonate, Rare Met., 2024, 43, 356–365 CrossRef CAS.
- F. Liu, Q. Tang, Y. Zhang and J. Zhu, Effect of Al content on mechanical properties and corrosion performance of novel Fe-Cr-Al alloy steels in a CO2-O2 coexistence environment, J. Alloys Compd., 2025, 1010, 177217 CrossRef CAS.
- K. Chen, Y. Pan, G. Tang, D. Liang, H. Hu, X. Liu and Z. Liang, Facile fabrication of TiN coatings to enhance the corrosion resistance of stainless steel, Surf. Coat. Technol., 2024, 494, 131450 CrossRef CAS.
- A. Fattah-alhosseini, S. Zaheri and M. Karbasi, A review of corrosion resistance in steel alloys with MOF-embedded coatings, Nano-Structures & Nano-Objects, 2024, 40, 101384 Search PubMed.
- Q. Wang, Y. Song, Y. Liu, J. Wang, C. Huang, Z. Xie and B. Liu, Composite fluorescence probe towards a smart coating for early corrosion detection of carbon steel, J. Electroanal. Chem., 2024, 972, 118641 CrossRef CAS.
- A. Suárez-Vega, C. Agustín-Sáenz, L. A. O'Dell, F. Brusciotti, A. Somers and M. Forsyth, Properties of hybrid sol-gel coatings with the incorporation of lanthanum 4-hydroxy cinnamate as corrosion inhibitor on carbon steel with different surface finishes, Appl. Surf. Sci., 2021, 561, 149881 CrossRef.
- Z. Zhang, D. Yan, X. Liu, W. Li, Z. Wang, Y. Wang, D. Song, T. Zhang, J. Liu and J. Wang, Self-healing nanocomposite coatings containing organic–inorganic inhibitors functionalized dendritic silica nanocontainers for synergistic corrosion protection of carbon steel, Colloids Surf., A, 2023, 656, 130430 CrossRef CAS.
- N. Faaizatunnisa, R. Ediati, E. N. M. D. Yusof, A. Fadlan, K. Karelius, U. Kulsum and M. N. Ariesta, The mixed-ligand strategy for structural modification of MOF materials to enhance the photocatalytic degradation and adsorption of organic pollutants: a review, Nano-Structures & Nano-Objects, 2024, 40, 101366 Search PubMed.
- M. Shahnawaz Khan, S. Zhu and S. Bor Chen, Metal–organic frameworks (MOFs) for oxo-anion removal in wastewater treatment: advancements and applications, Chem. Eng. J., 2024, 157396 CrossRef.
- V. Shukla, N. Haq and K. A. Siddiqui, Development of colorimetric probe using robust zinc-MOF and its consistent melamine sponge composite: instantaneous N3–, La3+ sensing and degradation applications, J. Mol. Struct., 2025, 1322, 140560 CrossRef CAS.
- Y. Zhao, F. Jiang, Y.-Q. Chen and J.-M. Hu, Coatings embedded with GO/MOFs nanocontainers having both active and passive protecting properties, Corros. Sci., 2020, 168, 108563 CrossRef CAS.
- T.-F. Chen, S.-Y. Han, Z.-P. Wang, H. Gao, L.-Y. Wang, Y.-H. Deng, C.-Q. Wan, Y. Tian, Q. Wang, G. Wang and G.-S. Li, Modified UiO-66 frameworks with methylthio, thiol and sulfonic acid function groups: the structure and visible-light-driven photocatalytic property study, Appl. Catal., B, 2019, 259, 118047 CrossRef CAS.
- L. Shen, R. Liang, M. Luo, F. Jing and L. Wu, Electronic effects of ligand substitution on metal-organic framework photocatalysts: the case study of UiO-66, Phys. Chem. Chem. Phys., 2015, 17, 117–121 RSC.
- J. Chen, J. Ouyang, W. Chen, Z. Zheng, Z. Yang, Z. Liu and L. Zhou, Fabrication and adsorption mechanism of chitosan/Zr-MOF (UiO-66) composite foams for efficient removal of ketoprofen from aqueous solution, Chem. Eng. J., 2022, 431, 134045 CrossRef CAS.
- W. Sun, H. Li, H. Li, S. Li and X. Cao, Adsorption mechanisms of ibuprofen and naproxen to UiO-66 and UiO-66-NH2: batch experiment and DFT calculation, Chem. Eng. J., 2019, 360, 645–653 CrossRef CAS.
- C. Gaudin, D. Cunha, E. Ivanoff, P. Horcajada, G. Chevé, A. Yasri, O. Loget, C. Serre and G. Maurin, A quantitative structure activity relationship approach to probe the influence of the functionalization on the drug encapsulation of porous metal–organic frameworks, Microporous Mesoporous Mater., 2012, 157, 124–130 CrossRef CAS.
- N. Amaka Chikaodili, C. Sunday Ume, P. Chukwudi Nnaji, N. Bartholomew Iroha, O. Dagdag, J. Okechukwu Ezeugo, A. Thakur, V. Chikaodili Anadebe and O. Dominic Onukwuli, Physisorption or chemisorption: insight from AI computing model based on DFT, MC/MD-simulation for prediction of MOF-based inhibitor adsorption on Cu in brine solution, Comput. Theor. Chem., 2024, 1238, 114730 CrossRef CAS.
- V. C. Anadebe, V. I. Chukwuike, K. Chandra Nayak, E. E. Ebenso and R. Chandra Barik, Combined electrochemical, atomic scale-DFT and MD simulation of nickel based metal organic framework (Ni-MOF) as corrosion inhibitor for X65 pipeline steel in CO2-saturated brine, Mater. Chem. Phys., 2024, 312, 128606 CrossRef CAS.
- A. Guendouz, W. Ettahiri, M. Adardour, J. Lazrak, E. H. E. Assiri, A. Taleb, B. Hammouti, Z. Rais, A. Baouid and M. Taleb, New benzimidazole derivatives as efficient organic inhibitors of mild steel corrosion in hydrochloric acid medium: electrochemical, SEM/EDX, MC, and DFT studies, J. Mol. Struct., 2025, 1321, 139901 CrossRef CAS.
- M. H. Rahmani, A. Dehghani, G. Bahlakeh and B. Ramezanzadeh, Introducing GO-based 2D-platform modified via phytic acid molecules decorated by zeolite imidazole ZIF-9 MOFs for designing multi-functional polymeric anticorrosive system; DFT-D computations and experimental studies, J. Mol. Liq., 2022, 364, 119945 CrossRef CAS.
- D. Majumdar, A. Chatterjee, M. Feizi-Dehnayebi, N. S. Kiran, B. Tuzun and D. Mishra, 8-Aminoquinoline derived two Schiff base platforms: synthesis, characterization, DFT insights, corrosion inhibitor, molecular docking, and pH-dependent antibacterial study, Heliyon, 2024, 10, e35591 CrossRef CAS PubMed.
- M. Jiang, X. Liu, Y. Xu, S. Li, X. Liu, X. Niu, L. Lu, X. Sun, Z. Xie, Z. Wang and S.-p. Cui, Corrosion inhibition effect and mechanism of eco-friendly corrosion inhibitors on mild steel in simulated concrete pore solution: experimental and theoretical studies, ACS Sustainable Chem. Eng., 2025, 13, 2411–2428 CrossRef CAS.
- L. Zhou, T. Zhao and Z. Li, Theoretical and experimental investigation of indazole derivatives as corrosion inhibitors for copper in acidic medium, Langmuir, 2024, 40, 27511–27522 CrossRef CAS PubMed.
- N. Keshmiri, P. Najmi, M. Ramezanzadeh, B. Ramezanzadeh and G. Bahlakeh, Ultrastable porous covalent organic framework assembled carbon nanotube as a novel nanocontainer for anti-corrosion coatings: experimental and computational studies, ACS Appl. Mater. Interfaces, 2022, 14, 19958–19974 CrossRef CAS PubMed.
- E. Berdimurodov, C. Verma, K. Berdimuradov, M. A. Quraishi, A. Kholikov, K. Akbarov, N. Umirov and B. Borikhonov, 8–Hydroxyquinoline is key to the development of corrosion inhibitors: an advanced review, Inorg. Chem. Commun., 2022, 144, 109839 CrossRef CAS.
- R.-M. Chang, Y.-Y. Zhang, G.-B. Zhang, X.-X. Zhang, A.-J. Chen, W. Zhang and Y. Li, Application of thermal alkaline hydrolysis technology to improve the loading and in vitro release of gallic acid in UiO-66, Food Chem., 2022, 391, 133238 CrossRef CAS PubMed.
- H. Tian, W. Li, A. Liu, X. Gao, P. Han, R. Ding, C. Yang and D. Wang, Controlled delivery of multi-substituted triazole by metal–organic framework for efficient inhibition of mild steel corrosion in neutral chloride solution, Corros. Sci., 2018, 131, 1–16 CrossRef CAS.
- R. Farahati, A. Ghaffarinejad, S. M. Mousavi-Khoshdel, J. Rezania, H. Behzadi and A. Shockravi, Synthesis and potential applications of some thiazoles as corrosion inhibitor of copper in 1 M HCl: experimental and theoretical studies, Prog. Org. Coat., 2019, 132, 417–428 CrossRef CAS.
- Z. U. Zango, A. Ramli, K. Jumbri, N. S. Sambudi, H. A. Isiyaka, N. H. H. Abu Bakar and B. Saad, Optimization studies and artificial neural network modeling for pyrene adsorption onto UiO-66(Zr) and NH2-UiO-66(Zr) metal organic frameworks, Polyhedron, 2020, 192, 114857 CrossRef CAS.
- S. Javanbakht, M. Shadi, R. Mohammadian, A. Shaabani, M. M. Amini, M. Pooresmaeil and R. Salehi, Facile preparation of pH-responsive k-carrageenan/tramadol loaded UiO-66 bio-nanocomposite hydrogel beads as a nontoxic oral delivery vehicle, J. Drug Delivery Sci. Technol., 2019, 54, 101311 CrossRef CAS.
- J. Choi, K. S. Yoo, D. Kim, J. Kim and M. R. Othman, Microporous Mo-UiO-66 metal–organic framework nanoparticles as gas adsorbents, ACS Appl. Nano Mater., 2021, 4, 4895–4901 CrossRef CAS.
- Y. Thi Dang, H. T. Hoang, H. C. Dong, K.-B. T. Bui, L. H. T. Nguyen, T. B. Phan, Y. Kawazoe and T. L. H. Doan, Microwave-assisted synthesis of nano Hf- and Zr-based metal–organic frameworks for enhancement of curcumin adsorption, Microporous Mesoporous Mater., 2020, 298, 110064 CrossRef.
- J. Ouyang, J. Chen, S. Ma, X. Xing, L. Zhou, Z. Liu and C. Zhang, Adsorption removal of sulfamethoxazole from water using UiO-66 and UiO-66-BC composites, Particuology, 2022, 62, 71–78 CrossRef CAS.
- Q. Al-Naddaf, M. Al-Mansour, H. Thakkar and F. Rezaei, MOF-GO hybrid nanocomposite adsorbents for methane storage, Ind. Eng. Chem. Res., 2018, 57, 17470–17479 CrossRef CAS.
- K. Chakarova, I. Strauss, M. Mihaylov, N. Drenchev and K. Hadjiivanov, Evolution of acid and basic sites in UiO-66 and UiO-66-NH2 metal–organic frameworks: FTIR study by probe molecules, Microporous Mesoporous Mater., 2019, 281, 110–122 CrossRef CAS.
- Y. Jiang, R. Zhou, B. Ye and Z. Hou, Acetalization of glycerol over sulfated UiO-66 under mild condition, J. Ind. Eng. Chem., 2022, 110, 357–366 CrossRef CAS.
- S. Han, Y. Song, S. Liu, L. Zhao and R. Sun, Dual responsive molecularly imprinted polymers based on UiO-66-DOX for selective targeting tumor cells and controlled drug release, Eur. Polym. J., 2022, 171, 111219 CrossRef CAS.
- W. Sąsiadek, I. Bryndal, T. Lis, M. Wandas and J. Hanuza, Synthesis and physicochemical properties of the methyl-nitro-pyridine-disulfide: X-ray, NMR, electron absorption and emission, IR and Raman studies and quantum chemical calculations, J. Mol. Struct., 2022, 1257, 132535 CrossRef.
- M. J. Anjum, J. Zhao, V. Zahedi Asl, G. Yasin, W. Wang, S. Wei, Z. Zhao and W. Qamar Khan, In situ intercalation of 8-hydroxyquinoline in Mg-Al LDH coating to improve the corrosion resistance of AZ31, Corros. Sci., 2019, 157, 1–10 CrossRef CAS.
- W. Dai, J. Hu, L. Zhou, S. Li, X. Hu and H. Huang, Removal of dibenzothiophene with composite adsorbent MOF-5/Cu(I), Energy Fuels, 2013, 27, 816–821 CrossRef CAS.
- A. Yurduşen and Y. Yürüm, A controlled synthesis strategy to enhance the CO2 adsorption capacity of MIL-88B type MOF crystallites by the crucial role of narrow micropores, Ind. Eng. Chem. Res., 2019, 58, 14058–14072 CrossRef.
- K. Cao, Z. Yu, D. Yin, L. Chen, Y. Jiang and L. Zhu, Fabrication of BTA-MOF-TEOS-GO nanocomposite to endow coating systems with active inhibition and durable anticorrosion performances, Prog. Org. Coat., 2020, 143, 105629 CrossRef CAS.
- Z. Mohammadpour and H. R. Zare, Fabrication of a pH-sensitive epoxy nanocomposite coating based on a Zn-BTC metal–organic framework containing benzotriazole as a smart corrosion inhibitor, Cryst. Growth Des., 2021, 21, 3954–3966 CrossRef CAS.
- H. Li, H. Li, S. Li and X. Cao, Adsorption mechanisms of ibuprofen and naproxen to UiO-66 and UiO-66-NH2: batch experiment and DFT calculation, Chem. Eng. J., 2019, 360, 645–653 CrossRef.
- J. Choi, K. S. Yoo, D. Kim, J. Kim and M. R. Othman, Microporous Mo-UiO-66 metal–organic framework nanoparticles as gas adsorbents, ACS Appl. Nano Mater., 2021, 4(5), 4895–4901 CrossRef CAS.
- H. Tian, W. Li, A. Liu, X. Gao and P. Han, Controlled delivery of multi-substituted triazole by metal–organic framework for efficient inhibition of mild steel corrosion in neutral chloride solution, Corros. Sci., 2018, 131, 1–16 CrossRef CAS.
- D. Cristina, M. Pina, A. Rossi, F. Maria and P. Alessandro, One-pot synthesis of polyaniline/Fe3O4 nanocomposites with magnetic and conductive behaviour. Catalytic effect of Fe3O4 nanoparticle, Synth. Met., 2012, 162(24), 2250–2258 CrossRef.
- D. Cunha, C. Gaudin, I. Colinet, P. Horcajada, G. Maurin and C. Serre, Rationalization of the entrapping of bioactive molecules into a series of functionalized porous zirconium terephthalate MOFs, J. Mater. Chem. B, 2013, 1(8), 1101–1108 RSC.
- R. Farahati, A. Ghaffarinejad, S. M. Mousavi-Khoshdel, J. Rezania, H. Behzadi and A. Shockravi, Synthesis and potential applications of some thiazoles as corrosion inhibitor of copper in 1 M HCl: experimental and theoretical studies, Prog. Org. Coat., 2019, 132, 417–428 CrossRef CAS.
- M. Albini, P. Letardi, L. Mathys, L. Brambilla, J. Schröter, P. Junier and E. Joseph, Comparison of a bio-based corrosion inhibitor versus benzotriazole on corroded copper surfaces, Corros. Sci., 2018, 143, 84–92 CrossRef CAS.
- J. Shen, D. Yang, L. Ma, Z. Gao, A. Yan and Q. Liao, Exploration of neonicotinoids as novel corrosion inhibitors for copper in a NaCl solution: experimental and theoretical studies, Colloids Surf., A, 2022, 636, 128058 CrossRef CAS.
- C. Jing, Z. Wang, Y. Gong, H. Huang, Y. Ma, H. Xie, H. Li, S. Zhang and F. Gao, Photo and thermally stable branched corrosion inhibitors containing two benzotriazole groups for copper in 3.5 wt% sodium chloride solution, Corros. Sci., 2018, 138, 353–371 CrossRef CAS.
- H. Huang and F. Bu, Correlations between the inhibition performances and the inhibitor structures of some azoles on the galvanic corrosion of copper coupled with silver in artificial seawater, Corros. Sci., 2020, 165, 108413 CrossRef CAS.
- M. Finšgar and D. K. Merl, 2-Mercaptobenzoxazole as a copper corrosion inhibitor in chloride solution: electrochemistry, 3D-profilometry, and XPS surface analysis, Corros. Sci., 2014, 80, 82–95 CrossRef.
- M. Finšgar, EQCM and XPS analysis of 1,2,4-triazole and 3-amino-1,2,4-triazole as copper corrosion inhibitors in chloride solution, Corros. Sci., 2013, 77, 350–359 CrossRef.
- F. Dou, J. Han, J. Li, H. Zhang, K. Qiao, J. Kan and J. Chen, Exploration of novel polyaspartic acid derivatives as fluorescent eco-friendly corrosion inhibitors for the carbon steel: electrochemical, surface analysis (SEM/XPS) and theoretical calculation, Colloids Surf., A, 2023, 658, 130606 CrossRef CAS.
- L. Wang, H. Wang, A. Seyeux, S. Zanna, A. Pailleret, S. Nesic and P. Marcus, Adsorption mechanism of quaternary ammonium corrosion inhibitor on carbon steel surface using ToF-SIMS and XPS, Corros. Sci., 2023, 213, 110952 CrossRef CAS.
- S. Wan, X.-Z. Ma, C.-H. Miao, X.-X. Zhang and Z.-H. Dong, Inhibition of 2-phenyl imidazoline on chloride-induced initial atmospheric corrosion of copper by quartz crystal microbalance and electrochemical impedance, Corros. Sci., 2020, 170, 108692 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |