
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOSMI-4-based PROTACs do not influence O-GlcNAc transferase and O-GlcNAcylation levels in cells†
Aleša Bricelj a,
Doroteja Novakab,
Lara Smrdela,
Christian Steinebachc,
Izidor Sosiča,
Marko Anderluh*a and
Martina Gobec*a
a,
Doroteja Novakab,
Lara Smrdela,
Christian Steinebachc,
Izidor Sosiča,
Marko Anderluh*a and
Martina Gobec*a
aFaculty of Pharmacy, University of Ljubljana, Aškerčeva Cesta 7, SI-1000 Ljubljana, Slovenia. E-mail: marko.anderluh@ffa.uni-lj.si; martina.gobec@ffa.uni-lj.si
bDepartment of Nuclear Medicine, University Medical Centre Ljubljana, Zaloška 7, SI-1000 Ljubljana, Slovenia
cPharmaceutical Institute, Department of Pharmaceutical & Medicinal Chemistry, University of Bonn, 53121 Bonn, Germany
First published on 18th July 2025
Abstract
The O-GlcNAc transferase (OGT) inhibitors disclosed thus far are burdened with certain shortcomings. We have incorporated OSMI-4, a nanomolar OGT inhibitor, into PROTACs. The prepared chimeras failed to induce effective OGT degradation or influence O-GlcNAcylation levels in MM.1S cells.
Glycosylation of proteins is the most abundant and diverse post-translational modification (PTM) required for the proper functioning of a myriad of biological cellular processes.1–3 One such process is the covalent binding of O-linked β-N-acetylglucosamine (O-GlcNAc) to serine and threonine residues of cytosolic, nuclear and mitochondrial proteins. O-GlcNAcylation is reversible and regulated by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), the former being responsible for catalysing the transfer of O-GlcNAc to target proteins, while the latter hydrolyses the bond formed.4 Similar to phosphorylation, cycling between attachment and removal of O-GlcNAc off proteins is rapid and dynamic, and the process interplays with many other PTMs that regulate cellular functions.5,6 Since the donor substrate of OGT, UDP–GlcNAc, is synthesized via the hexosamine biosynthesis pathway, O-GlcNAcylation acts as a nutrient sensor and responds to nutrient availability.5,7 Therefore, it is responsible for the proper functioning of nearly all key cellular processes, and perturbations of the OGT–OGA activity balance lead to various pathological conditions, most notably diabetes, cancer, neurodegenerative, and cardiovascular diseases, and possibly steers immune response.5,8–13 Furthermore, research points to OGT playing a role in tumorigenesis, as elevated OGT levels have been found in numerous cancers, such as breast,14,15 prostate,16 pancreatic,17 lung and colorectal cancer.18
In order to better understand the complex role of protein O-GlcNAcylation and to develop therapeutics to modulate its aberrant activity, numerous tools have been developed. In particular, OGT inhibitors are of interest, as reducing OGT activity leads to suppressed cancer proliferation.19 Among the first compounds used was uracil analogue alloxan (Fig. 1A), which exhibited nonspecific OGT inhibition with an unfavourably high IC50 of 100 μM and a short half-life.5,20 Subsequent research focused on the development of UDP and UDP–GlcNAc analogues and led to compound BADGP,20 and prodrugs Ac4-5S-GlcNAc,21 5SGlcNHex,22 and Ac4-ES1,23 which are converted to active drugs after intracellular deacetylation. Although these substrate analogues exhibited satisfactory binding affinities (IC50 in the low micromolar range), they did not allow for structural modifications to improve selectivity.5,24 Due to their mechanism of action, these compounds may also inhibit other GlcNAc transferases that use UDP–GlcNAc as their substrate.24 High-throughput or virtual screening efforts resulted in structurally distinct small-molecule OGT inhibitors such as BZX1 and its analogue BZX2,25,26 natural product L01,27 compound ST045849, OSMI derivatives and quinolone-based compound 6b (Fig. 1B).28–30 Despite the growing number of OGT inhibitors being developed, researchers in this field still face numerous challenges.4,5,19,24,31 Namely, the limited selectivity (BZX1 and BZX2 show off-target and toxic effects,19,26,31 specificity of OSMI-4 in cells is unknown and may cause adverse effects in cholesterol biosythesis31), poor aqueous solubility,24 insufficient cell permeability, and the lack of pharmacokinetic/pharmacodynamic data of the characterized compounds hinder their applicability in research, as does the fact that treatment of the cells with some of these inhibitors leads to an increase in OGT levels due to a compensatory feedback mechanism.5,19,24,29,31
 | ||
| Fig. 1 Some of the frequently used OGT inhibitors. (A) UDP and UDP–GlcNAc analogues: alloxan, BADGP, Ac4-5S-GlcNAc, 5SGlcNHex, Ac4-ES1; (B) other small molecules: BZX1, BZX2, L01, ST045849, OSMI-4 (ester) and compound 6b. | ||
Alternative modalities to classical inhibitors, which enable removal of disease-associated proteins, such as targeted protein degradation (TPD), have shown great promise for the development of chemical probes and therapies.32 Implementing TPD approaches to modulate OGT activity might circumvent the shortcomings of OGT inhibitors. Therefore, we set out to prepare bifunctional molecules with the aim to induce targeted OGT degradation.
We systematically designed three series of proteolysis-targeting chimeras (PROTACs). An established OGT inhibitor, OSMI-4, was selected as the target ligand due to its favourable physicochemical parameters (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D and TPSA), and its structure was modified accordingly to allow facile linker attachment. Our design relied on the fact that carboxylic acid of OSMI-4 was not essential for binding and that its ester derivative was even a slightly more potent OGT inhibitor, leaving the carboxylate as the plausible attachment point for a linker coupled with E3 ligase ligand (Fig. S1†).33 In each series, OSMI-4 analogue 37 (Scheme 1) was linked to a different E3 ligase ligand, i.e., cereblon (CRBN), von Hippel–Lindau (VHL), and inhibitor of apoptosis (IAP) protein (Table S1†).
D and TPSA), and its structure was modified accordingly to allow facile linker attachment. Our design relied on the fact that carboxylic acid of OSMI-4 was not essential for binding and that its ester derivative was even a slightly more potent OGT inhibitor, leaving the carboxylate as the plausible attachment point for a linker coupled with E3 ligase ligand (Fig. S1†).33 In each series, OSMI-4 analogue 37 (Scheme 1) was linked to a different E3 ligase ligand, i.e., cereblon (CRBN), von Hippel–Lindau (VHL), and inhibitor of apoptosis (IAP) protein (Table S1†).
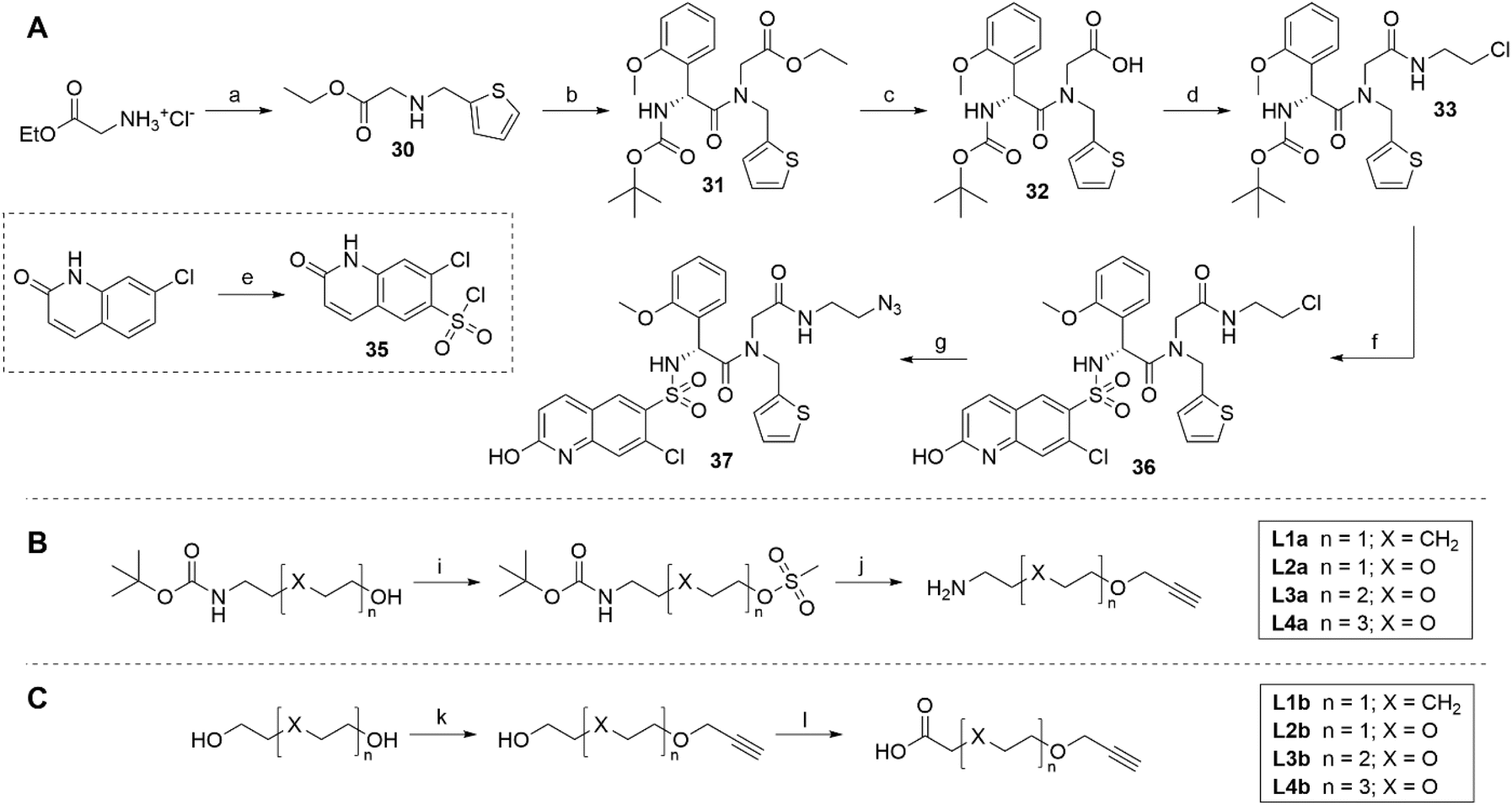 | ||
| Scheme 1 (A) Synthesis of OGT ligand 37. Reagents and conditions: (a) (i) Thiophene-2-carboxaldehyde, Et3N, MeOH, rt, 90 min; (ii) NaBH4, rt, 30 min; (b) (R)-2-((tert-butoxycarbonyl)amino)-2-(2-methoxyphenyl)acetic acid, HATU, DIPEA, DMF, rt, 18 h; (c) 1 M LiOH (aq.), THF, rt, 5 h; (d) 2-chloroethan-1-amine, HATU, DIPEA, DMF, rt, 18 h; (e) HSO3Cl, 160 °C, 4 h; (f) (i) 4 M HCl in dioxane, CH2Cl2, rt, 2 h; (ii) 35, DIPEA, DMF, rt, 18 h; (g) NaN3, DMF, 80 °C, 4 h; (B) synthesis of linkers L1a–L4a. Reagents and conditions: (i) MsCl, Et3N, CH2Cl2, rt, 3 h; (j) (i) propargyl alcohol, NaH, THF, rt, 18 h; (ii) 4 M HCl in dioxane, CH2Cl2, rt, 2 h; (C) synthesis of linkers L1b–L4b. Reagents and conditions: (k) 80% propargyl bromide in toluene, Bu4NHSO4, 50% NaOH (aq.), toluene, 0 °C to rt, 18 h; (l) BAIB, TEMPO, MeCN/H2O, rt, 18 h. | ||
First, we prepared two series of appropriately capped alkyne linkers, which enabled the assembly of final compounds by a straightforward click reaction with the synthesized azide-bearing OSMI-4 derivative 37 (Scheme 1A and S1†). The synthetic route towards OSMI-4 and derivative 37 had to be optimized, since the original procedure by Martin et al.29 led to the formation of a parent diketopiperazine.33 The two linker series were matched in length and composition. The amino group-capped linkers of the L1a–L4a series were used in CRBN-targeting compounds and were prepared from N-Boc-protected amino alcohols, which were first mesylated and then reacted with propargyl alcohol (Scheme 1B). The L1b–L4b linker series was prepared by etherification of diols with propargyl bromide, followed by BAIB/TEMPO-mediated oxidation to carboxylic acids (Scheme 1C).
CRBN-ligand – linker conjugates 38–41 were obtained by subjecting 4-fluorothalidomide (25) to nucleophilic aromatic substitution with linkers L1a–L4a, whereas the intermediates of the VHL- and IAP-series were prepared by coupling carboxylic acid linkers L1b–L4b with either VHL ligand 26 or Boc-protected IAP ligand 28, respectively (Scheme 2). Conjugates 38–49 were ‘clicked’ to OGT ligand 37 through a copper-catalysed azide–alkyne cycloaddition to obtain final PROTACs 1–8. The removal of the Boc-protecting group for the IAP series yielded the desired final compounds 9–12 (Scheme 2). The physicochemical properties of OSMI-4 and final heterobifunctional molecules are collected in the ESI (Table S4†). While the molecular weight of the prepared compounds increased, the lipophilicity of final PROTACs was generally decreased (except compound 1) in comparison to the parent OGT inhibitor (elogD7.4 from 2.3 to 2.6, versus 2.8 for OSMI-4).
 | ||
| Scheme 2 Synthesis of heterobifunctional compounds 1–12. Reagents and conditions: (a) linkers L1a–L4a (Table S2†), DIPEA, DMSO, 90 °C, 18 h; (b) 37, CuSO4, sodium ascorbate, THF, H2O, rt, 18 h; (c) linkers L1b–L4b (Table S3†), HATU, DIPEA, DMF, rt, 18 h; (d) linkers L1b–L4b (Table S3†), HATU, DIPEA, CH2Cl2, rt, 18 h; (e) 4 M HCl in dioxane, CH2Cl2, rt, 2 h. | ||
The library of bifunctional molecules was first assessed for target inhibitory potency and compared with that of OSMI-4, to confirm that the inhibition and therefore binding was not affected by linker attachment. The determined IC50 values are collected in Table 1 and show a comparable inhibitory potency and therefore affinity to the unmodified OGT binder. These values confirm our assumption that the carboxylate of OSMI-4 is a plausible point of attachment for a linker coupled with E3 ligase ligand, as it does not dramatically interfere with inhibition and therefore binding. Inhibition curves are provided in Fig. S2 in ESI.†
We then investigated the effects of chimeras on the protein levels of OGT by western blot analyses in MM.1S cells that express both OGT and E3 ligases of interest. After treatment with the 12 heterobifunctional molecules at 1.0 μM for 16 h, no significant reduction of OGT levels was observed (Fig. 2). We additionally screened the prepared compounds by treating MM.1S cells for 16 h at 0.1, 1.0 and 10 μM, however, no reduction of OGT levels was observed (representative figures and quantification of the western blots is collected in the ESI (Fig. S3–S5†)). IKZF1 levels were reduced in the CRBN series, indirectly proving that compounds do permeate cell membrane and induce degradation of CRBN neosubstrate IKZF1. Similarly, cIAP1 levels were significantly reduced after treatment with PROTACs 9–12, consistent with cIAP1 autoubiquitination triggered by engagement with IAP antagonists. Since cell permeability is confirmed, the lack of OGT depletion may stem from other factors, such as unproductive OGT:PROTAC:E3 ternary complexes formation, ineffective ubiquitination, or insufficient intracellular concentrations of the compounds.
 | ||
| Fig. 2 OGT, cIAP1 and IKZF3 protein levels in MM.1S cells after 16 h treatment with heterobifunctional compounds 1–12 at 1 μM concentration. Tubulin was used as loading control. | ||
To compare the proteome-wide effects of compound 12 versus the OGT inhibitor OSMI-4, we employed a diaPASEF-based mass spectrometry approach.34 MM.1S cells were treated with 1.0 μM of the selected compounds for three hours. The proteomics data confirmed previous literature reports that OGT inhibition by OSMI-4 leads to increased intracellular OGT levels (Fig. 3A).29 Interestingly, treatment with 12 seemingly counteracted this compensatory OGT overexpression (Fig. 3B). This observation points to a rather seldom documented rebound expression of proteins of interest, which can apparently hamper the effectiveness of PROTACs or other proximity-induced protein degraders, and we advise the scientific community to carefully assess this phenomenon when evaluating potential protein degraders.
 | ||
| Fig. 3 diaPASEF quantitative proteomics for (A) OSMI-4 and (B) PROTAC 12. MM.1S cells were treated with compound at 1.0 μM for 3 h. Bioconductor's limma package was used to perform statistical analysis of degrader treatment compared to DMSO vehicle treatment. The identified proteins were plotted as log2 fold change (PROTAC/DMSO) versus −log10 of p-value. Proteins with −log10 (p-value) > 3 (p-value < 0.001) and log2 fold change > 0.6 or < −0.6 (translating to 1.5-fold up- or downregulation) were considered to have significantly changed in abundance. Data are mean of biological duplicates. | ||
As observed in the proteomic analysis, additional western blot experiments confirmed that inhibition of OGT by OSMI-4 leads to its upregulation after 16 hours (Fig. 4A). Surprisingly, in a concentration-dependent experiment no effect of compound 12 on OGT expression was detected at any concentration tested (0.1 μM, 1 μM, or 10 μM). This finding also minimizes the possibility that a hook effect is preventing target degradation, which is occasionally observed with PROTACs. As observed previously, the efficiency of protein degradation by PROTACs can be influenced by the cellular microenvironment. Inflammatory conditions, in particular, can alter the proteome and impact degradation outcomes.35 To simulate an inflammatory environment, MM.1S cells were treated with TNFα (30 ng mL−1) in the presence or absence of compound 12. However, even under these conditions no improvements in OGT degradation were observed.
 | ||
| Fig. 4 (A) OGT protein levels under different conditions. MM.1S cells were subjected to treatment with either 10 μM OSMI-4 or PROTAC 12 at 0.1 μM, 1.0 μM, or 10 μM concentration for 16 h. If inflammatory microenvironment was mimicked, cells were first pre-treated for 1 h with 1.0 μM 12, and then TNFα (30 ng mL−1) was added for the subsequent 16 h. The number above OGT bands present the average protein level from two independent biological repeats. Tubulin was used as loading control. (B) Proliferation rate of MM.1S cells treated for 96 h with either 1.0 μM OSMI-4 or μM PROTAC 12, as determined by CFSE staining. Data represent the mean of three independent biological replicates. | ||
Given that OGT serves as a metabolic switch, we hypothesized that its modulation might manifest over a longer period of time. Namely, even though we have observed no significant difference in the OGT levels after 16 hours of incubation with 12, we hypothesised that 12 counteracted the compensatory OGT overexpression after 3 hours. The combination of these experiments indicates that OGT overexpression and equalization is not immediate. We questioned whether the potential effect of 12 on OGT degradation in the short post-treatment period (before 3 hours) was sufficient to trigger an effect on cell proliferation as a phenotypic response to lowered OGT levels. Therefore, we further investigated the potential effect of compound 12 on cell proliferation after 96 hours, using OSMI-4 for comparison. None of the compounds altered cell proliferation compared to control demonstrating that short-term inhibition or depletion of OGT has no long-term effects on cell proliferation (see Fig. 4B).
The lack of effects on cellular proliferation prompted us to reconsider our initial hypothesis, which proposed that compound 12 could counteract the compensatory overexpression of OGT. Alternatively, we explored the possibility that our compound might not interact with OGT in cells at all. Given that engagement of OSMI-4 leads to a significant reduction in O-GlcNAc levels, we performed additional western blot analyses on cells treated with OSMI-4 (at 1, 10, and 20 μM) and PROTAC 12 (at 0.1, 1, and 10 μM) for 16 h, assessing their impact on O-GlcNAcylation. The levels of O-GlcNAc were observed using pan-O-GlcNAc antibodies RL2 (Fig. 5A) and MultiMab (Fig. 5B). Treatment with OSMI-4 led to a significant inhibition of O-GlcNAcylation, as confirmed by detection with both antibodies. In contrast, this effect was largely absent after treatment with PROTAC 12, where O-GlcNAc levels remained similar to those in the untreated control even though 12 was shown to inhibit OGT and to penetrate cells. Furthermore, we observed a significant increase in OGT levels upon OSMI-4 binding, a response not seen with compound 12. These results suggest that the interaction of PROTAC 12 with OGT in cells is insufficient to elicit a response comparable to that of OGT inhibition by OSMI-4.
 | ||
| Fig. 5 Representative western blots depicting OGlcNAc ((A) RL2 and (B) MultiMab) and OGT protein levels in MM.1S cells after 16 h treatment with IAP-series PROTAC 12 at 0.1, 1, and 10 μM concentration and OSMI-4 at 1, 10, and 20 μM. β-actin was used as loading control. Data represents an average of two independent experiments. To ensure the equal loading of proteins, the membranes were stripped (100 mM 2-mercaptoethanol, 2% SDS, and 62.5 mM Tris/HCl, pH = 6.8) for 45 min at 50 °C and then washed (3 × 20 min) in 1× TTBS. Nonspecific binding sites were then blocked for 1 h at room temperature in 5% BSA in TTBS (Tris-buffered saline, 0.1% Tween) and the membranes were re-probed with appropriate antibodies under the same conditions as those described above. The stripping was performed on membranes depicted in (A) and (B), where proteins were first detected using O-GlcNAc MultiMab® Rabbit mAb mix antibody and anti-O-linked N-acetylglucosamine antibody [RL2]. The stripped membranes were then re-probed with antibodies for OGT and β-actin. | ||
In summary, we have synthesized a series of heterobifunctional compounds consisting of OSMI-4 as the OGT ligand coupled to a series of linkers and CRBN, VHL, and IAP E3 ligase ligands. Our attempts at constructing OGT degraders did not result in compounds able to potently lower cell OGT levels, despite of their proven cellular permeability assessed by the induced degradation of IKZF1 or cIAP1. Further investigation indicated that compound 12 might mitigate the compensatory overexpression of OGT observed after OSMI-4 treatment, as demonstrated in the diaPASEF quantitative proteomics experiment. However, the lack of response on MM.1S proliferation rate drove us to explore O-GlcNAcylation levels. The absence of significant downregulation of O-GlcNAc led us to conclude that the engagement of OGT by PROTAC 12 is insufficient to induce cellular effects. Although there was indirect evidence supporting cell permeability, the intracellular concentration of the prepared chimeric molecules may be too low to facilitate the formation of productive ternary complexes and/or inhibit OGT. We hypothesize that binary complexes between the E3 ligase and PROTAC may form preferentially, thereby hindering the engagement of OGT and preventing the cellular effects typically seen with OSMI-4.
Abbreviations
| BAIB | (Diacetoxyiodo)benzene |
| cIAP | Cellular IAP |
| CRBN | Cereblon |
| DIPEA | N,N-Diisopropylethylamine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| IAP | Inhibitor of apoptosis |
| IKZF1 | Zinc finger protein Ikaros |
| OGA | O-GlcNAcase |
| O-GlcNAc | O-Linked β-N-acetylglucosamine |
| OGT | O-GlcNAc transferase |
| PROTAC | Proteolysis targeting chimera |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| THF | Tetrahydrofuran |
| TNFα | Tumor necrosis factor alpha |
| TPD | Targeted protein degradation |
| TPSA | Topological polar surface area |
| UDP | Uridine diphosphate |
| VHL | von Hippel–Lindau |
Data availability
The data supporting this article (ESI figures, schemes and tables, overview on physicochemical properties of compounds, synthetic schemes and procedures; and 1H NMR, 13C NMR, and MS data) have been included as part of the ESI.†Author contributions
IS, MA, and MG conceptualized the project. AB, DN, LS, CS and MG curated the data. AB, LS, and DN performed the investigation. AB, DN, IS and MG performed the formal analysis. IS and MA acquired the funding. Methodology was planned out by IS, CS, and MG. The project was supervised by IS, MA, and MG. AB prepared the original draft; the manuscript was reviewed and edited with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Maja Frelih for the HRMS measurements. We also thank Eric Fischer and the Fischer Lab Degradation Proteomics Initiative for the collection of the global proteomics data supported by NIH CA214608 and CA218278. C. S. thanks Prof. M. Gütschow for support. This work received financial support from the European Union's Horizon 2020 program under the Marie Skłodowska-Curie grant agreement no. 765581 (project PhD4GlycoDrug; https://www.phd4glycodrug.eu) and the Slovenian Research Agency (grant P1-0208) is gratefully acknowledged. COST actions CA18103 (Innogly) and CA18132 (GLYCONanoPROBES) are also gratefully acknowledged.Notes and references
- K. T. Schjoldager, Y. Narimatsu, H. J. Joshi and H. Clausen, Global View of Human Protein Glycosylation Pathways and Functions, Nat. Rev. Mol. Cell Biol., 2020, 21(12), 729–749 CrossRef CAS PubMed.
- G. W. Hart, Nutrient Regulation of Signaling and Transcription, J. Biol. Chem., 2019, 294(7), 2211–2231 CrossRef CAS PubMed.
- K. W. Moremen, M. Tiemeyer and A. V. Nairn, Vertebrate Protein Glycosylation: Diversity, Synthesis and Function, Nat. Rev. Mol. Cell Biol., 2012, 13(7), 448–462 CrossRef CAS PubMed.
- S. S. Cheng, A. C. Mody and C. M. Woo, Opportunities for Therapeutic Modulation of O-GlcNAc, Chem. Rev., 2024, 124(22), 12918–13019 CrossRef CAS PubMed.
- J. Ma, C. Wu and G. W. Hart, Analytical and Biochemical Perspectives of Protein O-GlcNAcylation, Chem. Rev., 2021, 121(3), 1513–1581 CrossRef CAS PubMed.
- J. Shi, T. Tomašič, S. Sharif, A. J. Brouwer, M. Anderluh, R. Ruijtenbeek and R. J. Pieters, Peptide Microarray Analysis of the Cross-Talk between O-GlcNAcylation and Tyrosine Phosphorylation, FEBS Lett., 2017, 591(13), 1872–1883 CrossRef CAS PubMed.
- G. W. Hart, C. Slawson, G. Ramirez-Correa and O. Lagerlof, Cross Talk Between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease, Annu. Rev. Biochem., 2011, 80(1), 825–858 CrossRef CAS PubMed.
- J. R. Erickson, L. Pereira, L. Wang, G. Han, A. Ferguson, K. Dao, R. J. Copeland, F. Despa, G. W. Hart, C. M. Ripplinger and D. M. Bers, Diabetic Hyperglycaemia Activates CaMKII and Arrhythmias by O-Linked Glycosylation, Nature, 2013, 502(7471), 372–376 CrossRef CAS PubMed.
- F. Liu, K. Iqbal, I. Grundke-Iqbal, G. W. Hart and C.-X. Gong, O-GlcNAcylation Regulates Phosphorylation of Tau: A Mechanism Involved in Alzheimer's Disease, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(29), 10804–10809 CrossRef CAS PubMed.
- S. Yeluri, B. Madhok, K. R. Prasad, P. Quirke and D. G. Jayne, Cancer's Craving for Sugar: An Opportunity for Clinical Exploitation, J. Cancer Res. Clin. Oncol., 2009, 135(7), 867–877 CrossRef CAS PubMed.
- B. Laczy, B. G. Hill, K. Wang, A. J. Paterson, C. R. White, D. Xing, Y.-F. Chen, V. Darley-Usmar, S. Oparil and J. C. Chatham, Protein O-GlcNAcylation: A New Signaling Paradigm for the Cardiovascular System, Am. J. Physiol.: Heart Circ. Physiol., 2009, 296(1), H13–H28 CrossRef CAS PubMed.
- M. Weiss, M. Anderluh and M. Gobec, Inhibition of O-GlcNAc Transferase Alters the Differentiation and Maturation Process of Human Monocyte Derived Dendritic Cells, Cells, 2021, 10(12), 3312 CrossRef CAS PubMed.
- A. Saha and A. Fernández-Tejada, Chemical Biology Tools to Interrogate the Roles of O-GlcNAc in Immunity, Front. Immunol., 2023, 13, 1089824 CrossRef PubMed.
- S. A. Caldwell, S. R. Jackson, K. S. Shahriari, T. P. Lynch, G. Sethi, S. Walker, K. Vosseller and M. J. Reginato, Nutrient Sensor O-GlcNAc Transferase Regulates Breast Cancer Tumorigenesis through Targeting of the Oncogenic Transcription Factor FoxM1, Oncogene, 2010, 29(19), 2831–2842 CrossRef CAS PubMed.
- A. Krześlak, E. Forma, M. Bernaciak, H. Romanowicz and M. Bryś, Gene Expression of O-GlcNAc Cycling Enzymes in Human Breast Cancers, Clin. Exp. Med., 2012, 12(1), 61–65 CrossRef PubMed.
- T. P. Lynch, C. M. Ferrer, S. R. Jackson, K. S. Shahriari, K. Vosseller and M. J. Reginato, Critical Role of O-Linked β-N-Acetylglucosamine Transferase in Prostate Cancer Invasion, Angiogenesis, and Metastasis, J. Biol. Chem., 2012, 287(14), 11070–11081 CrossRef CAS PubMed.
- Z. Ma, D. J. Vocadlo and K. Vosseller, Hyper-O-GlcNAcylation is Anti-Apoptotic and Maintains Constitutive NF-KB Activity in Pancreatic Cancer Cells, J. Biol. Chem., 2013, 288(21), 15121–15130 CrossRef CAS PubMed.
- W. Mi, Y. Gu, C. Han, H. Liu, Q. Fan, X. Zhang, Q. Cong and W. Yu, O-GlcNAcylation is a Novel Regulator of Lung and Colon Cancer Malignancy, Biochim. Biophys. Acta, Mol. Basis Dis., 2011, 1812(4), 514–519 CrossRef CAS PubMed.
- H. M. Itkonen, M. Loda and I. G. Mills, O-GlcNAc Transferase – An Auxiliary Factor or a Full-Blown Oncogene?, Mol. Cancer Res., 2021, 19(4), 555–564 CrossRef CAS PubMed.
- E.-S. Kang, D. Han, J. Park, T. K. Kwak, M.-A. Oh, S.-A. Lee, S. Choi, Z. Y. Park, Y. Kim and J. W. Lee, O-GlcNAc Modulation at Akt1 Ser473 Correlates with Apoptosis of Murine Pancreatic β Cells, Exp. Cell Res., 2008, 314(11–12), 2238–2248 CrossRef CAS PubMed.
- H. C. Dorfmueller, V. S. Borodkin, D. E. Blair, S. Pathak, I. Navratilova and D. M. F. van Aalten, Substrate and Product Analogues as Human O-GlcNAc Transferase Inhibitors, Amino Acids, 2011, 40(3), 781–792 CrossRef CAS PubMed.
- T.-W. Liu, W. F. Zandberg, T. M. Gloster, L. Deng, K. D. Murray, X. Shan and D. J. Vocadlo, Metabolic Inhibitors of O-GlcNAc Transferase That Act In Vivo Implicate Decreased O-GlcNAc Levels in Leptin-Mediated Nutrient Sensing, Angew. Chem., Int. Ed., 2018, 57(26), 7644–7648 CrossRef CAS PubMed.
- M. Worth, C.-W. Hu, H. Li, D. Fan, A. Estevez, D. Zhu, A. Wang and J. Jiang, Targeted Covalent Inhibition of O-GlcNAc Transferase in Cells, Chem. Commun., 2019, 55(88), 13291 RSC.
- M. G. Alteen, H. Y. Tan and D. J. Vocadlo, Monitoring and Modulating O-GlcNAcylation: Assays and Inhibitors of O-GlcNAc Processing Enzymes, Curr. Opin. Struct. Biol., 2021, 68, 157–165 CrossRef CAS PubMed.
- B. J. Gross, B. C. Kraybill and S. Walker, Discovery of O- GlcNAc Transferase Inhibitors, J. Am. Chem. Soc., 2005, 127(42), 14588–14589 CrossRef CAS PubMed.
- J. Jiang, M. B. Lazarus, L. Pasquina, P. Sliz and S. Walker, A Neutral Diphosphate Mimic Crosslinks the Active Site of Human O-GlcNAc Transferase, Nat. Chem. Biol., 2012, 8(1), 72–77 CrossRef CAS PubMed.
- Y. Liu, Y. Ren, Y. Cao, H. Huang, Q. Wu, W. Li, S. Wu and J. Zhang, Discovery of a Low Toxicity O-GlcNAc Transferase (OGT) Inhibitor by Structure-Based Virtual Screening of Natural Products, Sci. Rep., 2017, 7(1), 12334 CrossRef PubMed.
- R. F. Ortiz-Meoz, J. Jiang, M. B. Lazarus, M. Orman, J. Janetzko, C. Fan, D. Y. Duveau, Z.-W. Tan, C. J. Thomas and S. Walker, A Small Molecule That Inhibits OGT Activity in Cells, ACS Chem. Biol., 2015, 10(6), 1392–1397 CrossRef CAS PubMed.
- S. E. S. Martin, Z.-W. Tan, H. M. Itkonen, D. Y. Duveau, J. A. Paulo, J. Janetzko, P. L. Boutz, L. Törk, F. A. Moss, C. J. Thomas, S. P. Gygi, M. B. Lazarus and S. Walker, Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors, J. Am. Chem. Soc., 2018, 140(42), 13542–13545 CrossRef CAS PubMed.
- M. Weiss, E. M. Loi, M. Sterle, C. Balsollier, T. Tomašič, R. J. Pieters, M. Gobec and M. Anderluh, New Quinolinone O-GlcNAc Transferase Inhibitors Based on Fragment Growth, Front. Chem., 2021, 9, 666122 CrossRef CAS PubMed.
- N. Zhang, H. Jiang, K. Zhang, J. Zhu, Z. Wang, Y. Long, Y. He, F. Feng, W. Liu, F. Ye and W. Qu, OGT as Potential Novel Target: Structure, Function and Inhibitors, Chem.-Biol. Interact., 2022, 357, 109886 CrossRef CAS PubMed.
- L. Zhao, J. Zhao, K. Zhong, A. Tong and D. Jia, Targeted Protein Degradation: Mechanisms, Strategies and Application, Signal Transduction Targeted Ther., 2022, 7(1), 113 CrossRef CAS PubMed.
- E. M. Loi, M. Weiss, S. Pajk, M. Gobec, T. Tomašič, R. J. Pieters and M. Anderluh, Intracellular Hydrolysis of Small-Molecule O-Linked N-Acetylglucosamine Transferase Inhibitors Differs among Cells and Is Not Required for Its Inhibition, Molecules, 2020, 25(15), 3381 CrossRef CAS PubMed.
- F. Meier, A.-D. Brunner, M. Frank, A. Ha, I. Bludau, E. Voytik, S. Kaspar-Schoenefeld, M. Lubeck, O. Raether, N. Bache, R. Aebersold, B. C. Collins, H. L. Röst and M. Mann, DiaPASEF: Parallel Accumulation–Serial Fragmentation Combined with Data-Independent Acquisition, Nat. Methods, 2020, 17(12), 1229–1236 CrossRef CAS PubMed.
- J. Nunes, G. A. McGonagle, J. Eden, G. Kiritharan, M. Touzet, X. Lewell, J. Emery, H. Eidam, J. D. Harling and N. A. Anderson, Targeting IRAK4 for Degradation with PROTACs, ACS Med. Chem. Lett., 2019, 10(7), 1081–1085 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02801a |
| This journal is © The Royal Society of Chemistry 2025 |