
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnodic oxidation of paraquat herbicide on BDD electrode: comparative evaluation of variable effects and degradation mechanisms
Nejmeddine Rabaaoui a,
Naoufel Ben Hamadib,
Mourad Cherifcd,
Ahlem Guesmib,
Wesam Abd El-Fattahb and
Houcine Naïli*a
a,
Naoufel Ben Hamadib,
Mourad Cherifcd,
Ahlem Guesmib,
Wesam Abd El-Fattahb and
Houcine Naïli*a
aLaboratory Physical-Chemistry of the Solid State, Chemistry Department, Faculty of Sciences of Sfax, University of Sfax, BP 1171, 3000 Sfax, Tunisia. E-mail: houcine_naili@yahoo.com; houcine.naili@fss.rnu.tn
bChemistry Department, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), P.O. Box 5701, Riyadh 11432, Saudi Arabia
cLaboratoire de Physico-Chimie des Matériaux, IPEST, BP51, 2070 La MARSA, Tunisia
dIPEIEM, Université de Tunis-El Manar, BP244, 2096, El Manar II, Tunisia
First published on 19th June 2025
Abstract
This work investigates the electrochemical degradation of paraquat (30 mg L−1) in aqueous medium using a boron-doped diamond (BDD) anode, with a graphite cathode. The influence of operational variables including the effect of the anode material, current density, type of supporting electrolyte, and initial pH was systematically examined. BDD electrodes exhibited the most efficient performance, achieving COD and TOC removal rates of 99% and 98.6%, respectively, under optimal conditions (15 mA cm−2, pH = 3, 50 mmol per L Na2SO4). Degradation followed pseudo-first-order kinetics (k = 3.14 × 10−2 s−1). While faradaic efficiency peaked at 70.14%, energy demand increased to 66 kWh m−3 over time. Analysis of reaction intermediates revealed the formation of aromatic and carboxylic acid by-products, which were tracked to elucidate a complete mineralization pathway. These findings demonstrate the potential of BDD driven electrochemical oxidation as a promising and sustainable technique for the treatment of persistent organic contaminants in water systems.
Introduction
Paraquat (Table 1) is a highly effective, non-selective contact herbicide widely used for controlling annual and perennial weeds in crops such as cotton, rice, and soybeans.1 Despite its agronomic advantages, paraquat is considered one of the most toxic agrochemicals, posing severe threats to human health and the environment.2,3 Its persistence in soil and high aqueous solubility (625 g L−1 at 25 °C) increase the risk of contamination in surface and groundwater sources, often exceeding regulatory safety thresholds such as the EU limit of 0.1 μg L−1 in drinking water.4 Due to its environmental persistence and acute toxicity, paraquat has been banned or restricted in several countries, including the European Union, Canada and China.5–7 However, it remains in use globally under various formulations.| Name | IUPAC name | Chemical formula | Structure |
|---|---|---|---|
| Paraquat | 1,1′-Dimethyl-4,4′-bipyridinium dichloride | C12H14N2Cl2 |  |
Once released into aquatic systems, paraquat can disrupt photosynthesis, reduce dissolved oxygen, and accumulate in sediments, creating long-term ecological risks.8 Its degradation is particularly challenging due to its quaternary nitrogen structure and stable aromatic rings.9
Conventional water treatment methods such as biological processes, coagulation, filtration, and adsorption have shown limited success in fully degrading such recalcitrant compounds.10–12 As a result, attention has turned to advanced oxidation processes, which are capable of mineralizing persistent organic pollutants into harmless end-products. Among AOPs, electrochemical advanced oxidation processes, and in particular anodic oxidation, have emerged as highly effective due to their operational simplicity, environmental compatibility, and ability to generate highly reactive hydroxyl radicals (˙OH) in situ.13–15
The efficiency of anodic oxidation is strongly influenced by the nature of the anode material. Boron-doped diamond (BDD) electrodes have gained prominence for their wide electrochemical potential window, high oxygen overpotential, and exceptional chemical inertness.16 These properties facilitate the generation of free ˙OH radicals with minimal side reactions, allowing for non-selective oxidation of a broad range of organic contaminants, including pharmaceuticals, dyes, and pesticides.17–24 In addition, recent advancements in nanostructured electrode design and electrochemical sensing technologies have demonstrated the significance of surface modification in enhancing electrochemical reactivity, particularly in biosensor and lab-on-a-chip systems.25–28 Although these approaches target analytical applications, the underlying principles of interface optimization and electrode surface engineering align with the electrochemical concepts employed in this work.
Although several studies have explored paraquat degradation using various AOPs including photo-Fenton, photocatalysis, and electrochemical methods most have focused primarily on partial decolorization or removal of parent molecules, without fully addressing mineralization efficiency, intermediate evolution, or energy metrics.13,14 In this regard, the present study provides a distinctive contribution by integrating multiple dimensions: (1) a comparative assessment of three electrode materials (BDD, PbO2, and Pt), (2) detailed identification of both aromatic and carboxylic acid intermediates, (3) monitoring of inorganic nitrogen by-products (NH4+ and NO3−), and (4) analysis of faradaic efficiency and specific energy consumption. This unified approach offers new insights into the sustainable design of electrochemical systems for the advanced treatment of persistent herbicides.
In this context, the present study aims to investigate the electrochemical degradation of paraquat using BDD, PbO2, and Pt anodes under various operating conditions. The influence of current density, initial pH, and supporting electrolyte type on COD and TOC removal is examined. Furthermore, the study evaluates kinetic behavior, faradaic efficiency, energy consumption, and the evolution of aromatic and carboxylic intermediates to propose a detailed degradation mechanism. By integrating performance, mechanism, and sustainability, this work contributes to the development of advanced electrochemical strategies for the treatment of toxic herbicides in wastewater.
Experimental section
Electrodes preparation
PbO2 was deposited galvanostatically on the pretreated lead substrate by electrochemical anodization of lead in oxalic acid solution (100 g L−1) at 25 °C. This acid solution was electrolyzed galvanostatically for 30 min at ambient temperature using an anodic current density of 100 mA cm−2. The cathode was stainless steel (austenitic type), the two electrodes were concentric with the lead electrode as axial. This arrangement gave the formation of a regular and uniform deposit.24BDD films were provided by CSEM and synthesized on a conductive p-Si substrate (1 mm, Siltronix) via a hot filament, chemical vapor deposition technique (HF-CVD). The temperature of the filament was from 2440 to 2560 °C and that of the substrate was monitored at 830 °C. The reactive gas used was 1% methane in hydrogen containing 1–3 ppm of trimethyl boron. The gas mixture was supplied to the reaction chamber at a flow rate of 5 mL min−1 to give a growth rate of 0.24 μm h−1 for the diamond layer. This procedure gave a columnar, randomly textured, polycrystalline diamond film, with a thickness of about 1 μm and a resistivity of 15 mΩ cm (±30%) onto the conductive p-Si substrate.29
Chemicals and electrolytes
Sodium sulfate (Na2SO4, ACS reagent, ≥99.0%), Orange G (99.8%), sodium chloride (NaCl, ≥99.5%), sodium hydroxide (NaOH, ≥98.0%) and methanol (CH3OH, ≥99.9%) were supplied by Sigma-Aldrich and were used as received without purification. Deionized water from a Millipore Milli-Q system (resistivity >18 MΩ cm) was used for the preparation of aqueous solutions.Monopyridone, dipyridone, 4-carboxy-1-methylpyridinium, 4-picolinic acid, 4,4′-bipyridyl, oxalic acid, malic acid, succinic acid, formic acid, citric acid, acrylic acid, glyoxylic acid and glycolic acid (as standards) were of analytical grade, and they were purchased from Sigma-Aldrich. These products were used to identify the by-products of the degradation of paraquat.
Electrolysis of paraquat herbicide solutions
Galvanostatic electrolysis experiments (Fig. 1) were conducted on paraquat herbicide solutions at a concentration of 30 mg L−1 in a single-compartment cylindrical cell, thermostated at 20 °C. | ||
| Fig. 1 The galvanostatic electrochemical setup used for paraquat degradation. | ||
The trials employed boron-doped diamond, platinum, and lead oxide anodes, each with dimensions of 2.5 cm × 4 cm, across current densities ranging from 0 to 100 mA cm−2. For clarity, the effective geometric surface area of each electrode was 10 cm2, calculated from its dimensions. This value was used consistently for current density calculations and maintained throughout all experiments to ensure valid comparisons and reproducibility. Specifically, the cathode used was a cylindrical graphite rod with a diameter of 0.8 cm and an immersed length of 4.0 cm, yielding a surface area approximately equal to that of the anode. This configuration ensured uniform current density across both electrodes during galvanostatic electrolysis. Electrolysis was performed in the presence of various electrolytes, each at a concentration of 50 mmol L−1, including NaCl, NaNO3 and Na2SO4, with pH values ranging from 3.0 to 11.0. The cathode used in all experiments was a 10 cm2 graphite bar, with an interelectrode gap of approximately 2 cm. The current and potential measurements were carried out using digital multimeter.
Analytical determinations
Solutions were prepared using distilled water. The temperature was maintained using a thermostatically controlled water bath. The current density was obtained using a Bench Power Supply GPC-3030D potentiostat–galvanostat. Chemical oxygen demand (COD) data were obtained with a Beckman UV/vis DU 800 spectrophotometer. The total organic carbon content (TOC) was obtained with a Shimadzu VCSH TOC analyzer. For this, samples were withdrawn and filtred through millipore syringe filter of 0.45 μm and aliquots of 50 μL were injected into the analyzer.The concentration of paraquat and reaction intermediates were determined by a Spectra SYSTEM HPLC-Thermo Fisher Scientific equipped with a diode array detector (DAD) detector. A ZORBAX Eclipse XDB-C18 reverse phase column (150 mm, 4.6 mm, 5 μm) was used for the quantification of paraquat and aromatic intermediates. HPLC measurements were conducted by injecting aliquots of 20 μL into the chromatograph. A mixture of water and methanol (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 v/v) was used as mobile phase at 0.8 mL min−1 as throughput in isocratic mode. For kinetic studies, the detection was performed at 230 nm. Quantitative analysis was conducted using external calibration curves established for paraquat and each intermediate, based on their peak areas at this wavelength. Calibration standards were injected under identical chromatographic conditions to ensure consistency and accuracy in quantification.
:30 v/v) was used as mobile phase at 0.8 mL min−1 as throughput in isocratic mode. For kinetic studies, the detection was performed at 230 nm. Quantitative analysis was conducted using external calibration curves established for paraquat and each intermediate, based on their peak areas at this wavelength. Calibration standards were injected under identical chromatographic conditions to ensure consistency and accuracy in quantification.
Results
Effect of experimental parameters on paraquat mineralization
 | ||
Fig. 2 Variation of chemical oxygen demand during anodic oxidation of 250 mL of 30 mg L−1 of paraquat in 50 mmol per L Na2SO4 at pH = 3.0 ( ) BDD, ( ) BDD, ( ) PbO2 and ( ) PbO2 and ( ) Pt anodes and a graphite cathode operating at 15 mA cm−2 and at 20 °C. ) Pt anodes and a graphite cathode operating at 15 mA cm−2 and at 20 °C. | ||
After 300 minutes of electrolysis, the BDD electrode achieved an outstanding COD removal efficiency of 99%, markedly outperforming PbO2 (88.5%) and Pt (79.5%). This superior electrochemical behavior of BDD is attributed to its capability to generate highly reactive hydroxyl radicals in a physisorbed form on its surface. This interpretation is supported by the observed high TOC and COD removal efficiencies, consistent pseudo-first-order degradation kinetics, and faradaic efficiency exceeding 80% under optimal conditions. These findings are in line with previous reports describing physisorbed ˙OH as the main oxidative species generated on BDD surfaces.30 Thereby maximizing their availability for non-selective oxidation reactions. While direct detection of ˙OH was not performed in this study, its generation under comparable BDD conditions is well documented in the literature. The observed rapid mineralization, high COD/TOC removal, and faradaic efficiency support the dominant role of physisorbed ˙OH in the oxidation mechanism. In contrast, PbO2 electrodes predominantly facilitate pollutant degradation via chemisorbed hydroxyl radicals, which inherently limits their oxidative potential.31 The Pt anode exhibited the lowest performance, due to its strong affinity for ˙OH radicals, which leads to surface passivation and reduced radical availability for organic oxidation. Beyond degradation efficiency, the structural durability of the anodes was evaluated through the measurement of average corrosion rate (CR), calculated via eqn (1):
| CR = Δm/At | (1) |
| Electrode | Average corrosion rate (g cm−2 h) |
|---|---|
| BDD | Negligible (<10−5) |
| PbO2 | 0.0335 |
While the Pt electrode maintained structural stability, its inferior catalytic activity limits its efficiency in persistent pollutant mineralization. Although long-term durability tests (e.g., ICP-OES analysis or weight loss measurement) were not conducted in this study, no visible degradation or mass loss of the Pt electrode was observed during the electrolysis experiments. Moreover, the current densities (10–20 mA cm−2) and nearly neutral pH values used in our conditions fall within the safe operational window reported in the literature for Pt electrodes. These observations support the short-term stability assumption made in our comparative analysis. Overall, the findings underscore the dual advantage of BDD electrodes in terms of both oxidative power and operational stability, making them highly suitable for advanced oxidation processes targeting refractory organic contaminants. Conversely, the moderate performance and durability of PbO2 and Pt electrodes suggest the need for further optimization such as surface modification, doping strategies, or integration into hybrid electrochemical systems to enhance their practical applicability.33
| j = I/A | (2) |
In BDD mediated anodic oxidation, the mineralization of organic pollutants such as paraquat is primarily driven by the electrochemical production of hydroxyl radicals via water discharge at the anode surface. This key reaction is represented by the following eqn (3):
| BDD + H2O → BDD(˙OH) + H+ + e− | (3) |
The generated ˙OH radicals are weakly adsorbed on the BDD surface, which enhances their reactivity and enables non-selective oxidation of organic compounds. The influence of current density on the anodic oxidation of paraquat using a BDD anode is depicted in Fig. 3.
 | ||
Fig. 3 Influence of applied current density on the evolution of DCO during anodic oxidation of 250 mL of 30 mg L−1 of paraquat in 50 mmol per L Na2SO4 at pH = 3.0 and at 20 °C on BDD anode, ( ) 10 mA cm−2, ( ) 10 mA cm−2, ( ) 15 mA cm−2 and ( ) 15 mA cm−2 and ( ) 20 mA cm−2. The inset shows the removal %. ) 20 mA cm−2. The inset shows the removal %. | ||
The results demonstrate that increasing the applied current density significantly improves degradation performance. At 20 mA cm−2, nearly complete COD removal was achieved within 300 minutes, clearly outperforming the rates observed at 15 and 10 mA cm−2. This enhancement is mainly attributed to the increased generation rate of ˙OH radicals, which intensifies the oxidative degradation of the herbicide.
However, excessive current densities may also promote undesired side reactions, particularly the oxygen evolution reaction, which competes with ˙OH generation and reduces the current efficiency. This can result in a diminished fraction of charge being effectively utilized for pollutant oxidation and may negatively impact the overall process selectivity. As shown in Fig. 3, the percentage of COD removal increases substantially with current density, confirming that higher current densities accelerate the degradation process and enable faster pollutant removal. Nevertheless, such operational advantages must be weighed against the associated energy demand. Although higher current densities achieve rapid degradation, they are typically linked to increased specific energy consumption, which can undermine the economic sustainability of the treatment process. From a practical standpoint, 15 mA cm−2 appears to represent the optimal operating condition, offering a favorable compromise between high degradation efficiency and moderate energy input. This balance is particularly crucial in large-scale applications, where energy costs often constitute a major portion of the total operational expenditure.24
 | ||
Fig. 4 Variation of chemical oxygen demand during anodic oxidation of 250 mL of 30 mg L−1 of paraquat in 50 mmol per L Na2SO4 on BDD anode at 15 mA cm−2 and at 20 °C ( ) pH = 3, ( ) pH = 3, ( ) pH = 7 and ( ) pH = 7 and ( ) pH = 11. ) pH = 11. | ||
This behaviour can be understood by considering the generation and stability of hydroxyl radicals, the state of the herbicide, and surface phenomena on the BDD electrode. In acidic conditions, although the hydroxide ion (OH−) concentration is low, BDD anodes facilitate the direct anodic oxidation of water molecules (H2O) and hydronium ions (H3O+), resulting in the formation of highly reactive physisorbed ˙OH radicals. The high oxygen evolution overpotential characteristic of BDD electrodes suppresses parasitic side reactions, thereby enhancing the degradation of organic pollutants under acidic conditions.35 Additionally, paraquat remains predominantly uncharged or weakly cationic at pH = 3, which enhances its interaction with the BDD surface, promoting direct or mediated electron transfer. These combined effects result in rapid and efficient mineralization of herbicide.
At neutral pH, the removal efficiency decreases, which may be attributed to a reduction in both radical generation and adsorption efficiency. Here, the system experiences a balance between ˙OH availability and competing processes such as radical recombination or secondary reactions with intermediate species. This intermediate behaviour aligns with the findings of Ambauen et al.,36 who noted moderate oxidation rates at neutral pH when treating similar quaternary ammonium compounds via BDD anodes. In contrast, the strongly alkaline environment at pH = 11 significantly impairs COD removal. This can be attributed to several interrelated factors: first, hydroxide ions HO− compete with water molecules at the electrode interface, suppressing the formation of ˙OH radicals. Second, the anionic form of paraquat becomes dominant at high pH, leading to electrostatic repulsion from the negatively polarized BDD surface, thereby reducing adsorption and reactivity. Third, alkaline conditions are known to promote the formation of polymeric by-products through side reactions, particularly when intermediate radicals undergo coupling or condensation pathways, resulting in electrode surface fouling a phenomenon previously reported by Rabaaoui et al.23 This process is further exacerbated by the persistence of aromatic intermediates under alkaline conditions, which are less efficiently oxidized due to lower ˙OH availability. These species are more prone to undergo radical–radical coupling or nucleophilic substitution, generating oligomeric or polymeric structures that contribute to electrode surface passivation. Additionally, the predominance of the oxygen evolution reaction (OER) in high pH media diverts the applied current away from useful oxidation, further lowering the faradaic efficiency. Such competition between target pollutant oxidation and OER has been well documented in electrochemical systems operating under alkaline environments37 in studies on anodic oxidation of aromatic pollutants. Furthermore, the prevalence of oxygen evolution reactions in alkaline media increases the parasitic consumption of electrons, further diminishing the current efficiency. These combined effects result in a passivated electrode surface, limited reactive species availability, and overall poor degradation performance. Taken together, these findings confirm that acidic conditions are most favorable for electrochemical oxidation using BDD electrodes, as they promote efficient radical generation, facilitate pollutant-electrode interactions, and minimize undesirable side reactions. From an application standpoint, pH adjustment can therefore serve as a key operational strategy for optimizing treatment outcomes, particularly in systems targeting the mineralization of recalcitrant organic contaminants such as paraquat.
 | ||
Fig. 5 Variation of chemical oxygen demand during anodic oxidation of 250 mL of 30 mg L−1 of paraquat in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C ( ) Na2SO4, ( ) Na2SO4, ( ) NaCl and ( ) NaCl and ( ) NaNO3. ) NaNO3. | ||
The results reveal a marked dependence of degradation efficiency on the electrolyte type. Among the tested media, NaCl led to the highest COD removal, followed by Na2SO4, while NaNO3 exhibited the lowest performance. This trend reflects the distinct oxidative pathways enabled by each anionic species under electrochemical conditions. In the case of NaCl, the enhanced efficiency is primarily attributed to the in situ generation of active chlorine speciessuch as molecular chlorine, hypochlorous acid, and hypochlorite ions through anodic oxidation of chloride ions. These species serve as potent secondary oxidants, extending the oxidative capacity of the system beyond the immediate vicinity of the electrode. Their longer lifetimes and greater diffusion range compared to hydroxyl radicals allow them to oxidize both bulk-phase and adsorbed contaminants, thus facilitating more complete mineralization. Similar enhancements in performance due to indirect chlorine-mediated oxidation have been reported in studies by Martínez-Huitle and Ferro.38 and more recently by by Santos et al.39 particularly in the treatment of quaternary ammonium herbicides. In contrast, the system employing Na2SO4 relies solely on direct oxidation by electro-generated ˙OH radicals, which, although highly reactive and non-selective, are short-lived and confined to the electrode surface. This limits their ability to oxidize pollutants in the bulk solution, reducing overall degradation efficiency. Nonetheless, sulfate remains a widely accepted electrolyte in EAOPs due to its stability, environmental compatibility, and lack of harmful by-product formation, as corroborated by Cai et al.40 On the other hand, the significantly lower performance observed with NaNO3 suggests that nitrate ions are electrochemically inert in generating reactive oxidants under the applied conditions. Rather than participating in oxidation, NO3− species may act as competitive electron acceptors, thereby reducing the charge available for hydroxyl radical formation or chlorine evolution. Furthermore, the absence of reactive nitrogen intermediates under mild conditions further limits the oxidative potential of nitrate-containing media. These observations align with findings by Oturan and Aaron,41 who noted the limited contribution of nitrate ions to radical-mediated degradation in low-voltage electrochemical systems. Collectively, these findings underscore the critical influence of the electrolyte matrix in shaping the dominant oxidation pathways and determining the overall efficiency of pollutant mineralization. Furthermore, since the supporting electrolytes (NaCl, Na2SO4, and NaNO3) do not contain any transition metal ions, their influence on the electrochemical oxidation process is purely due to the associated anions. This eliminates potential catalytic or inhibitory effects from metal species, ensuring a direct interpretation of anion-specific oxidation behavior. Chloride-containing electrolytes enable both direct and indirect oxidation mechanisms, resulting in superior degradation rates, while sulfate-based systems depend exclusively on direct ˙OH attack with moderate efficacy. In contrast, nitrate-based media offer minimal oxidative contribution and are thus suboptimal for the anodic treatment of refractory contaminants such as paraquat.24
Degradation kinetics and mineralization efficiency of paraquat
The kinetics of paraquat degradation via anodic oxidation using BDD anode were investigated under controlled experimental conditions. Fig. 6 displays the temporal evolution of paraquat concentration, illustrating a two-phase degradation pattern characterized by a rapid initial decline during the first 60 minutes, followed by a slower degradation rate in the later stages of electrolysis.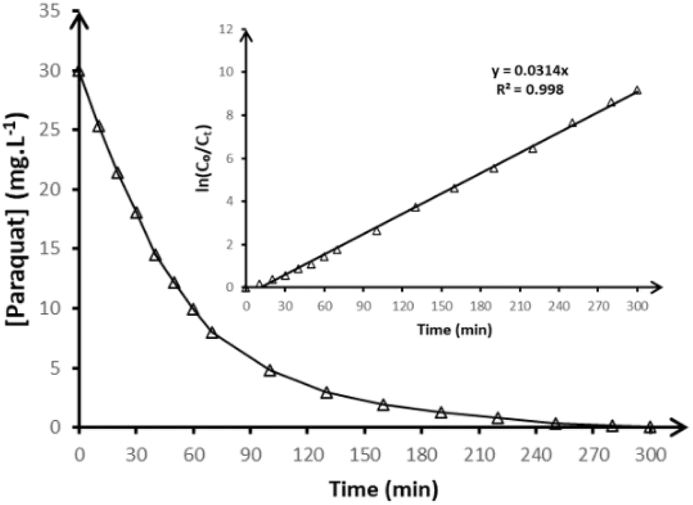 | ||
| Fig. 6 Variation of the concentration of paraquat during anodic oxidation of 250 mL in 50 mmol per L Na2SO4 in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C. The inset panel shows the kinetic analysis assuming a pseudo-first-order reaction. | ||
These results indicate an efficient oxidative attack during the initial phase, likely driven by the high availability of electrogenerated hydroxyl radicals known for their strong oxidizing power and non-selective reactivity. To quantify the degradation process, a kinetic analysis was performed using a pseudo-first-order model, as presented in the inset of Fig. 6 and described by the following eqn (4):
| ln(C0/Ct) = k × t | (4) |
While the degradation of the parent compound is rapid, the mineralization efficiency, as measured by total organic carbon removal, follows a significantly slower trajectory, as shown in Fig. 6. This divergence indicates the formation of persistent intermediate organic species, including carboxylic acids, quinones, and partially oxidized aromatic fragments. These intermediates are known to be more resistant to direct ˙OH attack and require extended electrolysis times for full conversion into CO2 and H2O. The progressive decrease in TOC, coupled with the detection of inorganic nitrogen species, supports the conclusion that most organic matter is ultimately converted into CO2 and H2O. Although water is not measured directly, its formation is intrinsic to the mineralization pathway and consistent with prior studies on electrochemical oxidation using BDD.
The relationship between faradaic efficiency (FE) and energy consumption (EC) is depicted in Fig. 7.
 | ||
| Fig. 7 Variation of faradaic efficiency (▲) and energy consumption (□) during anodic oxidation of 250 mL in 50 mmol per L Na2SO4 in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C. | ||
Initially, the FE is relatively high, indicating that much of the applied charge is effectively utilized for pollutant oxidation. However, as electrolysis proceeds, FE decreases progressively due to the increasing dominance of parasitic reactions primarily the oxygen evolution reaction which consume a significant portion of the applied current without contributing to organic degradation. This shift results in a concurrent rise in energy consumption, as a greater energy input is required to sustain the same level of oxidation.
This trade-off between efficiency and energy demand becomes particularly evident when compared with the slower TOC removal curve in Fig. 8.
 | ||
| Fig. 8 TOC removal with time during anodic oxidation of 250 mL of 30 mg L−1 of paraquat in 50 mmol per L Na2SO4 in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C. The inset shows the degradation %. | ||
The requirement for prolonged electrolysis to achieve complete mineralization exacerbates energy costs, which is a common challenge in advanced oxidation processes. Similar patterns have been reported in the degradation of pesticides and pharmaceuticals, as noted by Garcia-Segura et al.,42 who stressed the need for optimized operational windows to avoid excessive energy expenditure in low-efficiency phases.
To further quantify these observations, Table 3 summarizes key performance indicators across different electrolysis durations. After 220 minutes of treatment, a faradaic efficiency of 70.14% was recorded, corroborating the gradual decline observed in Fig. 7. Simultaneously, energy consumption rose to 66 kWh m−3, reflecting the energetic burden associated with extended oxidation. Despite the high spontaneity of the process, as indicated by the negative Gibbs free energy change (ΔrG = −366.25 kJ mol−1), the declining charge utilization efficiency over time highlights a fundamental limitation in prolonged electrolysis.
| Time (minutes) | TOC (mg L−1) | FE (%) | EEC (kWh m−3) | CEC (kWh m−3) | ΔrG (kJ mol−1) | TE (%) |
|---|---|---|---|---|---|---|
| 130 | 6.55 | 67.00 | 3.14 | 39.00 | −366.25 | 91.56 |
| 220 | 2.70 | 70.14 | 2.86 | 66.00 | ||
| 300 | 0.40 | 74.00 | 2.83 | 93.00 |
These findings underscore the need to establish an optimal electrolysis duration that balances three essential parameters: degradation efficiency, mineralization depth, and energy sustainability. In practical applications, exceeding this optimal duration may yield marginal gains in TOC removal while significantly increasing operational costs and electrode wear.
Thus, a comprehensive assessment of reaction kinetics, intermediate evolution, and energy parameters is crucial for the rational development of electrochemical treatment systems targeting persistent contaminants like paraquat.22
Identification and evolution of intermediates
The identification and monitoring of intermediate reaction during electrochemical oxidation are of paramount importance for both environmental safety and mechanistic understanding. Certain degradation by-products, though transient, may exhibit comparable or even higher toxicity than the parent compound. Therefore, tracking their formation and fate not only ensures that complete mineralization is achieved but also provides critical insights into the oxidative pathway, allowing for process optimization and mitigation of residual risk.In the case of paraquat degradation via anodic oxidation on a BDD anode, several aromatic intermediates were identified, primarily resulting from the electrophilic attack of hydroxyl radicals on the pyridine rings of the herbicide. As detailed in Table 4, key intermediates such as monopyridone, dipyridone, 4-carboxy-1-methylpyridinium, 4-picolinic acid, and 4,4′-bipyridyl were detected and confirmed through mass spectrometric analysis based on characteristic m/z values. These species represent successive oxidation states of the original molecule, and their detection aligns with degradation pathways reported in similar studies,43 validating the proposed sequence of transformation. The integration of HPLC and mass spectrometry analysis allowed us to identify key aromatic and carboxylic intermediates formed throughout the degradation process. These analytical insights were instrumental in elucidating the stepwise degradation mechanism of paraquat on BDD anodes, confirming ring-opening, hydroxylation, and final mineralization pathways. The corresponding chromatogram and mass spectrum (Fig. 9) are included in the manuscript to support this mechanistic interpretation.
| Compound | Molecular formula | Mass spectrometry (m/z) |
|---|---|---|
| Paraquat | C12H14N22+ | 93 [M/2]+ |
| Monopyridone(II) | C12H10N2O | 199 |
| Dipyridone(IV) | C12H9N2O2 | 214 |
| 4-Carboxy-1-methylpyridinium(VI) | C7H7NO2 | 138 |
| 4-Picolinic acid(VIII) | C6H5NO2 | 124 |
| 4,4′-Bipyridyl | C10H8N2 | 157 |
 | ||
| Fig. 9 HPLC and MS analyses confirming the identification of paraquat and key degradation intermediates. | ||
As shown in Fig. 10, the concentrations of these intermediates peaked between 30 and 60 minutes of electrolysis, marking a dynamic phase of molecular fragmentation and transformation. Their subsequent decline indicates further oxidative breakdown into low-molecular-weight species and eventual mineralization. By 150 minutes, most intermediates had substantially diminished, suggesting near-complete conversion of aromatic structures into final products such as CO2 and H2O.
 | ||
| Fig. 10 Time evolution of paraquat and its aromatic degradation intermediates detected during anodic oxidation of 250 mL in 50 mmol per L Na2SO4 in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C. | ||
This transition illustrates a typical two-phase mineralization pattern, beginning with oxidative cleavage of the parent molecule and followed by the gradual breakdown of intermediate species into simpler forms.
In the next oxidation phase, several carboxylic acids including oxalic, formic, acetic, succinic, citric, and acrylic acids were identified and monitored, as illustrated in Fig. 11.
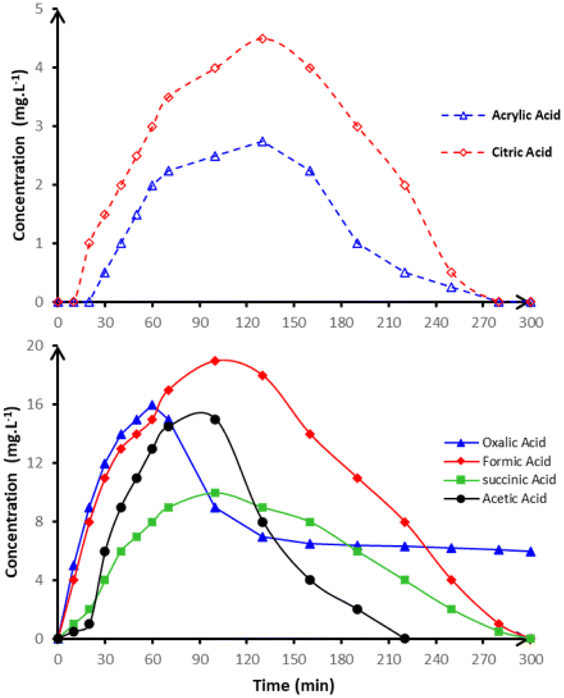 | ||
| Fig. 11 Time-course of the concentration of relevant carboxylic acids detected during anodic oxidation of 250 mL in 50 mmol per L Na2SO4 in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C. | ||
These compounds are well-known intermediates in the degradation of nitrogen-containing heterocycles. Notably, formic and acetic acids exhibited transient accumulation, reaching peak concentrations between 60 and 120 minutes before undergoing complete degradation, consistent with prior findings by Rabaaoui et al.23 who described similar profiles in the oxidative treatment of pesticides and pharmaceuticals. Oxalic acid, on the other hand, showed greater persistence, accumulating until around 60 minutes and declining slowly thereafter, indicating its relative resistance to ˙OH-mediated mineralization. This observation highlights the importance of sustaining adequate oxidant levels during the final stages of electrolysis to guarantee the full degradation of persistent intermediates.
Further confirmation of complete mineralization is provided by the analysis of inorganic ions released during the oxidation of paraquat, presented in Fig. 12. The gradual accumulation of ammonium and nitrate over 300 minutes reflects the progressive cleavage of organic nitrogen and Sulphur moieties. Notably, ammonium concentrations reached 12.3 mg L−1, significantly higher than those of nitrate (5.4 mg L−1), indicating that reduction pathways favoring ammonium formation predominate under the applied electrochemical conditions.
 | ||
Fig. 12 Accumulation of nitrate ( ) and ammonium ( ) and ammonium ( ) ions detected during anodic oxidation of 250 mL in 50 mmol per L Na2SO4 in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C. ) ions detected during anodic oxidation of 250 mL in 50 mmol per L Na2SO4 in pH = 3 on BDD anode at 15 mA cm−2 and at 20 °C. | ||
The combination of aromatic breakdown products, carboxylic acid intermediates, and inorganic ions collectively delineates the full mineralization pathway of paraquat under BDD-driven anodic oxidation. The system demonstrates strong oxidative performance across all stages from molecular fragmentation to final mineralization confirming its suitability for the treatment of persistent and toxic organic pollutants in aqueous environments.20
Reaction sequence for paraquat degradation
The anodic oxidation of paraquat using a BDD anode proceeds via a well-defined and multi-stage degradation pathway, predominantly driven by the action of electro-generated hydroxyl radicals. The proposed mechanism, illustrated in Fig. 13 and 14, reveals a systematic breakdown of the parent compound into aromatic intermediates, followed by carboxylic acid derivatives, and ultimately mineralization into inorganic end-products. The degradation initiates with the oxidative cleavage of the quaternary nitrogen linkage and electron-rich aromatic rings of the paraquat molecule (1,1′-dimethyl-4,4′-bipyridinium), leading to the formation of key intermediates such as monoquat, 4,4-bipyridyl, monopyridone, and dipyridone. These species arise via hydroxylation and N-demethylation reactions, which are facilitated by the high oxidative potential of ˙OH radicals on the BDD surface. | ||
| Fig. 13 Proposed reaction pathway for the mineralization of paraquat in aqueous acid medium by hydroxyl radicals generated in anodic oxidation process (Part I). | ||
 | ||
| Fig. 14 Proposed reaction pathway for the mineralization of paraquat in aqueous acid medium by hydroxyl radicals generated in anodic oxidation process (Part II). | ||
This pathway is consistent with prior mechanistic studies by Rabaaoui et al.,22 who documented similar intermediates during the electrochemical oxidation of nitrogenous herbicides. A critical turning point in the oxidative pathway involves ring-opening reactions of the substituted pyridine units, which initiate the fragmentation of the aromatic core. The formation of compounds such as 4-carboxy-1-methylpyridinium, 4-picolinic acid, and 4-carboxy-1-methylpyridine confirms the progressive oxidation of the heterocyclic moiety. These intermediates represent a transition from aromatic stability to open-chain reactivity, setting the stage for deeper oxidative attack. Subsequent oxidation steps produce low-molecular-weight carboxylic acids, including oxalic, formic, acetic, succinic, citric, and acrylic acids, as represented in Fig. 11. These acids accumulate transiently before degrading further into carbon dioxide and water.
Particularly, oxalic acid demonstrates greater persistence, likely due to its stable structure and resistance to attack by free ˙OH radicals. Concurrently, nitrogen atoms released from the heterocyclic rings are gradually transformed into NH4+ and NO3− ions, as shown in Fig. 12. The dominance of ammonium formation suggests a preferential reduction pathway under the applied conditions, which aligns with reports by Saad et al.,21 highlighting that electrochemical conditions often favor NH4+ over complete nitrogen oxidation in BDD-based systems. The final stage of the degradation sequence involves the complete oxidation of all intermediate organic species into inorganic end-products: CO2, H2O and NH4+. This full mineralization confirms the non-selective and deep oxidative capacity of the BDD anode, which can efficiently convert even the most persistent aromatic intermediates into harmless forms. The pathway thus reflects the hallmark features of advanced electrochemical oxidation: rapid initial attack, formation and degradation of stable intermediates, and eventual detoxification and mineralization. This mechanistic elucidation not only provides a molecular-level understanding of paraquat degradation but also reinforces the potential of BDD-based electrochemical advanced oxidation processes as a powerful and environmentally sustainable technology for the treatment of recalcitrant organic pollutants in industrial effluents. Such insights are essential for optimizing treatment conditions, minimizing by-product accumulation, and ensuring regulatory compliance in wastewater reuse or discharge scenarios.25
Conclusion
This study demonstrated the high effectiveness of boron-doped diamond electrodes in the anodic oxidation of paraquat, achieving COD and TOC removal rates of 99% and 98.6%, respectively, after 300 minutes. Kinetic modelling confirmed a pseudo-first-order degradation profile with a rate constant of 1.53 × 10−2 s−1, reflecting rapid and efficient oxidative transformation.Mechanistic analysis revealed a sequential degradation pathway involving aromatic cleavage, formation of carboxylic acids, and final mineralization into CO2, H2O, and NH4+, supported by spectrometric identification of intermediates. Despite the system's high initial faradaic Efficiency (70.14%), energy consumption increased to 66 kWh m−3 during extended electrolysis, emphasizing the importance of optimizing treatment duration for sustainability.
Overall, the findings confirm that BDD-based electrochemical oxidation is a robust and environmentally sound approach for treating persistent organic pollutants like paraquat. The integration of high degradation efficiency, deep mineralization, and clear mechanistic insight supports its application in advanced wastewater treatment and offers a solid foundation for further development in real-world scenarios. Moreover, the optimized operational parameters reported herein may serve as a practical baseline for future scaling efforts, enabling potential application of this process at larger treatment volumes or higher pollutant concentrations beyond the laboratory scale.
Data availability
The data that supported the findings of this study were available upon reasonable request.Author contributions
Nejmeddine Rabaaoui: software, formal analysis, writing – original draft; Naoufel Ben Hamadi: formal analysis, visualization; Mourad Cherif: investigation, validation; Ahlem Guesmi: data curation, investigation; Wesam Abd El-Fattah: formal analysis, validation; Houcine Naïli: validation, supervision, review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Funding
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-DDRSP2502).Notes and references
- P. Sillapawattana and P. Klungsupya, Ecotoxicity testing of paraquat metabolites degraded by filamentous fungi in model organism, Sci. Total Environ., 2022, 822, 105760, DOI:10.1016/j.scitotenv.2022.153631.
- L. Kumalayanti, J. Tabtamart, P. Kidkhunthod, N. Chankhunthod and S. Pinitsoontorn, Photocatalytic degradation study of paraquat dichloride by dumbbell-liked TiO2 capped gold nanorods under UV and NIR irradiation, Surf. Interfaces, 2025, 57, 105760, DOI:10.1016/j.surfin.2025.105760.
- I. Linares-Hernández, L. A. Castillo-Suárez, J. G. Ibanez, R. Vasquez-Medrano, B. M. López-Rebollar, F. Santoyo-Tepole, E. A. Teutli-Sequeira and I. G. Martínez-Cienfuegos, Degradation of commercial paraquat in a solar-Fenton pilot lagoon using iron oxalate as a chelating agent: Hydro-thermal analysis with CFD, J. Photochem. Photobiol., A, 2022, 429, 113914, DOI:10.1016/j.jphotochem.2022.113914.
- C. Marien, M. Pivert, A. Azaïs, I. M'Bra, P. Drogui, A. Dirany and D. Robert, Kinetics and mechanism of Paraquat's degradation: UV-C photolysis vs. UV-C photocatalysis with TiO2/SiC foams, J. Hazard. Mater., 2019, 370, 164–171, DOI:10.1016/j.jhazmat.2018.06.009.
- M. M. Desipio, R. Thorpe and D. Saha, Photocatalytic Decomposition of Paraquat Under Visible Light by Carbon Nitride and Hydrogen Peroxide, Optik, 2018, 172, 1047–1056, DOI:10.1016/j.ijleo.2018.07.124.
- P. Mantecca, S. Panseri, R. Bacchetta, C. Vismara, G. Vailati and M. Camatini, Histopathological effects induced by paraquat during Xenopus laevis primary myogenesis, Tissue Cell, 2006, 38, 209–217, DOI:10.1016/j.tice.2006.03.002.
- S. Vigneshwaran, J. Preethi and S. Meenakshi, Interface engineering of ultrathin multi-functional 2D draped chitosan for efficient charge separation on degradation of paraquat – A mechanistic study, J. Environ. Chem. Eng., 2020, 8, 104446, DOI:10.1016/j.jece.2020.104446.
- F. Zahedi, M. Behpour, S. M. Ghoreishi and H. Khalilian, Photocatalytic degradation of paraquat herbicide in the presence TiO2 nanostructure thin films under visible and sun light irradiation using continuous flow photoreactor, Sol. Energy, 2015, 120, 287–295, DOI:10.1016/j.solener.2015.07.010.
- S. Wongcharoen and G. Panomsuwan, Easy synthesis of TiO2 hollow fibers using kapok as a biotemplate for photocatalytic degradation of the herbicide paraquat, Mater. Lett., 2018, 228, 482–485, DOI:10.1016/j.matlet.2018.06.089.
- M. Jindakaraked, E. Khan and P. Kajitvichyanukul, Biodegradation of paraquat by Pseudomonas putida and Bacillus subtilis immobilized on ceramic with supplemented wastewater sludge, Environ. Pollut., 2021, 286, 117307, DOI:10.1016/j.envpol.2021.117307.
- D. Franco, J. Georgin, E. Lima and L. Silva, Advances made in removing paraquat herbicide by adsorption technology: A review, J. Water Process Eng., 2022, 49, 102988, DOI:10.1016/j.jwpe.2022.102988.
- C. Nguyen, D. Tungtakanpoung, V. Tra and P. Kajitvichyanukul, Kinetic, isotherm and mechanism in paraquat removal by adsorption process using corn cob biochar produced from different pyrolysis conditions, Case Stud. Chem. Environ. Eng., 2022, 6, 100248, DOI:10.1016/j.cscee.2022.100248.
- M. G. Sorolla, M. L. Dalida, P. Khemthong and N. Grisdanurak, Photocatalytic degradation of paraquat using nano-sized Cu-TiO2/SBA-15 under UV and visible light, J. Environ. Sci., 2012, 24, 1125–1132, DOI:10.1016/S1001-0742(11)60874-7.
- A. Dhaouadi and N. Adhoum, Degradation of paraquat herbicide by electrochemical advanced oxidation methods, J. Electroanal. Chem., 2009, 637, 33–42, DOI:10.1016/j.jelechem.2009.09.027.
- T. Xu, X. Tang, M. Qiu, X. Lv, Y. Shi, Y. Zhou, Y. Xie, M. Naushad, S. Lam, H. S. Ng, C. Sonne and S. Ge, Degradation of levofloxacin from antibiotic wastewater by pulse electrochemical oxidation with BDD electrode, J. Environ. Manage., 2023, 344, 118718, DOI:10.1016/j.jenvman.2023.118718.
- L. Saleh, M. Remot, Q. B. Remaury, P. Pardon, P. Labadi, H. Budzinski, C. Coutanceau and J. P. Croué, PFAS degradation by anodic electrooxidation: Influence of BDD electrode configuration and presence of dissolved organic matter, Chem. Eng. J., 2024, 489, 151355, DOI:10.1016/j.cej.2024.151355.
- R. Dhawle, A. Kajtazi, M. Sakellariou, Z. Frontistis, F. Lynen and D. Mantzavinos, Electrochemical oxidation of losartan on a BDD electrode: Influence of cathodes and electrolytes on the degradation kinetics and pathways, Water Resour. Ind., 2024, 31, 100240, DOI:10.1016/j.wri.2024.100240.
- Y. Wang, X. Xiao, J. Chen, D. Deng, D. Hu and X. Liang, Electrochemical advanced oxidation of biotreated dyeing and finishing wastewater by boron doped diamond electrode, Desa, Water Treat., 2024, 320, 100904, DOI:10.1016/j.dwt.2024.100904.
- Y. Gong, W. Jia, B. Zhou, K. Zheng, J. Gao, Y. Wu, S. Yu, Y. Xue and Y. Wu, Novel graphite-based boron-doped diamond coated electrodes with refractory metal interlayer for high-efficient electrochemical oxidation degradation of phenol, Sep. Purif. Technol., 2025, 355, 129550, DOI:10.1016/j.seppur.2024.129550.
- N. Rabaaoui, Y. Moussaoui, M. S. Allagui, B. Ahmed and E. Elaloui, Anodic oxidation of nitrobenzene on BDD electrode: Variable effects and mechanisms of degradation, Sep. Purif. Technol., 2013, 107, 318–323, DOI:10.1016/j.seppur.2013.01.047.
- M. K. Saad, N. Rabaaoui, E. Elaloui and Y. Moussaoui, Mineralization of p-methylphenol in aqueous medium by anodic oxidation with a boron-doped diamond electrode, Sep. Purif. Technol., 2016, 171, 157–163, DOI:10.1016/j.seppur.2016.07.018.
- A. Ouni, N. Rabaaoui, L. Mechi, N. Enaceur, A. K. D. AlSukaibi, E. M. Azzam, K. M. Alenezi and Y. Moussaoui, Removal of pesticide chlorobenzene by anodic degradation: Variable effects and mechanism, J. Saudi Chem. Soc., 2021, 25, 101326, DOI:10.1016/j.jscs.2021.101326.
- N. Rabaaoui, M. K. Saad, Y. Moussaoui, M. S. Allagui, A. Bedoui and E. Elaloui, Anodic oxidation of o-nitrophenol on BDD electrode: Variable effects and mechanisms of degradation, J. Hazard. Mater., 2013, 250–251, 447–453, DOI:10.1016/j.jhazmat.2013.02.027.
- N. Rabaaoui and M. S. Allagui, Anodic oxidation of salicylic acid on BDD electrode: Variable effects and mechanisms of degradation, J. Hazard. Mater., 2012, 243, 187–192, DOI:10.1016/j.jhazmat.2012.10.016.
- F. Yu-Jie and L. Xiang-yuan, Electro-catalytic oxidation of phenol on several metaloxide electrodes in aqueous solution, Water Res., 2003, 37, 2399–2407, DOI:10.1016/S0043-1354(03)00026-5.
- D. Dkhar, R. Kumari, S. Malode, N. Shetti and P. Chandra, Integrated lab-on-a-chip devices: Fabrication methodologies, transduction system for sensing purposes, J. Pharm. Biomed. Anal., 2023, 223, 115120, DOI:10.1016/j.jpba.2022.115120.
- D. Ilager, N. Shetti, Y. Foucaud, M. Badawi and T. Aminabhavi, Graphene/g-carbon nitride (GO/g-C3N4) nanohybrids as a sensor material for the detection of methyl parathion and carbendazim, Chemosphere, 2022, 292, 133450, DOI:10.1016/j.chemosphere.2021.133450.
- S. J. Malode, K. Prabhu, N. P. Shetti and K. R. Reddy, highly sensitive electrochemical assay for selective detection of Aminotriazole based on TiO2/poly (CTAB) modified sensor, Environ. Technol. Innovation, 2021, 21, 101222, DOI:10.1016/j.eti.2020.101222.
- S. Jianrui, L. Haiyan, L. Haibo, D. Lili, H. Weimin, L. Hongdong and C. Tian, Electrochemical oxidation of aqueous phenol at low concentration using Ti/BDD electrode, Sep. Purif. Technol., 2012, 88, 116–120, DOI:10.1016/j.seppur.2011.12.022.
- S. Hu, Y. Lv, X. Hou, J. Li, Y. Hou, X. Fu and T. Xu, BDD electrode pulsed alternating electrochemical oxidation of sulfamethazine in antibiotic wastewater: Process optimization and degradation mechanism, Environ. Res., 2025, 275, 121375, DOI:10.1016/j.envres.2025.121375.
- F. L. Migliorini, J. R. Steter, R. S. Rocha, M. R. V. Lanza, M. R. Baldan and N. G. Ferreira, Efficiency study and mechanistic aspects in the Brilliant Green dye degradation using BDD/Ti electrodes, Diamond Relat. Mater., 2013, 32, 54–60, DOI:10.1016/j.diamond.2015.12.013.
- F. Malaret, Exact calculation of corrosion rates by the weight-loss method, Exp. Results, 2022, 3, 1–12, DOI:10.1017/exp.2022.5.
- C. A. Martínez-Huitle, M. A. Rodrigo, I. Sirés and O. Scialdone, These findings reinforce the advantages of BDD electrodes not only in terms of oxidative performance but also in their durability and resistance to degradation, Appl. Catal., B, 2023, 328, 122430, DOI:10.1016/j.apcatb.2023.122430.
- O. T. Can, M. Bayramoglu, M. Sozbir and O. Aras, Mineralization of o-tolidine by electrooxidation with BDD, Ti/Pt and MMO anodes, Desalin. Water Treat., 2019, 141, 377–385, DOI:10.5004/dwt.2019.23311.
- C. F. Zorzo, L. Albornoz, A. M. Bernardes, V. Pérez-Herranz, F. H. Borba and S. W. da Silva, Electrochemical oxidation for the rapid degradation of emerging contaminants: Insights into electrolytes and process parameters for phytotoxicity reduction, Chemosphere, 2025, 377, 144363, DOI:10.1016/j.chemosphere.2025.144363.
- N. Ambauen, J. Muff, F. Tscheikner-Gratl, T. Trinh, C. Hallé and T. Meyn, Application of electrochemical oxidation in cold climate regions – Effect of temperature, pH and anode material on the degradation of Bisphenol A and the formation of disinfection by-products, J. Environ. Chem. Eng., 2020, 8, 104183, DOI:10.1016/j.jece.2020.104183.
- D. Dionisio, M. A. Rodrigo and A. J. Motheo, Electrochemical degradation of a methyl paraben and propylene glycol mixture: Interference effect of competitive oxidation and pH stability, Chemosphere, 2020, 287, 132229, DOI:10.1016/j.chemosphere.2021.132229.
- C. A. Martínez-Huitle and S. Ferro, Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes, Chem. Soc. Rev., 2006, 35, 1324–1340, 10.1039/B517632H.
- J. L. Santos, M. A. Gómez, D. C. Moura, M. Cerro-López, M. A. Quiroz and C. A. Martínez-Huitle, Removal of herbicide 1-chloro-2,4-dinitrobenzene (DNCB) from aqueous solutions by electrochemical oxidation using boron-doped diamond (BDD) and PbO2 electrodes, J. Hazard. Mater., 2021, 402, 123850, DOI:10.1016/j.jhazmat.2020.123850.
- J. Cai, M. Zhou, Y. Pan and X. Lu, Degradation of 2,4-dichlorophenoxyacetic acid by anodic oxidation and electro-Fenton using BDD anode: Influencing factors and mechanism, Sep. Purif. Technol., 2020, 230, 115867, DOI:10.1016/j.seppur.2019.115867.
- M. A. Oturan and J. J. Aaron, Advanced Oxidation Processes in Water/Wastewater Treatment: Principles and Applications: A Review, Environ. Sci. Technol., 2014, 44, 2577–2641, DOI:10.1080/10643389.2013.829765.
- S. Garcia-Segura, J. D. Ocon and M. Nan-Chong, Electrochemical oxidation remediation of real wastewater effluents — A review, Process Saf. Environ. Prot., 2018, 113, 48–67, DOI:10.1016/j.psep.2017.09.014.
- M. Samarghandi, A. Rahmani, M. Khazaei, A. Dargahi, A. Bahiraei and A. Shabanloo, Degradation and mineralization of diazinon pesticide by G/PbO2 anodic oxidation process, Case Stud. Chem. Environ. Eng., 2024, 29, 100685, DOI:10.1016/j.cscee.2024.100685.
| This journal is © The Royal Society of Chemistry 2025 |