
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the degree of long-range order/disorder in indaceno-based photovoltaic small molecules using data-driven machine learning analysis†
Hussein A. K. Kyhoiesh *ai,
Karrar H. Salemb,
Azal S. Waheebcj,
Riyam A. Hasand,
Hayder R. Salmane,
Ahmed A. Al-Kubaisif,
Ashraf Y. Elnaggarg,
Islam H. El Azabg and
Mohamed H. H. Mahmoudh
*ai,
Karrar H. Salemb,
Azal S. Waheebcj,
Riyam A. Hasand,
Hayder R. Salmane,
Ahmed A. Al-Kubaisif,
Ashraf Y. Elnaggarg,
Islam H. El Azabg and
Mohamed H. H. Mahmoudh
aNational University of Science and Technology, Nasiriyah, Dhi Qar 64001, Iraq. E-mail: hussein.k.sultan@nust.edu.iq; Tel: (+964)7807229491
bCollege of Medical and Health Technologies, Al-Zahraa University for Women, Karbala, Iraq
cDepartment of Chemistry, College of Science, Al-Muthanna University, AL-Muthanna, Iraq
dMazaya University College, Al-Zaytoun Street, Nasiriyah, Dhi-Qar 64001, Iraq
eCollege of Pharmacy, Al-Mustaqbal University, Babylon, 51001, Iraq
fDepartment of Medical Laboratories Techniques, College of Health and Medical Technology, University of Al Maarif, Al Anbar, 31001, Iraq
gDepartment of Food Sciences and Nutrition, College of Science, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia
hDepartment of Chemistry, College of Science, Taif University, P.O. Box 11099, Taif 2194, 4, Saudi Arabia
iRepublic of Iraq Ministry of Education, General Directorate of Education in Al-Muthanna Province, Samawah, Al-Muthanna, Iraq
jInorganic Chemistry Group, Scientific Research Center, Al-Ayen University, Thi-Qar, Iraq
First published on 1st July 2025
Abstract
Long-range order and disorder in small molecules significantly impact their physical and chemical properties, affecting their performance in photovoltaic devices. For the current study, a data-driven machine learning (ML) approach has been applied to explore the relationship between molecular structure and crystallinity in 480 indaceno-based small molecules. Three ML models, including support vector machines and random forest models, were trained to predict crystal propensity. A heatmap analysis revealed that 72.71% of the small molecules exhibit crystalline behavior, while the remaining 27.29% are non-crystalline. ML models achieved near-perfect accuracy (AUC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SVM-RBF = 0.999, RF = 0.998; MSE:RF = 0.00, SVM-RBF = 0.01). The predicted crystal propensity values showed high accuracy, with a mean squared error ranging from 0.0–0.64. Feature importance analysis using SHAP values identified Chi0v, kappa1, Chi1n, and NumRotatableBonds as the most contributing factors to crystal propensity. The synthetic accessibility score of the small molecules ranged from 0.02 to 0.12, providing insights for designing and optimizing indaceno-based small molecules with tailored crystallinity and photovoltaic properties. This study demonstrates the potential of ML approaches in guiding the development of high-performance small molecules for solar energy applications.
:SVM-RBF = 0.999, RF = 0.998; MSE:RF = 0.00, SVM-RBF = 0.01). The predicted crystal propensity values showed high accuracy, with a mean squared error ranging from 0.0–0.64. Feature importance analysis using SHAP values identified Chi0v, kappa1, Chi1n, and NumRotatableBonds as the most contributing factors to crystal propensity. The synthetic accessibility score of the small molecules ranged from 0.02 to 0.12, providing insights for designing and optimizing indaceno-based small molecules with tailored crystallinity and photovoltaic properties. This study demonstrates the potential of ML approaches in guiding the development of high-performance small molecules for solar energy applications.
Introduction
The importance of photovoltaic small molecules cannot be overstated in the wake of modern scientific developments.1 As the world grapples with the reality of an energy crisis, these materials have emerged as a promising solution to mitigate the current reliance on fossil fuels.2 The unique properties of photovoltaic small molecules, including their flexibility, lightweight nature, and low production costs, make them an attractive alternative to traditional solar panels.3 Furthermore, recent advancements in material science have led to the development of small molecules with enhanced power conversion efficiency, rivaling their inorganic counterparts.4 The applications of photovoltaic small molecules are vast and varied. One of the most promising areas is building-integrated photovoltaics (BIPV), where small molecules can be seamlessly integrated into building structures, generating electricity while serving as a building envelope.5 Additionally, the flexibility and lightweight nature of photovoltaic small molecules make them an ideal choice for powering wearable devices, such as smartwatches and fitness trackers.6 As research continues to advance, the importance of photovoltaic small molecules will only continue to grow.7The arrangement of small molecule chains, either in a crystalline or amorphous structure, significantly affects the materials with their mechanical strength, conductivity, and optical properties.8 In crystalline small molecules, long-range order is characterized by a repeating pattern of molecular arrangements, resulting in improved mechanical strength, thermal stability, and conductivity.9 On the other hand, amorphous small molecules exhibit a random, disordered structure, leading to increased flexibility, optical clarity, and solubility.10 The balance between long-range order and disorder is critical, as it directly influences the small molecules with their performance in various applications.11 Moreover, the degree of order and disorder can be influenced by factors such as molecular weight, temperature, and processing conditions, further emphasizing the need to evaluate these parameters before designing new small molecules.12 Understanding and controlling long-range order and disorder in small molecules is essential for optimizing their properties and performance.13 By elucidating the relationship between molecular structure and material properties, researchers can design small molecules with tailored properties for specific applications.14 Introducing disorder can improve the optical clarity and solubility of small molecules, making them ideal for biomedical and optoelectronic devices.15 Evaluating long-range order and disorder is thus a critical step in the design and development of new small molecules, enabling the creation of materials with optimized properties and performance.16
The ML has emerged as a powerful tool in materials science, enabling the prediction of unique material properties with accuracy.17 By analyzing complex datasets, ML algorithms can identify patterns and correlations to predict properties like mechanical strength, thermal conductivity, and optical properties.18 In small molecules, ML can predict crystal propensity features, critical for photovoltaic performance.19 This approach can accelerate the discovery of new small molecules with optimized photovoltaic features, enabling more efficient and sustainable energy harvesting systems.20 The current work applies ML analysis to a dataset of small molecules to evaluate crystal propensity features and predict photovoltaic performance. By identifying key structural features governing crystal propensity, a predictive model can be developed to forecast photovoltaic properties. This enables the design of new indaceno-based photovoltaic small molecules with optimized crystal propensity features, leading to enhanced photovoltaic performance and improved energy harvesting capabilities.
Methodology
Data collection
A dataset of 480 indaceno-based small molecule structures was curated from peer-reviewed literature and open-access chemical databases such as PubChem, the Cambridge Structural Database (CSD), the Harvard Organic Photovoltaic Database (https://opvdb.lbl.gov/) and the OMDB database (https://omdb.digital/). Search filters targeted the indaceno or similar indaceno-based core structures. For molecules not available in public databases, structures were manually drawn using ChemDraw and converted to SMILES format using the RDKit toolkit. All SMILES strings were standardized and validated using RDKit to ensure consistency and accuracy before descriptor calculation, and represented using Simplified Molecular Input Line Entry System (SMILES) notation (Fig. 1). The datasets were preprocessed by standardizing molecular representations and normalizing calculated descriptor values. The dataset was then divided into a training set (360 structures, 75%) and a test set (120 structures, 25%) to ensure model training and evaluation on separate data. All model performance metrics, including AUC and MSE, were calculated using the test set to evaluate the models' predictive power on unseen data. The datasets were preprocessed by normalizing the SMILES lengths to a common scale, preventing feature dominance and improving model performance. This setup enabled ML analysis to correlate SMILES length with photovoltaic performance, aiming to develop predictive models for these small molecule structures. Hyperparameters for SVM and Random Forest models were optimized via 5-fold cross-validation on the training set. All features were standardized using StandardScaler, and zero-variance features were removed. | ||
| Fig. 1 Distribution of SMILES string lengths for the 480 indaceno-based small molecules. | ||
Descriptor designing
A comprehensive set of molecular descriptors was generated from the collected indaceno-based structures using the RDKit toolkit.21 These descriptors encompassed various types, including electronic, topological, electrotopological, and molecular descriptors, which were designed to capture diverse aspects of the small molecule structures. This diverse set of descriptors allowed for a thorough characterization of the small molecules, enabling the identification of potentially relevant features that may influence their photovoltaic performance. The calculated descriptors that were correlated with target properties are provided in the ESI.†22–25Machine learning analysis
The latest Python package (Ver. 3.10.6) was used for all machine-learning operations.26 Advanced Python libraries such as NumPy, Pandas, SciPy, and Scikit-Learn were utilized for data processing, analysis, and visualization. Informative plots and graphs were created with Matplotlib to effectively illustrate the results. A robust and accurate machine learning model was developed to predict the crystal properties of indaceno-based small molecules, leveraging advanced algorithms and data analysis techniques, ultimately aiding in the identification of potential candidates for further experimental validation.Correlations and feature scores
The correlations and feature scores properties are provided in the ESI.†27Results and discussion
Model training
The ML model was evaluated using three different classification algorithms: Support Vector Machine (SVM)28 with a linear kernel, SVM with a radial basis function (RBF) kernel, and Random Forest (RF)29 Starting with the SVM (Linear) model, the obtained AUC score of 0.986 indicated that this model was performing quite well. Its AUC score close to 1 suggested that the model could be excellent at distinguishing between positive and negative classes. In the case of a linear kernel, the SVM model was trying to find a hyperplane that could separate the classes in a linearly separable way (Fig. 2). The high AUC score indicated that the model could be able to find a good separation between the classes, suggesting that the features used in the model are highly discriminative. Moving on to the SVM (RBF) model, the AUC score of 0.999 is extremely high, indicating almost perfect classification performance. This is not surprising, given that RBF kernels are more flexible than linear kernels and can handle non-linear relationships between features. The high AUC score suggested that the RBF kernel might have been able to capture complex patterns in the data that the linear kernel could be unable to capture. This might imply that the data could have non-linear relationships between features, and the RBF kernel has been able to model these relationships effectively. Finally, the RF model achieved an AUC score of 0.998, which is also considerably high. As RF model is an ensemble learning method to combines the predictions of multiple decision trees. Its high AUC score could indicate that the ensemble could have been able to capture the patterns in the data effectively. Also, the RF model is known for its ability to handle complex interactions between features and its robustness to overfitting.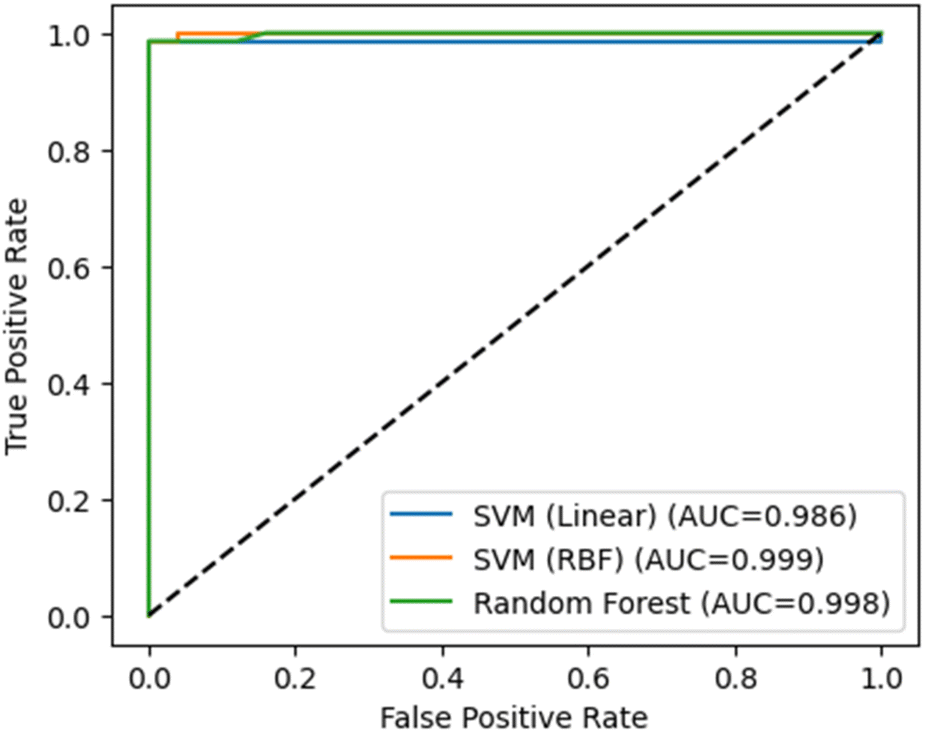 | ||
| Fig. 2 Receiver operating characteristic (ROC) curves for the three ML models (test set). AUC values: SVM-RBF = 0.999, RF = 0.998, SVM-Linear = 0.986. | ||
The high AUC score also suggested that the RF model could have been able to handle the interactions between features effectively and could have not overfitted to the training data. Overall, all three models have performed well which suggested that the features used in the model were highly discriminative, and the models could capture the underlying patterns in the data effectively. Their high performance might suggest that the data could have complex, non-linear relationships between features, and the models could have been able to capture these relationships effectively.
Crystal propensity range
Crystal propensity is defined as a binary classification label (crystalline/non-crystalline) based on experimental data from peer-reviewed literature. The crystal propensity analysis of all 480 compounds revealed a fascinating insight into their crystallinity and non-crystallinity characteristics. To visualize these results, a heatmap with a contour map was created to illustrate the degree of order (crystallinity) and disorder (non-crystallinity) among the compounds from their trained models. The heatmap was a 2D representation of the data, with Chi0v on the x-axis and NumRotableBonds on the y-axis. The x-axis ranged from 0–140, while the y-axis ranged from 0–90. The heatmap was divided into a grid of squares, each representing a unique combination of Chi0v and NumRotableBonds values (Fig. 3). The color scheme of the heatmap was important to understand their results. Its colors ranged from blue to red, with blue indicating its higher propensity for crystallinity and red indicating its higher propensity for non-crystallinity. The transition from blue to red was gradual, with intermediate colors indicating intermediate propensities for crystallinity. The heatmap exhibited a clear pattern, with the majority of the blue region concentrated in the upper-left quadrant. | ||
| Fig. 3 Heatmap of crystal propensity as a function of Chi0v (x-axis, valence connectivity index) and NumRotatableBonds (y-axis). Contour lines denote probability density (darker = higher density). Regions with Chi0v < 40 and NumRotatableBonds < 5 show maximal crystallinity (72.71% of molecules). | ||
This suggested that compounds with low Chi0v values and low NumRotableBonds values could be more likely to exhibit crystalline behavior. As Chi0v values increased and NumRotableBonds values decreased, the propensity for crystallinity decreased, and the color gradually shifted towards the red. Conversely, the red region was more prominent in the lower-right quadrant, indicating that compounds with high Chi0v values and high NumRotableBonds values could be non-crystalline. This made sense, as high Chi0v values and high NumRotableBonds values could often indicate a higher degree of molecular flexibility, which can hinder crystallization. The contour lines within the heatmap provide additional insight into the data. The lines represent regions of equal probability density, with darker lines indicating higher probability densities. The contour lines are denser in the blue region, indicating that the data is more concentrated around the crystalline region. The contour lines are less dense in the red region, suggesting that the data is more scattered around the non-crystalline region. By examining the heatmap, researchers can identify specific regions of high crystallinity or non-crystallinity, which can inform further research and development. For example, compounds with low Chi0v values and low NumRotableBonds values may be prioritized for crystallization attempts, while compounds with high Chi0v values and high NumRotableBonds values may require additional optimization to improve their crystallization properties.
The feature importance analysis revealed its fascinating insight into relationships between various molecular descriptors and crystal propensity. The highest correlation was observed between ESate_VSA8 and crystal propensity, with a correlation coefficient of 0.77. Being a topological polar surface area descriptor, ESate_VSA8 measures the surface area of a molecule that could be accessible to a solvent. In the context of crystal propensity, a high correlation with ESate_VSA8 suggested that small molecules with larger polar surface areas could be more likely to crystallize (Fig. 4). This made sense, as small molecules with larger polar surface areas could have stronger intermolecular interactions to facilitate their crystal formation. Its next features which included HeavyAtomMolwt, MolMr, MolWt, and ExactMolwt showed a correlation coefficient of 0.72 with crystal propensity. These features could be related to molecular weight, which is a fundamental property of small molecules. The correlation suggested that small molecules with higher molecular weights could be more likely to crystallize. This could be attributed to the fact that larger small molecules tend to have more pronounced intermolecular interactions, which could lead to their higher propensity for crystallization. The feature LabuteASA,30 which measures the accessible surface area of small molecules, also exhibited a high correlation with crystal propensity (0.72).
 | ||
| Fig. 4 Pearson correlation heatmap of top RDKit descriptors versus crystal propensity. Strong positive correlations (red) indicate higher crystallinity likelihood. | ||
That was consistent with the idea that small molecules with larger surface areas could likely crystallize, as they have more opportunities for their intermolecular interactions. The feature NumAromaticRings, counting the number of aromatic rings, showed a correlation coefficient of 0.71 with the crystal propensity. The aromatic rings are known to be planar and rigid to facilitate crystal formation by providing a flat surface for their intermolecular interactions. The correlation suggested that small molecules with more aromatic rings could be more likely to crystallize. The features Chi0v, Chi1, and HeavyAtomCount also showed a correlation coefficient of 0.71 with crystal propensity. Chi0v and Chi1 are topological indices that measure molecular connectivity and shape, while HeavyAtomCount measures the number of heavy atoms in small molecules. The correlation suggested that small molecules with more complex shapes and higher numbers of heavy atoms could be more likely to crystallize.
Feature-driven investigation
The observation that Chi0v produces a linear correlation with crystal propensity is a significant finding, as it suggested a direct relationship between the molecular descriptor Chi0v and the likelihood of crystallization. To further investigate this relationship, the distribution of NumRotatableBonds and Chi0v was evaluated, revealing that the range of NumRotatableBonds was up to 80, while Chi0v ranged up to 140 (Fig. 5). Notably, the majority of small molecules exhibited a relatively low number of rotatable bonds, with most having between 0 and 4 rotatable bonds. This suggested that the molecular flexibility of these small molecules is relatively low, which could facilitate crystallization by reducing the degree of molecular motion and increasing the likelihood of intermolecular interactions. On the other hand, Chi0v, which measures the molecular connectivity and shape, ranged from 20–65 for most small molecules. This range is interesting, as it suggested that the molecular shape and connectivity of these small molecules were relatively diverse. However, the fact that Chi0v correlates linearly with crystal propensity implied that there could be a specific range of Chi0v values that were conducive to crystallization. | ||
| Fig. 5 Distribution of (a) RotatableBonds (range: 0–80) and (b) Chi0v (range: 20–140) across the dataset. | ||
The combination of low rotatable bonds and specific Chi0v values may be indicative of small molecules that are more likely to crystallize. This is because small molecules with low rotatable bonds are more likely to adopt a rigid conformation, which can facilitate the formation of crystal lattices. Meanwhile, the specific range of Chi0v values may correspond to molecular shapes and connectivities that are more conducive to intermolecular interactions and crystal formation. The distribution of NumRotatableBonds and Chi0v highlighted the importance of molecular properties in determining crystal propensity. By understanding the relationships between these molecular descriptors and crystal propensity, researchers can design and optimize small molecules with improved crystallization properties. For instance, by targeting small molecules with low rotatable bonds and specific Chi0v values, it might be able to engineer small molecules with improved crystal structures and properties. The analysis of the top-ranked small molecules with the highest crystal probability reveals distinct patterns in their RotatableBondCount (RBC) and Chi0v values, which fall within narrow ranges (Fig. 6). The RotatableBondCount of these top-performing structures ranged from 26–28, indicating a relatively low degree of molecular flexibility. Meanwhile, the Chi0v values ranged from 58.52–59.71, suggesting that these small molecules have a specific range of molecular connectivity and shape that is conducive to crystallization. Notably, the top-ranked structures exhibited a strong correlation between RotatableBondCount and Chi0v values. Structures with RotatableBondCount values between 26–28 and Chi0v values between 58.52 and 59.71 were found to have the highest crystal probability, with values ranging from 0.97–0.99. These results suggested that small molecules with specific ranges of RotatableBondCount and Chi0v values might be more likely to crystallize, and therefore might be suitable targets for crystallization studies.
 | ||
| Fig. 6 Chemical structures of small molecules with the highest crystal probability along their RotatableBonds (RBC) and Chi0v. | ||
Regression analysis
The regression analysis performed on the trained models reveals a fascinating insight into the performance of different algorithms in predicting crystal propensity. The results show that Support Vector Regression (SVR) with a linear kernel achieved a mean squared error (MSE) of 0.64, while SVR with a radial basis function (RBF) kernel achieved an impressively low MSE of 0.01 (Fig. 7). This stark difference in performance suggested that the RBF kernel could be better suited for capturing the underlying relationships between the molecular descriptors and crystal propensity. The RBF kernel with its superior performance could be attributed to its ability to handle non-linear relationships between the inputs and outputs.31 In this case, the RBF kernel can model the complex interactions between the molecular descriptors and crystal propensity, resulting in a much lower MSE. This is in contrast to the linear kernel, which assumes a linear relationship between the inputs and outputs and is therefore limited in its ability to model complex interactions. An exceptional deviation is observed for one compound (blue point) in (Fig. 8). This outlier likely results from unique structural features or limitations in the model's representation of underrepresented molecular motifs. | ||
| Fig. 7 Regression plot of predicted vs. actual crystal propensity (test set). | ||
 | ||
| Fig. 8 SHAP value beeswarm plot of the collected dataset by the trained regression models. | ||
The RF model, on the other hand, achieved an astonishingly low MSE of 0.00, indicating that it has essentially perfectly predicted the crystal propensity of the small molecules. This is likely because RF is an ensemble learning method that combines the predictions of multiple decision trees, allowing it to capture complex interactions and non-linear relationships between the molecular descriptors and crystal propensity. The results of the regression analysis suggested that both SVR with an RBF kernel and RF could be suitable algorithms for predicting crystal propensity. However, the exceptionally low MSE of the RF algorithm could make it the most promising approach for this task. By using RF, researchers can accurately predict the crystal propensity of small molecules, which can facilitate the design and optimization of small molecules with improved crystal structures and properties.
Impact of model performance
The SHapley Additive eXplanation (SHAP)32,33 value beeswarm plot revealed that a select group of molecular descriptors had a disproportionately high impact on the model performance. Specifically, the top-ranked descriptors in terms of SHAP values were Chi0v, kappa1, Chi1n, NumRotatableBonds, Chi0n, and kappa3. These descriptors were found to have a significant influence on the model's ability to predict crystal propensity, with Chi0v and kappa1 emerging as the most important features. The high SHAP values for Chi0v and kappa1 suggested that these descriptors were capturing critical aspects of molecular structure and connectivity that were closely tied to crystal propensity (Fig. 8). The Chi0v, a topological index, was likely influencing the model predictions by encoding information about molecular shape and size. Meanwhile, kappa1, a kappashape index, is thought to be contributing to the model performance by capturing information about molecular flexibility and conformational entropy. The other top-ranked descriptors, including Chi1n, NumRotatableBonds, Chi0n, and kappa3, also appear to be playing important roles in the model predictions. Chi1n and Chi0n, as topological indices, are likely to encode information about molecular connectivity and branching patterns. NumRotatableBonds, which counts the number of rotatable bonds in small molecules, is expected to influence the model predictions by capturing information about molecular flexibility and conformational entropy. Finally, kappa3, as a kappashape index, is thought to be contributing to the model performance by capturing information about molecular shape and size. Overall, the SHAP value beeswarm plot highlighted the importance of these molecular descriptors in predicting crystal propensity and provided valuable insights into the underlying mechanisms that drive crystallization.34Data clustering
The t-SNE maps provided a fascinating visual representation of the high-dimensional molecular descriptor space. The map revealed that the first component, which captured the majority of the variation, spanned a range of values from −40 to 20 (Fig. 9). This component likely represented a combination of molecular descriptors that were highly correlated with crystal propensity. The clustering of data points along this component suggested that small molecules with similar crystal propensity values tend to have similar values for these descriptors. Furthermore, the fact that the component spans a range of negative to positive values implies that there may be a threshold or cutoff value beyond which crystal propensity increases or decreases significantly. In contrast, the second component, which captured a smaller but still significant portion of the variation, spanned a range of values from −20 to 30. This component may represent a different set of molecular descriptors that are also important for crystal propensity but are less correlated with the descriptors captured by the first component. The clustering of data points along this component suggested that small molecules with similar values for these descriptors tend to have similar crystal propensity values. The fact that the second component has a smaller range of values than the first component may indicate that the descriptors captured by this component have a smaller impact on crystal propensity. Overall, the t-SNE maps provide a valuable visualization of the relationships between molecular descriptors and crystal propensity. By identifying the most important descriptors and their relationships, researchers can gain a deeper understanding of the underlying mechanisms that drive crystal formation and develop more effective strategies for designing and optimizing small molecules with improved crystal properties.35,36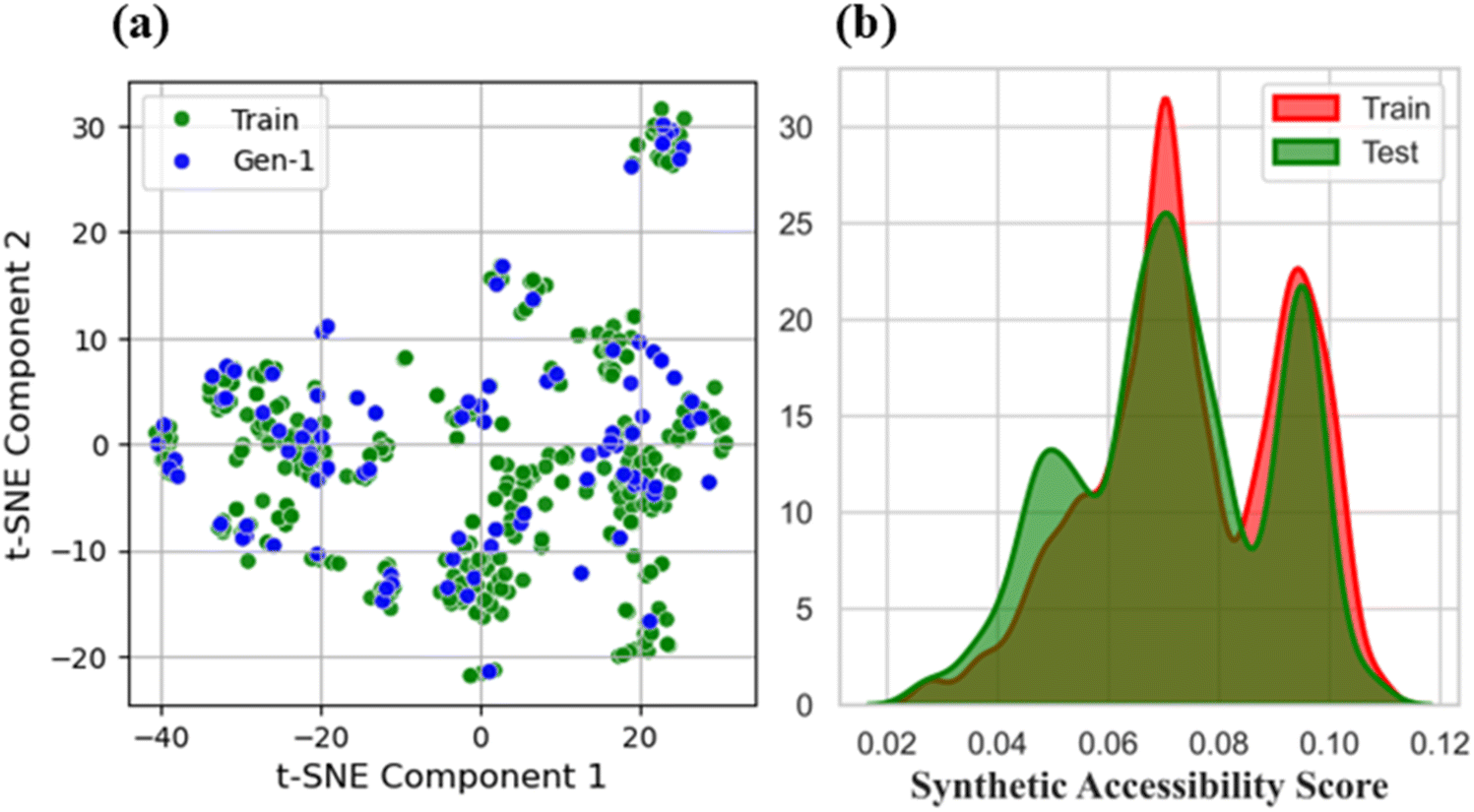 | ||
| Fig. 9 (a) t-SNE visualization of molecular descriptor clustering; (b) distribution of synthetic accessibility scores (0.02–0.12). | ||
Synthetic accessibility
The synthetic accessibility (SA) scores in this study were calculated using the method of Ertl et al.37 The score, ranging from 0.02 to 0.12, exhibits a high density of values clustering around 0.06 to 0.08, indicating a greater number of small molecules possessing scores within this range. This accumulation suggested a correlation between the synthetic accessibility score and the underlying structural or functional properties of the small molecules. A score of 0.06–0.08 might correspond to a moderate level of accessibility, making these small molecules more amenable to synthesis than those with lower scores. The highest synthetic accessibility (SA) scores were observed in small molecules 1 and 2, with scores of 0.109, accompanied by crystal propensity values of 0.987 and 0.991, respectively (Table 1).| Small molecules | SA score | Crystal propensity | Small molecules | SA score | Crystal propensity |
|---|---|---|---|---|---|
| 1 | 0.109 | 0.987 | 16 | 0.102 | 0.994 |
| 2 | 0.109 | 0.991 | 17 | 0.102 | 0.994 |
| 3 | 0.107 | 0.309 | 18 | 0.102 | 0.994 |
| 4 | 0.107 | 0.269 | 19 | 0.102 | 0.994 |
| 5 | 0.107 | 0.992 | 20 | 0.101 | 0.600 |
| 6 | 0.103 | 0.946 | 21 | 0.101 | 0.995 |
| 7 | 0.103 | 1.000 | 22 | 0.101 | 0.991 |
| 8 | 0.103 | 1.000 | 23 | 0.101 | 1.000 |
| 9 | 0.103 | 1.000 | 24 | 0.101 | 1.000 |
| 10 | 0.103 | 0.921 | 25 | 0.101 | 0.022 |
| 11 | 0.103 | 0.994 | 26 | 0.100 | 1.000 |
| 12 | 0.102 | 1.000 | 27 | 0.100 | 1.000 |
| 13 | 0.102 | 1.000 | 28 | 0.100 | 1.000 |
| 14 | 0.102 | 0.949 | 29 | 0.100 | 1.000 |
| 15 | 0.102 | 0.994 | 30 | 0.100 | 0.995 |
These small molecules exhibit a high degree of synthetic accessibility, suggesting that they can be readily synthesized. Furthermore, their high crystal propensity values indicate a strong tendency to form crystalline structures. The small molecules 5, 6, and 7 also demonstrated their high SA scores, ranging from 0.107–0.103, with crystal propensity values varying from 0.992 to 1.000. These small molecules display a moderate to high level of synthetic accessibility, and their crystal propensity values suggested a strong to absolute tendency to form crystalline structures. Notably, small molecules 12–15 and 17–20 exhibit SA scores ranging from 0.102–0.101, with crystal propensity values consistently above 0.99, indicating a high likelihood of crystallization. These small molecules demonstrate a moderate level of synthetic accessibility, making them amenable to synthesis, and their high crystal propensity values suggest that they may form crystalline structures with ease. The correlation between SA scores and crystal propensity values suggested that small molecules with higher SA scores tend to exhibit a greater propensity for crystallization. This relationship has significant implications for the design and synthesis of small molecules with specific structural and functional properties.
Conclusion
In this study, a machine learning (ML) approach to predicting the crystal propensity of indaceno-based photovoltaic small molecules has been applied. By analyzing the molecular structure of 480 small molecules, their key factors have been identified to contribute to crystallinity a predictive framework has been developed for their crystal propensity. The current results indicate that a crystallinity prevalence of 72.71% is linked to a low number of rotatable bonds (ranging from 0 to 4) and an optimal Chi0v value between 58.52 and 59.71. Near-perfect crystallinity prediction was achieved with an AUC of 0.999 and an MSE of 0.00 using RF/SVM-RBF models. Additionally, SA scores, ranging from 0.02 to 0.12, serve as an effective proxy for crystallinity, facilitating rapid screening. These results demonstrate that ML can accurately predict the crystal propensity of small molecules, paving the way for the design and optimization of high-performance candidates. The most fascinating aspect of this study is the potential for rapid screening and optimization of small molecules. By leveraging ML algorithms, a rapid evaluation can be made to predict the crystallinity (order/disorder) of thousands of small molecules to identify the most promising candidates for further development. This approach could lead to the discovery of novel small molecules with enhanced photovoltaic performance, revolutionizing the field of solar energy. Furthermore, this study opens up opportunities for future research. Integrating ML with experimental techniques (like spectroscopy and microscopy) can provide a deeper understanding of their structure–property relationships.Data availability
We confirm that the data collected and used in the present research is original and collected by the authors. It can be made public as per the requirement of the journal or may be provided upon a reasonable request to corresponding authors.Conflicts of interest
Authors declare no conflict of interest regarding the publication of this research in the form of a manuscript.Funding
This research was funded by Taif University, Saudi Arabia, Project No. (TU-DSPP-2024-93).Acknowledgements
Authors thank Taif University, Saudi Arabia, for financially supporting this research work by Project No. (TU-DSPP-2024-93)References
- S. K. Gaddam, R. Pothu and R. Boddula, Advanced polymer encapsulates for photovoltaic devices – A review, J Materiomics., 2021, 7, 920–928, DOI:10.1016/j.jmat.2021.04.004.
- Q. Hassan, P. Viktor, T. J. Al-Musawi, B. Mahmood Ali, S. Algburi, H. M. Alzoubi, A. Khudhair Al-Jiboory, A. Zuhair Sameen, H. M. Salman and M. Jaszczur, The renewable energy role in the global energy Transformations, Renew. Energy Focus, 2024, 48, 100545, DOI:10.1016/j.ref.2024.100545.
- M. R. Yazdani McCord, A. Seppälä, M. Pourakbari-Kasmaei, J. B. Zimmerman and O. J. Rojas, From low conductivity to high energy efficiency: The role of conductive polymers in phase change materials, Chem. Eng. J., 2025, 508, 160804, DOI:10.1016/j.cej.2025.160804.
- P. Rama Rao, Recent progress in the development of materials, Curr. Opin. Chem. Eng., 2014, 3, 13–17, DOI:10.1016/j.coche.2013.08.012.
- M. H. Alaaeddin, S. M. Sapuan, M. Y. M. Zuhri, E. S. Zainudin and F. M. AL- Oqla, Photovoltaic applications: Status and manufacturing prospects, Renew. Sustain. Energy Rev., 2019, 102, 318–332, DOI:10.1016/j.rser.2018.12.026.
- S. Kim, H. V. Quy and C. W. Bark, Photovoltaic technologies for flexible solar cells: beyond silicon, Mater. Today Energy, 2021, 19, 100583, DOI:10.1016/j.mtener.2020.100583.
- T. Kirchartz, G. Yan, Y. Yuan, B. K. Patel, D. Cahen and P. K. Nayak, The state of the art in photovoltaic materials and device research, Nat. Rev. Mater., 2025, 10, 335–354, DOI:10.1038/s41578-025-00784-4.
- Y.-Y. Peng, S. Srinivas and R. Narain, Chapter 2 - Nature and molecular structure of polymers. in Polymer Science and Nanotechnology, Narain, R., ed. Elsevier, 2020, pp. 13–19 Search PubMed.
- V. Podzorov, Long and winding polymeric roads, Nat. Mater., 2013, 12, 947–948, DOI:10.1038/nmat3790.
- J. H. Wendorff, The structure of amorphous polymers, Polymer, 1982, 23, 543–557, DOI:10.1016/0032-3861(82)90094-5.
- W. Zhang, C. Yang, W. Liu, H. Wang, S. Wei, J. Qi, P. Bai, B. Jin and L. Xu, Long-range order, short-range disorder: Engineering one-dimensional flow channel arrays with hierarchically porous reaction interfaces for electrocatalytic reduction of oxygen, Appl. Catal. B Environ., 2021, 293, 120199, DOI:10.1016/j.apcatb.2021.120199.
- S. B. Chun and C. D. Han, The Role of the Order–Disorder Transition Temperature of Block Copolymer in the Compatibilization of Two Immiscible Homopolymers, Macromolecules, 1999, 32, 4030–4042, DOI:10.1021/ma981665c.
- M. N. Pham, C.-J. Su, Y.-C. Huang, K.-T. Lin, T.-Y. Huang, Y.-Y. Lai, C.-A. Wang, Y.-K. Liaw, T.-H. Lin, K.-C. Wan, C.-T. He, Y.-H. Huang, Y.-P. Yang, H.-Y. Wei, U.-S. Jeng, J. Ruan, C. Luo, Y. Huang, G. C. Bazan and B. B. Y. Hsu, Forming Long-Range Order of Semiconducting Polymers through Liquid-Phase Directional Molecular Assemblies, Macromolecules, 2024, 57, 3544–3556, DOI:10.1021/acs.macromol.3c02188.
- Y. Amamoto, Data-driven approaches for structure-property relationships in polymer science for prediction and understanding, Polym. J., 2022, 54, 957–967, DOI:10.1038/s41428-022-00648-6.
- M. Gon, K. Tanaka and Y. Chujo, π-Conjugated polymers based on flexible heteroatom-containing complexes for precise control of optical functions, Polym. J., 2023, 55, 723–734, DOI:10.1038/s41428-023-00779-4.
- J. Cortese, C. Soulié-Ziakovic, M. Cloitre, S. Tencé-Girault and L. Leibler, Order–Disorder Transition in Supramolecular Polymers, J. Am. Chem. Soc., 2011, 133, 19672–19675, DOI:10.1021/ja209126a.
- Y. Zheng, Z. Tian, J. Chen, T. Jiang, L. Kong, H. Lu, D. Wang and J. Luo, A thermal history-based approach to predict mechanical properties of plasma arc additively manufactured IN625 thin-wall, J. Manuf. Process, 2025, 140, 91–107, DOI:10.1016/j.jmapro.2025.02.043.
- M. Aish, A. Ghafoor, F. Nasim, K. Ali, S. Akhter and S. Azeem, Improving Stroke Prediction Accuracy through Machine Learning and Synthetic Minority Over-sampling, J. Biomed. Inform., 2024, 07, 566 Search PubMed.
- Y. Jia, G. Chen and L. Zhao, Defect detection of photovoltaic modules based on improved VarifocalNet, Sci. Rep., 2024, 14(1), 15170, DOI:10.1038/s41598-024-66234-3.
- P. A. Beaucage, D. R. Sutherland and T. B. Martin, Automation and Machine Learning for Accelerated Polymer Characterization and Development: Past, Potential, and a Path Forward, Macromolecules, 2024, 57, 8661–8670, DOI:10.1021/acs.macromol.4c01410.
- G. Landrum, P. Tosco, B. Kelley, R. Rodriguez, D. Cosgrove, R. Vianello, S. Riniker, P. Gedeck, G. Jones, N. Schneider, E. Kawashima, D. Nealschneider, A. Dalke, M. Swain, B. Cole, S. Turk, A. Savelev, C. Tadhurst, A. Vaucher, M. Wójcikowski, I. Take, V. F. Scalfani, R. Walker, K. Ujihara, D. Probst, J. Lehtivarjo, H. Faara, G. Godin, A. Pahl and J. Monat, rdkit/rdkit: 2024_09_5 (Q3 2024) Release, 2025, https://zenodo.org/records/14779836 Search PubMed.
- A. L. Coutinho, R. Cristofoletti, F. Wu, A. Al Shoyaib, J. Dressman and J. E. Polli, Relative Performance of Volume of Distribution Prediction Methods for Lipophilic Drugs with Uncertainty in LogP Value, Pharm. Res., 2024, 41, 1121–1138, DOI:10.1007/s11095-024-03703-4.
- M. G. Brik and I. V. Kityk, Modeling of lattice constant and their relations with ionic radii and electronegativity of constituting ions of A2XY6 cubic crystals (A= K, Cs, Rb, Tl; X= tetravalent cation, Y= F, Cl, Br, I), J. Phys. Chem. Solids, 2011, 72, 1256–1260 CrossRef CAS.
- X. H. Li, A. F. Jalbout and M. Solimannejad, Definition and application of a novel valence molecular connectivity index, J. Mol. Struct., 2003, 663, 81–85, DOI:10.1016/j.theochem.2003.08.093.
- M. Müller, A. Hansen and S. Grimme, An atom-in-molecule adaptive polarized valence single-ζ atomic orbital basis for electronic structure calculations, J. Chem. Phys., 2023, 159, 164108, DOI:10.1063/5.0172373.
- P. Tosco, N. Stiefl and G. Landrum, The integration of Open3DTOOLS into the RDKit and KNIME, J. Cheminf., 2014, 6, P8, DOI:10.1186/1758-2946-6-S1-P8.
- K. Okoye and S. Hosseini, Correlation Tests in R: Pearson Cor, Kendall's Tau, and Spearman's Rho, in R Programming: Statistical Data Analysis in Research, Okoye, K. and Hosseini, S., ed. Springer Nature, Singapore, 2024, pp. 247–277 Search PubMed.
- S. Abimanyu, N. Bahtiar and E. A. Sarwoko, Implementasi Metode Support Vector Machine (SVM) dan t-Distributed Stochastic Neighbor Embedding (t-SNE) untuk Klasifikasi Depresi, Jurnal Masyarakat Informatika., 2023, 14, 146–158, DOI:10.14710/jmasif.14.2.59513.
- A. P. Marques Ramos, L. Prado Osco, D. Elis Garcia Furuya, W. Nunes Gonçalves, D. Cordeiro Santana, L. Pereira Ribeiro Teodoro, C. Antonio da Silva Junior, G. Fernando Capristo-Silva, J. Li, F. Henrique Rojo Baio, J. Marcato Junior, P. Eduardo Teodoro and H. Pistori, A random forest ranking approach to predict yield in maize with uav-based vegetation spectral indices, Comput. Electron. Agric., 2020, 178, 105791, DOI:10.1016/j.compag.2020.105791.
- P. Labute: Derivation and Applications of Molecular Descriptors Based on Approximate Surface Area, in Chemoinformatics: Concepts, Methods, and Tools for Drug Discovery, Bajorath, J., ed. Humana Press, Totowa, NJ, 2004, pp. 261–278 Search PubMed.
- M. Ring and B. M. Eskofier, An approximation of the Gaussian RBF kernel for efficient classification with SVMs, Pattern Recognit. Lett., 2016, 84, 107–113, DOI:10.1016/j.patrec.2016.08.013.
- A. U. Hassan, C. Güleryüz, I. H. El Azab, A. Y. Elnaggar and M. H. Mahmoud, Exploring the structural basis of crystals that affect nonlinear optical responses: an experimental and machine learning quest, Opt. Mater., 2025, 116783, DOI:10.1016/j.optmat.2025.116783.
- A. Koushik, M. Manoj and N. Nezamuddin, SHapley Additive exPlanations for Explaining Artificial Neural Network Based Mode Choice Models, Transp. in Dev. Econ., 2024, 10, 12, DOI:10.1007/s40890-024-00200-6.
- A. U. Hassan and M. J. Aljaafreh, A Machine Learning Study to Explore the Structural Basis of Non-Conjugated Compounds for Their Optical Activity Features, Adv. Theory Simul., 2025, e00140, DOI:10.1002/adts.202500140.
- C. Güleryüz, S. H. Sumrra, A. U. Hassan, A. Mohyuddin, A. S. Waheeb, M. A. Awad, A. R. Jalfan, S. Noreen, H. A. Kyhoiesh and I. H. El Azab, A machine learning and DFT assisted analysis of benzodithiophene based organic dyes for possible photovoltaic applications, J. Photochem. Photobiol. Chem., 2025, 460, 116157, DOI:10.1016/j.jphotochem.2024.116157.
- S. H. Sumrra, C. Güleryüz, A. U. Hassan, Z. A. Abass, T. M. Hanoon, A. Mohyuddin, H. A. Kyhoiesh and M. T. Alotaibi, Exploring structural basis of photovoltaic dye materials to tune power conversion efficiencies: A DFT and ML analysis of Violanthrone, Mater. Chem. Phys., 2025, 332, 130196, DOI:10.1016/j.matchemphys.2024.130196.
- P. Ertl and A. Schuffenhauer, Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions, J. Cheminf., 2009, 1, 8, DOI:10.1186/1758-2946-1-8.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02748a |
| This journal is © The Royal Society of Chemistry 2025 |