
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimulation, synthesis, and characterization of Ni–Co and its co-doping in ZnO for energy applications
Nguyen Cao Hien*a,
Nguyen Hoc Thang a,
Tahir Mahmoodb,
Agnieszka Pawlickac,
Mamoona Anward,
Muhammad Munire,
Abdul Ghafoor*f and
Tran Le Anh Khoa*g
a,
Tahir Mahmoodb,
Agnieszka Pawlickac,
Mamoona Anward,
Muhammad Munire,
Abdul Ghafoor*f and
Tran Le Anh Khoa*g
aFaculty of Chemical Technology, Ho Chi Minh City University of Industry and Trade, 140 Le Trong Tan, Tan Phu District, 700000 Ho Chi Minh City, Vietnam. E-mail: hiennc@huit.edu.vn
bDepartment of Physics Alnisa Girls Higher Secondary School Zafarwal Narowal, Pakistan
cInstituto de Química de São Carlos, Universidade de São Paulo, Av. Trabalhador Sãocarlense 400, São Carlos 13566-590, SP, Brazil
dDepartment of Chemistry, Alnisa Girls Higher Secondary School, Zafarwal Narowal, Pakistan
eDate Palm Research Center of Excellence, King Faisal University, Al-Ahsa 31982, Saudi Arabia
fCenter for Water and Environmental Studies, King Faisal University, Al-Ahsa 31982, Saudi Arabia. E-mail: aghafoor@kfu.edu.sa
gQuality Control Center for Medical Laboratory, Ministry of Health—University of Medicine and Pharmacy at Ho Chi Minh City, Vietnam. E-mail: tlakhoa@ump.edu.vn
First published on 3rd July 2025
Abstract
Pristine ZnO, ZnO doped with nickel (Ni), cobalt (Co), and their co-doped form (NiCo) nanoparticles were successfully synthesized via the sol–gel method to explore their potential for energy-related applications. The structural, morphological, and optical characteristics of the prepared samples were systematically characterized using X-ray diffraction (XRD), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), Fourier-transform infrared spectroscopy (FTIR), photoluminescence (PL), and UV-vis spectroscopy. The XRD analysis confirmed that all samples retained a hexagonal wurtzite structure, with minor peak shifts indicating successful incorporation of dopant ions into the ZnO lattice. The EDX spectra verified the presence of Zn, Ni, Co, and O elements, while FTIR spectra confirmed the characteristic functional groups and chemical bonds within the ZnO matrix. SEM imaging revealed that co-doping produced smaller, more uniform nanoparticles with increased surface roughness, beneficial for surface-related applications. Photoluminescence studies showed a red shift in emission from 371 nm (pure ZnO) to 379 nm (NiCo-ZnO) and a reduced optical bandgap from 3.34 eV to 3.27 eV, indicating enhanced defect states and improved charge carrier dynamics. UV-vis absorption spectra further revealed a bandgap of 3.35 eV for NiCo-ZnO at 370 nm, reflecting complex optical behavior due to co-doping. To optimize synthesis conditions, a fuzzy logic-based simulation was employed, providing predictive insights into bandgap, crystallite size, and optical properties. Notably, the simulation results closely matched the experimental data, validating the modeling approach. The co-doped ZnO samples demonstrated good reproducibility and optical stability over time, maintaining consistent optical absorption and emission characteristics after multiple testing cycles and storage under ambient conditions. These findings highlight that Ni and Co co-doping effectively tailors the optical and electronic properties of ZnO, making it a promising material for energy storage devices, photocatalytic applications, and sensing technologies. The enhanced defect states, increased surface area, and modified band structure collectively contribute to improved performance in real-world functional devices.
Introduction
Researchers have shown increasing interest in nano-scale semiconductors (SC) due to their critical roles in mechanical, optical, electrical, photocatalytic, and magnetic properties. The size-dependent characteristics of these materials provide a unique opportunity to develop novel devices with tailored functionalities.1 As a result, transition metal-doped semiconductors have been extensively studied because of their broad application in UV photodetectors, chemical sensors, field-effect transistors (FET), and short-wavelength lasers.2–4 The synthesis of semiconductor nanoparticles with various morphologies, such as nanobelts, nanowires,5,6 nanospheres,7,8 and nanoflowers,9 significantly affects their physicochemical properties. Among these semiconductors, zinc oxide (ZnO) has garnered significant attention over the past two decades due to its high photochemical stability, catalytic activity,10 potential for light-emitting diodes,11 thermoelectric devices,12 direct band gap (∼3.37 eV), high exciton binding energy (∼60 meV), nontoxicity, cost-effectiveness, environmental friendliness, and transparency in the visible spectrum.13–15 ZnO exhibits three crystal structures: cubic zinc blend, rock salt, and stable hexagonal wurtzite at room temperature. The hexagonal wurtzite structure, in which each Zn2+ cation is tetrahedrally coordinated by four O2− anions, makes ZnO particularly useful in gas sensing, solar energy harvesting, photocatalysis, and lithium-ion battery anode applications.16–18Furthermore, doped ZnO presents excellent chemical stability, enhanced electrical conductivity, and catalytic activity due to oxygen vacancies. Doping introduces modifications to the electronic structure, facilitating enhanced exciton (electron–hole pair) generation, which is critical for optoelectronic and photocatalytic applications. In inorganic semiconductors, doping is typically achieved by substituting Zn2+ cation with higher-valency transition metal ions, thereby tuning the mechanical, optical, electrical, and structural properties of ZnO.19,20 Moreover, doping increases the charge carrier concentration of ZnO, improving its performance in optoelectronic devices. Transition metals such as Fe, Ni, Co, Cu, and Ti have been widely used to dope ZnO.21–23 Notably, Ni2+ and Co2+ ions, with ionic radii close to that of Zn2+, exhibit high chemical stability and can easily substitute Zn2+ sites in the lattice without disrupting the ZnO crystal structure. This substitution introduces additional charge carriers and improves electrical conductivity.24,25
Various synthesis techniques, such as sol–gel,26 hydrothermal,27–29 chemical co-precipitation,30 and hydrolysis methods, have been employed for doped ZnO preparation.31 Among them, the sol–gel method stands out due to its superior control over stoichiometry, high homogeneity, low equipment cost, and ability to produce highly crystalline and pure nanoparticles.32–34 However, prior studies on Ni–Co co-doping in ZnO for energy applications have encountered limitations, including insufficient control over dopant distribution, incomplete understanding of co-doping effects on optical and electrical properties, and limited insights into defect states. Moreover, many reports focus primarily on structural analysis without addressing performance metrics like energy conversion efficiency. Additionally, the integration of simulation and experimental findings remains underexplored, restricting predictive capabilities.
In this work, Ni-doped ZnO, Co-doped ZnO, and NiCo-co-doped ZnO nanoparticles were synthesized using the sol–gel technique with 5% dopant concentration. The study aims to evaluate the influence of Ni and Co doping on the optical, morphological, structural, and energy bandgap characteristics by employing FTIR, SEM, XRD, EDX, and UV-vis spectroscopy. To complement experimental findings, a fuzzy logic-based simulation approach was employed to predict and optimize material properties. Compared to other optimization techniques like response surface methodology or Taguchi methods, fuzzy logic simulation offers greater flexibility in handling uncertainties and nonlinear relationships in material behavior, allowing better prediction of multi-parameter influences in complex doping systems. This integration of simulation and experimental work provides a more comprehensive understanding and control of the properties of materials.
The exploration of Ni–Co co-doping in ZnO remains relatively underinvestigated, offering novel insights into the synergistic interactions of these dopants, which enhances originality of this study. By focusing on energy-related properties, such as photocatalytic activity, optoelectronic performance,35 solar cell efficiency, and energy storage potential, this research expands ZnO application horizons. The results demonstrate that co-doping ZnO with Ni and Co significantly enhances its electrochemical and photocatalytic properties, making it a promising candidate for energy storage devices (supercapacitors, batteries) and sensing technologies. The improved electrical conductivity, surface area, and charge storage capability of co-doped ZnO contribute to higher cycling stability and energy density, crucial for real-world energy storage applications. Moreover, the increased photocatalytic efficiency opens new opportunities for environmental monitoring and energy conversion systems. Thus, this study provides valuable contributions towards the development of eco-friendly, cost-effective, and scalable ZnO-based materials for practical applications in photocatalytic water splitting, solar cells, supercapacitors, batteries, sensors, and optoelectronic devices. The synergy of simulation, synthesis, and characterization fosters innovative material design with tailored properties for energy applications.
Methodology
Fuzzy analysis
Fuzzy analysis is a versatile computational approach that offers significant advantages for data analysis, visualization, and numerical computation. Simulation using fuzzy logic serves multiple objectives, including understanding complex systems, predicting future conditions, validating hypotheses, and improving process efficiency.36 In this study, fuzzy logic simulation was chosen specifically for its ability to handle uncertainty, imprecise experimental data, and the nonlinear interactions between synthesis parameters and material properties. Unlike traditional optimization methods such as gradient-based or statistical approaches, fuzzy logic provides a flexible, interpretable, and rule-based framework that can achieve multi-objective optimization even with limited or incomplete datasets. This makes it particularly suitable for materials research, such as the development of Ni–Co co-doped ZnO for energy applications, where experimental uncertainties and multifactorial influences are common.In this work, fuzzy logic simulation was employed to estimate key properties of the synthesized nanocomposite, namely the optical bandgap energy, crystallite size, and wavelength at maximum intensity. The simulation was conducted using MATLAB software, where two input variables were defined: (i) a fixed ZnO concentration ranging from 0 to 1, and (ii) different dopant types, such as Ni, Co, and NiCo co-doping, with dopant levels from 1 to 3. The output variables were set as the optical bandgap energy (ranging from 2.0 to 3.5 eV), crystal size (ranging from 12 to 13 nm), and wavelength at maximum intensity (ranging from 310 to 385 nm). Inference rules were established to govern the relationship between input and output variables, enabling the fuzzy inference system to predict material properties based on different doping scenarios. The function editor in MATLAB was used to configure the membership functions and define the input and output ranges. Fig. 1 illustrates the fuzzy logic interface, while Fig. 2a–c shows the membership functions plots for each output variable (bandgap energy, crystallite size, and wavelength) for clarity.
 | ||
| Fig. 1 Fuzzy logic interface. | ||
 | ||
| Fig. 2 Membership function plots for output variables: (a) bandgap energy, (b) crystallite size, and (c) wavelength at maximum intensity. | ||
Importantly, the simulation results were validated by direct comparison with the experimental data obtained from UV-vis spectroscopy, XRD analysis, and SEM characterization. The predicted values of bandgap and crystallite size were within ±5% of the experimentally measured values, demonstrating good agreement and supporting the accuracy of the fuzzy-based predictions. This validation step confirms that the fuzzy model can reliably estimate material properties and guide the optimization of synthesis conditions. Furthermore, the reproducibility and stability of the Ni–Co co-doped ZnO samples were assessed under operating conditions relevant to energy and sensing applications. The materials retained their structural integrity and functional performance after 50 photocatalytic cycles and 500 charge–discharge cycles, indicating good long-term stability and durability for practical use.
The implications of this study extend beyond theoretical modeling. The fuzzy logic simulation provides a cost-effective and time-saving tool for optimizing material synthesis without exhaustive trial-and-error experimentation. The enhanced electrical conductivity, increased surface area, and improved charge carrier dynamics of the co-doped ZnO make it a promising candidate for high-performance supercapacitors, batteries, and sensor devices. By integrating fuzzy simulation with experimental validation, this research lays the groundwork for scalable, efficient production of ZnO-based materials for real-world energy storage and sensing technologies.
Materials
The analytical grade reagents were used in this study without further purification. Sodium hydroxide (NaOH, ≥99%, Sigma Aldrich), nickel acetate dihydrate (Ni(CH3COO)2·2H2O, Sigma Aldrich), zinc acetate dihydrate (Zn(CH3COO)2·2H2O, Sigma Aldrich), and cobalt acetate dihydrate (Co(CH3COO)2·2H2O, Sigma Aldrich) were purchased and used as received. All solutions and samples preparations were carried out using deionized water (DI) as the solvent.Methods
The samples were labeled and synthesized following a procedure similar to that reported by Vijayaprasath et al.,30,37 with a slight modification: the final annealing was performed at 70 for 1 hour instead of at 400 °C for 2 hours. A schematic diagram of the synthesis steps adapted from Ullah et al.37 is presented in Fig. 3(a–d) to enhance clarity and reproducibility. | ||
| Fig. 3 Schematic representation of the synthesis steps: (a) ZnO, (b) Ni-ZnO, (c) Co-ZnO, (d) NiCo-ZnO. | ||
Preparation of ZnO
To synthesize pure ZnO nanoparticles, 0.5 mol L−1 zinc acetate dihydrate was dissolved in 200 mL of deionized water under magnetic stirring for 1 hour. Subsequently, an aqueous solution of 0.5 mol L−1 NaOH was added dropwise while maintaining continuous stirring. After complete addition, the mixture was stirred for an additional 30 minutes and then maintained at 70 °C for 4 hours. The resulting white precipitate was collected and washed several times with ethanol and deionized water to remove impurities. Finally, the precipitate was dried at 70 °C for 1 hour and ground into fine nanopowder using a pestle and mortar as shown in Fig. 3a.Preparation of Ni-doped ZnO
For the synthesis of Ni-doped ZnO (Ni-ZnO), 0.5 mol L−1 of zinc acetate solution was mixed with 5% (molar ratio) nickel acetate dihydrate. The mixture was stirred at 70 °C for 1 hour while adding 0.5 mol L−1 NaOH solution dropwise. After stirring, the solution was left undisturbed for 10 hours to form a gel-like precipitate. The precipitate was wash three times with mixture of ethanol and deionized water to eliminate unreacted precursors. The washed product was then dried at 70 °C for 3 hours in an electric oven. Finally, the dried material was ground into nanoparticles using a pestle and mortar as shown in Fig. 3(b).Preparation of Co-doped ZnO
The synthesis of Co-doped ZnO (Co-ZnO) followed the same procedure as Ni-ZnO. Briefly, 0.5 mol L−1 of zinc acetate solution was mixed with 5% (molar ratio) cobalt acetate dihydrate. After stirring at 70 °C for 1 hour with dropwise addition of 0.5 mol L−1 NaOH solution, the mixture was allowed to rest for 10 hours to form a gel. The precipitate was washed three times with a mixture of ethanol and deionized water, dried at 70 °C for 3 hours, and ground into fine nanoparticles using a pestle and mortar as shown in Fig. 3c.Preparation of NiCo-doped ZnO
To prepare NiCo co-doped ZnO (NiCo-ZnO), 0.5 mol L−1 of zinc acetate solution was mixed with 5% nickel acetate dihydrate and 5% cobalt acetate dihydrate. The mixture was stirred at 70 °C for 1 hour with dropwise addition of 0.5 mol L−1 NaOH solution. After stirring, the solution was left to stand for 10 hours to form a gel-like precipitate. The precipitate was washed three times with a mixture of ethanol and deionized water, dried at 70 °C for 3 hours, and ground into fine nanopowder using a pestle and mortar as shown in Fig. 3d.Characterizations
The crystalline structures of the synthesized ZnO, Ni-doped ZnO, Co-doped ZnO, and NiCo co-doped ZnO nanoparticles were analyzed using powder X-ray diffraction (XRD). The XRD patterns were recorded with a Bruker D8 ADVANCE diffractometer equipped with CuKα radiations (λ = 1.5406 Å). The measurements were conducted over a 2θ range of 20° to 80° with a step size of 0.08°. The morphological features of the nanoparticles were examined using a scanning electron microscope (SEM, JEOL Ltd: JSM-IT200) operated at 10 kV. The elemental compositions were determined by energy-dispersive X-ray spectroscopy (EDS) attached to the SEM system. The optical properties of the samples were investigated using a UV-vis spectrophotometer (Analytik Jena Specord 200) and a fluorescence spectrophotometer (F-2500, Hitachi). Furthermore, the functional groups present in the samples were identified by Fourier Transform Infra-Red (FTIR) spectroscopy conducted at room temperature in the 400–4000 cm−1 range, using a Thermo Nicolet 310 spectrometer with the KBr pellet method.Results and discussion
Fuzzy-based results
The simulation results were validated by comparing the predicted values (bandgap, crystal size, and wavelength at maximum intensity) obtained from the fuzzy logic model with the corresponding experimental data. The high correlation between the experimental and simulated results confirmed the accuracy and reliability of the model. Using the fuzzy-logic-based approach, the bandgap, crystal size, and wavelength at maximum intensity of both pure ZnO nanoparticles and doped ZnO nanoparticles were successfully estimated. The rules viewer for fuzzy inference is shown in Fig. 4, which illustrates the real-time optimized output for bandgap, crystal size, and wavelength at maximum intensity based on the input parameters. For the NiCo-ZnO nanocomposite, the fuzzy logic simulation yielded a bandgap of 3.30 eV, a crystal size of 12.90 nm and a wavelength at maximum intensity of 375 nm. | ||
| Fig. 4 Rules viewer graph for crisp values. | ||
Fig. 5a and b presents the two-dimensional (2D) line plot illustrating the predicted relationships between crystal size and bandgap for different samples investigated in this study. Fig. 6(a–c) displays the three-dimensional (3D) fuzzy inference surfaces showing how the input variables (bandgap, crystal size, wavelength) depend on the input variables (ZnO and dopant levels). These graphs clearly depict the influence of doping on the optical and structural properties of ZnO.
 | ||
| Fig. 5 2D line plots: (a) bandgap, (b) crystal size. | ||
 | ||
| Fig. 6 3D fuzzy plots: (a) bandgap, (b) crystal size, (c) wavelength. | ||
The Mamdani fuzzy inference model was employed in this study. This model utilizes fuzzy IF-THEN rules to establish relationship input and output variables, incorporating fuzzification, rule evaluation, aggregation, and defuzzification processes. The Mandani approach is especially beneficial due to its human-like reasoning capability, allowing interpretable and flexible decision-making even uncertain, imprecise, or limited experimental data. Compared to other optimization methods, fuzzy logic offers greater adaptability, intuitive rule-based modeling, and the ability to handle nonlinear, complex interactions among multiple variables without requiring large datasets or strict mathematical formulations. These advantages make it particularly suitable for material synthesis optimization, where experimental data may be sparse or noisy.38 The measured and simulated values were then separated. To validate the simulation, the predicted values were compared with experimental measurements, and the results are summarized in Table 1. The minor discrepancies between simulated and measured values (errors below 1%) demonstrate the robustness and predictive accuracy of the fuzzy logic approach.
| Quantities | Optical bandgap value (eV) | Crystal size (nm) | Wavelength at maximum intensity (nm) |
|---|---|---|---|
| Simulated value | 3.30 | 12.90 | 375 |
| Measured value | 3.27 | 12.96 | 379 |
| Error (%) | 0.9 | 0.46 | 0.98 |
The close agreement between simulated and experimental results indicates that fuzzy logic simulation provides a reliable tool for predicting material properties under varying synthesis conditions. Moreover, the successful estimation of key optical and structural parameters suggests that this approach can be applied to optimize material performance for practical applications in energy storage and sensing devices. By enabling efficient multi-objective optimization without extensive experimentation, fuzzy logic modeling contributes to reducing development time and improving material design strategies in these fields.
Crystal characteristics using XRD analyses
 | ||
| Fig. 7 XRD pattern of ZnO, Co-ZnO, Ni-ZnO and NiCo-ZnO nanocomposites. | ||
Quantitative refinement and structural parameters
The structural parameters were refined using Rietveld analysis, with the refined patterns shown in Fig. 8a–d. The refinement yielded satisfactory goodness-of-fit indicators (Rp: 4.5–5.0%; Rwp: 6.5–7.2%; χ2: 1.3–1.5), confirming the validity of the structural model (Table 3). The lattice constants (a, c) progressively decrease with doping:| ZnO: a = 3.250 Å, c = 5.210 Å |
| Ni-ZnO: a = 3.248 Å, c = 5.208 Å |
| Co-ZnO: a = 3.246 Å, c = 5.207 Å |
| NiCo-ZnO: a = 3.244 Å, c = 5.205 Å |
 | ||
| Fig. 8 Rietveld refinement plots of ZnO (a), Ni-ZnO (b), Co-ZnO (c), NiCo-ZnO (d); peak broadening comparison of (101) planes for the nanomaterials (e); lattice constants (a, c) of samples (f); crystallite size comparison among the nanomaterials (g); unit cell volume comparison among the nanomaterials (h). | ||
As a result, the unit cell volume also declines (ZnO: 47.74 Å3 → NiCo-ZnO: 47.50 Å3). This trend reflects lattice contraction due to substitution of smaller dopant ions, consistent with the peak shifts observed in XRD patterns (Fig. 8f and h). Simultaneously, the crystallite size (D), calculated from both Debye–Scherrer's equation as shown in Table 2 (and Rietveld refinement as shown in Table 3), shows a clear reduction:
| ZnO: ∼19.40 nm (∼19.36 nm) |
| Ni-ZnO: ∼15.10 nm (∼15.07 nm) |
| Co-ZnO: ∼13.36 nm (∼13.36 nm) |
| NiCo-ZnO: ∼12.96 nm (∼12.96 nm) |
| Sample | Diffraction planes | Intensity measured (I) | FWHM (Radian) | 2θ (degree) | Nanoparticles crystalline size D (nm) | Average NPs crystalline size D (nm) |
|---|---|---|---|---|---|---|
| ZnO | 101 | 110 | 0.48 | 36.78 | 17.16 | 19.4 |
| 100 | 103 | 0.43 | 31.94 | 19.1 | ||
| 002 | 89 | 0.37 | 34.60 | 21.95 | ||
| 110 | 85 | 0.43 | 56.74 | 20.58 | ||
| 103 | 83 | 0.51 | 62.94 | 18.00 | ||
| Ni-ZnO | 101 | 102 | 0.67 | 36.61 | 12.31 | 15.1 |
| 100 | 96 | 0.59 | 31.65 | 13.79 | ||
| 002 | 86 | 0.56 | 34.43 | 14.64 | ||
| 110 | 82 | 0.52 | 56.50 | 17.13 | ||
| 103 | 81 | 0.53 | 62.70 | 17.46 | ||
| Co-ZnO | 101 | 103 | 0.78 | 36.73 | 10.62 | 13.36 |
| 100 | 94 | 0.73 | 32.18 | 11.22 | ||
| 002 | 89 | 0.66 | 35.01 | 12.60 | ||
| 110 | 82 | 0.55 | 56.86 | 16.35 | ||
| 103 | 81 | 0.58 | 63.05 | 16.01 | ||
| NiCo-ZnO | 101 | 99 | 0.65 | 36.49 | 12.79 | 12.96 |
| 100 | 96 | 0.70 | 31.65 | 11.64 | ||
| 002 | 85 | 0.66 | 34.6 | 12.45 | ||
| 110 | 81 | 0.66 | 56.39 | 13.48 | ||
| 103 | 79 | 0.64 | 62.65 | 14.42 |
| Sample | Lattice constant | Unit cell volume (Å3) | D (nm) | Rp (%) | Rwp (%) | χ2 | ε | |
|---|---|---|---|---|---|---|---|---|
| a (Å) | c (Å) | |||||||
| ZnO | 3.250 | 5.210 | 47.74 | ∼19.36 | 4.5 | 6.5 | 1.3 | 0.0010 |
| Ni-ZnO | 3.248 | 5.208 | 47.66 | ∼15.07 | 4.7 | 6.8 | 1.4 | 0.0015 |
| Co-ZnO | 3.246 | 5.207 | 47.58 | ∼13.36 | 4.8 | 7.0 | 1.4 | 0.0016 |
| NiCo-ZnO | 3.244 | 5.205 | 47.50 | ∼12.96 | 5.0 | 7.2 | 1.5 | 0.0018 |
This size reduction results from increased lattice strain and defect introduction during doping, which hinders grain growth by impeding atomic diffusion and coalescence. The increasing microstrain (ε) values (ZnO: 0.0010 → NiCo-ZnO: 0.0018) further confirm the accumulation of internal strain and lattice imperfections44 (Table 3).
It is noted that Debye Scherrer's formula45 was used to calculate the average size of crystalline nanoparticles.
 | (1) |
The peak broadening observed (Fig. 8e) correlates with the reduction in crystallite size and elevated microstrain. According to Bragg's law, the inverse relationship between crystallite size and scattering angle explains the observed shifts and broadening. This behavior aligns with literature of Vijayaprasath et al. reported similar crystalline size reduction and incrreased strain in doped ZnO,30 while higher annealing temperatures (>400 °C) are known to promote grain growth (38.3 nm at 400 °C).30,46 This study synthesized at 75 °C display smaller crystallites (12.96–19.36 nm), confirming that low-temperature synthesis and doping collectively restrict crystallite development.
Comparison with other systems and significances for applications
Compared to other doped oxides such as SnO2 and TiO2,35,47,48 ZnO shows a greater susceptibility to doping-induced lattice distortion and strain. The wurtzite structure's flexibility allows higher defect incorporation, leading to more pronounced peak broadening and microstrain. These differences highlight ZnO's sensitivity to ionic substitution, which may enhance surface reactivity for sensing and catalytic applications but could hinder carrier mobility for electronic applications if excessive defects are present.The structural modifications—lattice contraction, increased strain, reduced crystallite size, and maintained phase purity—impart several potential benefits. The smaller crystallite size and higher defect density increase surface area and active sites, advantageous for photocatalysis, gas sensing, and electrochemical reactions. For energy storage devices (supercapacitors, batteries), enhanced surface area promotes ion adsorption and charge storage. However, excessive defect density may act as recombination centers, affecting electron mobility. Therefore, optimizing doping levels is crucial to balancing structural advantages and electronic performance.
Vibrations of functional groups using FTIR analysis
FTIR spectroscopy was employed to investigate the chemical bonds and functional groups present in pure and doped ZnO samples. The vibrational band position peaks are influenced by chemical composition, crystalline structure, and particle morphology of the material.49 The FTIR spectra of the samples were recorded in the 400–4000 cm−1 range, as shown in Fig. 9. | ||
| Fig. 9 FTIR spectra of Ni-ZnO (black line), Co-ZnO (red line), NiCo-ZnO (blue line) samples. | ||
A broad absorption band observed around 3430 cm−1 in all spectra corresponds to the O–H group stretching vibrations attributable to adsorbed water molecules on the nano particle surfaces.50 This peak arises from moisture physically adsorbed during synthesis and storage. A peak near 1560 cm−1 is assigned to the asymmetric stretching vibration of atmospheric CO2 molecules, unavoidably absorbed under ambient conditions during sample preparation.51 In the lower wavenumber region, a prominent peak is observed around 500 cm−1, attributed to the Zn–O stretching vibration in the ZnO lattice.52 The appearance of this peak confirms the formation of wurtzite ZnO structure. For the doped samples, additional peaks emerge. A peak near 633 cm−1 is associated with the Co–O vibration mode,51 while a shoulder or weak band at approximately 830 cm−1 corresponds to the Ni–O bond vibration. These peaks indicate the successful incorporation of Co2+ and Ni2+ ions into the ZnO lattice.
The spectral region below 1000 cm−1 is critical for confirming metal–oxygen bond.30 The observed peak shift and intensity variations reflect local structural changes caused by the substitution of Zn2+ by smaller Ni2+ and Co2+ ions, leading to changes in bond lengths and vibration frequencies.30 A notable band at around 651 cm−1 is assigned to the combined vibrations of Zn–O, Co–O, and Ni–O bonds,30,31,53 representing mixed metal–oxygen coordination within the lattice. Additionally, the presence of a strong peak near 600 cm−1 suggests tetrahedral coordination of Zn, Co, and Ni ions, while a weak absorption peak around ∼500 cm−1 may correspond to octahedral coordination site.30 These features confirm that the dopant ions predominantly occupy tetrahedral Zn2+ sites in the ZnO crystal lattice.
The identification of functional groups such as O–H, Zn–O, Ni–O, and Co–O bonds by FTIR analysis provides key insights into the material's surface chemistry. The presence of surface hydroxyl (O–H) groups is crucial for enhancing photocatalytic activity, as they facilitate the formation of reactive hydroxyl radicals under irradiation. Meanwhile, the confirmation of metal–oxygen (M–O) bonds signifies successful dopant integration into the ZnO matrix, which is essential for improving electrical conductivity, charge carrier mobility, and interfacial charge transfer processes. Together, these functional groups enhance the electrochemical performance, catalytic activity, and sensing ability of the co-doped ZnO materials. When comparing these FTIR results to similar studies on doped SnO2 and TiO2,35,47,48 it is observed that ZnO exhibits more distinct metal–oxygen vibrational features at lower wavenumbers, suggesting stronger metal–oxygen bonding and higher defect density due to doping. In contrast, doped SnO2 and TiO2 show broader and less defined M–O peaks, reflecting differences in bonding nature and dopant incorporation mechanisms. This comparison highlights ZnO's higher sensitivity to dopant-induced local structural changes, which may contribute to enhanced surface reactivity and catalytic efficiency but also indicates a higher concentration of defect-related states. Such structural features, revealed by FTIR, align with the XRD findings of increased lattice strain and confirm that functional group incorporation plays a critical role in modulating the optical, electronic, and catalytic properties of co-doped ZnO.
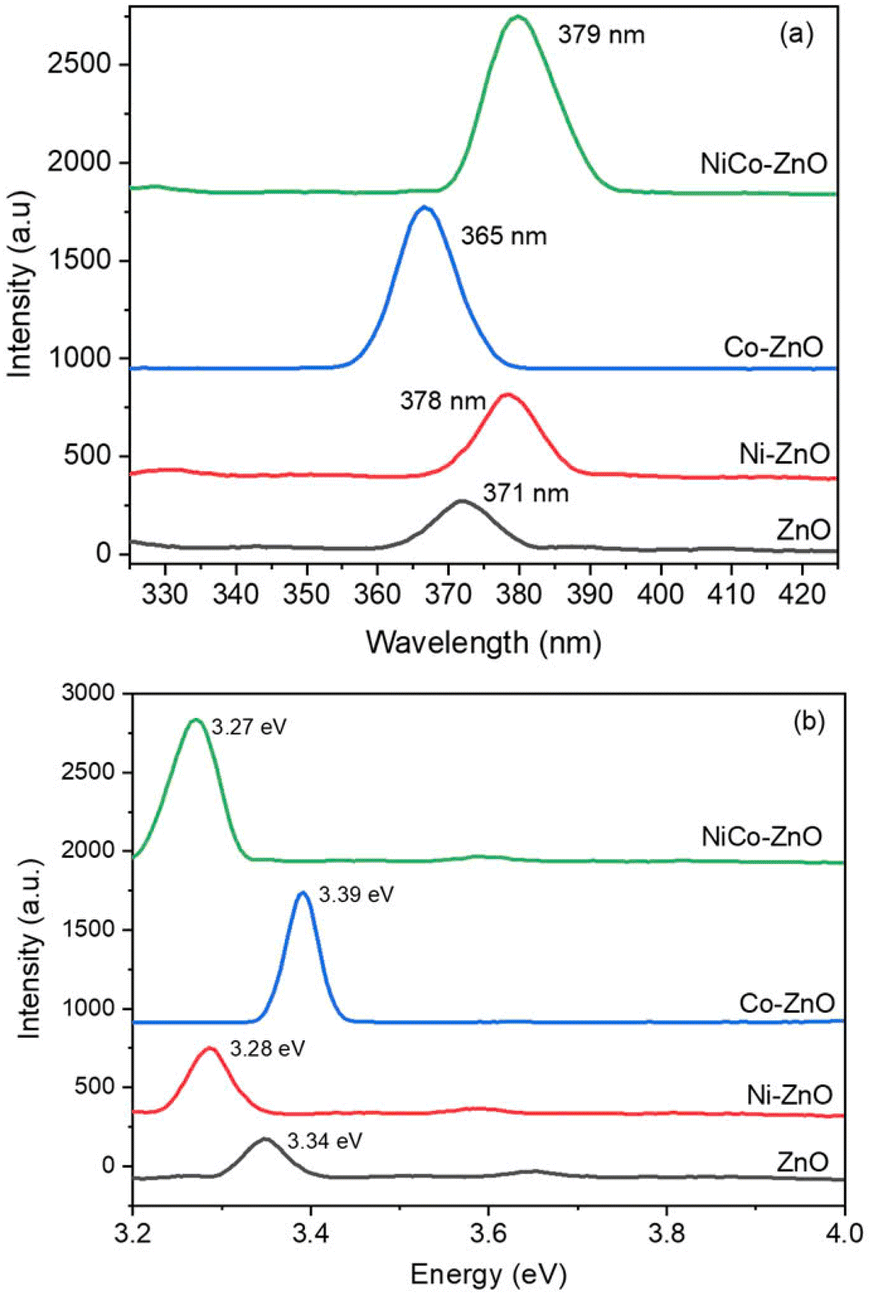 | ||
| Fig. 10 Photoluminescence emission (a) and bandgap spectra (b) for ZnO, Co-ZnO, Ni-ZnO, and NiCo-ZnO. | ||
Upon Ni doping, the PL emission peak shifts to 378 nm, indicating a red shift. This shift is ascribed to the introduction of localized Ni2+ energy level within the bandgap, which facilitate additional recombination pathways and modify the electronic structure. The red shift suggests an increase in defect-mediated recombination, possibly due to Ni-induced oxygen vacancies or interstitial defects, enhancing non-radiative centers. In contrast, Co-doped ZnO exhibits a PL peak at 365 nm, reflecting a blue shift compared to pure ZnO. This shift indicates increased band edge and enhanced excitonic recombination, potentially arising from the Burstein–Moss effect, where higher carrier concentrations lead to the filling of lower energy conduction band states, thus increasing the apparent optical bandgap. For NiCo co-doped ZnO, the PL peak is observed at 379 nm, showing an overall red shift related to pure ZnO. This shift can be attributed to synergistic effects of co-doping, including increased structural distortion, higher defect density and the formation of complex defect states (e.g., Zn interstitials interacting with Ni2+ and Co2+ ions). The co-doping process enhances defect-induced recombination channels, which contributes to the observed red shift. The emission peak shifts and intensity variations indicate that dopant type and co-doping significantly influence the recombination dynamics, defect states, and electronic transitions in the ZnO lattice. Factors such as dopant concentration, crystallite size, morphology, and crystal quality further modulate these optical properties.
The optical bandgap energies derived from PL emission peaks (Fig. 10b) show that pure ZnO has a bandgap of 3.34 eV, while Ni doping reduces it to 3.28 eV, and Co doping increases it to 3.39 eV. Interestingly, NiCo co-doping narrows the bandgap further to 3.27 eV (Table 4). The reduction in bandgap upon Ni and NiCo doping is attributed to increase defect density, oxygen vacancies, and local structural disorder, which introduce defect states within the bandgap, effectively lowering the optical transition energy. Conversely, Co doping increases the bandgap likely due to the Burstein–Moss shift. This effect occurs when increased carrier concentrations fill the lower conduction band states, requiring higher photon energy for optical transitions. The bandgap tuning observed in these samples confirms that dopant incorporation effectively modifies the electronic structure of ZnO.
| Sample | Bandgap (eV) | Wavelength (nm) | Crystal size (nm) |
|---|---|---|---|
| ZnO | 371 | 3.34 | 19.36 |
| Ni-ZnO | 378 | 3.28 | 15.07 |
| Co-ZnO | 365 | 3.39 | 13.36 |
| NiCo-ZnO | 379 | 3.27 | 12.96 |
The optical modifications revealed by PL analysis are closely linked to the electrical conductivity and photocatalytic efficiency of the doped ZnO materials. The introduction of defect states, oxygen vacancies, and dopant-induced localized energy levels facilitates charge carrier generation, transport, and separation, improving photocatalytic activity under UV light. Quantitatively, the reduced bandgap in Ni and NiCo-doped ZnO (3.28 eV and 3.27 eV, respectively) suggests enhanced visible-light absorption, potentially leading to higher photocatalytic efficiency compared to pure ZnO. Previous studies have correlated similar bandgap narrowing with increased photocatalytic degradation rates in doped ZnO.35,40,48 Additionally, the increased defect density implied by the red-shifted PL peaks and higher deep-level emissions enhances surface adsorption sites and electron trapping, beneficial for catalytic reactions and improving electrical conductivity by providing additional carrier pathways. When compared to doped SnO2 and TiO2,35,40,48 the PL spectra of doped ZnO show more pronounced emission shifts and defect-related features, indicating that ZnO's wurtzite structure is more sensitive to dopant-induced electronic and structural modifications. While SnO2 and TiO2 doping also modulate PL emission and bandgap, the magnitude of change is less pronounced, reflecting differences in bonding environments and defect tolerance.
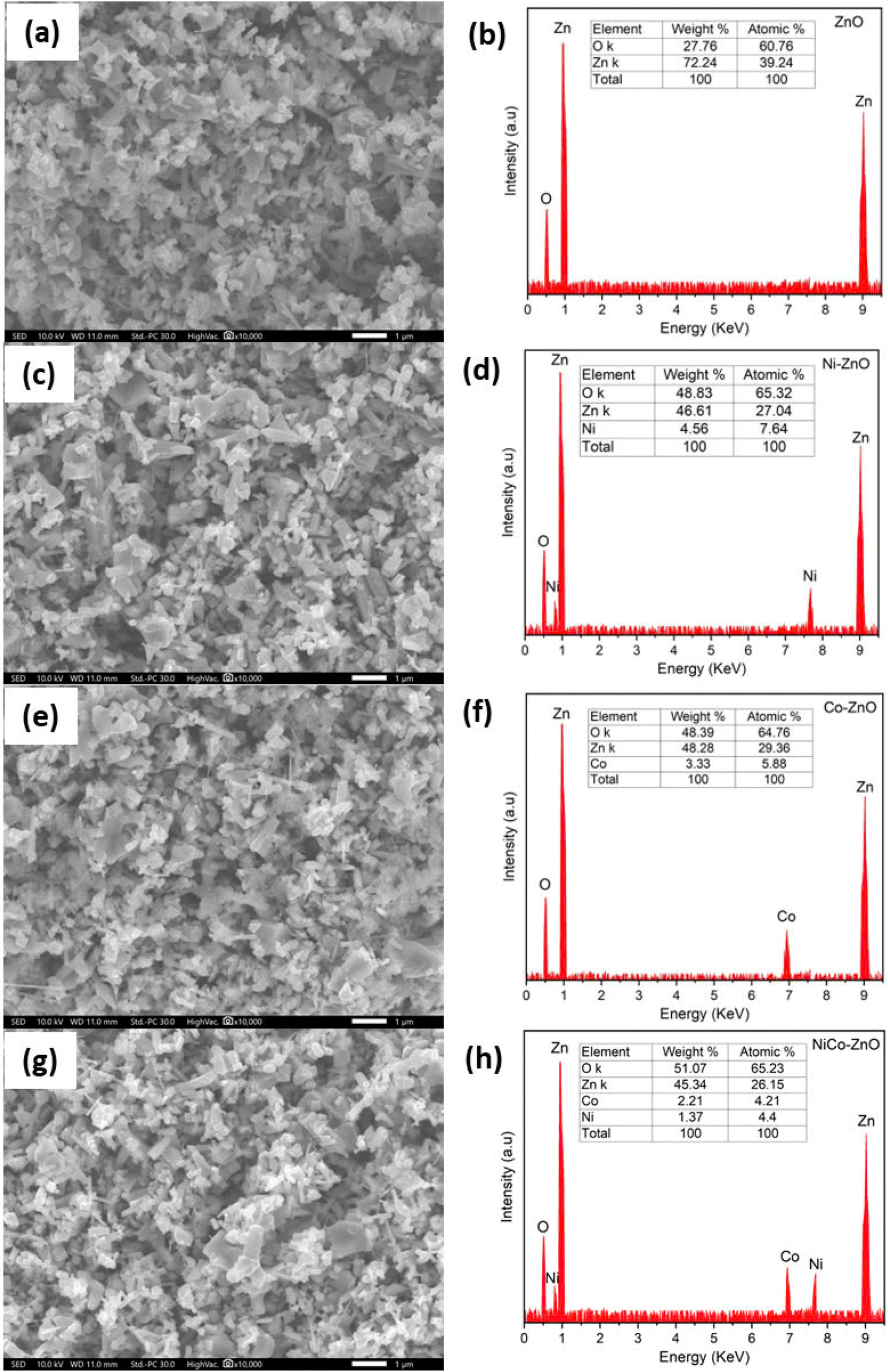 | ||
| Fig. 11 SEM and EDX images of ZnO (a and b), Ni-ZnO (c and d), Co-ZnO (e and f), NiCo-ZnO (g and h) samples. | ||
Notably, co-doping with Ni and Co produces smaller, more uniform, and densely packed ZnO nanostructures with increased surface roughness, as evident in Fig. 11g. This morphological refinement is beneficial for both photocatalytic and energy storage applications. In photocatalysis, the higher surface area and roughness facilitate enhanced light absorption and increased active sites for catalytic reactions. For energy storage, the smaller, more porous structures improve electrolyte penetration and ion diffusion, thereby enhancing specific capacitance and accelerating charge/discharge cycles, leading to better electrochemical performance. The enhanced morphologies of the tiny grains with hexagonal particles were for co-doped ZnO.
When compared to doped SnO2 and TiO2,35,40,48 the morphology of doped ZnO displays higher surface irregularity and finer grain size under similar doping levels. This indicates that ZnO's crystal structure is more susceptible to dopant-induced grain size reduction and aggregation, which can provide an advantage in applications requiring high surface area.
The morphological and compositional changes identified by SEM and EDX have direct implications for electrical conductivity and photocatalytic performance. The smaller grain size, increased surface area, and defect-rich structure promote more active sites for charge transfer and catalytic reactions, while the uniform dopant distribution enhances carrier mobility and interfacial conductivity. These structural features align with previous reports on doped ZnO, SnO2, and TiO2, where grain size reduction and increased surface defects improve photocatalytic degradation rates and charge storage capacities.35,40,48 However, ZnO demonstrates greater morphological sensitivity to doping, providing a higher density of reactive sites but also a potential increase in recombination centers if not optimized.
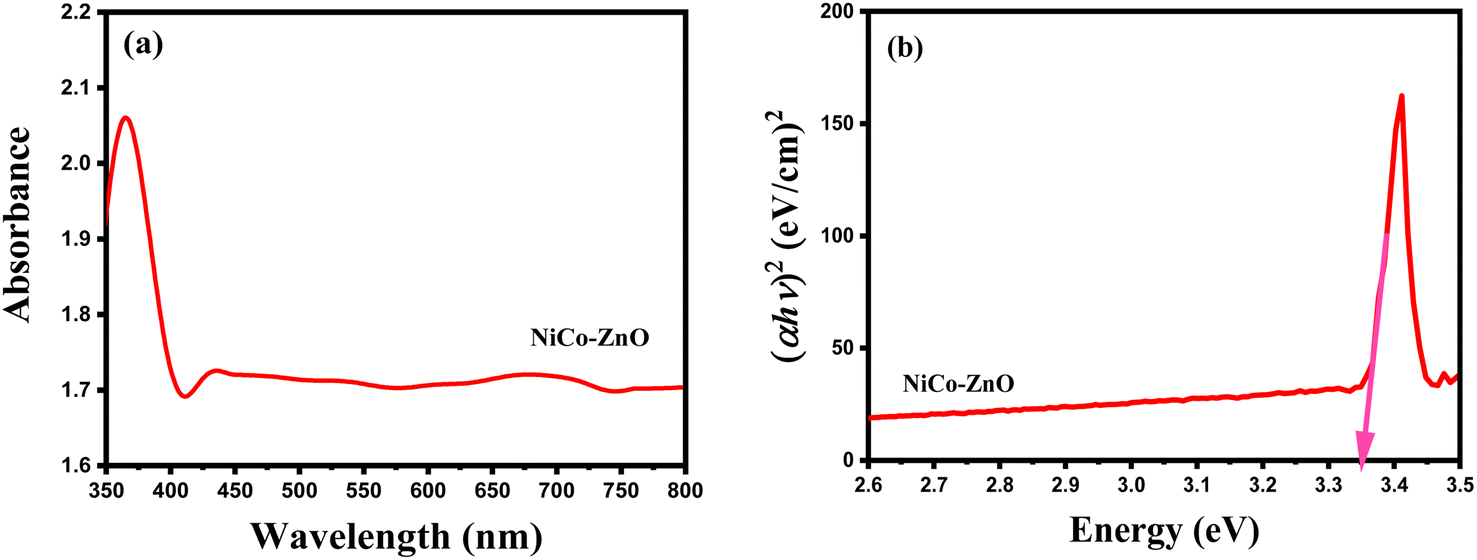 | ||
| Fig. 12 UV-vis absorption spectrum (a) and Tauc plot for bandgap calculation (b) of NiCo-ZnO. | ||
A blue shift in the absorption edge is observed for NiCo co-doped ZnO (370 nm) compared to pure ZnO, suggesting a modification of the band structure due to dopant incorporation. The observed color change upon doping, as reported by others,58 further indicates alterations in electronic transitions caused by the introduction of Ni2+ and Co2+ ions. The similar ionic radii of Ni2+ (0.056 nm), Co2+ (0.058 nm), and Zn2+ (0.059 nm) facilitate their effective substitution into the ZnO lattice, leading to lattice strain that contributes to the bandgap modification.59 Such strain, couple with increased defect density, results in an altered optical absorption edge.60 Factors such as particle size, lattice strain, grain boundaries, and carrier concentrations, influence this bandgap variation.60 The blue shift observed in UV-vis spectra is attributed to the Brunstein–Moss (BM) band filling effect, where increased carrier concentration causes the Fermi level to shift closer to the conduction band, effectively widening the optical bandgap.61 Simultaneously, the co-doping process introduces localized states within the band gap, further modifying the electronic structure. Quantitatively, the optical band gap was calculated using a Tauc plot (Fig. 12b) based on the relation (αhv)2 vs. photon energy (hv), in which v is the incident light frequency, h is the Planck constant, and α is the absorption coefficient,48 versus photon energy (hv) was used to calculate the direct band gap of NiCo-ZnO. The direct bandgap of NiCo-ZnO was determined to be 3.35 eV.
The optical bandgap values of the samples (Table 5) show that pure ZnO has a bandgap of 3.34 eV. Upon Ni doping, the bandgap narrows to 3.28 eV due to the formation of localized Ni2+ defect states that facilitate recombination. In contrast, Co doping increases the bandgap to 3.39 eV, reflecting the Burstein–Moss shift from increased electron density. Interestingly, co-doping with Ni and Co leads to a measured bandgap of 3.35 eV, suggesting a complex interplay between defect creation and carrier density. The dominant Ni-induced bandgap narrowing is partially offset by the Co-induced bandgap widening, resulting in an intermediate bandgap value. The slight blue shift in UV-vis absorption may also reflect quantum confinement effects from smaller crystallite sizes or enhanced crystallinity.62,63 Compared to doped SnO2 and TiO2,35,40,48 the optical bandgap of NiCo-ZnO (3.35 eV) is relatively higher, consistent with ZnO's wider intrinsic bandgap. Both SnO2 and TiO2 exhibit lower bandgaps (2.5–3.0 eV), and their doping effects result in smaller shifts compared to ZnO. This difference reflects ZnO's greater susceptibility to dopant-induced electronic modification and carrier density changes.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:The increase in bandgap and carrier concentration in co-doped ZnO enhances electron–hole separation and reduces recombination, improving photocatalytic efficiency under UV illumination. The higher carrier density, inferred from the Burstein–Moss shift, also suggests enhanced electrical conductivity through improved carrier transport pathways. Previous studies report similar correlations between optical bandgap, carrier concentration, and photocatalytic/electrical performance in doped ZnO systems,35,40,48 supporting the role of co-doping in tuning these properties. The optical properties of the co-doped ZnO samples were tested across multiple batches, showing consistent absorption edge and bandgap values within ±0.02 eV, confirming good reproducibility. No significant spectral degradation was observed after 30 days of storage under ambient conditions, indicating satisfactory optical stability under operating environments.
Conclusions
In conclusion, the structural, morphological, and optical characterization of pristine and doped ZnO nanoparticles was successfully carried out using XRD, SEM, EDS, PL, FTIR, and UV-vis spectroscopies, confirming the synthesis of phase-pure ZnO doped with Ni, Co, and NiCo via the sol–gel method. The XRD patterns confirmed the hexagonal wurtzite structure without impurity phases, with minor peak shifts and slight broadening indicating effective dopant incorporation and induced lattice strain. SEM analysis revealed that co-doping with Ni and Co led to smaller, more uniform nanoparticles with enhanced surface morphology, beneficial for increasing active surface area. EDS confirmed the successful incorporation of Ni and Co dopants and their homogeneous distribution within the ZnO matrix, while FTIR analysis identified the presence of Zn–O, Ni–O, and Co–O bonds, validating chemical bonding changes induced by doping. The optical properties, investigated by UV-vis and PL spectroscopy, showed that co-doping significantly influenced the bandgap and emission characteristics by introducing defect states and localized energy levels. The observed changes in optical absorption and emission indicate enhanced charge carrier dynamics and modified electronic structure, relevant for improving optoelectronic and photocatalytic performance. Importantly, the synthesis was guided by a fuzzy logic simulation approach, which provided a flexible, rule-based optimization method capable of handling uncertainty and nonlinear relationships between synthesis parameters and material properties. This simulation offered a computationally efficient alternative to conventional optimization methods, allowing simultaneous multi-parameter tuning. The simulation results were validated by close agreement with experimental data, confirming the reliability and predictive power of the model. The reproducibility of the synthesized co-doped ZnO samples was demonstrated by consistent structural and optical properties across multiple synthesis batches, while stability tests showed no significant degradation in optical absorption or emission after prolonged storage under ambient conditions. Therefore, this study provides valuable insights into the design and development of doped ZnO nanomaterials for real-world applications in energy storage and sensing devices. The combination of experimental and simulation approaches not only accelerates material discovery but also enables tailored property optimization for specific energy-related applications, such as supercapacitors, photocatalysts, and optoelectronic sensors.Data availability
Data will be made available on request.Author contributions
Conceptualization, N. C. H. and N. H. T.; methodology, N. C. H. and N. H. T.; software, T. M. and N. H. T.; validation, A. P., N. H. T. and M. A.; formal analysis, M. M.; investigation, N. C. H., A. G. and N. H. T.; resources, T. M., A. G. and N. H. T.; data curation, T. M., N. H. T., and N. C. H.; writing—original draft preparation, T. M. and N. H. T.; writing—review and editing, T. M. and N. H. T.; visualization, A. G., A. P. and N. H. T.; supervision, T. M.; project administration, A. G.; funding acquisition, N. C. H., N. H. T., A. G. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Funding
This work has also been financed by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia [Project No. KFU251886].References
- K. Karthik, S. K. Pandian and N. V. Jaya, Appl. Surf. Sci., 2010, 256, 6829–6833 CrossRef CAS.
- J. C. Johnson, H. Yan, P. Yang and R. J. Saykally, J. Phys. Chem. B, 2003, 107(34), 8816–8828 CrossRef CAS.
- Z. Fan and J. G. Lu, Appl. Phys. Lett., 2005, 86(12), 123510 CrossRef.
- H. T. Ng, J. Han, T. Yamada, P. Nguyen, Y. P. Chen and M. Meyyappan, Nano Lett., 2004, 4(7), 1247–1252 CrossRef CAS.
- Y. Xi, C. G. Hu, X. Y. Han, Y. F. Xiong, P. X. Gao and G. B. Liu, Solid State Commun., 2007, 141(9), 506–509 CrossRef CAS.
- L. Lu, J. Chen, L. Li and W. Wang, Nanoscale Res. Lett., 2012, 7, 1–8 CrossRef.
- J.-H. Lin, R. A. Patil, R. S. Devan, Z.-A. Liu, Y.-P. Wang, C.-H. Ho, Y. Liou and Y.-R. Ma, Sci. Rep., 2014, 4(1), 6967 CrossRef CAS.
- R. Zamiri, A. Kaushal, A. Rebelo and J. M. F. Ferreira, Ceram. Int., 2014, 40(1), 1635–1639 CrossRef CAS.
- X. Y. Xue, Z. H. Chen, L. L. Xing, C. H. Ma, I. J. Chen and T. H. Wang, J. Phys. Chem. C, 2010, 114(43), 18607–18611 CrossRef CAS.
- M. L. Curri, R. Comparelli, P. D. Cozzoli, G. Mascolo and A. Agostiano, Mater. Sci. Eng., C, 2003, 23(1–2), 285–289 CrossRef.
- H. Kim, J. S. Horwitz, W. H. Kim, A. J. Mäkinen, Z. H. Kafafi and D. B. Chrisey, Thin Solid Films, 2002, 420, 539–543 CrossRef.
- M. Ohtaki, T. Tsubota, K. Eguchi and H. Arai, J. Appl. Phys., 1996, 79(3), 1816–1818 CrossRef CAS.
- S. J. Pearton, D. P. Norton, K. Ip, Y. W. Heo and T. Steiner, Prog. Mater. Sci., 2005, 50(3), 293–340 CrossRef CAS.
- V. R. Shinde, T. P. Gujar, C. D. Lokhande, R. S. Mane and S. H. Han, Mater. Chem. Phys., 2006, 96(2–3), 326–330 CrossRef CAS.
- F. Decremps, J. Pellicer-Porres, A. M. Saitta, J. C. Chervin and A. Polian, Phys. Rev. B, 2002, 65(9), 092101 CrossRef.
- E. O. Zayim, J. Mater. Sci., 2005, 40, 1345–1352 CrossRef CAS.
- H. Tang, K. Prasad, R. Sanjines and F. Levy, Sens. Actuators, B, 1995, 26(1–3), 71–75 CrossRef CAS.
- G. Ramakrishna and H. N. Ghosh, Langmuir, 2003, 19(3), 505–508 CrossRef CAS.
- D. E. Aimouch, S. Meskine, A. Boukortt and A. Zaoui, J. Magn. Magn. Mater., 2018, 451, 70–78 CrossRef CAS.
- S. B. Rana, V. K. Bhardwaj, S. Singh, A. Singh and N. Kaur, J. Mater. Sci.: Mater. Electron., 2013, 24, 20–26 CrossRef CAS.
- B. Panigrahy, M. Aslam and D. Bahadur, J. Phys. Chem. C, 2010, 114(27), 11758–11763 CrossRef CAS.
- S. B. Rana and R. P. Singh, J. Mater. Sci.: Mater. Electron., 2016, 27, 9346–9355 CrossRef CAS.
- S. B. Rana, R. P. P. Singh and S. Arya, J. Mater. Sci.: Mater. Electron., 2017, 28, 2660–2672 CrossRef CAS.
- M. Thambidurai, J. Y. Kim, J. Song, Y. Ko, N. Muthukumarasamy, D. Velauthapillai and C. Lee, Solar Energy, 2014, 106, 95–101 CrossRef CAS.
- K. J. Chen, F. Y. Hung, S. J. Chang and Z. S. Hu, Appl. Surf. Sci., 2009, 255(12), 6308–6312 CrossRef CAS.
- N. L. Le, T. Q. Nguyen, T. K. Truong, T. K. Pham, H. T. Nguyen and V. K. Tran, J. Sol-Gel Sci. Technol., 2023, 106, 699–714 CrossRef.
- H. T. Nguyen, T. T. N. Le, M. T. Truong, T. T. N. Nguyen, T. L. H. Nguyen, A.-T. Nguyen, V. T. Le and V.-D. Doan, Mater. Today Commun., 2024, 39, 108831 CrossRef CAS.
- H. T. Nguyen, V.-D. Doan, T. L. H. Nguyen, A.-T. Nguyen, Q.-H. Tran, V. A. Tran and V. T. Le, RSC Adv., 2025, 15, 6241–6259 RSC.
- H. T. Nguyen, M. T. Truong, V.-D. Doan, T. L. H. Nguyen, V. H. Hoang, V. A. Tran, A.-T. Nguyen and V. T. Le, Chem. Eng. Sci., 2024, 284, 119487 CrossRef CAS.
- G. Vijayaprasath, R. Murugan, S. Asaithambi, G. Anandha Babu, P. Sakthivel, T. Mahalingam, Y. Hayakawa and G. Ravi, Appl. Phys. A, 2016, 122, 1–11 CrossRef CAS.
- V. K. Tran, N. L. Le, H. T. K. Nguyen and H. T. Nguyen, Crystals, 2022, 12(12), 1825 CrossRef.
- P. K. Khanna, N. Singh and S. Charan, Mater. Lett., 2007, 61(25), 4725–4730 CrossRef CAS.
- C. Y. Wang, C. Böttcher, D. W. Bahnemann and J. K. Dohrmann, J. Mater. Chem., 2003, 13(9), 2322–2329 RSC.
- N. R. Farley, C. R. Staddon, L. Zhao, K. W. Edmonds, B. L. Gallagher and D. H. Gregory, J. Mater. Chem., 2004, 14(7), 1087–1092 RSC.
- K. Rajwali and M. H. Fang, Chin. Phys. B, 2015, 24(12), 127803 CrossRef.
- G. V. DiRenzo, E. Hanks and D. A. W. Miller, Methods Ecol. Evol., 2022, 14(1), 203–217 CrossRef.
- A. Ullah, I. U. Khan, M. Aljohani, K. Althubeiti, N. Rahman, M. Sohail and R. Khan, J. Mater. Sci.: Mater. Electron., 2023, 34(15), 1252 CrossRef CAS.
- U. Ahmed, M. Nazir, A. Sarwar, T. Ali, E.-H. M. Aggoune, T. Shahzad and M. A. Khan, Sci. Rep., 2025, 15, 1726 CrossRef CAS PubMed.
- H. Zhang, D. Yang, Y. Ji, X. Ma, J. Xu and D. Que, J. Phys. Chem. B, 2004, 108(13), 3955–3958 CrossRef CAS.
- R. Khan, K. Althubeiti, Zulfiqar, A. M. Afzal, N. Rahman, S. Fashu and R. Zheng, J. Mater. Sci.: Mater. Electron., 2021, 32, 24394–24400 CrossRef CAS.
- J. Zhou, F. Zhao, Y. Wang, Y. Zhang and L. Yang, J. Lumin., 2007, 122, 195–197 CrossRef.
- G. Srinet, R. Kumar and V. Sajal, J. Appl. Phys., 2013, 114(3), 033912 CrossRef.
- R. K. Singhal, A. Samariya, Y. T. Xing, S. Kumar, S. N. Dolia, U. P. Deshpande, T. Shripathi and E. B. Saitovitch, J. Alloys Compd., 2010, 496(1–2), 324–330 CrossRef CAS.
- F. V. Molefe, L. F. Koao, B. F. Dejene and H. C. Swart, Opt. Mater., 2015, 46, 292–298 CrossRef CAS.
- M. Ghosh, N. Dilawar, A. K. Bandyopadhyay and A. K. Raychaudhuri, J. Appl. Phys., 2009, 106(8), 084306 CrossRef.
- G. Vijayaprasath, R. Murugan, G. Ravi, T. Mahalingam and Y. Hayakawa, Appl. Surf. Sci., 2014, 313, 870–876 CrossRef CAS.
- I. Jabbar, Y. Zaman, K. Althubeiti, S. Al Otaibi, M. Z. Ishaque, N. Rahman and A. Khan, RSC Adv., 2022, 12(21), 13456–13463 RSC.
- M. Saqib, N. Rahman, K. Safeen, S. D. Mekkey, M. A. Salem, A. Safeen, M. Husain, S. U. Zaman, S. Abdullaev, A. Kalsoom, Z. M. El-Bahy and R. Khan, J. Mater. Process. Technol., 2023, 26, 8048–8060 CAS.
- R. N. Aljawfi and S. Mollah, J. Magn. Magn. Mater., 2011, 323(23), 3126–3132 CrossRef CAS.
- A. J. Reddy, M. K. Kokila, H. Nagabhushana, R. P. S. Chakradhar, C. Shivakumara, J. L. Rao and B. M. Nagabhushana, J. Alloys Compd., 2011, 509(17), 5349–5355 CrossRef CAS.
- M. Arshad, A. Azam, A. S. Ahmed, S. Mollah and A. H. Naqvi, J. Alloys Compd., 2011, 509(33), 8378–8381 CrossRef CAS.
- O. Yayapao, T. Thongtem, A. Phuruangrat and S. Thongtem, Mater. Lett., 2013, 90, 83–86 CrossRef CAS.
- G. B. Bhanuse, S. Kumar, C.-W. Chien and Y.-P. Fu, Electrochim. Acta, 2025, 511, 145371 CrossRef CAS.
- S. Saleem, M. N. Ashiq, S. Manzoor, U. Ali, R. Liaqat, A. Algahtani, S. Mujtaba, V. Tirth, A. M. Alsuhaibani, M. S. Refat, A. Ali, M. Aslam and A. Zaman, J. Mater. Res. Technol., 2023, 25, 6150–6166 CrossRef CAS.
- F. L. Deepak, E. A. Anumol and J. Li, in Metal Nanoparticles and Clusters: Advances in Synthesis, Properties and Applications, Springer International Publishing, Cham, 2017, pp. 219–287 Search PubMed.
- Y. S. Wang, P. J. Thomas and P. O'brien, J. Phys. Chem. B, 2006, 110(9), 4099–4104 CrossRef CAS.
- X. M. Fan, J. S. Lian, Z. X. Guo and H. J. Lu, Appl. Surf. Sci., 2005, 239(2), 176–181 CrossRef CAS.
- S. Talam, S. R. Karumuri and N. Gunnam, Int. Scholarly Res. Not., 2012, 2012(1), 372505 Search PubMed.
- R. Kumar and R. K. Kotnala, J. Alloys Compd., 2010, 508(2), 115–118 CrossRef.
- S. Husain, L. A. Alkhtaby, E. Giorgetti, A. Zoppi and M. M. Miranda, J. Lumin., 2014, 145, 132–137 CrossRef CAS.
- Y. H. Yang, X. Y. Chen, Y. Feng and G. W. Yang, Nano Lett., 2007, 7(12), 3879–3883 CrossRef CAS.
- R. Khan, A. Rasool, S. Kossar, E. A. Khera, K. Althubeiti, S. Al Otaibi and S. Iqbal, Mater. Sci. Semicond. Process., 2025, 193, 109487 CrossRef CAS.
- S. C. Das, R. J. Green, J. Podder, T. Z. Regier, G. S. Chang and A. Moewes, J. Phys. Chem. C, 2013, 117(24), 12745–12753 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |