
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold(I) and gold(III) complexes of triazolyl-functionalised NHCs†
Hawraa S. Al-Buthabhak *a,
Karrar Al-Ameedab,
Yu Yucd,
Alexandre N. Soboleve,
Stephen A. Moggache,
Hani Al-Salamif,
Vito Ferrog and
Murray V. Baker*e
*a,
Karrar Al-Ameedab,
Yu Yucd,
Alexandre N. Soboleve,
Stephen A. Moggache,
Hani Al-Salamif,
Vito Ferrog and
Murray V. Baker*e
aDepartment of Chemistry, Faculty of Science, University of Kufa, P. O. Box 21, Najaf 54001, Iraq. E-mail: hawraas.dawood@uokufa.edu.iq
bDepartment of Engineering, University of Warith Al-Anbiyaa, Karbala, Iraq
cCurtin Medical School, Curtin Health Innovation Research Institute, Curtin University, Perth, WA 6102, Australia
dDivision of Obstetrics & Gynaecology, The University of Western Australia Medical School, Perth, WA 6009, Australia
eSchool of Molecular Sciences M310, The University of Western Australia, 35 Stirling Highway, Perth, WA 6009, Australia. E-mail: murray.baker@uwa.edu.au
fBiotechnology and Drug Development Research Laboratory, Curtin Medical School & Curtin Health Innovation Research Institute, Curtin University, Perth, WA 6102, Australia
gSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia
First published on 30th May 2025
Abstract
Novel triazolyl-functionalised imidazolium salts and their corresponding AuI/AuIII complexes were synthesised and characterised by spectroscopic techniques and X-ray diffraction studies. The reaction of (mono and bis carbene) AuI complexes with thionyl chloride gave AuIII complexes. These AuIII complexes are reduced to AuI complexes by DMSO dissolution, and react with silver nitrate to afford chelating complexes, where the triazolyl N atoms bind to gold. The triazolyl-functionalised AuI complexes showed potency against ovarian cancer cells (OVCAR-8 cells, IC50 < 15 μM).
Introduction
N-Heterocyclic carbenes (NHCs) are well known as ligands having strong donor properties akin to those of phosphines. Generally, however, highly functionalised NHCs are more readily accessible than phosphines, since they can be conveniently generated from air-stable azolium salts1,2 and an array of other stable precursor compounds.3–6 The ready synthetic accessibility of functionalised NHCs has meant that properties of metal–NHC complexes, such as lipophilicity and steric bulk around the metal, can easily be tailored to a broad range of applications, including catalysis,7–13 imaging,14–20 and chemotherapy.21–30 In cases where robust metal–NHC complexes are required, stability can be enhanced by incorporation of the NHC moiety into a multidentate or a pincer ligand.31–33An interesting type of NHC results from combining an imidazolium salt and a triazole in the same compound by applying an azide–alkyne cycloaddition or “click” reaction.34 One application of triazoles obtained by the click reaction is their use as ligands in metal complexes.35 1,2,3-Triazole ligands have been used previously for the generation of well-defined abnormal/mesoionic complexes of palladium, ruthenium, iridium, copper, silver, gold and platinum in which the triazolyl ligand binds to the metal centre as a carbene.33,35–40 1,2,3-Triazolyl ligands have also been used to form triazolyl–gold complexes in which the triazolyl unit binds to gold via C (as an abnormal carbene) or N, and these complexes have been tested for their catalytic activity.33,39,41–43 The triazolyl–Au complexes or triazolyl–Au complexes having hemilabile ligands have proven to be efficient catalysts for enone formation from propargylic esters and alcohols,42 for intermolecular hydroamination of amines with alkynes,41 for formation of oxazolines by condensation of aldehydes and isocyanides,39 and for synthesis of γ-substituted γ-butyrolactones.40
Combining triazolyl-type ligands with NHCs in the same ligand framework could lead to interesting properties and coordination modes of the resulting complexes.38,44 For example, chelating triazolyl-functionalised NHC ligands (Tz-NHCs) have been used in combination with ruthenium in the transfer hydrogenation of ketones.38 Similar ligand structures have been used in medicine,45 metallopolymers,44 and supramolecular chemistry.46 Gold complexes derived from imidazolium salts that contain triazolyl units attached at the imidazolyl C4 or C5 positions have been reported.45,47 Tubaro and coworkers have reported Au–NHC complexes of Tz-NHC ligands where the Tz units are pendant from the N of the NHC ring, but the Tz-NHC ligands did not chelate the Au centre.48,49 In this work the synthesis of both neutral and cationic AuI complexes of two new Tz-NHCs is reported. The AuI complexes were then oxidised to give the corresponding AuIII complexes. The anticancer activity of some of these complexes was investigated in OVCAR-8 (ovarian cancer) cells. The new Tz-NHC ligands include a bromine atom to allow the possibility of using the NanoSIMS technique to track the fate of Au complexes in cells in future imaging studies.50
Results and discussion
Synthesis of triazolyl-functionalised imidazolium salts
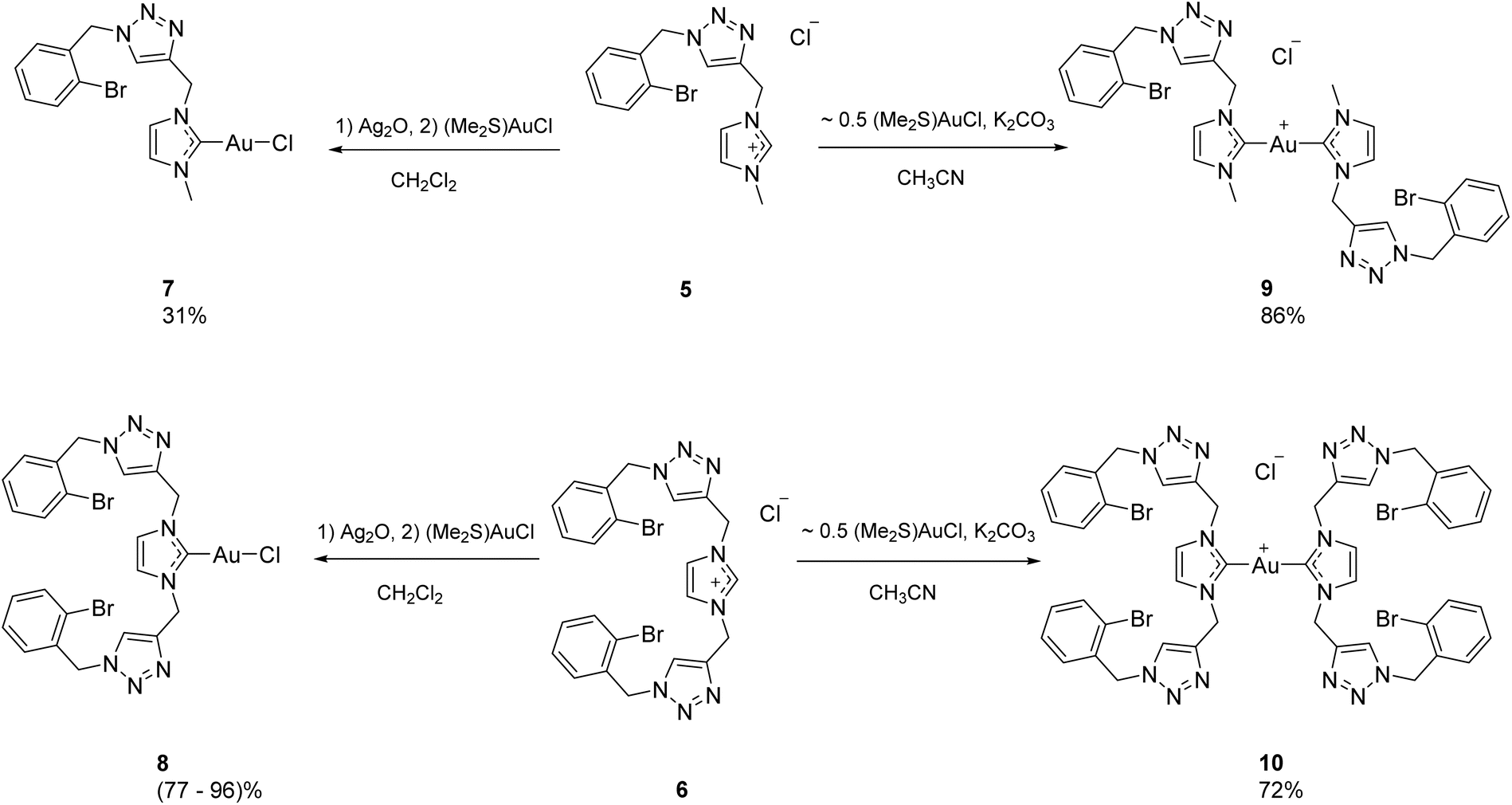
The synthetic pathway to the triazolyl-functionalised imidazolium salts is shown in Scheme 1. It relies on applying the copper catalysed azide–alkyne cycloaddition reaction to appropriate azide and alkyne coupling partners. The known azide 2 was obtained in high yield (83%) from 2-bromobenzyl bromide 1 by treatment with sodium azide at room temperature in dimethylformamide.51 Azide 2 was then reacted with 1-methyl-3-propargylimidazolium chloride 3 by adapting a literature procedure,38 using catalytic amounts of a Cu/CuSO4 mixture under heating at reflux. After workup, which included filtration through a column of silica gel, the triazolylimidazolium salt 5 was obtained as a brown oil in 45% yield. Using a similar procedure, reaction of azide 2 with 1,3-dipropargylimidazolium chloride 4 gave the new triazolylimidazolium salt 6 as a tan solid in 77% yield. | ||
| Scheme 1 Synthesis of triazolylimidazolium chloride salts 5 and 6. | ||
Synthesis of Au–NHC complexes having pendant triazolyl groups
Two neutral AuI–NHC complexes 7 and 8 were then prepared by reacting imidazolium salts 5 and 6 with silver oxide according to a well-known literature procedure (Scheme 2).52 The presumed intermediate Ag–NHC complexes derived from 5 and 6 are treated in situ with dimethylsulfide gold chloride [(Me2S)AuCl] in one-pot reactions. Complex 7 was obtained as an off-white powder in relatively low yield (31%). Significant losses occurred due to the need for the material to be recrystallised twice, initially from acetonitrile/diethyl ether, and then from ethyl acetate at 0 °C, to achieve satisfactory purity. On the other hand, complex 8 was obtained as a white powder in 77–96% yield by filtration either through a plug of silica (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM/MeCN) or Celite, followed by precipitation with DCM/hexanes.
:1 DCM/MeCN) or Celite, followed by precipitation with DCM/hexanes.
 | ||
| Scheme 2 Synthesis of the AuI–NHC complexes having pendant triazolyl groups. | ||
The cationic bis-NHC AuI complexes 9 and 10 were prepared by reaction of the salts 5 and 6 and with ∼0.5 equivalents of (Me2S)AuCl in the presence of potassium carbonate53 (8–10 equiv.) in acetonitrile (Scheme 2). Workup of 9 was straightforward and the compound was obtained as an off-white powder in 86% yield. Compound 10 was poorly soluble in acetonitrile, so during workup the acetonitrile was evaporated and the residue dissolved in ethanol. Filtration through Celite followed by evaporation of the solvent and washing with diethyl ether then gave 10 as a white powder in 72% yield.
In terms of solubility, it should be noted that both salts 5 and 6 are soluble in H2O but differ in their solubility profile for less polar solvents, with salt 6 being soluble in DCM, while salt 5 is not. The neutral complexes 7 and 8 and the cationic complex 9 are all soluble in DCM but are insoluble in water. The cationic complex 10 is the only complex to show good solubility in water, but it is not soluble in DCM.
Oxidation of the triazolyl-functionalised AuI–NHC complexes
:1 respectively, and the reaction had gone to completion after heating at reflux for a further 22 h (total time at reflux ∼48 h). Removal of volatiles under vacuum and washing of the residue with diethyl ether left the AuIII complex 12 as a yellow powder, in excellent yield (97%) and purity that is completely free from 8 (Fig. S5†). The choice of SOCl2 oxidizing agent is vital, the ESI† describes complications encountered upon employment of KAuCl4 and I2.
 | ||
| Scheme 3 Oxidation of AuI–NHC complex 8 to AuIII–NHC complex 12 by SOCl2. | ||
While the NMR spectrum of trichloride 12 (Fig. S5†) in CD2Cl2 suggests a single species in solution, 12 converts to the monochloride 8 in DMSO-d6. For example, the 1H NMR spectrum of 12 in DMSO-d6 showed 12 as the primary complex present immediately after preparation, but with trace 8 also detected (Fig. S7, top spectrum†). After 44 h, about a third of complex 12 had been reduced back to 8, and after 25 days, the sample contained only 8 (Fig. S7, bottom spectrum†). The ESI† describes even greater apparent solution instability of related iodo analogues.
Curiously, 12 showed similar stability in acetone-d6 and DMSO-d6, even though DMSO is a more strongly coordinating solvent. A sample prepared by dissolving 12 in acetone-d6 was monitored by 1H NMR spectroscopy (Fig. S8†). After 4 days, the sample contained 8 and 12 in a ratio of ∼1:1. In this experiment, a drop of water was added to the sample after 5 days, to see if water made the reversion of 12 to 8 occur more rapidly. The 1H NMR spectrum recorded immediately after addition of water showed a similar ratio of 8 and 12 seen in the 1H NMR spectrum recorded a day previously, indicating that addition of water did not immediately cause 12 to be reduced to 8, but nevertheless after 13 days, no 12 remained in the sample.
In view of the encouraging results obtained using thionyl chloride for the oxidation of AuI–NHC complex 8 to AuIII complex 12, similar oxidations were explored for AuI complexes of the form [Au(NHC)2]+, 9 and 10. Complex 9 was oxidised to AuIII complex 13 by treatment with excess thionyl chloride in DCM under reflux for 4 days. After workup complex 13 was obtained as a pale yellow powder in 90% yield (Scheme 4).
 | ||
| Scheme 4 Oxidation of bis AuI–NHC complexes by SOCl2. | ||
In the same way, oxidation of the bis AuI–NHC 10 with excess thionyl chloride gave the AuIII complex 14 as a yellow powder in 86% yield (Scheme 4). In this case, however, the starting material 10 was poorly soluble in DCM. When 10 was stirred in DCM a cloudy suspension was obtained, but when SOCl2 was added a clear solution formed. Interestingly, 10 is soluble in thionyl chloride, so the oxidation of 10 to 14 could be achieved simply by addition of neat reagent without any DCM, and the yield of 14 was >80% in both variations.
The pure compounds 13 and 14 are not soluble in DCM or acetone, and while 13 has only very limited solubility in acetonitrile, 14 is insoluble in this solvent. Fortunately, 13 and 14 were both soluble and sufficiently stable in DMSO-d6 that they could be characterised by NMR spectroscopy in freshly-prepared DMSO-d6 solutions. Nevertheless, the complexes slowly reverted to their AuI precursors in DMSO-d6, and for 14, NMR spectra of even freshly prepared solutions showed signals due to traces of 10. Elemental analyses of both 13 and 14 indicated that both complexes were pure, suggesting that the traces of 10 seen in NMR spectra of 14 were due to reduction of 14 by reaction with the solvent.
Another important finding here is that the choice of solvent is significant because AuIII complexes 11–14 can be easily reduced, especially 11, which is reduced immediately in DMSO-d6. By avoiding DMSO-d6 and acetone-d6 and utilising less polar and non-coordinating solvents such as dichloromethane-d2, reduction of AuIII to AuI is minimised. This finding is consistent with reports by Hirtenlehner et al.58 of the ease of reduction of AuIII–NHC complexes to AuI–NHC complexes, although those studies were primarily focused on photo-reductions and there was no comment on DMSO as a potential reductant. The present study emphasizes the importance of solvent on stability of AuIII–NHC complexes, even though some AuIII–NHC complexes are stable DMSO-d6.58
It is not surprising that 11–14 are easily reduced to their AuI counterparts as previous studies have shown that AuIII–NHCs can be easily reduced to AuI–NHCs by such reducing agents as phosphines, sulfides59 or when chloropyridine is utilised as the solvent of the reaction.55,56,60 In addition, it has been reported that when some AuIII–NHC complexes are utilised as precatalysts for hydration of alkynes, reduction to AuI–NHC complexes occurred.61
Studies of Au–NHC complexes by NMR spectroscopy
1H NMR spectroscopy was the primary tool used to monitor reactions and identify products of reactions explored in this work. 1H and 13C NMR spectra of the new Tz-Im salts 5 and 6 and the corresponding AuI and AuIII complexes are consistent with the structures determined by X-ray crystallography. 1H and 13C NMR chemical shifts seen for 5 and 6 and the corresponding AuI and AuIII complexes are in the ranges seen for similar compounds reported in the literature.6,22,30,56,58,62–64 The NMR signals associated with the imidazolyl and triazolyl moieties of the imidazolium salts and NHC complexes (e.g., Scheme 5) are particularly informative. Changes in signals associated with imidazolyl and triazolyl moieties were used as indicators of coordination of these moieties to Au. Signals due to other protons and carbons showed little variation in splitting patterns or chemical shifts whether they arose from an imidazolium salt, an AuI complex, or an AuIII complex. | ||
| Scheme 5 Informative 1H and 13C environments for characterisation. | ||
Consider, for example, the 1H NMR spectra for the Tz-Im salt 6 and the corresponding AuI complex 8 and AuIII complex 12 shown in Fig. 1. The 1H NMR spectrum of the salt 6 in DMSO-d6 shows three downfield singlets, due to the imidazolium H2 proton (9.36 ppm), the triazole ring proton (8.27 ppm) and the H4/H5 protons (7.77 ppm). These signals are in the regions expected based on previous reports for similar compounds.38 Signals for the protons of the aromatic ring are seen in the region 7.0–7.6 ppm and are well-resolved, and the protons of the two methylene groups occur as singlets at 5.7 ppm (C9 methylene, between the triazolyl and phenyl rings) and 5.5 ppm (C6 methylene, between triazolyl and imidazolyl rings). When 6 was treated with Ag2O followed by (Me2S)AuCl, formation of the corresponding AuI complex 8 was indicated by the disappearance of the 1H NMR signal at 9.36 ppm due to the imidazolyl proton (Fig. 1(b)). The other significant differences between the 1H NMR spectra of 6 and 8 were the chemical shifts due to the H4/H5 protons (7.49 ppm, shifted upfield from 7.77 ppm for 6) and C6 methylene protons (5.46 ppm, shifted upfield from 5.5 ppm for 6). These upfield shifts can be attributed to the decrease in the Lewis acidity65 of the substituent at the imidazolyl C2 position, H in 6 compared to AuI in 8. When the Lewis acidity of the substituent on C2 decreases, there is less withdrawal of the electron density from the imidazolyl ring, resulting in increased electronic shielding of the H4/H5 protons and the C6 methylene protons. The other protons in 8, being more remote from C2 and the effects of the substituent on C2, have chemical shifts quite close to their counterparts in 6.
 | ||
| Fig. 1 1H NMR spectra (DMSO-d6) of: (a) the imidazolium salt 6 showing the characteristic downfield signal for the H2 proton at 9.36 ppm; (b) the AuI–NHC complex 8; and (c) the AuIII–NHC complex 12. Spectra (a) and (b) were recorded at 500 MHz, and spectrum (c) was recorded at 600 MHz. | ||
In the 1H NMR spectrum of 12 (Fig. 1(c)), the chemical shifts of the H4/H5 and C6 methylene protons are shifted significantly downfield of their counterparts in 8, and slightly further downfield than their counterparts in 6. These shifts for 12 can be attributed to the AuIIICl3 substituent at the imidazolyl C2 being a substantially stronger Lewis acid than the AuICl substituent in 8, due to the electron withdrawing effects of AuIII and the three attached electronegative chlorine atoms. Again, the other protons in 12, being more remote from C2 and the effects of the substituent on C2, show little difference in chemical shifts to their counterparts in 6 or 8. Similar trends in 1H NMR chemical shifts were seen for the key protons in the other Au complexes discussed herein, and are summarized in Table S2.†
In the 13C NMR spectra of compounds 5 and 6 and the corresponding AuI and AuIII complexes, for most carbons there is little variation in chemical shifts amongst the series imidazolium salt/AuI–NHC complex/AuIII–NHC complex etc. For example, amongst all of compounds 5 and 6 and their corresponding AuI and AuIII complexes, the 13C chemical shifts of the triazolyl C8 carbons all fall within the range 122–126 ppm and the 13C chemical shifts of the imidazolyl C4/C5 carbons all fall within the range 121–126 ppm (Table S3†). The C2 (carbene) carbons, however, show huge differences in chemical shifts within the series imidazolium salt/AuI–NHC complex/AuIII–NHC complexes and interesting trends are evident (Table S3†). The chemical shifts of the carbene carbons fall into narrow but very distinct ranges depending on the class of compound: imidazolium salts ∼ 136 ppm; (NHC)AuICl ∼ 169 ppm; (NHC)AuIIIX3 140–141 ppm; AuI(NHC)2 ∼ 183 ppm; AuIII(NHC)2Cl2 151–152 ppm. These ranges show that oxidation state of Au, as well as the ligands attached to Au, cause significant differences in the chemical shift of the carbene carbon. In all cases, the carbene chemical shift for AuIII complexes is about 30 ppm upfield of the carbene chemical shift for AuI complexes, which has been attributed to the increased Lewis acidity of AuIII compared to AuI causing increased delocalisation of electron density from the imidazolyl C4–C5 double bond to the carbene carbon via the imidazolyl aromatic system.56
Syn and anti isomerism of AuIII complex 13
Oxidation of the AuI complex 9 by excess of thionyl chloride in DCM led to formation of the AuIII complex 13 (Scheme 4). The 1H NMR spectrum of 13 (Fig. S6†) showed two distinct sets of signals, indicating the presence of two species in a ratio of 62:38, but elemental analysis was consistent with the elemental composition expected for 13 (with traces of occluded CH2Cl2). When the temperature was increased, various signals for the two species broadened and coalesced, resulting in an “averaged” spectrum at high temperature (Fig. S10†), indicating that the two species interconvert on the NMR timescale. The two species detected by NMR spectroscopy are therefore assigned as syn and anti conformations of 13 that differ by the orientation of the NHC moieties about the C–Au–C axis (Scheme 6).
 | ||
| Scheme 6 Syn and anti conformers of AuIII complex 13. | ||
The existence of two observable conformations of 13 presumably results from the square planar geometry of AuIII complex 13 and steric bulk provided by the chlorido ligands on Au, which can restrict the rotation of NHC ligands around the C–AuIII–C axis. Two conformations are not seen in the case of the AuI complex 9 because, without the bulk provided by additional chlorido ligands on the Au centre, there is no substantial barrier to rotation of the NHC ligands about the C–AuI–C axis. This sort of conformational isomerism has been reported previously for other square planar complexes of the form M(NHC)2X2.1,58,66,67 Intuitively, the dominant conformation is expected to be anti-13, since the syn conformation would be destabilised by unfavourable steric interactions between the bromobenzyltriazolyl moieties. The 1H–1H ROESY NMR spectrum for a sample containing anti-13 and syn-13 showed a correlation between the signals due to the CH3 and H6 protons of the major isomer, but not for the minor isomer. The observed correlation is consistent with the assignment of the major isomer as anti-13, since in this conformation the CH3 protons in one NHC ligand will be close in space to the H6 protons of the other NHC ligand. In syn-13, the CH3 protons in one NHC ligand are remote from both the H6 protons in the same NHC ligand and the H6 protons in the other NHC ligand.
The variable temperature NMR study of 13 in DMSO-d6 (Fig. S10†) shows various signals broadening and coalescing as the temperature is raised. This behaviour is reversible, as expected for an NMR exchange process involving interconversion of conformations. Two sets of signals, for the CH3 groups and for the imidazolyl H4 protons in the syn and anti conformations, are sufficiently well-separated that they can be integrated in the room temperature spectrum. Integration of these signals indicated that ratio of the populations of nuclei in the anti and syn conformations is 62:38 respectively. At room temperature, the chemical shift separation for the signals due to the CH3 protons of the two conformations is 50.0 Hz, and coalescence of those signals occurs at ∼338 K (Fig. S10†). Similarly, the chemical shift separation for the signals due to the imidazolyl H4 protons of the two conformations is 30.0 Hz, and coalescence of those signals occurs at ∼328 K (Fig. S10†).
For an exchange between equally populated sites, the rate constant k at the coalescence temperature (the temperature at which signals of the exchanging nuclei are just merged together) can be estimated as k = 2.22Δν, where Δν is the chemical shift separation (in Hz) of the nuclei in the absence of exchange.68 In the case of the interconversion of anti-13 and syn-13, the populations of exchanging nuclei are unequal (see ESI†).
The results in (Table S1†) appear to be reasonable. The rate constants increase with temperature as expected, and the relative size of the rate constants for the anti → syn and the syn → anti processes are consistent with the observed ratio of anti-13:syn-13 in solution. In principle, it should be possible to calculate values for ΔS‡ and ΔH‡69 from the results in (Table S1†), but the uncertainty in the coalescence temperature is probably too large for such calculations to be meaningful.
The results in (Table S1†) are in the range seen for the results of previous studies for the interconversion of syn and anti conformations of AuIII–NHC complexes. Hirtenlehner et al.58 used quantum chemical calculations to estimate the barrier to rotation for interconversion of syn and anti conformations of a [(NHC)2AuIIIBr2]+ complex as ∼60 kJ mol−1, but noted that the result was not consistent with their 1H NMR study (200 MHz, DMSO-d6). In their NMR study, 1H NMR signals separated by ∼40 Hz at room temperature were not near coalescence even when the temperature was raised to 90 °C, indicating a high rotational barrier, expected to be significantly greater than 60 kJ mol−1. Huynh and Wu70 estimated the rotational barrier for syn/anti interconversion in a [trans-(NHC)2PdBr2] complex to be 74 kJ mol−1, from 1H NMR experiments in which the coalescence occurred at high temperature (380 K). None of these studies appear to have taken into account of the population difference between anti and syn conformations in their analyses.
Computational conformational analysis of 13 was also used to explore the experimental observation of two species in a ratio 62:38 in the NMR spectrum of 13 for anti:syn isomers, respectively. This suggests a significant preference for the anti-conformation under the studied conditions. Computational calculations support this finding, indicating that the anti isomer is more stable by 2.0 kcal mol−1 compared to the syn form (see Fig. 3). While this energy difference is not exceptionally large, it is sufficient to largely suppress the population of the syn isomer at equilibrium. The reported ΔE was used only to qualitatively support the observation that the anti isomer is energetically favored, rather than to quantitatively reproduce the NMR-derived syn:anti ratio. According to Boltzmann distribution principles, even differences as small as 1–2 kcal mol−1 can result in a dramatic shift in population at room temperature.71 This has been demonstrated in several experimental and computational studies. For example, in a study on azomethine derivatives, the anti isomer was favoured by only ∼1.8 kcal mol−1.72 Similarly, in a study on losartan isomers, the anti conformation was more stable and dominant in NMR spectra.73 These findings underscore that even moderate energy differences can dictate the dominant species in solution. Therefore, while 2.0 kcal mol−1 might seem modest, it is more than sufficient to explain why the anti conformer is the dominant species form the ratio in the NMR spectrum of 13.
Like the AuIII complexes 11 and 12, the AuIII complex 13 was unstable in DMSO. For example, in DMSO-d6 solution, ∼50% of the AuIII complex 13 had been reduced to the corresponding AuI complex 9 after 4 days, and little 13 remained after 25 days (Fig. S9†).
Halide abstraction reactions of AuIII complexes of Tz-NHC ligands: the quest for chelate AuIII–NHC complexes
AuIII–NHC complexes in which the Au centre is chelated by a functionalised NHC ligand are interesting for their potential use as catalysts.74 Chelation should stabilise the AuIII centre, allowing such complexes to participate in reactions as catalysts without the AuIII centre reverting to AuI. In order to chelate the AuIII centre in complexes such as 12 and 13 to the N atoms of triazolyl rings, experiments to abstract chloride from the AuIII centres were explored. The goal was to facilitate displacement of the two chloride ligands of 12 and 13 with intramolecular coordination of the pendant triazolyl groups to from the proposed chelate complexes 15 and 16 (Scheme S3†).Reaction of the (NHC)AuCl3 complex 12 with AgNO3
When complex 12 was treated with AgNO3 in DMSO-d6 solution in an NMR tube, a white precipitate (presumably AgCl) formed immediately. A new AuIII–NHC complex was formed and was assigned the structure 15, in which the metal centre is chelated by the NHC unit and the two triazolyl units in the organic ligand (Scheme 7). When only 2.2 equivalents of AgNO3 were used, 15 was formed as a mixture with the AuI–NHC complex 8 in a ratio of 19:1, respectively. The two complexes were separated during workup based on their solubilities (15 was insoluble in CH2Cl2 so 8 was easily removed by a simple washing step with CH2Cl2) and pure 15 was obtained as a white powder in 56% yield. Interestingly, when 3.3 equivalents of AgNO3 was used (again in DMSO-d6 in an NMR tube), 15 was the only complex formed. It may be that with excess Ag+, abstraction of chlorido ligands from 12 occurs rapidly, so that the AuIII centre is quickly stabilised by chelation by the triazolyl moieties, whereas when less Ag+ is used, the chelation occurs more slowly, allowing some time for 12 to be reduced to 8 as discussed above. Furthermore, the fact that the same product (15) was formed when 12 was treated with 2.2 or 3.3 equivalents of AgNO3 indicates that Ag+ is only able to abstract two chlorido ligands from 12. Evidently 15 is resistant to abstraction of the final chlorido ligand from the Au centre. It may be that the result of such an abstraction, an AuIII centre chelated by an NHC and two triazolyl units, with the remaining coordination site either vacant or occupied by nitrate or DMSO, is unstable and so cannot be formed, or it may be that chelation provides kinetic stability to 15.
 | ||
| Scheme 7 Abstraction of chlorido ligands from 12 by Ag+, to form 15. | ||
It is interesting to compare these results with those of Hirtenlehner et al.,58 who treated complexes of form (NHC)AuBr3 and [(NHC)2AuBr2]+ with Ag+ in the presence of various anionic ligands L−. In that study, the NHC ligand was 1,3-dibenzylimidazol-2-ylidene, which has no ability to chelate the AuIII centre. In every case, the starting AuIII–NHC complexes decomposed to form complicated mixtures containing AuI species such as (NHC)AuL and [(NHC)2Au]+, in addition to Au(0) and imidazolium salts as the major products.
Unfortunately, attempts to obtain single crystals of 15 were not successful, so structure determination was based on the results of elemental analysis and NMR studies. Elemental analysis indicated an empirical formula C23H20AuBr2ClN10O6·(CH2Cl2)1.2, which is consistent with the structure proposed for 15 with some entrapped CH2Cl2 (used during workup). The 1H NMR spectrum (Fig. 2) was also consistent with the structure 15 in terms of number of signals seen, their chemical shifts, and their relative integrals. Key signals are a 2H singlet at 9.03 ppm (assigned to the triazolyl protons), a 2H singlet at 7.99 ppm (assigned to the imidazolyl H4/H5 protons), and two 4H singlets at 6.1 and 5.8 ppm, assigned to the methylene groups adjacent to the bromophenyl and imidazolyl groups, respectively. Assignments of all 1H NMR signals for 15 were carried out with the aid of 1H–13C HSQC and HMBC experiments. The number of signals (one signal for the H4/H5 protons, one for the two triazolyl protons, two signals for the four methylene groups, and one set of signals for the two bromophenyl groups) is consistent with a structure that has a plane of symmetry, as in the chelate complex 15 where both triazolyl groups are bound directly to the Au centre. The possibility of one or both triazolyl groups being bound as an abnormal carbene (C–Au bond) can be discounted by the presence of the 2H signal for the triazolyl protons.
 | ||
| Fig. 2 1H NMR spectra (600 MHz, DMSO-d6) of chelate complex 15 (top spectrum), and AuIIICl3 complex 12 (bottom spectrum). | ||
Comparison of the 1H NMR spectra of 12 and 15 (both recorded in DMSO-d6) is informative. The most significant difference between the two spectra is the chemical shift of the signal for the triazolyl protons (H8 in Fig. 2), which is shifted from ∼8 ppm in 12 to ∼9 ppm for 15, consistent with the triazolyl N donating electron density to Au centre in 15, leaving less electron density in the ring to shield the triazolyl proton. Both methylene signals are also shifted downfield in the spectrum of 15, again consistent with coordination of the triazolyl groups to Au, resulting in electrons being withdrawn from the triazolyl ring causing deshielding of the methylene groups relative to the situation in 12.
In the 13C NMR spectrum of 15, the key signals are those for the Au-bound carbon and the triazolyl carbon. In DMSO-d6 solution the imidazolyl C2 carbon appears at 138.0 ppm and the triazolyl carbons (C8) appear at 128.1 ppm. These chemical shifts are quite close to those of the corresponding signals for 12, for which C2 occurs at 140.9 ppm and C8 occurs at 125.1 ppm, but the comparison is problematic because the instability of 12 in DMSO made it necessary to record the 13C NMR spectrum in CD2Cl2. Assignments of all 13C and 1H NMR signals for 15 were aided by 1H–13C HSQC and HMBC experiments.
To explore the stability of complex 15, an NMR sample of 15 prepared by adding 3.3 equivalents of AgNO3 to 12 in DMSO-d6 was monitored for change over the course of one week (Fig. S11†). 1H NMR spectra showed new signals emerging that did not match the chemical shifts of AuI complex 8, although new signals associated with protons of the bromophenyl moiety did appear in the region where the corresponding signals for 8 occurred. These observations suggest that decomposition might involve dissociation of one (or both) triazolyl groups from the Au centre, but not simply via some reduction process to form the AuI complex 8. Excess Ag+ was present in the sample and may have been involved in additional reactions with 15. Nevertheless, 15 was quite stable in the solution, and much more stable than its non-chelate counterpart 12. Integration of the signal for the triazolyl proton (using the DMSO-d6 solvent signal as an internal standard) showed that 86% of 15 remained after 22 h, and 55% remained after 7 days.
Furthermore, the origin of the pronounced stability observed for the closed form of the compound 15 was investigated, density functional theory (DFT) calculations were performed to compare two forms of compound 15: tridentate N,N,C-Tz-NHC AuIII monochloride complex (closed form) versus hypothetical monodentate Tz-NHC AuIII monochloride complex (open form). In the proposed open structure, the compound adopts a linear coordination geometry, while the experimentally observed compound 15 exhibits a square planar coordination involving intramolecular interactions with nitrogen atoms from the adjacent five-membered rings. The calculations in Fig. 3 indicate that the closed form is energetically favored by 70.4 kcal mol−1 relative to the open form. This substantial energy difference not only underscores the thermodynamic preference for the closed conformation but also provides a compelling justification for why the open form has not been experimentally isolated. The strong stabilization arising from the Au–N chelation likely plays a pivotal role in stabilizing the closed geometry of compound 15.
 | ||
| Fig. 3 The calculated relative stability of the complexes 13, 15 and the attempted 16: the atomic colors: gray; carbon, blue; nitrogen, green; chlorine, brown; bromide, yellow; gold, white; hydrogen. | ||
Reaction of the [(NHC)2AuCl2]+ complex 13 with AgNO3
In preliminary attempts to abstract chlorido ligands from 13 to form chelate complexes, KPF6 was used as the chloride abstracting agent. However, several attempts to abstract chloride by treating 13 with KPF6 in DMSO-d6, at room temperature and at 50 °C, were unsuccessful. No evidence of chloride abstraction was seen and no precipitate was formed, indicating that the salt KCl (which would form if abstraction occurred) was not present in the mixtures. The only change seen in the samples was partial reduction of 13 to the AuI complex 9.After these unsuccessful experiments to abstract the chlorido ligands by KPF6, AgNO3 was tested as the abstracting agent. When 3.4 equivalents of AgNO3 was added to a solution of 13 in DMSO-d6 in an NMR tube, a white precipitate (presumably AgCl) formed immediately. The 1H NMR spectrum of the sample showed that none of 13 remained in the sample—both the syn and anti conformations of 13 had been consumed, and a complex mixture of products was formed (Fig. S12†).
Sadly, there were no 1H NMR signals seen that could be confidently attributed to the desired product, the chelate complex 16 (Scheme S3†). Nevertheless, the 1H NMR spectrum showed signals that are tentatively assigned to a chelate ligand in a species such as 17 (Scheme 8). One of the signals of interest here is a singlet at 9.11 ppm, which is tentatively assigned to the triazolyl proton H8 of a chelating triazolyl moiety, based on its strongly downfield chemical shift. The triazolyl H8 protons of the chelate complex 15 had a similar chemical shift. The other signals of interest are a group in the region 5.9–6.1 ppm. This group has the appearance of two doublets, but with small shoulders on either side of the group, suggesting that they may actually be two AB doublets. These signals are tentatively assigned to the C6 and C9 methylene protons, one AB pattern to each group. The integrals of the signal assigned to H8 and the C6 + C9 group are in the ratio 1:4 respectively, consistent with the tentative assignment.
 | ||
| Scheme 8 Abstraction of Cl− from AuIII complex 13 to form AuIII complex 17, followed by reduction to form AuI complex 9. | ||
The AB patterns arise because the two protons in each methylene group are rendered inequivalent by the asymmetry in 17 associated with the orientation of the non-chelating NHC ligand with respect to the C–Au–C axis. Interestingly, when the sample was heated (Fig. S13†), the two AB patterns collapsed to two singlets, which is consistent with interconversion of two isomeric conformations of 17 by rotation of the non-chelating NHC ligand about the C–Au–C axis (Scheme 8). Unfortunately, 17 was not stable at elevated temperatures, a significant proportion being decomposed when the sample was heated to 318 K and none remaining after the sample had been heated to 378 K (Fig. S13†), so the behaviour of 17 could not be explored in detail.
The species 17 is interesting as a potential precursor of 16 (Scheme S3†). Unfortunately, the species was formed as part of a complex mixture. The 1H NMR spectrum of the mixture containing 17 and other products formed by treatment of 13 with AgNO3 shows an envelope of signals at 5.5–5.9 ppm. These signals fall into the range seen for the methylene signals for anti- and syn-13 (Fig. S6 and S10†), and are tentatively assigned to methylene signals associated with NHC ligands in which the triazolyl groups are not involved in chelation of the Au centre. Comparison of integrals for these signals and the signals assigned to the methylene groups of the chelating NHC ligand in 17 suggests that about 46% of 13 was converted to 17 and the remaining 54% was converted to other unidentified products. The mixture slowly underwent further change over time, to form the AuI complex 9. As noted above, formation of 9 was evident in the VT NMR study (Fig. S13†), but 9 was also formed in significant quantities in samples left at room temperature. Similarly, Hirtenlehner et al.58 reported that attempts to abstract bromido ligands from [(NHC)2AuIIIBr2]+Br (NHC = 1,3-dibenzylimidazolin-2-ylidene) by AgNO3 resulted in partial or complete reduction to AuI.
A comparative computational analysis of compound 13 with the attempted compound 16 is illustrated in Fig. 3. Although compound 16 has not been experimentally observed, the reaction energy calculations indicate a striking difference of over 500 kcal mol−1 in favour of compound 13. This substantial stability suggests that compound 13 occupies a deep thermodynamic minimum, likely due to favourable electronic and structural factors that are absent in the structure of 16.
X-ray diffraction studies
The structures of the imidazolium salt 6·2H2O, its (NHC)AuICl derivative 8·0.5Et2O, its (NHC)AuIIIClI2 derivative 11 (see ESI†) and its (NHC)AuIIICl3 derivative 12 were characterised by X-ray crystallographic studies. Crystals suitable for X-ray analysis were grown by diffusion of vapours between pentane and a dichloromethane solution of the compound (for 6, 11, and 12) or diethyl ether and a solution of the compound in acetone (for 8).In the solid-state structure of 6·2H2O, the cation was associated with one chloride counterion and two molecules of water (see Fig. S14,† which includes the atom numbering). The chloride ion shows weak interactions with an imidazolyl hydrogen (Cl(1)⋯H(3) 2.769 Å) and the triazolyl hydrogen (Cl(1)⋯H(25) 2.764 Å), smaller than the sum of the van der Waals radii as calculated by Bondi (2.95 Å)75 and Alvarez (3.02 Å).76 One water is involved in hydrogen bonding with an imidazolyl hydrogen (O(2)⋯H(4) 2.159 Å) and the other water is involved in hydrogen bonding with the triazolyl hydrogen (O(1)⋯H(55) 2.512 Å), both O⋯H distances being smaller than sum of the van der Waals radii as calculated by Bondi and Alvarez (2.72 and 2.70 Å respectively).75,76 Interestingly, there appear to be no hydrogen bonding interactions involving the imidazolium N–CH–N hydrogen. The cation is in a trans-extended zig-zag conformation, with the three methylene carbons approximately coplanar, and the five rings being oriented approximately perpendicular to that plane (Fig. S15†). The cations are stacked into columns, perhaps stabilised by π-stacking interactions; the inter-centroid distance between opposing rings in adjacent cations is 4.467 Å (Fig. 4). Such arrangements can indicate weak parallel π–π interactions.77
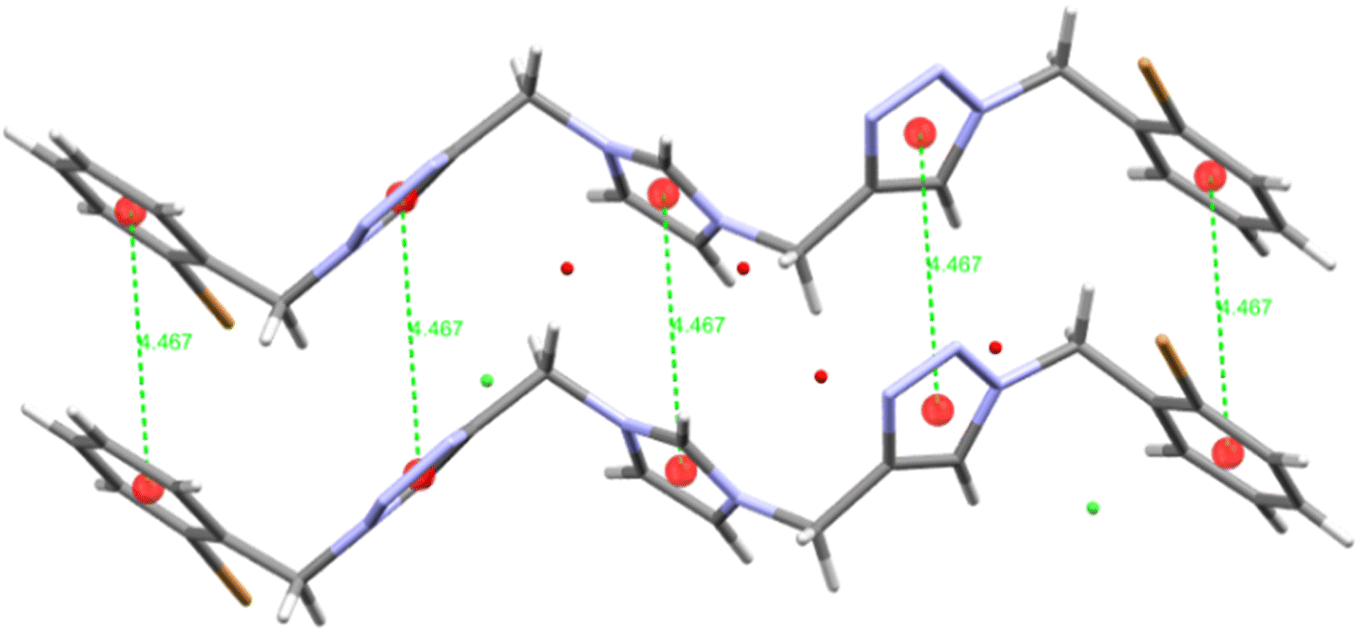 | ||
| Fig. 4 Two adjacent cations in 6·2H2O, showing the inter-centroid spacings (4.467 Å) between opposing rings. | ||
The structure of 8·0.5Et2O contains two independent molecules of 8, one with disorder associated with one of its phenyl rings (Fig. 5). The Au–C distances are (Au(1)–C(11) 2.04(2), Au(2)–C(21) 2.03(2) Å), and Au–Cl distances are (Au(1)–Cl(1) 2.295(5), Au(2)–Cl(2) 2.294(5) Å). Coordination about Au is approximately linear (C(11)–Au(1)–Cl(1) 179.0(6)°, C(21)–Au(2)–Cl(2) 178.8(7)°). These distances and angles are in the ranges typically seen for (NHC)AuICl complexes.64,78–80 The N-substituents on the NHC moiety are oriented anti to one another, with their N–C bonds in planes approximately orthogonal to the imidazolyl plane (Fig. 6).
 | ||
| Fig. 5 Crystal structure of the 8·0.5 Et2O (50% probability level for the thermal displacement of ellipsoids). Selected bond distances (Å) and angles (°). Molecule 1: Au(1)–C(11), 2.04(2); Au(1)–Cl(1), 2.295(5); C(11)–Au(1)–Cl(1), 179.0(6); molecule 2: Au(2)–C(21), 2.03(2); Au(2)–Cl(2), 2.294(5); C(21)–Au(2)–Cl(2), 178.8(7). | ||
 | ||
| Fig. 6 One of the (NHC)AuCl molecules in 8·0.5Et2O, viewed close to the NHC plane, showing the pendant triazolyl groups with their triazolyl planes approximately orthogonal to the imidazolyl plane. | ||
In the structures of both 11 and 12 there are two independent molecules and the coordination modes for Au in the (NHC)AuIIIX3 seen in the structures of 11 and 12 are similar (see ESI, Fig. S16–S18†). The Au–Ccarbene and Au–X bond distances (see figure captions and Table S4†) are in the ranges seen in related compounds.56,58,81–84 In 12, the chlorido ligands trans to the NHC group has a longer Au–Cl bond (Au–Cltrans 2.3078, 2.3215 Å) than for the chlorido ligands cis to the NHC group (Au–Clcis 2.2851, 2.2828, 2.2835, 2.2806 Å),83 as expected due to the strong trans effect of the NHC ligand. In 12, the Au is in a square planar coordination motif, with angles Ccarbene–Au–Cltrans ∼ 176° and Ccarbene–Au–Clcis in the range 87–89°. The Clcis–Au–Clcis angles are in the range 175.7–176.9°, with the chloride atoms pointing towards the carbene carbon. It has been suggested that this arrangement may be a consequence of an interaction between a chloride lone pair and the empty p-orbital on the carbene carbon.81,85 Consistent with this suggestion, structures show Ccarbene⋯Cl contacts (C⋯Clcis = 2.975–3.003 Å) that are substantially less than the sum of the van der Waals radii as calculated by Bondi (C⋯Cl 3.45 Å)75 and Alvarez (C⋯Cl 3.59 Å).76 The suggestion should be treated with some scepticism though, since the contacts between the Cl atom trans to the carbene C and the adjacent Cl atoms (Cltrans⋯Clcis = 3.279–3.318 Å) are also less than the sum of the van der Waals radii as calculated by Bondi (Cl⋯Cl 3.50 Å)75 and Alvarez (Cl⋯Cl 3.64 Å).76
In 12, the triazolyl pendants are mutually anti about the imidazolin-2-ylidene moiety, creating a groove within which the Clcis–Au–Clcis moiety is located. The plane of the imidazolin-2-ylidene ring is inclined at an angle to the CAuCl3 plane (θ in Fig. 7). The angle of inclination for the (NHC)AuCl3 complex 12 (∼69°).
 | ||
| Fig. 7 View of the structure of the cation in 12 along the CltransAu–Ccarbene axis. The plane defined by the imidazolyl ring in each case is horizontal and perpendicular to the page with the Cltrans ligand in front of the page. The coordination plane for Au (defined by the Au centre and the Cltrans, Ccarbene) is also perpendicular to the page and inclined at an angle θ with respect to the plane of the imidazolyl ring. In 12, θ = 68.68° (molecule 1) and 68.87° (molecule 2). | ||
Activity of triazolyl-functionalised Au–NHC complexes against OVCAR-8 cells
Gold-based compounds, especially Au–NHC complexes, are receiving much attention due to their promising anti-cancer activity against a wide variety of cancer cell lines.26,86,87 The dominant proposed mechanism for the activity of Au–NHCs is that Au inhibits the activity of mitochondrial thioredoxin reductase (TrxR) by binding to the soft Se and S atoms in the cysteine and selenocysteine residues at the active site.24,26 TrxR is an important enzyme that regulates the cellular redox system and inhibition of TrxR can lead to a build-up of reactive oxygen species, which sets off a cascade of processes that leads to apoptosis (controlled cell death).26,87,88 In this study, Au–NHC complexes were tested against OVCAR-8 ovarian cancer cells, a cisplatin-resistant cell line. Previous genetic profiling on OVCAR-8 suggested that it resembles high-grade serous ovarian carcinoma in patients (PMID 23839242), which is the most common histological subtype of ovarian cancer. A few classes of Au complexes, including auranofin and [Au(dppe)2]ClO4,89 and imidazole-based complexes90 have been tested in OVCAR-8 cells previously.The activity of the triazolyl functionalised Au–NHC complexes 7–9 against OVCAR-8 cells is summarised in Table 1 and Fig. 8. The AuIII complexes 12 and 14 were not tested due to their instability in DMSO. The chelate AuIII complex 15 was not tested as it was only synthesised in small amounts, insufficient for biological testing.
| Compound | IC50 (μM) ± SEMc | n |
|---|---|---|
| a OVCAR-8 cells were exposed to test compounds during incubation for 72 h and activity of the compounds on cell viability was evaluated using an MTT assay. Data are shown as mean ± SEM of at least 3 independent experiments (n) each of which used 2 to 4 replicates per concentration for the relevant test compound.b Imidazolium salt 6 used as a negative control.c IC50 refers to the concentration of the drug that is required to inhibit the growth of the cancerous cell by 50%. | ||
| 7 | 7.9 ± 1.3 | 4 |
| 8 | 13.2 ± 1.7 | 4 |
| 9 | 6.5 ± 0.1 | 3 |
| 6b | >100 | 3 |
 | ||
| Fig. 8 The inhibition activity of the triazolyl-functionalised Au–NHC complexes against OVCAR-8 cells, measured by MTT assay after 72 h of treatment. The imidazolium salt 96 was used as negative control. Data are shown as mean ± SEM of at least 3 independent experiments, each of which used 2 to 4 replicates per concentration for the relevant test compounds. | ||
The triazolyl functionalised Au–NHCs were active against the OVCAR-8 cells but showed higher IC50 values than the (NHC)Au(SCOR) complexes and [(Pr2Im)2Au]Br. From the IC50 values, the cationic bis-NHC complex 9 and the least hindered neutral (NHC)AuCl complex 7 are the most potent inhibitors, the more hindered neutral (NHC)AuCl complex being somewhat less active. Several factors likely contribute to activity in this series—the cationic nature of 9 (which would help it to target mitochondria), the hydrophilicity of the triazolyl groups, and different steric bulk around Au. The hydrophilic characteristic of the triazole moiety may decrease inhibitory activity, as Au compounds with greater hydrophobicity are typically more active than comparable hydrophilic ones.24 In 8, the steric bulk of the two bromophenylmethyltriazolyl groups may shield the Au centre from its biological targets, leading to decreased inhibitory activity of 8 compared to 7; this shielding effect has been suggested previously.23,91 Further work and a larger library of compounds are needed to provide further insights into the relative activities within the triazolyl-functionalised Au–NHC complexes.
Conclusions
The CuAAC click reaction of two propargyl-functionalised imidazolium salts with 2-bromobenzyl azide proceeded smoothly to form the corresponding triazolyl-functionalised imidazolium salts. The triazolyl-functionalised imidazolium salts were in turn converted into stable AuI complexes of form (NHC)AuCl (e.g., 8) or [(NHC)2Au]Cl (e.g., 10). The triazole-functionalised (NHC)AuCl complexes could be oxidised by thionyl chloride to yield AuIII complex (NHC)AuCl3. The complex was relatively stable in CD2Cl2 solution, but when dissolved in DMSO-d6 the (NHC)AuCl3 complex was reduced to the corresponding (NHC)AuICl complex, further studies of AuIII–NHC complexes focussed on chlorides rather than iodides.Oxidation of [(NHC)2Au]Cl complexes with thionyl chloride gave the corresponding [(NHC)2AuIIICl2]Cl complexes, for example 13, which was obtained as a mixture of syn and anti isomers. The [(NHC)2AuIIICl2]Cl complexes could be characterised by NMR spectroscopy in fresh DMSO-d6 solutions, but were slowly reduced to [(NHC)2AuI]Cl, and poor solubility issues prevented their study in other solvents. The stability of AuIII–NHC complexes was improved by chelation. Treatment of the (NHC)AuIIICl3 complex 12 with AgNO3 in DMSO-d6 resulted in abstraction of the chloride ligands and formation of the chelated complex 15, which showed good stability in DMSO-d6.
The chelate compound 15 is a novel compound and it would be worthwhile to confirm the structure by X-ray diffraction. Further work could also be devoted to achieving the synthesis of the chelate complex 16, which so far remains elusive. Efforts to react the AuIII–NHC complex 13 with AgNO3 to form the doubly-chelate complex 16 have so far only led to a product tentatively assigned as 17. Longer reaction times, more forcing reaction conditions, or the use of other chloride abstraction agents may permit 16 to be obtained.
Experimental section
General procedures
NMR spectra were recorded using Bruker AV500 III HD (500.13 MHz for 1H, 125.77 MHz for 13C, 202.45 MHz for 31P), Bruker AV600 III HD (600.13 MHz for 1H, 150.90 MHz for 13C), or Bruker AV NEO (400.15 MHz for 1H) spectrometers at ambient temperature, unless otherwise indicated. 1H and 13C NMR chemical shifts were referenced to the residual signal of the solvent (DMSO-d6: 1H 2.50 ppm; 13C 39.52 ppm; CD2Cl2: 1H 5.32 ppm; 13C 53.8 ppm; acetone-d6: 1H 2.05 ppm; 13C 29.84 ppm). Assignments of signals were confirmed with the aid of 1H–13C HSQC (heteronuclear single quantum coherence) and 1H–13C HMBC (heteronuclear multiple bond correlation) spectra when necessary. Microanalyses were performed by Microanalytical Service, School of Chemistry & Molecular Biosciences, The University of Queensland, Australia. High resolution mass spectra were measured using Agilent LCMS 6510 Q-TOF and Waters LCT Premier XE spectrometers, using the APCI or ESI methods, with 9:1 CH3CN:H2O as solvent. All the reactions were performed under a nitrogen atmosphere, in oven dried glassware. (Me2S)AuCl was prepared via the literature method.92 Gold complexes were wrapped in aluminium foil to exclude light. Solvents were of analytical or reagent grade and used without further purification.
Computational methodology
All structures were fully optimized using Density Functional Theory (DFT) with the unrestricted hybrid B3LYP functional93,94 and the all-electron def2-TZVP basis set,95 as implemented in the Gaussian 16 software package.96 Grimme's D3 dispersion correction with Becke–Johnson damping (D3-BJ)97 was applied to account for long-range dispersion interactions. Geometry optimizations were followed by analytical frequency calculations to confirm that the stationary points correspond to true minima on the potential energy surface, as indicated by the absence of imaginary frequencies in the Hessian matrix.Single X-ray crystal diffraction
Crystallographic data were measured at 100(2) K on an Oxford Diffraction Gemini or an Oxford Diffraction Xcalibur X-ray diffractometer using Mo Kα or Cu Kα radiation. Following analytical absorption corrections and solution by direct methods, the structures were refined against F2 with full-matrix least-squares using the SHELX program suite.98 Unless stated differently below, all hydrogen atoms were added at calculated positions and refined by use of riding models.Anti-cancer activity
Synthesis
:1 H2O/tert-butanol (5 mL each). The mixture was heated at reflux for 4 h, then cooled to room temperature and the solvents were evaporated to dryness. The residue was dissolved in 1:3 EtOH/MeOH (200 mL) and filtered through a plug of silica. The filtrate was evaporated to dryness and to the oily residue was added EtOH. The resulting precipitate was filtered off and the green filtrate was evaporated to give a yellow/brown oil (210 mg, 45%) which was dried under a flow of N2. 1H NMR (500 MHz, DMSO-d6): δ 9.24 (br s, 1H, H2), 8.29 (s, 1H, H8), 7.76 (1H, apparent t, splitting 1.7 Hz, H4), 7.71–7.69 (m, 2H, H5 and H14), 7.42 (1H, apparent td, “triplet” splitting 7.5 Hz, 4JH,H = 1.2 Hz, H12), 7.33 (1H, apparent td, “triplet” splitting 7.7 Hz, 4JH,H = 1.8 Hz, H13), 7.24 (1H, dd, 3JH,H = 7.5 Hz, 4JH,H = 1.7 Hz, H11), 5.71 (s, 2H, H9), 5.55 (s, 2H, H6), 3.80 (s, 3H, 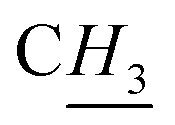 ). 13C NMR (125.7 MHz, DMSO-d6): δ 140.6 (C7), 136.7 (C2), 134.5 (C10), 133.0 (C14), 130.7 (C11), 130.6 (C13), 128.3 (C12); 125.0 (C8), 123.8 (C5), 123.0 (C15), 122.4 (C4), 53.3 (C9), 43.5 (C6), 35.8 (
). 13C NMR (125.7 MHz, DMSO-d6): δ 140.6 (C7), 136.7 (C2), 134.5 (C10), 133.0 (C14), 130.7 (C11), 130.6 (C13), 128.3 (C12); 125.0 (C8), 123.8 (C5), 123.0 (C15), 122.4 (C4), 53.3 (C9), 43.5 (C6), 35.8 (![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H3). Microanalysis: found: C, 39.74; H, 4.82; N, 16.02%. C14H15BrClN5 (H2O)3.2 requires C, 39.44; H, 5.06; N, 16.43. HRMS (ESI+): calcd for C14H15BrN5 [M − Cl+], m/z 332.0511. Found, m/z 332.0507.:1 H2O/tert-butanol (4 mL each), followed by addition of CuSO4 (16.0 mg, 0.10 mmol) and copper powder (2.70 mg, 0.044 mmol). The mixture was heated at reflux for 4 h, then cooled to room temperature and the solvent evaporated under vacuum. The residue was dissolved in DCM (40 mL) and filtered; the filtrate then passed through plug of silica eluting with 4:1 DCM/MeOH. The solvents were evaporated and the sticky brown residue that was obtained was washed with diethyl ether (3 × 30 mL) and precipitated from DCM/hexane with cooling (ice bath) to leave a tan powder (350 mg, 77%). 1H NMR (500 MHz, DMSO-d6): δ 9.36 (1H, br t, 4JH,H = 1.6 Hz, H2), 8.27 (s, 2H, H8), 7.77 (2H, d, 4JH,H = 1.6 Hz, H4/H5), 7.70 (2H, dd, 3JH,H = 7.9 Hz, 4JH,H = 1.3 Hz, H14), 7.41 (2H, apparent td, “triplet” splitting 7.5 Hz, 4JH,H = 1.3 Hz, H12), 7.33 (2H, apparent td, “triplet” splitting 7.7 Hz, 4JH,H = 1.7 Hz, H13), 7.22 (2H, dd, 3JH,H = 7.5 Hz, 4JH,H = 1.8 Hz, H11), 5.71 (s, 2H, H9), 5.55 (s, 2H, H6). 13C NMR (125.7 MHz, DMSO-d6): δ 140.4 (C7), 136.3 (C2), 134.5 (C10), 132.9 (C14), 130.7 (C11), 130.6 (C13), 128.3 (C12); 125.1 (C8), 123.0 (C15), 122.8 (C4/C5), 53.1 (C9), 43.6 (C6). Microanalysis: found: C, 42.61; H, 3.85; N, 17.07% C21H23Br2ClN8·(H2O)2.7 requires C, 42.28; H, 4.07; N, 17.15%. HRMS (ESI+): calcd for C23H21Br2ClN8 [M − Cl+], m/z 567.0256. Found, m/z 567.0277.
H3). Microanalysis: found: C, 39.74; H, 4.82; N, 16.02%. C14H15BrClN5 (H2O)3.2 requires C, 39.44; H, 5.06; N, 16.43. HRMS (ESI+): calcd for C14H15BrN5 [M − Cl+], m/z 332.0511. Found, m/z 332.0507.:1 H2O/tert-butanol (4 mL each), followed by addition of CuSO4 (16.0 mg, 0.10 mmol) and copper powder (2.70 mg, 0.044 mmol). The mixture was heated at reflux for 4 h, then cooled to room temperature and the solvent evaporated under vacuum. The residue was dissolved in DCM (40 mL) and filtered; the filtrate then passed through plug of silica eluting with 4:1 DCM/MeOH. The solvents were evaporated and the sticky brown residue that was obtained was washed with diethyl ether (3 × 30 mL) and precipitated from DCM/hexane with cooling (ice bath) to leave a tan powder (350 mg, 77%). 1H NMR (500 MHz, DMSO-d6): δ 9.36 (1H, br t, 4JH,H = 1.6 Hz, H2), 8.27 (s, 2H, H8), 7.77 (2H, d, 4JH,H = 1.6 Hz, H4/H5), 7.70 (2H, dd, 3JH,H = 7.9 Hz, 4JH,H = 1.3 Hz, H14), 7.41 (2H, apparent td, “triplet” splitting 7.5 Hz, 4JH,H = 1.3 Hz, H12), 7.33 (2H, apparent td, “triplet” splitting 7.7 Hz, 4JH,H = 1.7 Hz, H13), 7.22 (2H, dd, 3JH,H = 7.5 Hz, 4JH,H = 1.8 Hz, H11), 5.71 (s, 2H, H9), 5.55 (s, 2H, H6). 13C NMR (125.7 MHz, DMSO-d6): δ 140.4 (C7), 136.3 (C2), 134.5 (C10), 132.9 (C14), 130.7 (C11), 130.6 (C13), 128.3 (C12); 125.1 (C8), 123.0 (C15), 122.8 (C4/C5), 53.1 (C9), 43.6 (C6). Microanalysis: found: C, 42.61; H, 3.85; N, 17.07% C21H23Br2ClN8·(H2O)2.7 requires C, 42.28; H, 4.07; N, 17.15%. HRMS (ESI+): calcd for C23H21Br2ClN8 [M − Cl+], m/z 567.0256. Found, m/z 567.0277.Crystals suitable for X-ray diffraction studies were grown by diffusion of vapours between diethyl ether and a solution of the compound in acetonitrile.
 ). 13C NMR (125.7 MHz, DMSO-d6): δ 169.0 (C2), 142.4 (C7), 134.7 (C10), 132.8 (C14), 130.4 (C11), 130.4 (C13), 128.3 (C12), 124.5 (C8), 123.0 (C4), 122.8 (C15), 121.6 (C5), 52.9 (C9), 45.4 (C6), 37.7 (H3). Microanalysis: found: C, 30.32; H, 2.72; N, 11.89% C14H14AuBrClN5 (CH3CH2OCOCH3)0.2 requires C, 30.53; H, 2.70; N, 12.03%.
). 13C NMR (125.7 MHz, DMSO-d6): δ 169.0 (C2), 142.4 (C7), 134.7 (C10), 132.8 (C14), 130.4 (C11), 130.4 (C13), 128.3 (C12), 124.5 (C8), 123.0 (C4), 122.8 (C15), 121.6 (C5), 52.9 (C9), 45.4 (C6), 37.7 (H3). Microanalysis: found: C, 30.32; H, 2.72; N, 11.89% C14H14AuBrClN5 (CH3CH2OCOCH3)0.2 requires C, 30.53; H, 2.70; N, 12.03%. ). 13C NMR (125.70 MHz, DMSO-d6): δ 183.1 (C2), 142.8 (C7), 134.5 (C10), 132.9 (C14), 130.5 (C11), 130.4 (C13), 128.2 (C12), 124.3 (C8), 123.4 (C4), 122.9 (C15), 122.2 (C5), 53.0 (C9), 45.1 (C6), 37.5 (H3). Microanalysis: found: C, 35.48; H, 3.32; N, 14.66% C28H28 AuBr2ClN10·(H2O)3 requires C, 35.37; H, 3.60; N, 14.73%. HRMS (ESI+): calcd for C28H28Br2N10 Au [M − Cl+], m/z 589.0531. Found, m/z 589.0594.
). 13C NMR (125.70 MHz, DMSO-d6): δ 183.1 (C2), 142.8 (C7), 134.5 (C10), 132.9 (C14), 130.5 (C11), 130.4 (C13), 128.2 (C12), 124.3 (C8), 123.4 (C4), 122.9 (C15), 122.2 (C5), 53.0 (C9), 45.1 (C6), 37.5 (H3). Microanalysis: found: C, 35.48; H, 3.32; N, 14.66% C28H28 AuBr2ClN10·(H2O)3 requires C, 35.37; H, 3.60; N, 14.73%. HRMS (ESI+): calcd for C28H28Br2N10 Au [M − Cl+], m/z 589.0531. Found, m/z 589.0594. anti), 3.97 (s, 4H,
anti), 3.97 (s, 4H,  ). 13C NMR (150.90 MHz, DMSO-d6) Two isomers: δ 151.84 (C2, syn), 151.76 (C2, anti), 141.71 (C7, anti), 141.5 (C7, syn), 134.5 (C10), 132.9 (C14), 130.5 (C13), 130.4 (C11), 128.1 (C12), 125.4 (C5, syn), 125.1 (C5, anti), 125.0 (C8, syn), 124.8 (C8, anti), 124.5 (C4, anti), 124.1 (C4, syn); 122.8 (C15), 53.1 (C9, anti), 44.5 (C6, anti), 44.3 (C6, syn), 37.5 (H3, anti), 37.3 (H3, syn).
). 13C NMR (150.90 MHz, DMSO-d6) Two isomers: δ 151.84 (C2, syn), 151.76 (C2, anti), 141.71 (C7, anti), 141.5 (C7, syn), 134.5 (C10), 132.9 (C14), 130.5 (C13), 130.4 (C11), 128.1 (C12), 125.4 (C5, syn), 125.1 (C5, anti), 125.0 (C8, syn), 124.8 (C8, anti), 124.5 (C4, anti), 124.1 (C4, syn); 122.8 (C15), 53.1 (C9, anti), 44.5 (C6, anti), 44.3 (C6, syn), 37.5 (H3, anti), 37.3 (H3, syn).Microanalysis: found: C, 33.88; H, 2.90; N, 13.78% C28H28AuBr2C13N10. (CH2Cl2)0.5 requires C, 33.89; H, 2.89; N, 13.87%.
:4 MeCN/DCM (500 mL). The filtrate was evaporated, the product precipitated by dissolving it in ∼2 mL DCM and excess of hexanes. The product was collected and dried under vacuum to afford a white powder (105 mg, 96%). 1H NMR (500 MHz, DMSO-d6): δ 8.19 (s, 2H, H8), 7.68 (2H, dd, 3JH,H = 7.8, 4JH,H = 1.2 Hz, H14), 7.49 (s, 4H, H4/H5), 7.39 (2H, apparent td, “triplet” splitting 7.5 Hz, 4JH,H = 1.2 Hz, H12), 7.30 (2H, apparent td, “triplet” splitting 3JH,H = 7.8 Hz, 4JH,H = 1.7 Hz, H13), 7.16 (2H, dd, 3JH,H = 7.5 Hz, 4JH,H = 1.7 Hz, H11), 5.67 (s, 4H, H9), 5.42 (s, 4H, H6). 13C NMR (125.7 MHz, DMSO-d6): δ 169.1 (C2), 142.2 (C7), 134.7 (C10), 132.8 (C14), 128.2 (C11), 130.4 (C13), 128.4 (C12), 124.5 (C8), 122.8 (C15), 121.9 (C4/C5), 53.0 (C9), 45.6 (C6). Microanalysis: found: C, 34.42, H, 2.32, N, 13.60% C23H20AuBr2ClN8 requires C, 34.50, H, 2.52, N, 13.99%. Crystals suitable for X-ray diffraction studies were grown by diffusion of vapours between pentane and a solution of the complex in dichloromethane.Also, the work up can be by filtration through Celite using dichloromethane as a solvent to afford a white powder 85%.
Method 2: the triazolyl-functionalised (NHC)AuCl complex 10 (15.8 mg, 11.5 μmol) was dissolved in thionyl chloride (3 mL) and the mixture heated at reflux for 5 days under N2. The solvent was removed under vacuum, the residue was stirred with diethyl ether, and the diethyl ether decanted off, to leave the product as a yellow solid (15 mg, 86%). 1H NMR (600 MHz, DMSO-d6) NHC–AuIII: δ 8.21 (s, 4H, H8), 7.82 (s, 4H, H4/H5), 7.64 (4H, br d, 3JH,H = 7.5 Hz, H14), 7.29 (8H, m, H12 and H11), 7.08 (4H, br d, 3JH,H = 7.3 Hz, H13), 5.80 (s, 8H, H6), 5.64 (s, 8H, H9). 13C NMR (150.9 MHz, DMSO-d6) NHC–AuIII. δ 151.7 (C2), 141.3 (C7), 134.4 (C10), 132.9 (C14), 130.4 (C11), 130.4 (C13), 128.2 (C12), 124.9 (C8), 124.5 (C4/C5), 122.8 (C15), 53.0 (C6), 44.6 (C9). Microanalysis: found: C, 34.07; H, 2.85; N, 12.76% C46H40AuBr4Cl3N16. (CH2Cl2)3.5 requires C, 34.23; H, 2.73; N, 12.90%.
:31:25 respectively.(i) Synthesis using three equivalents of AgNO3. A solution of AgNO3 (3.20 mg, 18.7 μmol) in DMSO-d6 (0.25 mL) was added to a solution of the AuIII complex 12 (5.00 mg, 5.70 μmol) in DMSO-d6 (0.25 mL) in an NMR tube. A white precipitate (presumed to be AgCl) formed immediately. The compound remaining in solution was identified as by its 1H and 13C NMR spectrum.
(ii) Synthesis using two equivalents of AgNO3. A solution of AgNO3 (3.40 mg, 19.98 μmol) in DMSO-d6 (0.25 mL) was added to a solution of the AuIII complex 12 (8.00 mg, 9.18 μmol) in DMSO-d6 (0.25 mL) in an NMR tube. A white precipitate (presumed to be AgCl) formed immediately and the presence of AuIII complex 15 in solution was confirmed by 1H NMR spectroscopy. The clear solution was separated from the precipitate using a pipette and the solvent was evaporated under a flow of N2. The solid was swirled with DCM (3 mL), the DCM was drawn off by pipette and the process was repeated. The white powder that remained was dried under vacuum. Yield: 4.8 mg, 56%. 1H NMR (500 MHz, DMSO-d6): δ 9.03 (s, 2H, H8), 7.99 (s, 2H, H4/H5), 7.75 (2H, dd, 3JH,H = 8.0 Hz, 4JH,H = 1.0 Hz, H14), 7.54–7.49 (4H, m, H11 and H12), 7.40 (2H, ddd, 3JH,H = 8.0 Hz 3JH,H = 7.1 Hz, 4JH,H = 2.1 Hz, H13), 6.08 (s, 4H, H9), 5.82 (s, 4H, H6). 13C NMR (125.7 MHz, DMSO-d6): δ 137.9 (C2), 133.2 (C14), 132.2 (C7), 132.0 (C10), 131.9 (C11), 131.6 (C13), 128.5 (C12), 128.1 (C8), 123.8 (C15), 123.7 (C4/C5), 56.5 (C9), 44.5 (C6). Microanalysis: found: C, 28.09; H, 2.21; N, 13.98%. C23H20AuBr2ClN10O6. (CH2Cl2)1.2 requires C, 28.31; H, 2.20; N, 13.64%.
Data availability
The data supporting this article have been included as part of the ESI.† More raw data are available from the corresponding author on reasonable request.Author contributions
Hawraa S. Al-Buthabhak: synthesis and characterisation, formal analysis; investigation; methodology; writing – original draft. Karrar Al-Ameed: computational calculations; formal analysis; investigation Yu Yu: methodology. Alexandre Sobolev: data curation; formal analysis; investigation. Stephen Moggach: data curation; formal analysis; investigation. Hani Al-Salami: resources. Vito Ferro: supervision; writing – review and editing. Murray V. Baker: supervision; resources; writing – review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the facilities and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, the University of Western Australia. Hawraa S. Al-Buthabhak thanks the Higher Committee for Education Development in Iraq (HCED) for financial support. Al-Salami is supported by the European Union Horizon 2020 Research Project and Innovation Program under the Marie Skłodowska-Curie Grant Agreement No. 872370, Curtin Faculty ORS-WAHAI Consortium, and the Australian National Health and Medical Research Council (NHMRC, APP9000597). Stephen A. Moggach thanks the Australian Research Council (ARC) for a Future Fellowship (FT200100243). We thank the University of Western Australia and the University of Queensland for a UWA-UQ Partnership Research Collaboration Award (to VF and MVB).References
- W. Zhao, V. Ferro and V. M. Baker, Coord. Chem. Rev., 2017, 339, 1–16 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- R. Jothibasu and H. V. Huynh, Chem. Commun., 2010, 46, 2986–2988 RSC.
- J. Turek, I. Panov, P. Svec, Z. Ruzickova and A. Ruzicka, Dalton Trans., 2014, 43, 15465–15474 RSC.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708–3714, 10.1039/b602560a.
- N. Hamdi, I. Slimani, L. Mansour, F. Alresheedi, N. Gürbüz and I. Özdemir, New J. Chem., 2021, 45, 21248–21262 RSC.
- A. Martínez, J. L. Krinsky, I. Peñafiel, S. Castillón, K. Loponov, A. Lapkin, C. Godard and C. Claver, Catal. Sci. Technol., 2015, 5, 310–319 RSC.
- B. Jacques, D. Hueber, S. Hameury, P. Braunstein, P. Pale, A. Blanc and P. de Frémont, Organometallics, 2014, 33, 2326–2335 CrossRef CAS.
- M. Bouhrara, E. Jeanneau, L. Veyre, C. Coperet and C. Thieuleux, Dalton Trans., 2011, 40, 2995–2999 RSC.
- A. S. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhelmsen, M. Rudolph and F. Rominger, Chem.–Eur. J., 2013, 19, 1058–1065 CrossRef CAS PubMed.
- A. S. K. Hashmi, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8247–8249 CrossRef CAS PubMed.
- A. G. Nair, R. T. McBurney, M. R. D. Gatus, S. C. Binding and B. A. Messerle, Inorg. Chem., 2017, 56, 12067–12075 CrossRef CAS PubMed.
- C. Hemmert and H. Gornitzka, Dalton Trans., 2016, 45, 440–447 RSC.
- P. J. Barnard, L. E. Wedlock, M. V. Baker, S. J. Berners-Price, D. A. Joyce, B. W. Skelton and J. H. Steer, Angew. Chem., Int. Ed., 2006, 45, 5966–5970 CrossRef CAS PubMed.
- Ö. Karaca, S. M. Meier-Menches, A. Casini and F. E. Kühn, Chem. Commun., 2017, 53, 8249–8260 RSC.
- T. Zou, C. T. Lum, S. S. Chui and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930–2933 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- L. E. Wedlock and S. J. Berners-Price, Aust. J. Chem., 2011, 64, 692–704 CrossRef CAS.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- S. Y. Hussaini, R. A. Haque and M. R. Razali, J. Organomet. Chem., 2019, 882, 96–111 CrossRef CAS.
- H. S. Al-Buthabhak, Y. Yu, A. Sobolev, H. Al-Salami and M. V. Baker, J. Inclusion Phenom. Macrocyclic Chem., 2021, 101, 227–242 CrossRef CAS.
- T. Zou, C. N. Lok, P. K. Wan, Z. F. Zhang, S. K. Fung and C. M. Che, Curr. Opin. Chem. Biol., 2018, 43, 30–36 CrossRef CAS PubMed.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS PubMed.
- G. Achar, S. A. Patil, J. G. Małecki and S. Budagumpi, New J. Chem., 2019, 43, 1216–1229 RSC.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 CrossRef CAS PubMed.
- G. Moreno-Alcántar, P. Picchetti and A. Casini, Angew. Chem., Int. Ed., 2023, 62, e202218000 CrossRef PubMed.
- B. S. Murray and P. J. Dyson, Curr. Opin. Chem. Biol., 2020, 56, 28–34 CrossRef CAS PubMed.
- G. Augello, A. Azzolina, F. Rossi, F. Prencipe, G. F. Mangiatordi, M. Saviano, L. Ronga, M. Cervello and D. Tesauro, Pharmaceutics, 2023, 15, 466 CrossRef CAS PubMed.
- H. S. Al-Buthabhak, V. Falasca, Y. Yu, A. N. Sobolev, B. W. Skelton, S. A. Moggach, V. Ferro, H. Al-Salami and M. V. Baker, Appl. Organomet. Chem., 2022, 38, e6645 CrossRef.
- M. G. Gardiner and C. C. Ho, Coord. Chem. Rev., 2018, 375, 373–388 CrossRef CAS.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS.
- M. Navarro, A. Tabey, G. Szalóki, S. Mallet-Ladeira and D. Bourissou, Organometallics, 2021, 40, 1571–1576 CrossRef CAS.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- M. Albrecht, Adv. Organomet. Chem., 2014, 62, 111–158 CrossRef CAS.
- M. Delgado-Rebollo, D. Canseco-Gonzalez, M. Hollering, H. Mueller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462–4473 RSC.
- M. Hollering, M. Albrecht and F. E. Kühn, Organometallics, 2016, 35, 2980–2986 CrossRef CAS.
- D. Canseco-Gonzalez, A. Petronilho, H. Mueller-Bunz, K. Ohmatsu, T. Ooi and M. Albrecht, J. Am. Chem. Soc., 2013, 135, 13193–13203 CrossRef CAS PubMed.
- P. Font, H. Valdés, G. Guisado-Barrios and X. Ribas, Chem. Sci., 2022, 13, 9351–9360 RSC.
- H. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2009, 131, 12100–12102 CrossRef CAS PubMed.
- D. Wang, Y. Zhang, A. Harris, L. N. S. Gautam, Y. Chen and X. Shi, Adv. Synth. Catal., 2011, 353, 2584–2588 CrossRef CAS.
- Y. Yang, W. Hu, X. Ye, D. Wang and X. Shi, Adv. Synth. Catal., 2016, 358, 2583–2588 CrossRef CAS.
- C. Lang, K. Pahnke, C. Kiefer, A. S. Goldmann, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2013, 4, 5456–5462 RSC.
- M. Baron, S. Bellemin-Laponnaz, C. Tubaro, M. Basato, S. Bogialli and A. Dolmella, J. Inorg. Biochem., 2014, 141, 94–102 CrossRef CAS PubMed.
- V. Lewe, M. Preuss, E. A. Woźnica, D. Spitzer, R. Otter and P. Besenius, Chem. Commun., 2018, 54, 9498–9501 RSC.
- M. Monticelli, S. Bellemin-Laponnaz, C. Tubaro and M. Rancan, Eur. J. Inorg. Chem., 2017, 2017, 2488–2495 CrossRef CAS.
- M. Monticelli, M. Baron, C. Tubaro, S. Bellemin-Laponnaz, C. Graiff, G. Bottaro, L. Armelao and L. Orian, ACS Omega, 2019, 4, 4192–4205 CrossRef CAS PubMed.
- A. Longhi, M. Baron, M. Rancan, G. Bottaro, P. Sgarbossa and C. Tubaro, Molecules, 2019, 24, 2305 CrossRef CAS PubMed.
- T. Eybe, T. Bohn, J. N. Audinot, T. Udelhoven, H. M. Cauchie, H. N. Migeon and L. Hoffmann, Chemosphere, 2009, 76, 134–140 CrossRef CAS PubMed.
- D. Rodríguez-Hernández, A. J. Demuner, L. C. A. Barbosa, L. Heller and R. Csuk, Eur. J. Med. Chem., 2016, 115, 257–267 CrossRef PubMed.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- A. Collado, A. Gomez-Suarez, A. R. Martin, A. M. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541–5543 RSC.
- M. Pažický, A. Loos, M. J. Ferreira, D. Serra, N. Vinokurov, F. Rominger, C. Jäkel, A. S. K. Hashmi and M. Limbach, Organometallics, 2010, 29, 4448–4458 CrossRef.
- S. Zhu, R. Liang, L. Chen, C. Wang, Y. Ren and H. Jiang, Tetrahedron Lett., 2012, 53, 815–818 CrossRef CAS.
- P. de Fremont, R. Singh, E. D. Stevens, J. L. Petersen and S. P. Nolan, Organometallics, 2007, 26, 1376–1385 CrossRef CAS.
- A. H. Mageed, B. W. Skelton and M. V. Baker, Dalton Trans., 2017, 46, 7844–7856 RSC.
- C. Hirtenlehner, C. Krims, J. Holbling, M. List, M. Zabel, M. Fleck, R. J. Berger, W. Schoefberger and U. Monkowius, Dalton Trans., 2011, 40, 9899–9910 RSC.
- S. Ahmad, A. A. Isab, H. P. Perzanowski, M. S. Hussain and N. M. Akhtar, Transit. Met. Chem., 2002, 27, 177–183 CrossRef CAS.
- S. Zhu, R. Liang and H. Jiang, Tetrahedron, 2012, 68, 7949–7955 CrossRef CAS.
- H. Ibrahim, P. de Frémont, P. Braunstein, V. Théry, L. Nauton, F. Cisnetti and A. Gautier, Adv. Synth. Catal., 2015, 357, 3893–3900 CrossRef CAS.
- R. Jothibasu, H. V. Huynh and L. L. Koh, J. Organomet. Chem., 2008, 693, 374–380 CAS.
- H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591–4600 CrossRef CAS.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625–5635 CAS.
- W. A. Herrmann, O. Runte and G. Artus, J. Organomet. Chem., 1995, 501, C1–C4 CrossRef CAS.
- Y.-P. Huang, C.-C. Tsai, W.-C. Shih, Y.-C. Chang, S.-T. Lin, G. P. A. Yap, I. Chao and T.-G. Ong, Organometallics, 2009, 28, 4316–4323 CrossRef CAS.
- H. V. Huynh, C. H. Yeo and Y. X. Chew, Organometallics, 2010, 29, 1479–1486 CrossRef CAS.
- H. Günther, NMR Spectroscopy, Basic Principles, Concepts, and Applications in Chemistry, Wiley, 2nd edn, 1995, vol. 343 Search PubMed.
- K. D. Zimmer, R. Shoemaker and R. R. Ruminski, Inorg. Chim. Acta, 2006, 359, 1478–1484 CrossRef CAS.
- H. V. Huynh and J. Wu, J. Organomet. Chem., 2009, 694, 323–331 CrossRef CAS.
- P. W. Atkins and J. de Paula, Physical Chemistry, Oxford University Press, 2010 Search PubMed.
- M. Hricovíni, J. Asher and M. Hricovíni, RSC Adv., 2020, 10, 5540–5550 RSC.
- J. Kujawski, K. Czaja, T. Ratajczak, E. Jodłowska and M. K. Chmielewski, Molecules, 2015, 20(7), 11875–11190 CrossRef CAS PubMed.
- M. Boronat, A. Corma, C. González-Arellano, M. Iglesias and F. Sánchez, Organometallics, 2010, 29, 134–141 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- D. Piovesan, G. Minervini and S. C. Tosatto, Nucleic Acids Res., 2016, 44, W367–W374 CrossRef CAS PubMed.
- P. de Fremont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef CAS.
- M. V. Baker, P. J. Barnard, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2005, 37–43, 10.1039/b412540a.
- H. M. J. Wang, C. Y. L. Chen and I. J. B. Lin, Organometallics, 1999, 18, 1216–1223 CrossRef CAS.
- H. Sivaram, J. Tan and H. V. Huynh, Organometallics, 2012, 31, 5875–5883 CrossRef CAS.
- B. Jacques, J. Kirsch, P. de Frémont and P. Braunstein, Organometallics, 2012, 31, 4654–4657 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2010, 29, 394–402 CrossRef CAS.
- M. Baron, C. Tubaro, M. Basato, A. Biffis, M. M. Natile and C. Graiff, Organometallics, 2011, 30, 4607–4615 CrossRef CAS.
- H. V. Huynh, Y. Han, J. H. H. Ho and G. K. Tan, Organometallics, 2006, 25, 3267 CrossRef CAS.
- M. Porchia, M. Pellei, M. Marinelli, F. Tisato, F. Del Bello and C. Santini, Eur. J. Med. Chem., 2018, 146, 709–746 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, C. N. Lok, J. J. Zhang and C. M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and M. P. Rigobello, Free Radic. Biol. Med., 2007, 42, 872–881 CrossRef CAS PubMed.
- J. H. Kim, E. Reeder, S. Parkin and S. G. Awuah, Sci. Rep., 2019, 9, 12335 CrossRef PubMed.
- P. C. Kunz, M. U. Kassack, A. Hamacher and B. Spingler, Dalton Trans., 2009, 37, 7741–7747 RSC.
- X. Zhou and L. Zhou, Theor. Chem. Acc., 2016, 135, 30 Search PubMed.
- M.-C. Brandys, M. C. Jennings and R. J. Puddephatt, J. Chem. Soc., Dalton Trans., 2000, 24, 4601–4606 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Wallingford, CT, 2016 Search PubMed.
- H. Schröder, A. Creon and T. Schwabe, J. Chem. Theory Comput., 2015, 11, 3163–3170 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- T. Mosmann and J. lmmun, Methods, 1983, 65, 55–63 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2416356–2416359. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra02655e |
| This journal is © The Royal Society of Chemistry 2025 |