DOI:
10.1039/D5RA02555A
(Paper)
RSC Adv., 2025,
15, 17706-17710
Synthesis of an amphiphilic bi-prodrug based on gossypol and cytarabine for drug self-delivery†
Received
12th April 2025
, Accepted 14th May 2025
First published on 27th May 2025
Abstract
Drug–drug conjugates have become a research hotspot in nanomedicine. An amphiphilic assembled drug–drug conjugate can achieve drug self-delivery without any carriers and change the distribution of free drugs. Herein, we synthesized a pH-responsive bi-prodrug of gossypol (GP) and cytarabine (Ara-C) for cancer therapy. FT-IR, UV-vis, TLC and MS confirmed that GP-Ara-C was successfully synthesized. The nanoparticle size of GP-Ara-C is approximately 100 nm, and the stability of GP-Ara-C is maintained for 6 days in pH 7.4 phosphate-buffered saline (PBS). HeLa cell viability results showed that GP-Ara-C exhibited anticancer ability similar to that of free GP. Cellular uptake demonstrated that GP-Ara-C could be distributed in cell nuclei at 7 h. Altogether, this amphiphilic GP-Ara-C could be assembled into nanomedicine in an aqueous environment and could be a candidate for cancer therapy in clinics.
Introduction
Combination chemotherapy is the common method for treating cancer in human beings.1 It can achieve synergistic effects, increase the therapeutic index, decrease single drug dose with fewer side effects and prevent drug resistance.2,3 However, it cannot change the kinetic parameters of a single drug and simultaneously distribute two drugs into tumor tissues in vivo.3,4 Using amphiphilic bi-prodrugs could be an alternative method for addressing the drawbacks of combination chemotherapy. Thus far, a few drug–drug conjugates5–11 have been synthesized and used for cancer therapy. For example, Yan and co-workers synthesized an amphiphilic drug–drug conjugate based on the hydrophilic drug irinotecan and the hydrophobic drug chlorambucil to achieve higher anticancer activity. This conjugate could be assembled into a nanoparticle and showed a longer half-life and higher distribution in tumor tissues compared with a single drug.12 Construction of assembled drug–drug conjugates in an aqueous environment should involve careful selection of hydrophobic and hydrophilic single drugs. Ni and co-workers selected the stronger hydrophilic drug gemcitabine compared with other anticancer drugs to prepare a glutathione-responsive camptothecin-ss-1,2,3-triazole-gemcitabine conjugate for combination chemotherapy.13 To endow the amphiphilic drug–drug conjugates with targeting ability, the superstrong hydrophilic molecule of lactose with hepatocyte-targeting ability was conjugated with the hydrophobic drug doxorubicin via a pH-responsive hydrazone bond to treat tumors.14 Based on these drug–drug conjugates, we found that hydrophobic and hydrophilic drugs are crucial factors for constructing assembled nanomedicine.
Herein, we selected the hydrophobic drug gossypol (GP) and the hydrophilic drug cytarabine (Ara-C) to prepare amphiphilic GP-Ara-C nanomedicine for combination chemotherapy. GP is a natural molecule obtained from cottonseed and has been used as a potent antifertility drug in China. Meanwhile, the antiproliferative activities of GP have been investigated in cancers such as breast, colon, pancreatic, ovarian, and prostate cancer.15,16 Moreover, it exhibits an antiproliferative effect via many mechanisms, such as cell cycle arrest at the G0/G1 phase,17 inhibition of DNA polymerase alpha and topoisomeraseII,18 dual-targeting mouse double minute 2 (MDM2) and vascular endothelial growth factor (VEGF),19 and inhibition of Bcl family proteins.20 The hydrophilic drug Ara-C is a nucleoside analog approved by the Food and Drug Administration in 1969.21 Its active form—cytarabine triphosphate (Ara-CTP)—is incorporated into DNA and induces single-strand breaks, leading to cell death.22 Based on the different mechanisms of GP and Ara-C for cancer therapy, this amphiphilic GP-Ara-C conjugate shows synergistic anticancer ability. Moreover, the pH-responsive imine bond in the GP-Ara-C conjugate reacted with the aldehyde group in GP, and the amino group in Ara-C was controllably broken in the acidic tumor microenvironment. Then, two free anticancer drugs were simultaneously released into the tumor tissue to achieve authentic combination chemotherapy.
Experimental
Materials and instruments
Cytarabine hydrochloride (purity: 99%) was purchased from Bidepharm Biocompany (Shanghai, China). Acetate gossypol (purity: 98%) was purchased from Huateng Pharma Company. (Hunan, China). Fetal bovine serum (FBS), DID (fluorescent probe), trypsin, cell counting kit-8, culture media, other bioagents and consumables were supplied by Beyotime company (Jiangsu, China). All other organic solvents and common consumables were purchased from Jinbaiao Biotechnology Company (Changzhi, China).
The structure of GP-Ara-C was characterized by Fourier Transform Infrared Spectrometer (FT-IR, Tensor 27, Bruker, Germany), Ultraviolet-visible Spectroscopy (UV-vis, TU-1901, Puxi company, Beijing, China) and Thermo Scientific Hybrid Quadrupole-Orbitrap Mass Spectrometers (Thermo, America). The purity of GP-Ara-C was confirmed by High Performance Liquid Chromatography (HPLC, Thermo Fisher/UltiMate 3000, Flow rate: 1 mL min−1, and Mobile phase: methanol/water = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). The particle size was measured by applying dynamic light scattering (Zetasizer Nano ZSE, Malvern) and a Transmission Electron Microscope (TEM, FEI Tecnai F30, America). Cell viability was evaluated by applying a Microplate Reader (Infinite 200Pro, TECAN), and the distribution of GP-Ara-C in cells was monitored using a Confocal Laser Scanning Microscope (LEICA/SP8, Germany).
:20). The particle size was measured by applying dynamic light scattering (Zetasizer Nano ZSE, Malvern) and a Transmission Electron Microscope (TEM, FEI Tecnai F30, America). Cell viability was evaluated by applying a Microplate Reader (Infinite 200Pro, TECAN), and the distribution of GP-Ara-C in cells was monitored using a Confocal Laser Scanning Microscope (LEICA/SP8, Germany).
Synthesis of amphiphilic GP-Ara-C
Acetate gossypol (0.2 mmol, 116 mg) and cytarabine hydrochloride (0.2 mmol, 56 mg) were added to a flask with 20 mL of anhydrous acetone. Then, a molecular sieve and stirrer were added to the flask. The flask was placed into a 50 °C water bath to react for 48 h. The reaction solution was centrifuged to obtain the supernatant. Then, the supernatant was concentrated by applying a rotary evaporator. The concentrated supernatant was purified using a silica gel column. (Vdichloromethane:Vtetrahydrofuran = 50:1) (Yield: 20%).
Characterization of amphiphilic GP-Ara-C
The pure amphiphilic bi-prodrug of gossypol-cytarabine conjugate was characterized by thin-layer chromatography (TLC), HPLC, FT-IR, UV-vis and MS. The free GP, free Ara-C and GP-Ara-C conjugate were monitored by TLC during reaction in 48 h. A new dot (Rf = 0.6) appeared on the TLC. Afterward, the powder of free GP, free Ara-C and GP-Ara-C was mixed with KBr to obtain the FT-IR spectra. Meanwhile, the UV-vis spectra were measured in acetone. Finally, the MS results of GP-Ara-C were obtained using a quadrupole-Orbitrap mass spectrometer.
Nanoparticle size of GP-Ara-C and its stability
The GP-Ara-C conjugate was dissolved in pH 7.4 phosphate-buffered saline (PBS). Then, the nanoparticle size of GP-Ara-C was measured using DLS and TEM. Meanwhile, GP-Ara-C was dissolved in pH 6.0 and pH 7.4 PBS. Afterward, the stability of GP-Ara-C was evaluated using DLS at different times.
Cell viability assays
The HeLa cell suspension was seeded into a 96-well plate for 12 h (8000 cells per well). Then, free GP and GP-Ara-C in cultural media with different concentrations (5 μg mL−1, 15 μg mL−1, 30 μg mL−1, 50 μg mL−1, 100 μg mL−1) were replaced with the old media in a 96-well plate. Afterward, CCK-8 solution (20 μL) was added into a 96-well plate at 24 h. One hour later, the 96-well plate was measured under a microplate reader at 450 nm. Finally, the cell morphology of the HeLa cells treated with free GP and GP-Ara-C was observed under a microscope.
Cellular uptake
The HeLa cell was seeded into confocal dishes for 12 h. Then, free DID and DID encapsulated GP-Ara-C with culture media were placed into confocal dishes. Eventually, the images at 1 h, 3 h and 7 h were captured using a confocal laser scanning microscope.
Preparation of DID@GP-Ara-C: DID and GP-Ara-C were simultaneously dissolved in DMSO. Then, the DMSO solution was dripped into PBS to obtain the solution of DID@GP-Ara-C. Finally, the DID@GP-Ara-C solution was diluted in the culture media for cellular uptake experiments.
Results and discussion
Synthesis of the GP-Ara-C conjugate
The product of GP-Ara-C is difficult to obtain because GP and Ara-C cannot simultaneously dissolve in CHCl3, which does not trigger the aldehyde-lactol tautomerization of GP.23 Moreover, we found that GP-Ara-C could not be obtained in DMSO. We speculated that GP and Ara-C could dissolve in DMSO simultaneously and that GP-Ara-C was saturated in DMSO with 17% aldehyde groups in GP. Studies confirmed that the characteristic proton peak of the 100% aldehyde group in GP appears in CDCl3, 53% in CD3OD and 17% in DMSO-d6.24 Furthermore, the tautomerization rate changes over time.25 Unfortunately, Ara-C could not dissolve in CHCl3. Therefore, the reactions of GP and Ara-C cannot be easily carried out in CDCl3. We chose acetone as the solvent to conduct the reaction because it does not affect the aldehyde group during the reaction.26 The GP can dissolve in acetone, and Ara-C only slightly dissolves in acetone. Consequently, the yield and reaction velocity were limited. Finally, a small amount of GP-Ara-C was synthesized by the reaction of the aldehyde group in GP and the amino group in Ara-C. The reaction mechanism of the imine bond in GP-Ara-C has been explained by previous researchers.27
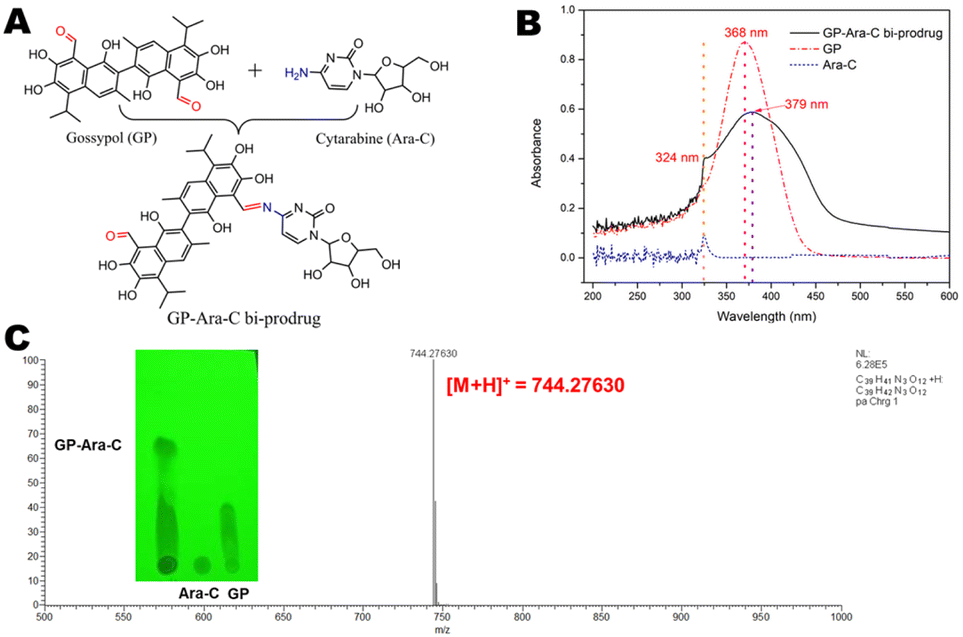
Characterization of the GP-Ara-C conjugate
The structure of GP-Ara-C is shown in Fig. 1A. The synthesis result of GP-Ara-C was confirmed by TLC, HPLC, FT-IR, UV-vis and MS. We clearly observed a new dot (GP-Ara-C) in the TLC image (Fig. 1C). Furthermore, the FT-IR spectra (Fig. 2) showed that the characteristic peaks of new bond (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–, 1629 cm−1) in GP-Ara-C, alkyl (–CH2– and –CH3), hydroxyl (1271 cm−1) and Ph–OH (1286 cm−1 and 1269 cm−1) appeared in the spectrum of GP-Ara-C. Moreover, we discovered that the characteristic peaks (324 nm and 368 nm) of Ara-C and GP in acetone were simultaneously observed in the product of GP-Ara-C in the UV-vis curve; meanwhile, the largest absorbance peak in GP at 368 nm was shifted toward 379 nm in GP-Ara-C (Fig. 1B). Based on the most important MS results, we found that the [M + H]+ ion is at m/z 744.2763. The molecular formula of GP-Ara-C is C39H41N3O12 (MW = 743.756), which is consistent with the MS results. These results confirmed that GP-Ara-C was successfully synthesized. To further confirm the structure of GP-Ara-C from the NMR data, we measured the NMR spectra of GP and Ara-C as control spectra. From the NMR data of GP in DMSO-d6, we observe that there are no peaks in the range of 5.5–6.5 ppm. Interestingly, we observe new peaks in the range of 5.5–6.5 ppm in DMSO-d6 in the structure of GP-Ara-C. This means that the new peaks in the range of 5.5–6.5 ppm in GP-Ara-C come from Ara-C (Fig. S1†). More importantly, the 1H NMR, 13C NMR and C–H COSY spectra of GP-Ara-C in CDCl3 in Fig. S2† showed that the integrals of characteristic peaks (n and a positions) are 0.95 and 1.05, respectively. Combining the MS result (m/z = 744.27630) with the ratio (1:1) of the n position (means GP) and a position (means Ara-C), we conclude that one molecule GP was conjugated with one molecule Ara-C. Additionally, the elution times of GP, Ara-C and GP-Ara-C measured by HPLC showed that the product of GP-Ara-C was successfully purified by silica gel column (Fig. S3†).
N–, 1629 cm−1) in GP-Ara-C, alkyl (–CH2– and –CH3), hydroxyl (1271 cm−1) and Ph–OH (1286 cm−1 and 1269 cm−1) appeared in the spectrum of GP-Ara-C. Moreover, we discovered that the characteristic peaks (324 nm and 368 nm) of Ara-C and GP in acetone were simultaneously observed in the product of GP-Ara-C in the UV-vis curve; meanwhile, the largest absorbance peak in GP at 368 nm was shifted toward 379 nm in GP-Ara-C (Fig. 1B). Based on the most important MS results, we found that the [M + H]+ ion is at m/z 744.2763. The molecular formula of GP-Ara-C is C39H41N3O12 (MW = 743.756), which is consistent with the MS results. These results confirmed that GP-Ara-C was successfully synthesized. To further confirm the structure of GP-Ara-C from the NMR data, we measured the NMR spectra of GP and Ara-C as control spectra. From the NMR data of GP in DMSO-d6, we observe that there are no peaks in the range of 5.5–6.5 ppm. Interestingly, we observe new peaks in the range of 5.5–6.5 ppm in DMSO-d6 in the structure of GP-Ara-C. This means that the new peaks in the range of 5.5–6.5 ppm in GP-Ara-C come from Ara-C (Fig. S1†). More importantly, the 1H NMR, 13C NMR and C–H COSY spectra of GP-Ara-C in CDCl3 in Fig. S2† showed that the integrals of characteristic peaks (n and a positions) are 0.95 and 1.05, respectively. Combining the MS result (m/z = 744.27630) with the ratio (1:1) of the n position (means GP) and a position (means Ara-C), we conclude that one molecule GP was conjugated with one molecule Ara-C. Additionally, the elution times of GP, Ara-C and GP-Ara-C measured by HPLC showed that the product of GP-Ara-C was successfully purified by silica gel column (Fig. S3†).
 |
| | Fig. 1 (A) Structure of GP-Ara-C, (B) UV-vis curves of GP, Ara-C and GP-Ara-C in acetone, and (C) TLC and mass spectrum of GP-Ara-C. | |
 |
| | Fig. 2 IR spectra of Ara-C, GP and GP-Ara-C. | |
Nanoparticle size of GP-Ara-C and its stability
Assembled GP-Ara-C was measured using TEM and DLS. The TEM image showed that the assembled nanoparticle of GP-Ara-C (1 mg mL−1) is around 30 nm derived from a dried state with significant shrinkage compared with a hydrodynamic size (Fig. 3A). When GP-Ara-C was dissolved in pure water, we observed a distinguishable red-light beam (Fig. 3B). Moreover, the DLS results showed that the assembled nanoparticle of GP-Ara-C (1 mg mL−1) was around 100 nm with the hydrodynamic size (Fig. 3B) and stable for 6 days in pH 7.4 PBS (Fig. 3C). The same assembled nanoparticle of GP-Ara-C was quickly disrupted in hours in pH 6.0 PBS (Fig. 3D). These results confirmed that the pH-responsive bond of imine in GP-Ara-C was easily broken down in pH 6.0 PBS compared with pH 7.4 PBS, resulting in the disruption of nanoparticles, which could achieve the controllable disruption of assembled nanoparticles in the acidic microenvironment in tumor tissues.
 |
| | Fig. 3 Nanoparticle sizes of GP-Ara-C measured through (A) TEM and (B) DLS, (C) their stability in pH 7.4 PBS for 6 days and (D) their pH-responsive disruption in pH 6.0 PBS in 6 hours. | |
Cell viability assays
The HeLa cell viability of GP-Ara-C was evaluated using CCK-8 assays. The IC50 values of free acetate GP and GP-Ara-C were 50 μg mL−1 and 70 μg mL−1 at 24 h, respectively. Meanwhile, we observe cell proliferation at different concentrations of free acetate GP and GP-Ara-C. As depicted in Fig. 4, the cell proliferation was significantly inhibited at the concentration of acetate GP 50 μg mL−1, and the drug efficacy of GP-Ara-C was similar to that of acetate GP for HeLa cells, demonstrating that the modification with Ara-C by imine bond could not affect the anticancer ability of acetate GP. To obtain observable images of HeLa cell morphology, we particularly captured images of acetate GP and GP-Ara-C from the 96-well plate used in the CCK-8 assays. The morphology of the HeLa cells showed that the intact cell membranes were damaged, and numerous intact cells were significantly decreased in a drug concentration-dependent mode (Fig. 5).
 |
| | Fig. 4 HeLa cell viabilities of acetate GP and GP-Ara-C with different concentrations of GP at 24 h. | |
 |
| | Fig. 5 Morphology of HeLa cells treated with different concentrations of acetate GP and GP-Ara-C (GP's concentration) from the 96-well plate in CCK-8 assays at 24 h. | |
Cellular uptake
To monitor the distribution of GP-Ara-C in HeLa cells, we prepared the DID encapsulated GP-Ara-C to observe the distribution of GP-Ara-C. Meanwhile, the free DID (red fluorescent probe) was used as the control group. We clearly observe that the free DID is completely distributed into the cell nuclei (blue color) in the merged image at 7 h. The blue color was not completely overlapped by red color (DID@GP-Ara-C) at 7 h (Fig. 6). These results confirmed that the assembled GP-Ara-C could affect the distribution of free DID in cells, meaning that the red color (DID fluorescent probe in GP-Ara-C) authentically delegated the position of assembled GP-Ara-C in cells. These results confirmed that the pathways of endocytosis were different between free DID and DID@GP-Ara-C. The authentic pathways of assembled GP-Ara-C could probably be clathrin-coated pit-mediated endocytosis (CME)28 because the features of the HeLa cell and particle size of GP-Ara-C (100 nm) were consistent with the classical size of the endocytic vesicle phenomenon.28 The different mechanisms of CME of GP-Ara-C and simple diffusion of DID probably could explain the different distribution of cell viability between free DID (or free GP) and GP-Ara-C.
 |
| | Fig. 6 Distribution of DID@GP-Ara-C in HeLa cells for 1 h, 3 h and 7 h monitored by CLSM (scale: 20×). | |
Conclusions
In this study, we prepared a pH-responsive amphiphilic GP-Ara-C nanomedicine based on GP and Ara-C. The FT-IR, UV-vis, HPLC, TLC and MS results confirmed that the product of GP-Ara-C was successfully synthesized and purified. The TEM and DLS results demonstrated that assembled GP-Ara-C could be stable in pH 7.4 PBS for 6 days and disrupted in pH 6.0 PBS for hours. The cell viabilities showed that the anticancer abilities of free GP and GP-Ara-C were similar. Furthermore, GP-Ara-C was observed in the HeLa cell nuclei at 7 h in the cellular uptake experiment. Briefly, this simple and effective pH-responsive amphiphilic GP-Ara-C nanomedicine is a candidate for cancer therapy in clinics.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Conceptualization: Ling-na Han and Xiao Duan; Data collection and analysis: Jianzhong Li, Qiang Wang, Hailong Liu, and Xinping Liu; funding acquisition: Xiao Duan and Ling-na Han; supervision: Xiao Duan; writing, review & editing: Jiangzhong Li, Xiao Duan and Ling-na Han.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82204327), the Research Project Supported by Shanxi Scholarship Council of China (No. 2024-136), the Program of Scientific and Technological Activities for Returned Scholars in Shanxi Province (No. 20230043), and the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi, China (No. 2019L0676).
Notes and references
- S. Li, M. Jiang, L. Wang and S. Yu, Biomed. Pharmacother., 2020, 129, 110389 CrossRef CAS PubMed.
- X. Pei, Z. Zhu, Z. Gan, J. Chen, X. Zhang, X. Cheng, Q. Wan and J. Wang, Sci. Rep., 2020, 10, 2717 Search PubMed.
- Z. Zhang, Y. Yue, L. Xu, Y. Wang, W. Geng, J. Li, X. Kong, X. Zhao, Y. Zheng, Y. Zhao, L. Shi, D. Guo and Y. Liu, Adv. Mater., 2021, 33, 2007719 CrossRef CAS PubMed.
- J. Chen, Y. Zhang, Z. Meng, L. Guo, X. Yuan, Y. Zhang, Y. Chai, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2020, 11, 6275–6282 RSC.
- M. Hu, P. Huang, Y. Wang, Y. Su, L. Zhou, X. Zhu and D. Yan, Bioconjugate Chem., 2015, 26(12), 2497–2506 CrossRef CAS PubMed.
- J. Zuo, X. Gao, J. Xiao and Y. Cheng, Chin. Chem. Lett., 2023, 34, 107827 CrossRef CAS.
- M. Sun, Q. Qian, L. Shi, L. Xu, Q. Liu, L. Zhou, X. Zhu, J. Yue and D. Yan, Sci. China:Chem., 2020, 63, 35–41 CrossRef CAS.
- H. Qu, L. Li, H. Chen, M. Tang, W. Cheng, T. Lin, L. Li, B. Li and X. Xue, J. Controlled Release, 2023, 363, 361–375 CrossRef CAS PubMed.
- Y. Bai, C. Liu, J. Yang, C. Liu, Q. Shang and W. Tian, Colloids Surf., B, 2022, 217, 112606 CrossRef CAS PubMed.
- J. Xi and H. Liu, Adv. Ther., 2020, 3, 1900107 CrossRef.
- J. Xiang, X. Liu, G. Yuan, R. Zhang, Q. Zhou, T. Xie and Y. Shen, Adv. Drug Delivery Rev., 2021, 179, 114027 CrossRef CAS PubMed.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136(33), 11748–11756 CrossRef CAS PubMed.
- S. Dong, J. He, Y. Sun, D. Li, L. Li, M. Zhang and P. Ni, Mol. Pharm., 2019, 16(9), 3770–3779 CrossRef CAS PubMed.
- Q. Mou, Y. Ma, X. Zhu and D. Yan, J. Controlled Release, 2016, 238(28), 34–44 CrossRef PubMed.
- Y. Huang, L. Wang, H. Chang, W. Ye, M. K. Dowd, P. J. Wan and Y. C. Lin, Anticancer Res., 2006, 26(3A), 1925–1933 CAS.
- H. Cao, K. Sethumadhavan, F. Cao and T. T. Y. Wang, Sci. Rep., 2021, 11, 5922 CrossRef CAS PubMed.
- J. Jiang, V. Slivova, A. Jedinak and D. Sliva, Clin. Exp. Metastasis, 2012, 29, 165–178 CrossRef CAS PubMed.
- R. C. Stein, A. E. A. Joseph, S. A. Matlin, D. C. Cunningham, H. T. Ford and R. Charles Coombes, Cancer Chemother. Pharmacol., 1992, 30, 480–482 CrossRef CAS PubMed.
- J. Xiong, J. Li, Q. Yang, J. Wang, T. Su and S. Zhou, Breast Cancer Res., 2017, 19, 27 CrossRef PubMed.
- Y. Liu, L. Wang, L. Zhao and Y. Zhang, Nat. Prod. Rep., 2022, 39, 1282 Search PubMed.
- J. Song, J. Yu, L. S. Jeong and S. K. Lee, Cancer Lett., 2021, 497, 54–65 CrossRef CAS PubMed.
- L. Hruba, V. Das, M. Hajduch and P. Dzubak, Biochem. Pharmacol., 2023, 215, 115741 CrossRef CAS PubMed.
- B. Marciniak, G. Schroeder, H. Kozubek and B. Brzezinski, J. Chem. Soc., Perkin Trans. 1, 1991, 2, 1359–1362 RSC.
- B. Brzezinski, J. Olejnik, S. Paszyc and T. F. Aripov, J. Mol. Struct., 1990, 220, 261–268 CrossRef CAS.
- L. Wang, Y. Liu, Y. Zhang, A. Yasin and L. Zhang, Molecules, 2019, 24, 1286 CrossRef PubMed.
- V. T. Dao, C. Gaspard, M. Mayer, G. H. Werner, S. N. Nguyen and R. J. Michelot, Eur. J. Med. Chem., 2000, 35, 805–813 CrossRef CAS PubMed.
- E. Matamoros, P. Cintas and J. C. Palacios, Org. Biomol. Chem., 2019, 17, 6229–6250 RSC.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a