DOI:
10.1039/D5RA02549D
(Review Article)
RSC Adv., 2025,
15, 15052-15085
Recent advances in the use of N-heterocyclic carbene adducts of N, P, C elements as supporting ligands in organometallic chemistry
Received
12th April 2025
, Accepted 29th April 2025
First published on 8th May 2025
Abstract
With the rapid development of N-heterocyclic carbene (NHC) coordination chemistry, the potential of NHCs as strong electron-donating ligands has been fully explored. At the same time, an increasing number of p-block element NHC adducts, due to their possession of lone pairs or polarized carbon-element π-bonds, have gradually become important ligands of significant interest in coordination chemistry. Among these, N-heterocyclic imine (NHI), N-heterocyclic phosphinidene (NHCP) and N-heterocyclic olefin (NHO) adducts, formed by linking the 2-position of the N-heterocycle with nitrogen, phosphorus, and carbon elements, respectively, have been widely applied in both main-group and transition metal chemistry. The efficient stabilization of positive charges by the N-heterocycle allows the exocyclic atoms to accumulate negative charges, thereby significantly enhancing the basicity and nucleophilicity of the exocyclic elements. The improvement in nucleophilicity has facilitated the successful synthesis of numerous complexes with unique element–metal bonds. Additionally, by modifying the N-substituents, the kinetic stabilization requirements of highly reactive species can be fulfilled. This review aims to summarize the latest research progress on NHIs, NHCPs, and NHOs in main-group and transition metal compounds. It is important to emphasize that this review focuses on the commonly used five-membered N-heterocyclic carbene adducts, including imidazolin-2-ylidenes, imidazolidin-2-ylidenes, and benzimidazolin-2-ylidenes.
1. Introduction
Since the first isolation and structural characterization of N-heterocyclic carbenes (NHCs) by Arduengo in 1991,1 this field has garnered widespread attention.2 The cyclic framework and presence of nitrogen atoms in these carbenes confer unexpectedly high stability, enabling them to form strong metal–carbon σ-bonds as ancillary ligands,3 demonstrating greater bond strength than corresponding phosphine complexes.4 By linking an exocyclic atom X to the 2-position of the N-heterocycle, the corresponding derivative ligands can be prepared. These adducts often possess polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds5 or exocyclic atoms carrying lone pairs of electrons, facilitating the transfer of nucleophilicity from the carbene carbon to the exocyclic moiety. The enhanced π-accepting ability of the carbene fragment provides opportunities for constructing unique element–metal bonds. Currently, the widely applied ligand groups mainly include N, P, C element adducts: N-heterocyclic imines (NHIs), N-heterocyclic phosphinidenes (NHCPs) and N-heterocyclic olefins (NHOs). The electron density on the exocyclic atom is directly influenced by the σ-donating and π-accepting properties of the N-heterocyclic carbenes (NHCs). Due to the excellent stabilization of positive charges by the imidazoline ring,6 the exocyclic CN, CP, and CC double bonds in NHIs, NHCPs and NHOs are significantly polarized, allowing the exocyclic atoms to accumulate negative charges. This significantly enhances their basicity and nucleophilicity. These ligands are best illustrated by the charge-separated resonance forms shown in Scheme 1.7
X bonds5 or exocyclic atoms carrying lone pairs of electrons, facilitating the transfer of nucleophilicity from the carbene carbon to the exocyclic moiety. The enhanced π-accepting ability of the carbene fragment provides opportunities for constructing unique element–metal bonds. Currently, the widely applied ligand groups mainly include N, P, C element adducts: N-heterocyclic imines (NHIs), N-heterocyclic phosphinidenes (NHCPs) and N-heterocyclic olefins (NHOs). The electron density on the exocyclic atom is directly influenced by the σ-donating and π-accepting properties of the N-heterocyclic carbenes (NHCs). Due to the excellent stabilization of positive charges by the imidazoline ring,6 the exocyclic CN, CP, and CC double bonds in NHIs, NHCPs and NHOs are significantly polarized, allowing the exocyclic atoms to accumulate negative charges. This significantly enhances their basicity and nucleophilicity. These ligands are best illustrated by the charge-separated resonance forms shown in Scheme 1.7
 |
| | Scheme 1 Canonical resonance structures of neutral N-heterocyclic imines (NHIs), N-heterocyclic phosphinidenes (NHCPs) and N-heterocyclic olefins (NHOs). | |
Nitrogen, as a highly electronegative element, has attracted considerable attention for its prominent role as a strong electron-donating atom in ligand systems. By introducing an exocyclic NH group at the 2-position of the NHC ring, N-heterocyclic imines (NHIs) with significantly enhanced nucleophilicity and basicity have been developed. In the imidazoline ring, the electron-rich π-system reduces the electrophilicity of the imine carbon atom, while the formation of a zwitterionic structure disperses electron density from the heterocycle to the exocyclic nitrogen atom, enhancing its electron-donating ability. In resonance structure Ib, the exocyclic nitrogen atom bears a negative charge, while structure Ic represents partial N-heterocyclic carbene (NHC) character (Scheme 1(I)). Meanwhile, the resonance form with two negative charges demonstrates the ability to coordinate as a 2σ, 4π electron donor (Scheme 2).7a,8 Metal complexes formed with NHI ligands typically exhibit short M–N bonds, elongated C–N distances, and large M–N–C angles (160–180°), indicating strong metal–nitrogen interactions and significant metal–2-aza-allyl or metal–imine behavior.7a Furthermore, since NHI ligands and the cyclopentadienyl (Cp) group both possess a set of 2-electron frontier orbitals, these two ligand systems can be considered isolobal, with electron-donating capabilities comparable to those of the cyclopentadienyl (Cp) and phosphinimine (R3PN) groups.9
 |
| | Scheme 2 Donor functions of anionic N-heterocyclic imines (NHIs), N-heterocyclic phosphinidenes (NHCPs) and N-heterocyclic olefins (NHOs). | |
N-heterocyclic phosphinidenes (NHCPs) can be regarded as reverse-polarized phosphinidenes.5,10 This conclusion is supported by 31P-NMR spectroscopic data, which show high electron density on the phosphorus atom, and the degree of polarization of the phosphorus–carbon double bond can serve as an indicator of the π-accepting properties of the corresponding NHC.11 Resonance structures IIa and IIb (Scheme 1(II)) each possess one or two lone pairs of electrons on phosphorus available for metal coordination, forming mono- or bidentate complexes. The availability of two lone pairs was demonstrated for the first time by isolating the bis(borane) adduct [{(IMes)PPh}(BH3)2].12 The presence of the nitrogen atom adjacent to the PC double bond leads to the accumulation of π-electron density on the terminally coordinated phosphorus atom, similar to the isolobal N-heterocyclic imines (NHIs) (Scheme 2).
Regarding N-heterocyclic olefins (NHOs), they can be viewed as the coupling product of a terminal methylene unit and an N-heterocyclic carbene (NHC). They are often referred to as deoxy-Breslow intermediates,13 ylidic olefins,14 or nitrogen-based N-heterocyclic ketenimines (HKAs).15 Since 1993, Kuhn and colleagues described the first isolable example of an NHO, NHOs have attracted significant attention in modern chemistry.16 Due to the tendency of the N-heterocyclic moiety to accommodate positive charges, N-heterocyclic olefins (NHOs) possess an electron-rich double bond. As described by the resonance forms (Scheme 1(III)), the exocyclic CC bond is highly polarized, with excess electron density concentrated on the exocyclic carbon atom (Cexo), rendering it highly nucleophilic and strongly basic. The nucleophilicity of NHOs is significantly stronger than that of commonly used Lewis bases such as DMAP (4-(dimethylamino)pyridine) but weaker than that of their NHC analogs, as verified by kinetic studies of the interaction between different NHOs and electrophiles.17 Notably, NHOs typically require storage under inert conditions at low temperatures, which is closely related to their strong basicity. The proton affinity (PA) of NHOs lies significantly at the upper end of the superbasic range.7c,17b,18 The π* orbital of the exocyclic CC double bond in NHOs overlaps minimally with the d orbitals of metal centers, resulting in little to no backbonding. Therefore, NHOs primarily act as strong σ-donors, capable of transferring significant electron density to the metal center (Scheme 2). Upon coordination, the exocyclic carbon atom (Cexo) adopts the expected sp3 hybridization.
The unique electronic and steric properties of these Lewis base ligands constitute the fundamental principle for stabilizing highly reactive elemental species and organometallic fragments through strong coordination interactions, significantly expanding the research scope of traditionally considered “transient” species. Simultaneously, they serve as highly efficient catalytic ligands widely applied in both metal-based and metal-free homogeneous catalysis systems. The integration of metal–organic frameworks (MOFs) with these ligands also holds promise for establishing novel photoredox-organocatalytic synergistic systems, paving new avenues for developing highly efficient and selective heterogeneous catalysts.19 By selecting different substituents, the steric bulk of the imidazoline ring can be conveniently modified to meet the specific requirements for the kinetic stabilization of highly reactive species. The common method for obtaining these ligands involves the deprotonation of the corresponding pre-salts using strong bases such as potassium hydride (KH) or organolithium reagents (RLi). However, this method is often limited by the steric hindrance of N-substituents. Starting from N-heterocyclic carbenes (NHCs), reacting with appropriate nitrogen, phosphorus, and carbon sources to obtain the desired ligands has proven to be a feasible approach.8c,18a,20
In this article, we focus on discussing the coordination chemistry of NHIs, NHOs and NHCPs ligands in recent years. It should be noted that while the contributions of Ghatak,8d,21 Inoue,7a and Naumann,18a among others,8c,20,22–24 who have previously summarized various aspects of NHIs, NHCPs and NHOs chemistry, our work aims to provide a more targeted and updated perspective. Notably, Tamm and colleagues have also provided a review of N-heterocyclic carbene main-group element adducts and their applications in transition metal complexes, which will be partially discussed in this review.25 Importantly, the review will concentrate on NHIs, NHOs, and NHCPs derived from imidazolin-2-ylidenes, imidazolidin-2-ylidenes, and benzimidazolin-2-ylidenes.
2. Mono(N-heterocyclic carbenes) as ligands
2.1. Group 13 element complexes
In 1993, the Kuhn group conducted pioneering work exploring the use of NHOs as ligands to stabilize electron-deficient boron centers.16 Subsequently, boron complexes of NHCPs and NHIs have also been reported successively.12,26 Currently, the coordination chemistry of the three ligands related to boron and aluminum has been extensively studied. Notably, in recent years, research has expanded to include the heavier group 13 elements (gallium, indium, thallium), with preliminary studies revealing promising developments in the area.
2.1.1. Boron complexes. To prepare novel borane species, cross-metathesis reactions have been applied multiple times to boron-based compounds. Treating germanium halide RIPrCH(GeCl3) (R = Me(1a), H(1b); RIPr = [(RCNDipp)2C])27 with Li[BH4] in Et2O or MeIPrCH(SiMe3) (2)27 mixed with THF·BH3 in toluene yields the diborane complex [RIPrCH(BH2)2(μ-H)] (R = Me(3a), H(3b)) (Scheme 3).28 The molecular structure of [IPrCH(BH2)2(μ-H)] (3b) shows a sharp B–C–B angle of 72.0(2)°. Interestingly, the exocyclic C–C bond length in 3b (1.434(2) Å) is shorter than the terminal NHO C–C bond in the noncyclic B2H5+ complex [(IPrCH2)BH2(μ-H)BH2(IPrCH2)]+ (1.467(2) Å) but longer than the terminal CC double bond (1.332(4) Å) in IPrCH2. DFT calculations support the explanation that there is no B–B bonding interaction. The structural parameters of the B2H5 complexes 3a and 3b are similar to those of the diborane [(SPPh2)2C(BH2)2(μ-H)]Li(OEt2) by Mézailles group reported,29 which also suggests that the [IPrCH]− unit acts as a four-electron donor in 3b. This intramolecular frustrated Lewis pair based on boron holds promise as an effective catalyst for the hydroboration and hydrosilylation of ketone substrates. Surprisingly, it was found that the NHOs themselves, MeIPrCH2 and IPrCH2, are effective organic catalysts for the hydroboration of ketones and aldehydes. For example, the reduction of MesCHO with IPrCH2 as the organocatalyst achieved a turnover frequency (TOF) of 99 h−1, in contrast to only 0.7 h−1 when using IPr under identical conditions. This indicates the great potential of NHOs as organic catalysts in hydroboration and hydrosilylation.
 |
| | Scheme 3 Synthesis of NHO-stabilized diborane complexes 3a and 3b. | |
Also, 3a can also be obtained from (MeIPrCH)2Zn (4) with the borane adduct H3B·SMe2.30 Additionally, mixing 4 with catecholborane (HBcat) in toluene, obtaining the product (MeIPrCH)Bcat (5) from the soluble fraction (Scheme 4). Subsequently, (4) was mixed with ArBH2 to yield (MeIPrCH)B(Ar)H (Ar = Mes(6a), Trip(6b); Trip = 2,4,6-iPr3C6H2) (Scheme 4). The C–B bonds in products 5–6b are shorter than those in BPh3 (average 1.689(6) Å),31 indicating the presence of some C–B π character. DFT calculations support the interpretation that C–B π interactions exist in 5 and 6b (WBIC–B = 1.14(5), 1.23(6b)). Mixing 6a with trityl triflate [Ph3C]OTf yielded an unusual triflate salt [(MeIPrCHMes)B(THF)(OTf)H][OTf] (7) (Scheme 5) rather than the expected bidentate boronium ion [MeIPrCH–B–Mes]+. The process maybe undergoes 1,2-Mes migration to generate a borane-type radical.
 |
| | Scheme 4 Synthesis of NHO-stabilized boranes by 4. | |
 |
| | Scheme 5 Reaction of 6a with [Ph3C]OTf. | |
Similarly, [(MeIPrCH)In]4 (8) can also be used as transmetalation reagent to yield corresponding boron complexes (6b) (Scheme 6).32 Mixing 4 and HBpin (pin = diphenylmethylidene) in toluene did not yield the expected pinacolborane derivative (MeIPrCH)Bpin, presumably due to the reduced electrophilicity of the boron center in HBpin (relative to HBcat). Notably, compound [(MeIPrCH)Li]2 did not react with HBpin either. However, Rivard and coworkers obtained (MeIPrCH)Bpin (9) by mixing 8 with HBpin (Scheme 6). This indicates that 8 has a higher propensity for B–H bond activation/ligand transfer.
 |
| | Scheme 6 Synthesis of NHO-stabilized boranes by 8. | |
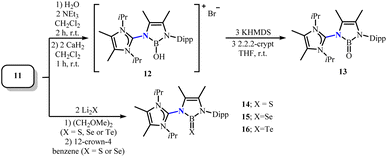
In 2020, Frank and coworkers isolated a complete series of Lewis acid-free neutral chalcogenoboranes with BX structures (X = O, S, Se, Te) by using novel ligand systems (10, HAmIm).33 The corresponding oxoboranes and telluroboranes were reported for the first time in a non-Lewis acid form. Adding hexane solution of 10 to BBr3 solution in the same solvent and then decomposing it with AgBF4 can obtain the suitable precursor 11 for preparing BX species (Scheme 7). Subsequently, the precursor 11 reacted with an equimolar amount of water in the presence of triethylamine to yield the ionic hydroxyborane 12. Deprotonation with KHMDS in the presence of 2,2,2-cryptand isolated oxoborane 13 (Scheme 8). The precursor 11 further converted with lithium chalcogenides Li2X in 1,2-dimethoxyethane to obtain the other BX species (X = S, Se, Te), with the thioborane (14) and selenoborane (15) requiring treatment with the complexing agent 12-crown-4 to remove the byproduct LiBr (Scheme 8). The BO bond length in 13 (1.353(9) Å) is very short, which may result from the absence of oxygen-coordinating Lewis acids strengthens the BO double bond character. The Wiberg bond indices of compounds 13–16 are 1.86, 1.75, 1.69 and 1.69, respectively, indicating that the novel series of chalcogenoboranes exhibit significant double bond character. Meanwhile, the decrease in WBI values indicates a weakening of π–π interactions between the boron atom and the heavier chalcogen atoms. At the same time, the chemical behavior of the BO bond in oxoboranes is similar to the classical carbonyl reactivity of CO bonds.
 |
| | Scheme 7 Preparation of 11 by HAmIm 10. | |
 |
| | Scheme 8 Synthesis of a series of neutral chalcogenoboranes with BX structures 13–16. | |
To isolate borasilene, Rivard group reacted dichloroborane 17 (ref. 34) with potassium hypersilanide (KSi(SiMe3)3) to obtain the precursor (Me3Si)3SiB(Cl)NHI 18. Treatment of 18 with KOtBu is postulated to form the elusive borasilene INT(19). Further isolation of the N-heterocyclic carbene (NHC)-stabilized borasilene adduct 20 was achieved (Scheme 9).35 The B–Si bond in 20 exhibits zwitterionic character, with the positive charge localized at the trigonal-planar boron atom and the negative charge at the silicon atom, which marks the apex of a trigonal pyramid. The conversion of 20 with pinacolborane (HBpin) results in the cleavage of the B–Si bond, yielding (NHI)BH2(NHC) and (Me3Si)2Si(Bpin)2, consistent with the polarized nature of the B–Si bond in 20.
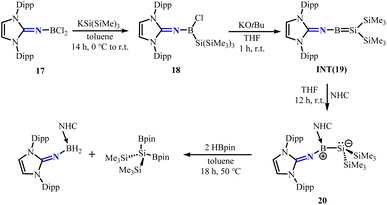 |
| | Scheme 9 Synthesis of borasilene 20 and its reaction with HBpin. NHC = 1,3,4,5-tetramethylimidazol-2-ylidene. | |
Using a similar ligand support approach, the separation of alkyl borane 21 supported by N-heterocyclic imine (NHI) and NHC ligands is achieved through dehydrohalogenation/NHC coordination (Scheme 10).36 The effective weakening of the B–C bond strength in 21 by σ- and π-electron donors allows for the synthesis of novel organoboron compounds through unconventional transformations. Compound 21 reacts with pyridine-N-oxide, sulfur, and selenium to form 1,3-dioxa-2,4-diboracyclobutane 22, thioxoborane 23, and selenoborane 24, respectively (Scheme 11). Additionally, the reaction of 21 with CO2 and CS2 yields borenium/fluorenide 25 and dithiaboretane 26 (Scheme 11). Notably, 25 rapidly converts into transient oxoborane and imidazolium enolate under heating conditions. These reactions demonstrated the oxidative functionalization, insertion, and boron-Wittig reactions of alkyl boranes.
 |
| | Scheme 10 Synthesis of alkyl borane 21. | |
 |
| | Scheme 11 Reactions of 21 with pyridine-N-oxide, sulfur, selenium, CO2 and CS2. NHC = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene. | |
2.1.2. Aluminium complexes. The use of N-heterocyclic imine-based ligands has enabled the synthesis of a variety of aluminum hydrides. Utilizing a previously established strategy,37 the reaction of {IMesNAlH2}2 (27)38 with Me3SiOTf yielded the novel dimeric aluminum hydride {IMesNAl(H)OTf}2 (28) (Scheme 12).39 Unlike the bulkier analogue {IDippNAl(H)OTf}2, where the triflate substituents adopt a cis configuration, the triflate groups in 28 are arranged in a trans configuration on the central four-membered Al2N2 ring. Moreover, N-heterocyclic imine-supported aluminum hydride compounds can serve as catalysts for the hydroboration of pinacolborane, CO2 reduction, and amine–borane dehydrocoupling, demonstrating broad catalytic applications.40 By modifying the substituents and stoichiometry, the conversion of (benzo)imidazolin-2-imine ligands with AlMe3 enables the preparation of a series of mononuclear, binuclear, and trinuclear structural aluminum complexes 29–35 (Scheme 13).41 These complexes are capable of effectively catalyzing the ring-opening polymerization of ε-caprolactone, yielding polycaprolactones. Among them, mononuclear complex 29c achieved near-quantitative monomer conversion (>99%) within 1 hour, while its dinuclear analogue 31b yielded 44% poly(ε-caprolactone) under identical conditions. Both catalysts showed significantly higher activity than AlMe3 reference (21% yield). Moreover, the reaction of [(SIDipp)NH] with AlH3·NMe2Et allows for the isolation of the dimeric aluminum dihydride {μ-LAlH2}2 (36) (Scheme 14), which can undergo substitution reactions with various small molecules.42
 |
| | Scheme 12 Synthesis of NHI-stabilized dimeric aluminum hydride {IMesNAl(H)OTf}2 (28). | |
 |
| | Scheme 13 Synthesis of a series of mononuclear, binuclear, and trinuclear structural aluminum complexes 29–35. | |
 |
| | Scheme 14 Synthesis of NHI-stabilized dimeric aluminum hydride {μ-LAlH2}2 (36). | |
In 2019, three novel NHO–trialkylaluminum complexes (37a–38) were reported.43 At room temperature, combining MeIPrCH2 with one equivalent of AlR3 in toluene yielded the monoadducts MeIPrCH2·AlR3 (R = Me(37a), Et(37b)) (Scheme 15). XRD analysis of 37a showed that the CNHO–Al bond length of 2.1198(13) Å is longer than the coordinating NNHI–Al interaction in the NHI·AlMe3 complex by Masuda group reported (1.9648(19) Å).44 This observation follows the general rule that the ligand–element bond in N-heterocyclic imine (NHI) adducts is shorter than that in NHO–element bonds. Combining MeIPrCH–CHCH2 with AlMe3 yielded MeIPrCH–CHCH2·AlMe3 (38) (Scheme 15) coordinated through the terminal exocyclic carbon atom rather than the hoped-for product coordinated to the proximal exocyclic carbon, which may be due to the large steric hindrance near the heterocycle and the more Lewis basic terminal coordination site. Interestingly, it was found that MeIPrCH2·AlMe3 (37a) exhibited FLP-type behavior in THF, leading to the polymerization of several Michael-type monomers under mild conditions.
 |
| | Scheme 15 Synthesis of NHO-stabilized trialkylaluminum complexes 37–38. | |
Combining 4 with diisobutylaluminum hydride (DIBAL–H, [iBu2AlH]2) via transmetallation yields the dialane product [MeIPrCH–(AliBu2)2(μ-H)] (39) (Scheme 16).30 The adjacent CNHO–Al bonds in 39 (2.0513(17) and 2.0328(15) Å) are shorter than the CNHO–Al bond length in 37a. The bridging Al–H bond lengths in 39 are obtained as 1.662(18) and 1.753(18) Å, similar to the average bridging Al–H bond length in α-AlH3 (1.715 Å).45
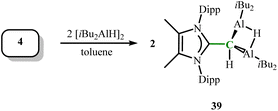 |
| | Scheme 16 Synthesis of NHO-stabilized dialane complexes 39. | |
Similarly, the monoaluminum complexes [{(IDipp)PH}AlMe3] and [{(IMes)PH}AlMe3] (R = Dipp(40a), Me(40b)) were synthesized by the transformation of (IDipp)PH and (IMes)PH with AlMe3, respectively (Scheme 17).46 However, (MeIMes)PH reacted with AlMe3 (two equivalents) to yield the dialuminum complex [{(MeIMes)PH}(AlMe3)2] (41) (Scheme 17). The P–Al–C angles in 40a ranged from 89.02(5)° to 116.77(5)°; the Al–P–C angle in 40a was 124.98(4)°, indicating that the Al atom is in a distorted tetrahedral environment. The P–C bond lengths of 40a–41 were slightly longer than their corresponding reactants, and even more so in the bis(trimethylaluminum) complex 41, which is a result of the coordination of the AlMe3 groups. These structural features suggest that the P–Al interactions in complexes 40a–41 enhance the polarization of the carbon–phosphorus double bond towards the P atom. Interestingly, it was found that the monoaluminum complexes 40a and 40b could initiate the ring-opening polymerization of racemic lactide, yielding poly(racemic lactide) with high meso selectivity, whereas the dialuminum complex 41 produced an almost atactic polymer.
 |
| | Scheme 17 Synthesis of NHCP-stabilized monoaluminum complexes 40a and 40b and dialuminum complex 41. | |
Hänisch group reported the synthesis of novel alane complexes utilizing the precursor (SIMes)PK (42a).47 The potassium salt 42a was prepared through the deprotonation of the saturated N-heterocyclic carbene-stabilized phosphinidene SIMesPH with benzyl potassium. Interestingly, converting 42a with tBu2AlCl yielded the novel cage compound [K(SIMesP)3AltBu] (43) (Scheme 18). The three phosphorus atoms in 43 are not in a planar environment, with their angles summing to 306.6(3)°. The K–P distance (375.0(1) pm) is longer than other reported potassium counterion-containing anionic Al–P compounds, such as K[Al4(PPh2)7PPh] (318.2(1) pm).48 The K–C distances are 359.6(1) pm (K–Cipso) and 311.2(1) pm (K–Cortho), suggesting that the potassium atom in 43 interacts with the aromatic system of the methylphenyl substituents. The structure of compound 43 can be inferred as an ion pair consisting of an aluminate anion and a potassium cation coordinated by an aryl group.
 |
| | Scheme 18 Synthesis of NHCP-stabilized aluminum complexes 43 and 44 by the precursor 42. | |
Subsequently, the same group reacted AlCl3 with identical precursors (SIMes)PK (42a) and (SIDipp)PK (42b) in a saturated toluene solution to obtain the cyclic dimers [(NHC)PAlCl2]2 (NHC = SIMes(44a), SIDipp(44b)) (Scheme 18).49 The structures of compounds 44a and 44b were characterized. Compound 44a formed a butterfly-shaped Al2P2 ring (internal angles: 83.9(1)–86.5(1)°) with the NHC ligands in a cis orientation; whereas compound 44b formed an almost planar square Al2P2 ring (internal angles: 86.8(1)–93.2(1)°) with the NHC ligands in a trans orientation.
2.1.3. Gallium, indium and thallium complexes. Treatment of (SIDipp)PK (42b) with one equivalent and two equivalents of GatBu2Cl yielded the mixed bridging system [(SIMes)P(GatBu2)2Cl] (45) and the four-membered ring compound [SIMesPGatBu2]2 (46), respectively (Scheme 19).47 XRD analysis showed that 46 has an almost planar coordination environment, with the angles on the phosphorus atoms sum to 355.6(2)°. The Ga–P bond lengths in 45 (239.34(4) and 240.84(4) pm) are shorter than those in 46 (246.21(7)–248.12(7) pm), indicating that the steric hindrance in the structure of 45 is less than that in 46. Similarly, mixing the precursors 42a and 42b with GaCl3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio yielded the dimers [(NHC)PGaCl2]2 (NHC = SIMes(47a), SIDipp(47b)) (Scheme 19).49 Similar to 44a and 44b, the shape of the central Ga2P2 ring (butterfly-shaped or nearly planar square) depends on the steric hindrance of the NHC ligands.
:1 ratio yielded the dimers [(NHC)PGaCl2]2 (NHC = SIMes(47a), SIDipp(47b)) (Scheme 19).49 Similar to 44a and 44b, the shape of the central Ga2P2 ring (butterfly-shaped or nearly planar square) depends on the steric hindrance of the NHC ligands.
 |
| | Scheme 19 Synthesis of NHCP-stabilized gallium complexes 45–47. | |
In 2021, the first five-membered ring triel(I) carbene analogues E(AmIm) (E = Ga, In, Tl; AmIm = aminoimidazolin-2-imine) were reported.50 The potassium salt of HAmIm 48 was prepared via benzyl potassium and reacted with GaCp* to obtain 49 (Scheme 20). In(I) or Tl(I) species 50 and 51 were obtained by conversion of the lithium salt of HAmIm 48 with InCp or TlBF4, respectively (Scheme 20). In compounds 49–51, the imidazolin-2-imine part and the phenyl part are arranged vertically, with the exocyclic N–C bond distance increased compared to 48, indicating a weakening of the double bond character. The pentagonal ring structures of compounds 49–51 all belong to planar geometry. Furthermore, the bond angles between N–E–N decreased from 79.67(7)° to 70.41(5)°, consistent with the regular increase of longer E–N bonds in the heavier triel.
 |
| | Scheme 20 Synthesis of five-membered ring triel(I) carbene analogues 49–51. | |
N-Heterocyclic imine-based bis-gallium(I) carbene analog 52 can be synthesized through the reaction of (IDipp)NLi, tBuOK, and Cp*Ga (Scheme 21).51 Compound 52 features a four-membered Ga2N2 ring structure, with the HOMO primarily dominated by non-bonding lone pairs of gallium atoms. Moreover, compound 52 demonstrates excellent nucleophilicity and potential as an imino-group transfer reagent, as evidenced by its addition reactions with MeOTf, diphenyl disulfide, and 9,10-phenanthrenequinone, as well as its reactions with ECl2 (E = Ge or Sn) to form chlorogermylene dimers and chlorostannylene dimers.
 |
| | Scheme 21 Synthesis of NHI-stabilized gallium complex 52. | |
Mixing [(MeIPrCH)Li]2 (53) with CpIn in toluene can isolate [(MeIPrCH)In]4 (8) (Scheme 22).32 The In–In–In angles in 8 [59.112(13) to 61.104(13)°] are close to the tetrahedral arrangement. The CC bonds in the [MeIPrCH] ligands of 8 (average 1.345(5) Å) are similar in length to those in free MeIPrCH2 (1.3489(18) Å),52 revealing that the C–C double bond character is retained. Combining 8 with B(C6F5)3 yielded (MeIPrCH)In(THF)·B(C6F5)3 (54) (Scheme 22), indicating that 8 can serve as a feasible source of monomeric RIn: units. The In(I) centers in 8 can also activate the B–H bonds in boranes, facilitating rapid H/NHO ligand exchange on boron (see above). To further expand the neutral indium imine species, treatment of 8 with the sterically hindered diphenyl azide ArDippN3 (ref. 53) to form indium imine (MeIPrCH)In–NArDipp (55) (Scheme 22). AIM analysis showed that the delocalization index δ(In, N) for the InN unit in 55 is 1.13, which is notably lower than the δ(In, N) values for HNInH (1.73) and PhNIn(HCC(NHCH)2) (1.39), indicating weak In–N π-bonding. Also, NBO analysis indicated that there is only one σ-bonding orbital in 55 which may result from the In–N bonding in 55 is highly polarized/weak π-bonding.
 |
| | Scheme 22 Synthesis of NHO-stabilized indium complex 8 and its reactions with B(C6F5)3 and ArDippN3. | |
2.2. Group 14 element complexes
The trimethylsilyl adducts of NHIs, NHCPs and NHOs have been widely demonstrated to be highly efficient synthons, useful for preparing a variety of ligands and serving as transmetalation reagents for the synthesis of main-group element and transition metal complexes.7b,27,54 In recent years, these ligands have also proven to be highly effective in stabilizing group 14 elements, particularly achieving significant progress in the synthesis of low-coordinate group 14 alkenes, showcasing their exceptional ability to stabilize low-coordinate species.
2.2.1. Silicon complexes. To obtain novel silaalkenes, Rivard and coworkers designed to prepare stable acyclic silaalkenes (R2Si:) through the steric effects of the vinyl N-heterocyclic olefin ligands and the electron-donating ability of the hypersilyl group [Si(SiMe3)3].55 By conversion of MeIPrCH2 with SiBr4 to obtain the precursor (MeIPrCH)SiBr3 (56). Further, reacting 56 with hypersilyl potassium [K(THF)2][Si(SiMe3)3] yielded the first bicoordinated acyclic silaalkene stabilized by a carbene ligand, (MeIPrCH)Si{Si(SiMe3)3} (57) (Scheme 23). The 29Si NMR spectrum shows a signal at 432.9 ppm, within the range of known bicoordinated acyclic silylkenes.56 Additionally, the vinyl C–C bond in 57 (1.406(3) Å) is longer compared to that in the free NHO MeIPrCH2 (1.3489(18) Å),52 presumably due to the transfer of partial π-electron density from the vinyl group to silicon. The C–Si–Si angle of 101.59(7) is consistent with the high s-character of the silicon lone pair, further validating the formation of the silaalkene. Surprisingly, compound 57 also exhibited high reactivity towards small molecules, capable of activating strong homonuclear and heteronuclear bonds such as B–H, P–P and C–H at room temperature.
 |
| | Scheme 23 Synthesis of NHO-stabilized acyclic silaalkenes 57. | |
In 2021, the same group reported the synthesis of the first bicoordinated acyclic silaalkene without heteroatom donor binding.57 By treatment of [K(THF)2][Si(SiMe3)3] and SiBr4 with MeIPr obtaining the degradation-resistant Si(II) precursor MeIPr·SiBr2 (58). Further mixing 58 with [(MeIPrCH)Li]2 (53) in toluene yielded the target product divinylsilylene (MeIPrCH)2Si: (59) (Scheme 24). The C–Si–C bond angle in 59 (100.58(8)°) is in line with the mixed s/p character of the Si lone pair, which validates the formation of the silylene. The 29Si NMR chemical shift (272 ppm) is in the highly shielded region compared to 59 (433 ppm). Notably, the initial half-wave potential E1/2 of compound 59 is −1.35 V, making it a potential strong reductant. Combining 59 with an equimolar amount of [Pd3(dba)2] (dba = dibenzylideneacetone) yielded (MeIPrCH)2Si(dba) (60) (Scheme 24), indicating that the Si(II) center in 59 has both electrophilic and nucleophilic properties.
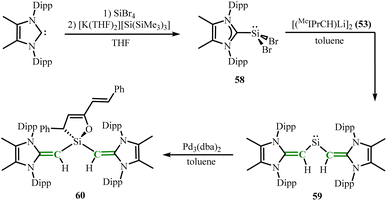 |
| | Scheme 24 Synthesis of bicoordinated acyclic silaalkenes 59 and its reaction with [Pd3(dba)2]. | |
In recent years, the isolation of multiple silicon seven-membered rings formed through the intramolecular insertion of acyclic silylenes into aromatic CC bonds has been reported.56c,58–60 Treating IPrNSiBr3(61)61 with reagents bearing different silyl ligands yields the corresponding silylenes (62) (Scheme 25). These silylenes are stabilized by NHI as a π-donor and silyl groups as σ-donors, and they can reversibly transform into silepins (63), showing the facile interconversion between Si(II) and Si(IV) (Scheme 25). Among these, the modification of different silyl ligands significantly influences the σ-donating and π-accepting abilities of the silylenes. Such silepins have been proven to function as “masked” silylenes for small molecule activation, exhibiting superior catalytic potential.
 |
| | Scheme 25 Synthesis of silylenes 62/silepins 63. (i) For 62a: KSiTMS3; for 62b: [Na·(THF)2]SitBu3; for 62c: KSi(TMS)2Si(iPr)3; for 62d: KSi(TMS)2SiPh3. | |
Furthermore, exposing 63a and 63b to an atmosphere of N2O led to the isolation of the first acyclic, neutral, three-coordinate silanones 64 (Scheme 26).58 In 64b, hyperconjugation exists between the Si–C σ-bond of the silyl group to the π*-bonding orbital of SiO moiety. Concurrently, the SiO π-bond in 64b mixes with the nonbonding orbital of the exocyclic nitrogen atom, consistent with the shorter Si–N distance (1.646(3) Å), demonstrating the electron-donating effects of the NHI ligand and the silyl group. Silanones 64 converted with CO2 and methanol to yield the corresponding silicon carbonate complexes 65 and silanol 66, respectively (Scheme 27). Notably, 64a can undergo a 1,3-silyl migration in THF-d8 to generate the intermediary disilene INT(67) (Scheme 27), which is subsequently trapped by reacting with ethylene and IMe4. It is worth mentioning that the application of the Wittig reaction to silanone 64b was reported in 2019, contributing to the extension of the synthetic methods for heavier alkenes.62 Interestingly, 64b undergoes thermal rearrangement in C6D6 or THF-d8 solution to form N,O-silylene 68 (Scheme 27). Compound 68 demonstrates diversified oxidation reactions, successfully activating small molecules such as P4, NH3, CO2 and N2O.63
 |
| | Scheme 26 Synthesis of silanones 64. | |
 |
| | Scheme 27 Reaction of silanones 64 with CO2 and MeOH, isomerization pathway of silanone 64a to INT(67) and selective transformation of silanone 64b to 68. | |
Using the same strategy, the methylated backbone N-heterocyclic iminosilicon tribromide 69 was converted with the silanide complex [Na·(THF)2]SitBu3 to yield silylene 70 (Scheme 28).64 Unlike the above silylenes 62, the silylene center in compound 70 did not insert into the aromatic C–C bonds of the Dipp substituent to form a silepin structure, which can be a result of the steric hindrance of the methylated backbone. Silylene 70 can react with benzene and its derivatives or N-heteroarenes, inserting into aromatic C–C and C–N bonds to form corresponding seven-membered rings. Moreover, silylene 70 had been proven to activate various small molecules and undergo metathesis type process with aryl isocyanides, demonstrating higher reactivity compared to the silylenes 62.65,66
 |
| | Scheme 28 Synthesis of silylenes 70. | |
Similarly, the NHCP-stabilized silylene–phosphinidene 72 was prepared by treatment of KSiTMS2SiTol3 (Tol = p-Tolyl) with IDippPSiBr3 (71) in toluene (Scheme 29).67 The 29Si NMR chemical shift (455.0 ppm, 1JSi–P = 187.5 Hz) and 31P NMR chemical shifts (269.4 ppm) are in the significantly downfield, which may due to the polarization of the P–Si bond. The angles Si–Si–P (97.99(3)°) and Si–P–C (101.43(6)°) showed a bent geometry in 72, indicating the presence of lone pairs on both silicon and phosphorus. Additionally, DFT calculations support the multiple bond character between Si and P exist in 72 (WBI = 1.47; MBO = 1.53). Therefore, compound 72 can be inferred as a rare NHC-stabilized phosphosilaalkyne. Similar to the reactivity of silylenes,65,68 the silicon center in 72 allows for oxidative addition to various unsaturated small molecules such as CO2, ethylene and acetylene, and coordination with iron tetracarbonyl to yield relevant compound 73–76 (Scheme 30), showing a highly nucleophilic silicon center. Further investigation into the mechanism of ammonia activation by compound 72. Unlike known tetrahedral center ammonia activation, after forming the silylene–ammonia adduct, further conversion to a Meisenheimer-type complex followed by deprotonation of ammonia to yield 77 (Scheme 30).69 This series of reactions led to intermolecular 1,5-hydroamination with dearylation on the NHC flank. Notably, compound 72 can isomerize to the corresponding phosphasilenylidene 78, similar to the isomerization rearrangement from nitriles to isonitriles. Compound 78 can also via conversion with acetylene and coordinate with iron tetracarbonyl to yield corresponding compound 79 and 80, exhibiting similar reactivity to 72 (Scheme 31).
 |
| | Scheme 29 Synthesis of NHCP-stabilized silylene–phosphinidene 72. | |
 |
| | Scheme 30 Reactions of 72 with small molecules and coordination with iron tetracarbonyl. | |
 |
| | Scheme 31 Isomerization of 72 to 78 and its reaction with acetylene and iron tetracarbonyl. | |
Mo group successfully isolated an N-heterocyclic imino-stabilized silylene [LSi(NHI)] (81) (L = PhC(NtBu)2) with strong σ-donor property and significant steric hindrance through reacting [LSiCl] with [NHILi] (Scheme 32).70 Notably, utilizing silylene 81 can isolate rare disilicon(0) complex 82 and ditin(0) complex 83 (Scheme 33).70,71 The complexes 82 and 83 can be further transformed into a series of unique silicon and tin complexes, demonstrating the remarkable potential of silylene 81 in stabilizing low-valent element complexes.
 |
| | Scheme 32 Synthesis of silylenes 81. | |
 |
| | Scheme 33 Disilicon(0) complex 82 and ditin(0) complex 83. | |
Recently, the synthesis of a silyl radical 86 stabilized by an NHI ligand and two silyl substituents has been reported (Scheme 34).72 The synthesis of silyl radical 86 can be achieved intentionally through the reaction of the conversion of ItBuNSiBr3 (84) with three equivalents of NaSitBu2Me or one equivalent of NaSitBu2Me with imino(silyl)disilene (85). The central silicon atom of compound 86 resides in a slightly pyramidalized geometry, with a bond angle sum of 353.51°, slightly less than that reported for several other silyl radicals (close to 360°).73 Moreover, the Mulliken spin population of 0.922 at the central silicon atom, further confirming the formation of a silicon-centered radical.
 |
| | Scheme 34 Synthesis of silyl radical 86. | |
2.2.2. Germanium, tin and lead complexes. In 2019, the synthesis of NHCPs stabilized group 14 heavier tetrelenes 87a–87c via salt elimination reactions were reported.74 Using the deprotonated compound (SIDipp)PK (42b) as a precursor, they reacted it with (SIMes)MX2 (0.5 equivalents, M = Ge, Sn, Pb; X = Cl or Br) in toluene to obtain single crystals of [(SIDipp)P]2M(M = Ge(87a), Sn(87a), Pb(87c)) (Scheme 35), which were deep purple crystals. The deep color of compounds 87a–87c initially revealed π–π interactions. The 31P NMR spectra (145.2 ppm, 87a; 121.4 ppm, 87b; 116.8 ppm, 87c) showed upfield shifts compared to other corresponding phosphinimine complexes, indicating multiple bond character between phosphorus and group 14 atoms. The P–M–P angles in compounds 87a–87c were 87.4(1)°, 85.8(1)°, and 84.6(1)°, respectively, which were lower than those in other known corresponding compounds ([(Dipp)2P]2Ge (107.40(4)°),75a [(Dipp)2P]2Sn (106.20(3)°),72 and [{(Tripp)(tBu)(F)Si}(iPr3Si)P]2Pb (97.88(4)°)75b), which could attributed to steric hindrance from large substituents. Additionally, conversion of 42b with (SIMes)MX2 (M = Ge, Sn, Pb; X = Cl or Br) (one equivalent) only successfully isolated [(SIDippP)SnCl]2 (88a) and [(SIDippP)PbBr]2 (88b) (Scheme 35). Both compounds 88a and 88b did not show π–π interactions, demonstrating the significant ability of bis(phosphinimine) coordination to stabilize group 14 atoms. Recently, the isolation of NHCP–silyl-substituted stannylene (IDipp)PSn(SiTMS2SiTol3) along with NHCP–silyl- and aryl-substituted plumbylenes (IDipp)PPb(SiTMS2SiTol3) and (IDipp)PPb(MesTer) (MesTer = 2,6-Mes2C6H3) has opened new avenues for developing mono-NHCP plumbylene systems featuring Pb–P partial multiple bond character with aryl/silyl substituents.76
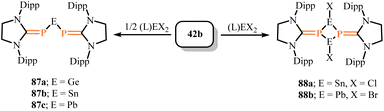 |
| | Scheme 35 Synthesis of NHCP-stabilized heavier tetrelenes 87 and 88, L = SIMes. | |
Gudat group reported the reaction of imidazolium phosphinides with organotin chlorides.77 Conversion of imidazolium phosphinides 89a,b with organotin chlorides Me3SnCl and Ph3SnCl in the presence of tertiary amines did not yield the expected substitution reactions but instead produced tin-based (oligo)phosphides and imidazolium salts without phosphorus, along with some intractable solids. Treatment of imidazolium phosphinides with diorganotin dichlorides produced Lewis adducts 90a,b and 91a,b (Scheme 36), which underwent base-induced dechlorination reactions in the presence of excess base and electrophilic reagents to generate cationic imidazolium bis(stannyl)phosphides chloride complexes 92a,b and 93a,b (Scheme 36). These studies showed that product selectivity was influenced by the electrophilicity of the Lewis acids and can clearly distinguish the following three different species: (a) weak electrophiles: for example, R3SnCl, which will form unstable Lewis adducts; (b) moderate strength electrophiles: such as R2SnCl2, which can form stable Lewis adducts; (c) strong electrophiles: for example, PCl3, which can produce Lewis pairs with sufficient PH acidity and transfer protons to the auxiliary base.
 |
| | Scheme 36 Reaction of NHCP 89 with diorganotin dichlorides. | |
Utilizing the strong electron-donating properties of N-heterocyclic imine ligands and reacting the trimethylsilyl phosphinidene complex (IDipp)PSiMe3 (94) with the germylene and stannylene chloride dimers [MesTerECl]2 to isolate the MesTerEP(IDipp) (E = Ge(95a), Sn(95b)) (Scheme 37).78 The group 14 metals exhibit sharp bond angles (89.56(5)° at Ge, 86.69(6)° at Sn), similar to those in (amino)tetrylenes (MesTer)(NHDipp)Ge: (88.6(2)°)79a and (MesTerNH)2E: (88.6(2)° at Ge, 87.07(8)° at Sn).79b The E–P bond distances in 95a and 95b (2.2364(6) Å and 2.4562(7) Å) suggest the presence of multiple bond character in the E–P bonds. The complex formed by the reaction of compound 95b with tris(pentafluorophenyl)borane (BCF) features a very short Sn–P bond, revealing a unique “push–pull” zwitterionic structure. Notably, compound 95b exhibited high catalytic activity in the hydroboration of aldehydes and ketones. As exemplified by acetophenone reduction, complete and selective conversion is achieved within 30 minutes using only 0.05 mol% catalyst loading, highlighting its significant potential for practical synthetic applications.
 |
| | Scheme 37 Synthesis of NHCP-stabilized germanium and tin complex 95a and 95b. | |
The transmetallation reagent (MeIPrCH)2Zn (4) can also be applied to group 14 halides. Mixing 4 with one equivalent of GeCl2 dioxane formed the germylene–zinc chloride adduct (MeIPrCH)2Ge·ZnCl2 (96) (Scheme 38).30 In compound 96, the germanium and zinc centers adopt a trigonal planar geometry, which is a rare Ge–Zn bonded species involving a tricoordinated germanium center. When 4 was reacted with two equivalents of ECl2 dioxane (E = Ge, Sn), it formed the halogen-bridged products, the binuclear propeller-shaped cations [(MeIPrCHE)2(μ-Cl)]2[Zn2Cl6] (E = Ge(97a), Sn(97b)) (Scheme 38). Compounds 97a and 97b both contain a C2E2Cl propeller-shaped core structure, which is a significant feature of their cationic components.
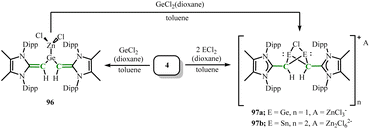 |
| | Scheme 38 Reaction of the transmetallation reagent 4 with ECl2 dioxane (E = Ge, Sn). | |
Rivard group first used N-heterocyclic olefin ligands to isolate divinylgermylene in 2017.27 Subsequently, the same group in 2021 synthesized a series of N-heterocyclic olefin stabilized group 14 heavier tetrelenes (MeIPrCH)2E: (E = Ge(98a), Sn(98b), Pb(98c)) by deprotonating vinyl lithium reagents (MeIPrCH)Li (53) with Cl2Ge·dioxane, SnCl2, and PbBr2 in Et2O (Scheme 39).57 The C–Sn–C bond angle in compound 98b [90.83(14)°] validated the existence of the Sn lone pair. The C–Pb–C bond angle in compound 98c (88.6(2)°) was sharper than that in the first known diarylplumbylene (94.5(1)°),80 showing its structural uniqueness and differences.
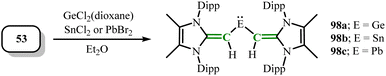 |
| | Scheme 39 Synthesis of NHO-stabilized disilylenes heavier tetrelenes 98a, 98b and 98c. | |
Inoue group reported the synthesis of the dimeric cyclopentadienyl(imino)stannylene [(η1-Cp)SnNIPr]2 (99) through the conversion of (IDipp)NLi with Cp2Sn.81 The central Sn2N2 ring of stannylene 99 adopts a nearly planar structure. Unlike similar stannylenes ([(η3-Cp)SnNC(NMe)2]2),82 compound 99 did not exhibit an equilibrium between cis and trans Cp isomers in solution. Moreover, stannylene 99 can undergo substitution reactions with CH2Cl2 or BrCH2CH2Br to yield the corresponding halogenated compounds [ClSnNIPr]2 (100a) and [BrSnNIPr]2 (100b) (Scheme 40). In contrast to the fluxional behavior of the Cp ligands in stannylene 99, the halogen groups in compounds 100a and 100b adopt a trans configuration relative to the Sn2N2 ring.
 |
| | Scheme 40 Synthesis of halogenated compounds 100a and 100b. (i) For 100a: CH2Cl2; for 100b: BrCH2CH2Br. | |
Subsequently, the same group achieved various N-heterocyclic imine stabilized group 14 heavier tetrelenes (100a, 101–104) through reacting (IDipp)NLi with corresponding chloride (Scheme 41).83–86 Similar to 100a, the Ge–Cl bonds in compound 101 adopt a trans configuration relative to the Ge2N2 ring. Furthermore, conversion of compounds 100a and 101 with Na2Fe(CO)4 yielded Fe(CO)4-bridged tin and germanium complexes 105a and 105b. Notably, a reversible formation exists between the monocoordinated and bridged species (105a and 105b) and their corresponding monocoordinated counterparts (106a and 106b) (Scheme 42).83 Interestingly, compound 100a can convert with M[Fe(CO)2(η5-C5H5)] (M = Li, K) to form the Li/Sn/Fe trimetallic complex 107. The electrophilicity of complex 107 is demonstrated by its reaction with LiI, affording the iodo-substituted tin complex 108 (Scheme 43).83 The 119Sn{1H} NMR spectrum of 108 (61.6 ppm) shows a significant upfield shift compared to that of 107 (314.0 ppm), which can be attributed to the higher electron-donating ability of the iodide ligand.
 |
| | Scheme 41 Synthesis of tetrelenes 100a, 101–104. MesTer = 2,6-Mes2C6H3. | |
 |
| | Scheme 42 Exchange between 105a and 105b to 106a and 106b. | |
 |
| | Scheme 43 Reaction of the Li/Sn/Fe trimetallic complex 107 with LiI. | |
By treatment of the bis(imino)germylene 102 (ref. 61) with gaseous N2O, a rare three-coordinate germanone [IPrN]2GeO (109) was isolated (Scheme 44),84 with the germanium center situated in a trigonal planar environment. Germanone 109 exhibits shorter Ge–N bonds (1.7819(12) Å and 1.7825(12) Å) and longer exocyclic N–C bonds (1.2872(18) Å and 1.2914(18) Å), compared to that in germylene 102 (Ge–N: 1.8194(15) Å; N–C: 1.273(2) Å), demonstrating partial metalimide character. In addition, germanone 109 displays rich reactivity toward small molecules, highlighting its polarized Geδ+–Oδ− reactivity and the ability to mimic of nucleophilic transition-metal oxides. Regarding the heteroleptic stannylene 103, it has been demonstrated to effectively activate and catalytically reduce CO2 through ligand assistance.85
 |
| | Scheme 44 Synthesis of germanone 109. | |
Recently, the synthesis of novel N-heterocyclic imine stabilized group 14 heavier tetrelenes has been reported.87 Through a ligand exchange reaction, Inoue group successfully synthesized an isostructural amino stannylene compound (TMS2N)(ItBuN)Sn: (110) by treatment of (ItBuN)H with stannylene (TMS2N)2Sn (Scheme 45).88 Additionally, when reacting ECl2·dioxane (E = Ge, Sn) with two equivalents of lithiated ligand (ItBuN)Li, obtaining homostructural tetrelenes (ItBuN)2E: (E = Ge(111a), Sn(111b)) (Scheme 46). Compound 111a, in toluene-d8 solvent, reaches a dynamic equilibrium between monomer and dimer forms with temperature changes, tending to exist as a monomer at room temperature. Compound 111b, whether in solution or in the solid state, stably exists as a monomeric molecular structure without showing changes in aggregation state. Notably, 111a and 111b exhibit high selectivity in the conversion reactions with N2O or CO2, which expanding the application scope of homostructural tetrelenes in chemical synthesis, and providing new perspectives and tools for studying small molecule activation mechanisms.
 |
| | Scheme 45 Synthesis of NHI-stabilized stannylene 110. | |
 |
| | Scheme 46 Synthesis of NHI-stabilized homostructural tetrelenes 111a and 111b. | |
2.3. Group 15 element complexes
Among the pnictogen elements, the combination of NHIs, NHCPs and NHOs ligands with nitrogen facilitates the creation of novel ligand systems, expanding the field of sterically hindered ligands and providing more possibilities for applications in organometallic chemistry and catalytic chemistry.7a For phosphorus, the three ligands have demonstrated even broader utility, with significant developments in stabilizing phosphinyl radicals and constructing new ligand systems.7a,22–23
2.3.1. Nitrogen complexes. Rivard group isolated compound NHO (IPrCH)NMe2 (112) by reacting Eschenmoser's salt [H2CNMe2]I89 with two equivalents of the carbene DippIPr.90 Notably, research found that the amine donor attached to this neutral N-heterocyclic olefin primarily coordinates with AuCl (113) through the exocyclic olefin part rather than the terminal N-based center (Scheme 47).
 |
| | Scheme 47 Synthesis of (IPrCH)NMe2 (112) and its reaction with AuCl. | |
N-heterocyclic olefins react with N2O in acetonitrile, where the N–O bond breaks to form NHO dimers (114a–114c) connected by azo-bridge (Scheme 48).91 These dimers, as emerging superelectron donors, exhibit significant characteristics, with their first oxidation potential (E1/2 = −1.34) indicating the strong reducing ability. Additionally, these dimers can undergo selective single-electron or double-electron oxidation processes, respectively converting into stable radical cations or di-cationic forms of imidazolium salts. Surprisingly, finding the formation of diazene (115a–115d) from the treatment of NHOs with N2O (Scheme 48),92 where the nature of the solvent is a key factor in product selectivity. Like azo compounds, compounds 115a–115d are easily decomposed under ultraviolet light but exhibit significant thermal stability. N-heterocyclic diazenes possess high charge density, showing strong ylide characteristics. Notably, N-heterocyclic diazenes can serve as strong C-donor ligands, combining with transition metal and main group metal complexes to separate diazene complexes 116–119 without releasing dinitrogen (Scheme 49).92 Additionally, N-heterocyclic diazene can undergo (2 + 3) cycloaddition reactions with CS2, dimethyl acetylenedicarboxylate, N-phenylmaleimide, and tetracyanoethylene (TCNE) to yield compound 120–123 (Scheme 49);92 in methanol, it undergoes head-to-tail dimerization through a (3 + 3) cycloaddition reaction to produce strongly reducing quinone-type tetrazines (124a–124c) (Scheme 49),93 demonstrating rich reactivity and broad application prospects. Furthermore, neutral group 14 diazoolefin complexes 125 (M = Ge(125a), Sn(125b)) can be isolated starting from 115a (Scheme 50). Interestingly, the reaction of 125a with potassium hypersilanide (KSiTMS3) yields the cyclic bis-vinyl germylene 126, which features an aromatic five-membered ring structure.94
 |
| | Scheme 48 Reaction of NHOs with N2O. | |
 |
| | Scheme 49 Reaction of N-heterocyclic diazene 115a. | |
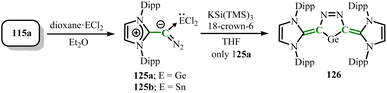 |
| | Scheme 50 Synthesis of neutral group 14 diazoolefin complexes 125 and cyclic bis-vinyl germylene 126. | |
Similarly, Hansmann group recently prepared novel benzimidazole-derived diazoalkene 127 through an NHC/N2 ligand exchange reaction.95 Compound 127 can react with DAC, releasing N2 and generating a new polarized carbodicarbene 128 (Scheme 51). This metal-free N2/DAC exchange strategy enables the free carbodicarbene ligand to react with any chosen metal. By conversion with (dms)AuCl or [RhCl(CO)2]2, the corresponding metal complexes 129 and 130 were generated (Scheme 51), respectively, validating the effectiveness of this approach.
 |
| | Scheme 51 Reaction of benzimidazole-derived diazoalkene 127 with DAC and coordination reaction of 128 with metal. | |
2.3.2. Phosphorus complexes. In 2019, a new method to synthesize heteroleptic bis(NHC) complexes diphosphines and diarsines was reported.96 By treatment of the anionic N-heterocyclic carbenes 131a and 131b with (IDipp)PSiMe3(73), obtaining the monochlorides (WCA–IDipp)E(Cl)P(IDipp) (E = P(132a), As(132b)) (Scheme 52). Subsequently, dechlorination with GaCl3 yielded the mono-cationic diphosphine and diarsine species [(WCA–IDipp)EP(IDipp)][GaCl4] (E = P(133a), As(133b)) (Scheme 53). Conversion of compounds 132a and 132b with 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine yielded the neutral radical species 134a and 134b (Scheme 53). The X-band EPR spectrum of compound 134a exhibited a broad doublet of doublets (g = 2.0087), accompanied by two distinct hyperfine coupling constants of A(31P1) = 53.5 G and A(31P2) = 35.9 G. In contrast, the spectrum of compound 134b showed a broad multiplet (g = 2.0249), with hyperfine coupling constants A(75As1) = 26.8 G and A(31P2) = 33.9 G, revealing a high degree of asymmetry in the spin density distribution in 134a and 134b. This method provides the possibility to prepare various other types of hetero-diatomic species.
 |
| | Scheme 52 Synthesis of the monochlorides (WCA–IDipp)E(Cl)P(IDipp) 132a and 132b. | |
 |
| | Scheme 53 Synthesis of the mono-cationic and neutral heteroleptic diphosphine and diarsine 133 and 134. | |
Similarly, Weigand group reduced (ClImDipp)PP(Dipp)[OTf] (135[OTf]) or (ClImDipp)PP(Cl)(Dipp) (136) with cobaltocene (CoCp2) to yield novel mixed-substituted neutral diphosphinyl radical (137).97 The X-band EPR spectrum of 137 showed a hyperfine doublet signal generated by the interaction of the unpaired electron with two inequivalent phosphorus nuclei (A(31P1) = 15.1 G and A(31P2) = 70.5 G, g = 2.0095). This EPR data is similar to 134a, revealing that the spin density is mainly concentrated on the two phosphorus atoms. Interestingly, 137 reacts with P4 to yield P8 compound 138 (Scheme 54). Additionally, this process can also be achieved by adding P4 after the formation of 136. The P4 core of compound 138 is configured in a butterfly shape, with the P2 units arranged in an exo, exo-configuration on either side, and the Dipp substituents on the phosphorus atoms pointing in opposite directions.
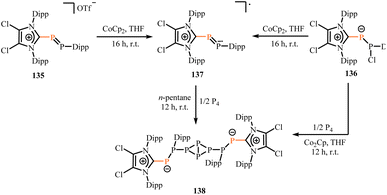 |
| | Scheme 54 Synthesis of the neutral diphosphinyl radical 137 and its reaction with P4. | |
By abstracting chloride ions from the corresponding phosphoryl chlorides, two oxophosphonium ions [139]+ and [140]+, stabilized by bulky imidazolin-2-imine and imidazolin-2-olefin substituents on phosphorus, can be prepared (Scheme 55).98 Due to the differing donor capabilities of N-heterocyclic olefins and N-heterocyclic imines, the electrophilicity of the phosphorus center can be effectively modulated, with [139]+ exhibiting significantly stronger Lewis acidity compared to [140]+.
 |
| | Scheme 55 NHI, NHO-stabilized oxophosphonium salts 139[X] and 140[X]. [X] = [OTf]− or [BArF]−. | |
Dielmann group reported rare Lewis base-free phosphonium dication salts 141 and 142 (Scheme 56),99 in which the phosphonium dications adopt a trigonal planar geometry, and the electron-deficient phosphorus centers maintain a suitable distance from the anions. In 2023, the same group further isolated the first terminal methylene phosphonium ions 143 and 144 (Scheme 56),100 which can serve as suitable precursors for generating mono-substituted phosphine carbenes.
 |
| | Scheme 56 NHI-stabilized phosphonium salts 141–144. For 141: [X] = [B(C6F5)4]− or [BArF]−; for 142: [X] = [B(C6F5)4]−. | |
Interestingly, the generation of electron-rich phosphines with photo-switchable electronic properties is achieved by utilizing the NHI substituents linked to dithienylethene (DTE).101 This is accomplished through the deprotonation of imidazolium salt 145 using n-butyllithium, followed by a reaction with the corresponding chlorophosphine to yield photo-switchable electron-rich phosphines PS–IAPs (R = iPr(146a–o), tBu(146b–o), Ph(146c–o)) with varying donor capabilities (Scheme 57), where the notation o indicates the open-ring form of the DTE unit and c indicates the close-ring form of the DTE unit. Through measuring the TEP values of the corresponding nickel complexes [(PS–IAP)Ni(CO)3] (R = iPr(147a), tBu(147b), Ph(147c)), showing that under UV and visible light irradiation, the photochromic DTE unit undergoes a reversible photocyclization reaction, thereby changing the electron-donating ability of the phosphine.
 |
| | Scheme 57 Synthesis of NHI-stabilized electron-rich phosphines 146 and its interconversion under different light conditions. The linkage of NHOs with a P-substituted indenyl heterocycle. | |
In 2024, a series of P(III) and P(V) bis(azido)phosphines and bis(azido)phosphine chalcogenides were reported (148–150) (Scheme 58).102 These bis(azido)phosphines can undergo chemoselective Staudinger reactions with tertiary or secondary phosphines, generating modular mono(azido) P-chiral phosphines (151–153) (Scheme 59), demonstrating the stabilizing effect of NHI ligands on storable bis(azido)phosphines.
 |
| | Scheme 58 Synthesis of P(III) and P(V) bis(azido)phosphines and bis(azido)phosphine chalcogenides 148–150. | |
 |
| | Scheme 59 Synthesis of mono(azido) P-chiral phosphines 151–153. | |
Very recently, a open-shell singlet diphospha-indenylide system featuring imidazole-based N-heterocyclic olefin with terminal CH2 donor groups has been reported (Scheme 60).103 The imidazolyl group can undergo thermal rotation around the central C–C bond connecting it to the phosphorus-substituted indenyl ring, thereby modulating the charge separation and diradical character. For compounds 154a–154c, the LUMO is predominantly localized on the indenyl ring, whereas in compound 154d, the LUMO is mainly situated on the NHO moiety. This localization makes the perylene segment in 154d more susceptible to reduction, leading to the formation of a magnesium salt byproduct containing a radical anion during the synthesis of 154d.
 |
| | Scheme 60 Synthesis of the linkage of NHOs with a P-substituted indenyl heterocycles 154a–154d. | |
In recent years, similar to N elements, the combination of NHC ligands with P-based donors to prepare novel large-bulky phosphine ligands has attracted increasing attention. In early research reports, Beller group revealed that imidazolium-alkylphosphines (155) can generate highly active catalysts under the synergistic effect of Pd(II) sources and bases, promoting the hydroxylation of aryl halides, and applied to the construction of C–N bonds in Buchwald–Hartwig coupling and C–C bonds in Sonogashira coupling, fully demonstrating the great potential of such ligands.104
Rivard group isolated the target product (IPrCH)PR2 (R = iPr(156a); Ph(156b)) by mixing IPrCH2 with ClPR2 in THF (Scheme 61).100 The C(sp2)–P bond in 156a (1.780(3) to 1.788(2) Å) are shorter compared to the C(olefin)–P bond in cis-Ph2P–CHCH–PPh2 (1.817(3) and 1.825(3) Å),105 which may be due to the increased C(π) → P–C(σ*) hyperconjugation effect. The donor capabilities of the new phosphines NHOPs 156a and 156b were verified through coordination of the terminal P atoms with Lewis acids BH3 and Me2S·AuCl (Scheme 62).
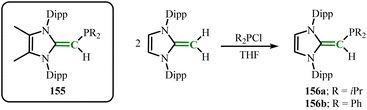 |
| | Scheme 61 Synthesis of (IPrCH)PR2 (R = iPr(156a); Ph(156b)) and imidazolium-alkylphosphines 155. | |
 |
| | Scheme 62 Reaction of 156a and 156b with BH3, Me2S·AuCl. | |
Subsequently, the same group further expanded their research on the coordination chemistry of N-heterocyclic olefin phosphine ligands.106 By mixing 156b with [AuCl(SMe2)] in toluene, isolating the target bis(gold(I)) chloride adduct [(IPrCH)PPh2(AuCl)2] (159) (Scheme 62), which confirmed that the P and C donor sites in 156a,b can be simultaneously utilized. In compound 159, the length of the exocyclic C–C bond (1.477(14) Å) is significantly longer than the corresponding C–C bond in compound 158b (1.381(4) Å), indicating a weakening of the double bond character in compound 159. Additionally, the AuCl units in compound 159 are arranged in a trans configuration, and no significant Au⋯Au interactions were observed.
Recently, corresponding ruthenium complexes using N-heterocyclic olefin–phosphine ligands (NHOP) were synthesized.107 By treatment of the N-heterocyclic olefin with PPh2Cl, followed by reaction with [Ru(p-cymene)Cl2]2 yielded the half-sandwich ruthenium complexes [{(160a/160b)}Ru(p-cymene)Cl2]I (161) (Scheme 63). Notably, 161b can be obtained directly from N-benzyl benzamide derivatives and internal alkynes in a one-pot two-step sequence to yield cyclized and olefinated isoquinolinones, showing great potential as catalysts for C–H and N–H functionalization reactions.
 |
| | Scheme 63 Synthesis of NHOP 160a and 160b and its ruthenium complexes 161a and 161b. | |
To further expand phosphine carbene complexes, a series of monodentate or bidentate phosphine ligands modified with N-heterocyclic imine groups have be synthesized recently (Scheme 64).108 These ligands exhibit strong electron-donating capabilities, even surpassing those of alkyl phosphines and classical NHC ligands. Moreover, Dielmann group has reported the synthesis and characterization of a novel class of NHI-substituted phosphino(silyl)carbenes.109 Conversion of the phosphonium chloride salts 167a,b (ref. 110) with lithium-(trimethylsilyl)diazomethane, obtaining compound 168a,b. Further, 168a,b were formed into room temperature stable carbenes 169a,b by irradiation with 365 nm ultraviolet light or sunlight in n-hexane solution (Scheme 65). Notably, compared to other phosphine carbenes,111 169a (132°) and 169b (135°) exhibited significantly smaller carbene bond angles, which would be beneficial for them to coordinate with transition metals. Access to the complex 170 is granted by conversion of 169a with AuCl(tht) (tht = tetrahydrothiophene), while complex 171 can be similarly obtained by reacting 169b with CuOtBu (Scheme 65). These results demonstrate the significant function of such phosphine carbenes in stabilizing transition metals.
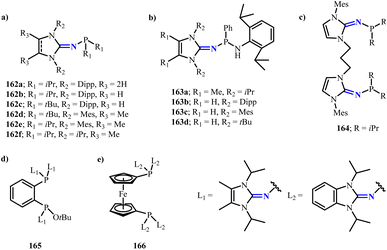 |
| | Scheme 64 Phosphine carbene complexes bearing N-heterocyclic imine group 162–166. | |
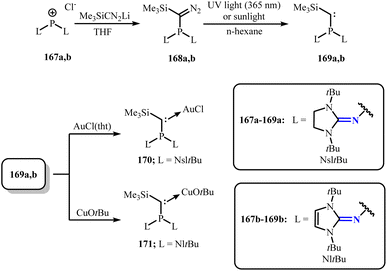 |
| | Scheme 65 Synthesis of phosphino(silyl)carbenes 169a and 169b and its reaction with AuCl(tht) (tht = tetrahydrothiophene) and CuOtBu. | |
2.4. Transition element complexes
Over the past two decades, the coordination chemistry of NHIs, NHCPs and NHOs ligands has experienced extensive development, encompassing nearly all transition metals. The following section will briefly document the latest advancements in the chemistry of transition metals and rare earth metals featuring the NHIs, NHCPs and NHOs as supporting ligands in recent years.
Conversion of the tris(aminobenzyl) rare earth complexes RE(CH2C6H4NMe2-o)3 with imidazolin-2-imines at room temperature can obtain a series of imidazolin-2-imine-based rare earth alkyl complexes 172a–172d (Scheme 66).112 DFT calculations support the existence of a significant strong interaction between the N atom of the NHI and the scandium(III) ion in 172a (WBISc–N = 1.36). Further MBO decomposition analysis found that the formation of the Sc–N bond benefited from the contributions of one σ orbital and two π orbitals, revealing the ability of the N atom to act as a 2σ, 4π-electron donor.
 |
| | Scheme 66 Synthesis of NHI-stabilized rare earth alkyl complexes 172a–172d. | |
Recently, a series of tetravalent cerium(IV) complexes 173–175 using imidazolin-2-iminato ligands as auxiliary ligands were reported (Scheme 67).113 Studies have shown that the 5d orbitals play a crucial role in the bonding process, and the strong electron-donating properties of the imidazolin-2-iminato ligands significantly enhance the energy-level matching between the Ce(IV) 5d orbitals and the alkyl, aryl and alkynyl groups. This discovery further validates the broad application potential of imidazolin-2-iminato ligands in rare-earth metal complexes.
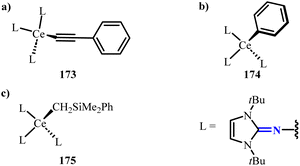 |
| | Scheme 67 Synthesis of NHI-stabilized cerium complexes 173–175. | |
Xu and coworkers reported the combination of unsaturated N-heterocyclic olefins or N-heterocyclic carbenes with rare earth aryloxide complexes and their synergistic reactivity. By combining the unsaturated NHC(ItBu) with rare earth aryloxide complexes RE(OAr)3, obtaining unusually bound NHC complexes (RE = La(176a), Sm(176b), Y(176c)) (Scheme 68),114 and no expected product was obtained for the complex of the smallest atom, Sc. Although the reaction of NHO (4,5-dimethyl-2-isopropyl imidazole) with rare earth aryloxide complexes did not yield the corresponding metal–NHO adducts, studies showed that the Lewis acid–base pairs formed by the combination of RE(OAr)3 with NHO and RE/aNHC could achieve synergistic H2 activation (177) (Scheme 68). Interestingly, the Lewis acid–base pair formed by La/NHO could convert with (trimethylsilyl)diazomethane to generate isocyanide trimethylsilyl lanthanum amine complex (178) (Scheme 68). At the same time, it could also via conversion with α, β-conjugated imines to produce La/C 1,4-addition products (179) (Scheme 68).
 |
| | Scheme 68 Unusually NHC-stabilized rare earth aryloxide complexes 176a–176c, NHO with activated hydrogen 177, isocyanide trimethylsilyl lanthanum amine complexes 178 and La/C 1,4-addition products 179. | |
A series of mono- and bis-benzimidazolin-2-imino titanium complexes (181–182) were prepared by treatment of N-silylated benzimidazolin-2-imine ligands (180) with TiCl4 (Scheme 69).115 The structures of 181a, 181c and 182a were characterized, showing distorted tetrahedral geometries. The C–N(CN)–Ti bond angles were nearly linear (171.7(2)°, 181a; 175.93(17)°, 181c; 170.1(3)° and 167.5(3)°, 182a). Additionally, the CN and N(CN)–Ti bond distances and cone angles were smaller than those of other analogues,116 indicating stronger electron-donating ability and larger coordination space. These complexes demonstrate promising catalytic performance in both ethylene homopolymerization and α-olefin copolymerization, producing polymers with narrow molecular weight distributions (PDI = 1.2–2.8) while maintaining good polymerization activity even under elevated temperatures (90 °C).
 |
| | Scheme 69 Synthesis of NHI(180)-stabilized titanium complexes 181 and 182. | |
To further prepare titanium(IV) complexes formed by the combination of asymmetric imidazolin-2-imine ligands, reacting the silylated imines 183a and 183b with titanium chloride precursors, removing Me3SiCl, obtaining a series of mono- and bis(imidazolin-2-imino) titanium complexes 184–186 (Scheme 70).117 Similar to 181a and 181c, 184a and 184b showed distorted tetrahedral geometries. The Ti–N–C angle in 184a was 162.58(5)°, significantly deviating from linearity, which may due to the different steric hindrance of the methylphenyl and adamantyl substituents. The titanium complexes 184–186 all had short Ti–N distances and large Ti–N–C angles (162°–173°), revealing that these systems can effectively provide electrons to form stable chemical bonds with the central metal ions as ligands.
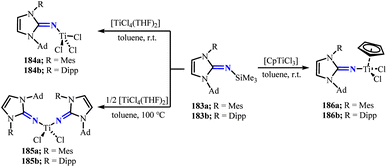 |
| | Scheme 70 Synthesis of NHI(183)-stabilized titanium complexes 184–186. | |
In 2022, Beckhaus group obtained the deprotonated product 188 by reacting the bistitanium complex (187) with ImMe4CH2 (Scheme 71).118 The corresponding anionic N-heterocyclic olefin unit (ImMe4CH−) added to the Ti center in the reaction without the formation of by-products. The structure of complex 188 showed a trigonal pyramidal geometry around the central titanium atom. DFT calculations indicated that the HOMO contains a polarized titanium–carbon π bond, which is distributed over the Ti–C–C motif. This polarized titanium–carbon π bond indicates the presence of delocalized π bonding along the Ti–C–C axis. The Wiberg bond indices for exocyclic C–C and Ti–C (WBIC–C = 1.49, WBITi–C = 1.02) further indicate the partial π bond character along the Ti–C–C axis.
 |
| | Scheme 71 Synthesis of NHO-stabilized titanium complexes 188. | |
Subsequently, novel NHO–transition metal complexes (MeIPrCH)2ECl2 (E = Ti(189a), Zr(189b), Hf(189c)) were reported (Scheme 72),119 which were prepared by the salt exchange reaction of (MeIPrCH)Li with group 4 tetrachlorides. DFT calculations found that the most red-shifted HOMO to LUMO transition in 189a was calculated to be 550 nm, close to the experimental value of 520 nm, involving the overlap of the ligand CC π and metal Ti(d) orbitals. This calculation result validated the deep color characteristics of the complex 189a. It is speculated that the NHO–metal interaction is stronger in its heavier homologues, leading to blue shifts in absorption and downfield shifts in the 1H NMR CCH resonances. Interestingly, mixing 189b with excess sodium metal yielded the rare arene-masked zirconium complex (MeIPrCH)2Zr (190) (Scheme 72).119
 |
| | Scheme 72 (MeIPrCH)2ECl2 (E = Ti(189a), Zr(189b), Hf(189c)) and arene-masked zirconium complex 190. | |
Mono-(imidazolin-2-iminato) hafnium(IV) complexes 191–194 (R = iPr(191), tBu(192), Mes(193), Dipp(194)) can be prepared by reacting imidazolin-2-imines with hafnium tetrabenzyl complex (HfBn4) (Scheme 73).120 Among these, complex 191 adopts a dimeric structure, which can be attributed to the smaller steric hindrance around the Hf center. Furthermore, by reacting with different equivalents of iPrOH and BnOH, tri-isopropoxide complex 195 and dimeric complexes 196–198 can be obtained (Scheme 74). Notably, these hafnium complexes effectively catalyze the block copolymerization of ε-caprolactone with rac-lactide. Particularly, complexes 195 and 197 exhibit outstanding performance with TOF of 140 × 105 h−1 and 130 × 105 h−1, respectively, highlighting their significant potential for oxygen-containing substrate transformations.
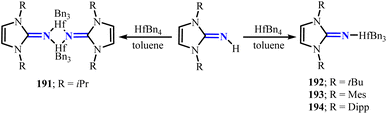 |
| | Scheme 73 Synthesis of NHI-stabilized hafnium complexes 191–194. | |
 |
| | Scheme 74 Reactions of 194 with different equivalents of iPrOH and BnOH. | |
The rare half-sandwich imidazolin-2-imine iron and cobalt complexes were reported (Scheme 75).121 The  distance of the “pogo stick” structure iron complex [Cp′Fe(NImDipp)] (199a) is 1.90 Å, which was comparable to the values of related Fe(II) high-spin species (1.88–2.05 Å) and consistent with the high-spin system of 4e−.122 The high-spin state of the Fe center was initially revealed, which was further confirmed by intermediate paramagnetic relaxation in the Fe Mössbauer spectrum. Similar cobalt complex (199b) represent a rare single-legged piano stool species. The spin state of the cobalt center in 199b exhibited temperature-dependent spin crossover. These half-sandwich imidazolin-2-imine metal complexes all showed short M–N bond lengths, which can be attributed to the multiple bond character induced by the strong 2σ, 4π electron-donating effect of the imidazolin-2-imine ligand.
distance of the “pogo stick” structure iron complex [Cp′Fe(NImDipp)] (199a) is 1.90 Å, which was comparable to the values of related Fe(II) high-spin species (1.88–2.05 Å) and consistent with the high-spin system of 4e−.122 The high-spin state of the Fe center was initially revealed, which was further confirmed by intermediate paramagnetic relaxation in the Fe Mössbauer spectrum. Similar cobalt complex (199b) represent a rare single-legged piano stool species. The spin state of the cobalt center in 199b exhibited temperature-dependent spin crossover. These half-sandwich imidazolin-2-imine metal complexes all showed short M–N bond lengths, which can be attributed to the multiple bond character induced by the strong 2σ, 4π electron-donating effect of the imidazolin-2-imine ligand.
 |
| | Scheme 75 Half-sandwich imidazolin-2-imine iron and cobalt complexes 199a and 199b and NHCP-stabilized 3d metal(II) complexes 200a, 200b and 200c. | |
In 2021, the first open-shell, tricoordinated 3d metal(II) phosphinimine adduct complexes [M{PH(SIDipp)}{N(SiMe3)2}2] (M = Mn(200a), Fe(200b), Co(200c)) were reported,123 which were prepared by treatment of [M{N(SiMe3)2}2] (M = Mn–Co) with [(SIDipp)PH] (Scheme 75). The M–P distances in the complexes (2.6033(10) Å (200a), 2.5185(6) Å (200b), and 2.4572(8) Å (200c)) were significantly longer than those in other similar adducts such as [(NHC)P(H)Fe(CO)4] (1.828(3) Å).124 Additionally, these complexes showed a Y-shaped geometry with M–P–C angles (about 135°) larger than those in [(NHC)PH] or [(NHC)PPh] type ligands (about 110°),124–126 indicating weaker σ and π electron-donating interactions between the metal and the phosphinimine, which may due to steric repulsion between the Dipp groups of the NHC framework and the silylamine ligands.
Lin group reported a series of three-coordinate terminal halide complexes of first-row transition metals 201 supported by imidazolin-2-iminato ligand.127 Furthermore, salt elimination reactions between these halide complexes and methyl transfer reagents (LiCH3 and CH3MgCl) yielded methyl complexes 202 and 203, adopting bridging (Cr) and terminal (M = Mn, Fe, Co) coordination modes, respectively (Scheme 76). Antiferromagnetic coupling was observed between the metal ions in these methyl complexes. Moreover, 201d and 202 demonstrated competent catalytic activity in Kumada cross-coupling reactions.
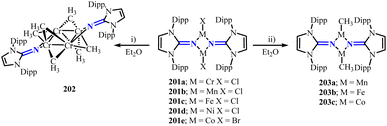 |
| | Scheme 76 Synthesis of NHI-stabilized methyl complexes of transition metals 202 and 203. (i) For Cr: 3 equivalents CH3Li in DME. (ii) For Mn, Fe: 2 equivalents CH3MgCl in THF; for Co: 3 equivalents CH3Li in DME. | |
The conversion of (IDipp)PPh (204) with the dimer [M(μ-Cl)(COD)]2 (M = Rh, Ir, COD = 1,5-cyclooctadiene) led to the isolation of the corresponding complexes 205 and 206 (Scheme 77). Following a similar approach, (IDipp)PSiMe3 (94) reacted with the dimer [Ir(μ-Cl)(COD)]2 to yield [Ir(COD)Cl{(IDipp)PSiMe3}] (207) (Scheme 77). Furthermore, exposing complex 205 to CO gas resulted in the formation of the rhodium–carbonyl complex 208 (Scheme 77).126 The P–Ccarbene bond lengths in 205–207 (1.817(3) to 1.828(3) Å) are significantly longer than those in (IDipp)PPh (149) (1.7658(10) Å) and (IDipp)PSiMe3 (94) (1.7800(13) Å), indicating single-bond character. Notably, the phosphorus atoms in 205–207 exhibit a trigonal pyramidal coordination geometry, consistent with the presence of a lone pair on the central phosphorus atom, suggesting potential for further coordination.
 |
| | Scheme 77 Synthesis of NHCP-stabilized rhodium and iridium complexes 205–208. | |
Tamm group reported the synthesis of iridium(I) catalysts with phosphine-imidazolin-2-imine ligands.128 Combining the novel bidentate P,N ligand (209) with [Ir(COD)Cl]2 (COD = 1,5-cyclooctadiene). Then, adding sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, NaBArF and lithium tetrakis(nonafluoro-tert-butoxy)aluminate, Li[Al{OC(CF3)3}4], respectively, to obtain complexes 210a and 210b (Scheme 78). Placing complex 210b in a dihydrogen atmosphere to obtain the diiridium tetrahydride (211b) (Scheme 78). 211b formed an Ir2H4 core with two bridging and two trans-oriented terminal hydrogen atoms, featuring the rare structural characteristic of terminal hydride ligands with empty coordination sites on the opposite side. Notably, 211a and 211b showed similar catalytic performance, exhibiting significant performance for aromatic substrates. In particular, these two catalysts could efficiently perform ortho-selective deuteration and tritium labeling of phenylacetic acid esters and amides, which are of significant pharmacological importance.
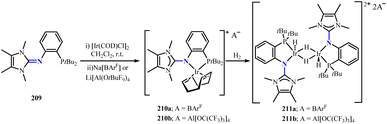 |
| | Scheme 78 Synthesis of NHI-stabilized iridium complexes 210 and 211. | |
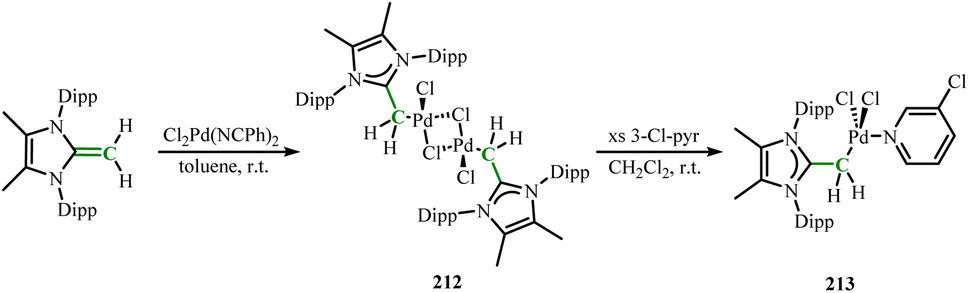
In 2019, a series of structurally different Pd(II)–NHO complexes was reported.129 By combining MeIPrCH2 with trans-[Cl2Pd(NCPh)2] in toluene, obtaining the dimer [(MeIPrCH2)PdCl(m-Cl)]2 (212). Adding 3-chloropyridine to 212 further yielded [(MeIPrCH2)PdCl2(3-Cl–pyr)] (213) (Scheme 79). The exocyclic C–C distance in 212 (1.453(3) Å) was longer than the CC bond in the free NHO ligand (1.3489(18) Å),52 indicating π electron transfer to the C–Pd bond. Subsequently, using different NHO ligands to obtain complexes 214, and 215 through the same procedure (Scheme 80). Notably, using NaOtBu as a base and combining the framework-methylated NHO with [Pd(cinnamyl)Cl]2, obtaining the system with the highest catalytic activity in promoting C–N bond formation between hindered arylamines and aryl halide substrates.
 |
| | Scheme 79 Synthesis of NHO-stabilized palladium complexes 212 and 213. | |
 |
| | Scheme 80 NHO-stabilized palladium complexes 214 and 215. | |
Recently, novel linear-coordinated copper(I) complexes were reported, and further synthesis of challenging copper complexes with the AlCp* ligand was achieved.130 Reacting [(CF3SO3Cu)2(C6H6)] with 1,3-bis(2,6-diisopropylphenyl)-2-(trimethylsilylimino)imidazoline (DippImTMS) and 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-imine (DippImH) to obtain [Cu(DippImSiMe3)(OTf)] (216) and [Cu(DippImH)2][OTf] (217), respectively (Scheme 81). The Cu–N bond lengths (1.884 Å (216), 1.842 Å (217)) showed shorter distances, revealing strong interactions between copper and the NHI ligand. Both compounds 216 and 217 showed a linear coordination environment centered on copper, with N–Cu–O (174.93° (216), 179.83° (217)). Conversion of the air-stable compound 217 with NaBArF in dichloromethane yielded [Cu(DippImH)2][BArF] (218). Further, conversion of [Cu(DippImH)2][BArF] (218) with AlCp* (four equivalents) to obtain the rare homonuclear CuAl4 cationic complex [Cu(AlCp*)4][BArF] (219) (Scheme 81). Compound 219 has equidistant Cu–Al bonds (2.266 Å) and exhibits a nearly perfect tetrahedral geometry. The molecular structure is similar to [Ni(AlCp*)4], but the average Al–Cp* center distance (1.858 Å) is shorter than that in [Ni(AlCp*)4] (1.933 Å). This contration may result from the enhanced electrophilicity of the cationic copper center compared to its neutral nickel analogue, attributable to its positive charge and distinct electronic configuration. Interestingly, upon isolating the mixed products from the reaction of imidazolin-2-imine acid with copper(I) chloride, the triangular cluster compound [Cu3(DippIm)2Cl] (220) was obtained (Scheme 81). In compound 220, the three copper atoms showed a triangular configuration, with Cu–Cu distances (2.470–2.522 Å) all within the effective range of copper affinity interactions (below 2.8 Å).
 |
| | Scheme 81 Synthesis of NHI-stabilized copper complexes 216–220. | |
Tamm group obtained monometallic complexes [{(IMes)PH}MCl] (M = Cu(221a), M = Ag(221b)) and [{(IDipp)PH}MCl] (M = Cu(222a), M = Ag(222b)) via conversion of (NHC)PH derivatives (IMes)PH and (IDipp)PH with copper(I) or silver(I) chlorides (one equivalents) (Scheme 82).131 The molecular structure of 221a showed a trimer form, with each molecule having a six-membered Cu3P3 ring, and 222a formed a dinuclear silver complex [{(IMes)PH}(AgCl)2] at low temperature. This can be attributed to the smaller steric hindrance of (IMes)PH, which tends to form multinuclear structures, while the larger steric hindrance of (IDipp)PH allows the formation of stable mononuclear complexes. It is noteworthy that the reaction of (IMes)PH and (IDipp)PH with two equivalents of copper(I) chloride led to the isolation of copper complexes 223a and 223b, which contain phosphonium ion [(NHC)PH2]+ as a byproduct (Scheme 82). 223b contains two linear and two triangular planar copper sites, forming a Cu2(μ2-Cl)2 core, with a nearly planar four-membered Cu2Cl2 ring, and a folding angle between the CuCl2 planes of 3.93(5)°.
 |
| | Scheme 82 Synthesis of NHCP-stabilized copper and silver complexes 221–223. | |
Under different reaction conditions, the reaction of (benzo)imidazolin-2-imine ligands with ZnEt2 can yield corresponding dimeric or trimeric zinc complexes (224–226) (Scheme 83).41 Due to the smaller steric hindrance of N,N′-diisopropylbenzimidazolin-2-imine, a trinuclear complex is formed after the reaction, in which two ligands bridge three zinc atoms and another ligand bridges two zinc atoms. Additionally, when the metal alkyl precursor is replaced with Mg(n-Bu)2, a dimeric magnesium complex similar to 224 can be obtained.
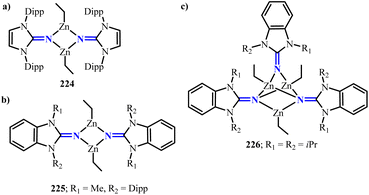 |
| | Scheme 83 Synthesis of NHI-stabilized zinc complexes 224–226. | |
Conversion of actinide complexes 227a and 227b, as well as metallocenes 228a and 228b, with equimolar neutral imidazolin-2-imine ligands ImRNH to obtain mono(imidazolin-2-imine) actinide(IV) complexes [(ImAd,RN)An{N(SiMe3)2}3] (R = Mes) (229a–230b) and (231a–232b) in high yield (Scheme 84).132 The An–N–C angles (175.9(3)°, 229a; 162.7(6)°, 229b; 178.5(3)°, 230a; 162.0(3)°, 230b) showed nearly linear configurations, while the An–Nimido bond lengths (2.177(3) Å, 229a; 2.191(7) Å, 229b; 2.119(4), 230a; 2.123(5) Å, 230b; 2.195(3) Å, 231a; 2.218(10) Å, 231b; 2.117(3) Å, 232a; 2.258(11) Å, 232b) were all shorter, indicating high double bond character between the An–Nimido bonds. Studies revealed that these complexes all exhibited catalytic activity in the transfer hydrogenation of aldehydes, ketones, and 2-propanol. Notably, the thorium complexes exhibit significantly superior catalytic performance compared to the uranium complexes. Case studies show that with 1 mol% catalyst loading and within 5 hours of reaction time, thorium complex 229b achieves a high conversion rate of 75%, while structurally similar uranium complex 230b yields only 33% conversion under identical conditions, clearly demonstrating the activity gradient between thorium complexes (moderate-to-high efficiency) and uranium complexes (low-to-moderate efficiency).
 |
| | Scheme 84 Synthesis of NHI-stabilized actinide complexes 229–232. | |
3. Bis(N-heterocyclic carbenes) as ligands
3.1. Bis(NHIs) as ligands
Bis(imidazolin-2-imine) ligands often serve as highly effective electron-donating ligands that coordinate with main group elements and transition metals. When the bridging unit contains additional functional groups, a tridentate or multidentate ligand system is formed. The effective charge delocalization of the guanidine CN3 unit enhances the basicity and nucleophilicity within the system, enabling it to effectively stabilize positive charges. These biguanide ligands can be regarded as superbases. Primary bis-NHIs ligands (233–241) shown in Scheme 85, the related synthesis methods and researchs have been reviewed elsewhere.25,133 These guanidine ligands have broad applications as superbases “proton sponges” and in metal coordination.134
 |
| | Scheme 85 Primary bis(NHIs) ligands 233–241. | |
The stabilization of electron-deficient three-coordinate boron dicationic species was achieved through the use of a strongly electron-donating system with ligand 233b. The reaction of 233b with phenyl dibromoborane led to the isolation of a mononuclear boron(III) dicationic salt 242. Further, conversion of 233b with (B(Cl)NMe2)2 followed by the addition of SbCl5 resulted in the isolation of a dinuclear, three-coordinate diboron(II) dication 244. Both boron centers exhibited a trigonal planar coordination environment. The hydrogen transfer reactions of 242a with LiBH4 to form the boronium cation 243 and of 244 with LiAlH4 to form the dialuminum hydride cation 245 validated the Lewis acidity of the boron centers in 242 and 244 (Scheme 86).135
 |
| | Scheme 86 Synthesis of bis(NHI)-stabilized borane 242 and 244 and its reaction with metal hydrides. | |
The isolation of chlorotetryliumylidenes stabilized by bis-NHIs ligands 246, 247a and 247b, which was achieved through the conversion of ligand 233b with ECl2 (E = Si, Ge, Sn) (Scheme 87).136,137 The reactivity of the lone pair electrons of the siliconylidene 246 was validated through coordination with heavier chalcogens and group 11 metals (Scheme 88). Notably, a zwitterionic heterobimetallic siliconylidene complex 250, which is overall neutral, was isolated via a double anion exchange reaction between siliconylidene 246 and PPh3AuCl and K2Fe(CO)4 (Scheme 88). Complex 250 exhibits an almost linear Si–Au–Fe structure (173.65(3)°), with a short distance between the gold atom and the meso-methyl ligand, indicating rare weak interactions. The Au d(xz) and d(yz) orbitals exhibit significant π-backbonding capability of the siliconylidene ligand, and there is a coordination interaction between the silicon lone pair and the gold atom.136
 |
| | Scheme 87 Synthesis of bis(NHI)-stabilized tetryliumylidenes 246–247. | |
 |
| | Scheme 88 Reaction of 246 with heavier chalcogens, group 11 metals and coordinate with PPh3AuCl and K2Fe(CO)4. | |
Further treatment of 247a and 247b with TMSOTf resulted in the isolation of the trifluoromethanesulfonates 247a[OTf] and 247b[OTf]. In 247b[SnCl3], tin center is in a trigonal pyramidal coordination environment, with nearly perpendicular Cl–Sn–N bonds (93.42° and 89.23°), confirming the formation of the tin(II) cation 247a+. Both 247a[OTf] and 247b[OTf] exhibit similar 1H NMR spectra and bonding situations, with only the germylidene cationic unit and stannylidene cationic unit observed. Additionally, the outer ring N–C bonds are slightly elongated, and the inner ring N–C bonds are slightly shortened, indicating electron transfer from the ligand to the metal center. Studies have shown that the electron-rich 247a[OTf] and 247b[OTf] also exhibit a tendency to undergo metal transfer reactions. In the case of treatment with LiAlH4, metal transfer products were generated. However, the conversion with the weaker reducing agent NaBH4 exhibited different results. The reaction of 247b[OTf] with NaBH4 yielded the expected metal transfer product, while the reaction of 247a[OTf] with NaBH4 yielded a push–pull stabilized hydridogermylidene complex (Schemes 89 and 90).137
 |
| | Scheme 89 Reaction of 247a with metal hydrides. | |
 |
| | Scheme 90 Reaction of 247b with metal hydrides. | |
Subsequently, Inoue group further explored the application of bis-NHI ligands 233c, 236 and 237 in stabilizing tetryliumylidenes.138 Stannyliumylidenes (255a[A], 256a[A], 257a[A]) and germyliumylidenes (255b[A], 256b[A], 257b[A]) (A = ECl3−, OTf−) (Scheme 91) were obtained via the same reaction pathway as described above. Due to the lack of an extended conjugated π-system and the more rigid o-phenylene geometry in 257a+, the 119Sn NMR chemical shifts of 257a+ showed resonances at higher fields (−232.53 ppm, 255a+; −154.49 ppm, 256a+ and −261.25 ppm, 257a+). However, the corresponding strategy for silyliumylidenes could only be prepared as 257c[Cl], which can be attributed to the greater steric demands of bis-NHI ligands 233c or 236. Interestingly, 255c[SnCl3] was successfully prepared through transmetalation of 255a[SnCl3] (Scheme 91). The transmetalation of the synthesized stannyliumylidenes with LiAlH4 yielded the corresponding aluminum complexes.
 |
| | Scheme 91 Synthesis of bis(NHI)-stabilized tetryliumylidenes 255–257, A = ECl3−, OTf−. | |
The bis-NHI ligand 233b reacting with AuCl(SMe2) to obtain the dinuclear gold(I) complex 1,2-(IMesN–AuCl)2–C2H4 (258) was reported (Scheme 92).139 The N–Au–Cl angles were 176.71(7) and 178.45(7)°, respectively, showing a linear coordination environment around the Au centers, with the coordinating nitrogen atoms in the trans position. Compound 258 mainly exhibited gold–arene interactions (Au⋯C (2.946(2) Å and 2.999(2) Å)), and no interactions were observed between the two Au center atoms. Additionally, the high stability of complex 258 in aqueous solution was verified, with its 1H NMR spectrum showing no significant decay recorded within 24 hours. Notably, complex 258 exhibited high selectivity and significant antiproliferative activity against A549 lung carcinoma cells, which may provide a new effective means for cancer treatment.
 |
| | Scheme 92 Synthesis of bis(NHI)-stabilized gold complex 258. | |
As early as 2010, a series of Mn–Zn complexes using bis-NHIs ligand 233a were published.140 Using the same strategy, a series of complexes [{fc(NIm)2}MCl2] (M = Mn, Fe, Co, Ni, Cu, Zn, Pd) (259a–259g) (Scheme 93) were obtained vis conversion of bis-NHIs ligand 235 with MCl2 salts.141,142 Both the zinc and palladium complexes (259f and 259g) were confirmed to be diamagnetic. Due to insufficient quantities, NMR analysis of the copper complex has not yet been performed. Similar to the values established in ethylene-bridged bis(imidazolin-2-imine) zinc(II) complexes, the 1H NMR spectrum of the zinc complex 259f shows a septet at 5.53 ppm and a doublet at 1.48 ppm. Notably, the palladium complex 259g exists in a slightly distorted square planar environment. The 1H NMR spectrum reveals a mixture of two diamagnetic species, with one set of signals showing a small chemical shift difference (Δδ = 0.13 ppm) between the ferrocene α- and β-protons, indicating the formation of a Fe–metal bond, demonstrating the ability of the ferrocene moiety in the ligand to act as a strong donor in metal–metal bonding.143 Except for the palladium complex 259f, all other complexes exhibit a slightly distorted tetrahedral geometry. Additionally, the reaction of ligand 235 with Cu(I) salts yielded dimeric (CuCl, CuBr) or polymeric (CuI) structures (260a, 260b and 261) (Scheme 93).
 |
| | Scheme 93 Synthesis of bis(NHI)-stabilized manganese, iron, cobalt, nickel, copper zinc and palladium complexes 259–261. | |
3.2. Bis(NHCPs) as ligands
NHC-stabilized phosphinidenes, as inversely polarized phosphoalkenes, each phosphorus atom hold two lone pairs of electrons. This allows the corresponding chelating bis-phosphinidene ligands to possess multiple binding sites, potentially forming chelate complexes accommodating more metal centers. In recent years, this class of ligands has been preliminarily explored. Currently, there are two main synthetic routes for chelating bis-phosphinidene ligands: (1) reducing bis-phosphine precursors with N-heterocyclic carbenes (NHCs);144,145 (2) starting from bis(NHC)s with triphosphiranes, which can conveniently yield the corresponding chelating bis-phosphinidenes.146,147 Using these strategies, chelating bis(NHCP)s (262–265) (Scheme 94) have been synthesized and utilized as ligands to form multiple metal complexes.
 |
| | Scheme 94 Primary bis(NHCPs) ligands 262–265. | |
The coordination chemistry of compound 262 was probed through the addition of Fe, Zn, Ge, Sn, and Cu halides.144 In reactions with ZnI2, FeCl2, GeCl2, and SnBr2, corresponding cationic complexes (266–269) containing tetravalent elements were obtained (Scheme 95). Among these, the Ge and Sn complexes tended to form ion-separated pairs, manifesting as germylenes and stannylenes salts. However, due to the excessive binding angles on the opposite side of the phosphinimine ligand, no expected bimetallic species were obtained when more EX2 halides (E = Fe, Zn, Ge, Sn) were added. When compound 262 reacted with CuCl, a cationic Cu3P3 cyclic complex (270) was obtained (Scheme 95), where three P centers bridged two Cu centers, forming a six-membered ring structure, revealing the ability of 262 as a tetradentate chelating ligand. Similarly to compound 262, 263 yielded corresponding stannylenes salts (271–272) upon reaction with SnX2 complexes (X = Cl, Br, I, OTf) (Scheme 96),145 where the Sn(II) cation exhibited a trigonal pyramidal coordination environment. Interestingly, stannylene salt 271b was demonstrated to be capable of transferring the [SnCl]+ unit from a bis-NHCP system to a bis-NHI system in a THF-d8 solution. Additionally, compound 263a was shown to be capable of stabilizing Si(IV) dications, with the isolated SiH2 dications 275 and 276 exhibiting high Lewis acidity and thermal stability (Scheme 96).148
 |
| | Scheme 95 Synthesis of bis(NHCP)-stabilized germanium, tin, iron, copper and zinc complexes 266–270. | |
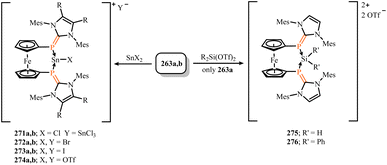 |
| | Scheme 96 Synthesis of bis(NHCP)-stabilized silicon and tin complexes 271–276. | |
In 2023, Hering-Junghans group employed strategy (2) to synthesize novel bis(NHC)-bridged bis(NHCP) ligands 264 and 265.146,147 Starting from 264, further synthesizing the complexes 277 and 278 (Scheme 97). The Fe(II) center in 277 showed a distorted tetrahedral coordination environment, with only one pair of lone pairs on each P atom participating in coordination in the complex. One of the C–H bonds of the bridging N–CH2–N part in 277 and 278 face towards the metal atom (CH⋯Fe 2.5408(5) Å, H⋯Rh 2.6094(5) Å, C⋯Rh 3.4885(47) Å), indicating the presence of weak interactions. Subsequently, the coordination chemistry of ligand 265 with GaI3 and GeCl2(dioxane) has been reported (Scheme 98). In 265a, the two MesP units are trans-arranged on either side of the benzene bridge. Interestingly, the MesP in 279 units are cis-arranged on the same side of the benzene plane, indicating the flexibility of the ligand framework. The Ga–I bond in 279 is coplanar with the central benzene group, and the Ga⋯C distance is short (3.127(4) Å), suggesting an aromatic ring interaction between the Ga atom and the central benzene bridge. Compounds 280a and 280b both showed three resonances for the benzene bridge in the 1H NMR spectra, indicating Cs symmetry in solution. The angles around the germanium atoms are 267.661(17)° (280a) and 264.55(3)° (280b), indicative of the trigonal coordination geometry for germanium. No interactions with the trichlorogermanate anion in 280a indicate the presence of charge-separated ion pairs. The charge transfer from the ligands to the germanium atoms in the germaniumium subunits 280a (−1.054 e−) and 280b (−1.082 e−) is significant, showing strong polarization of the P–Ge bonds.
 |
| | Scheme 97 Synthesis of bis(NHCP)-stabilized iron and rhodium complexes 277 and 278. | |
 |
| | Scheme 98 Synthesis of bis(NHCP)-stabilized gallium and germanium complexes 279 and 280. | |
3.3. Bis(NHOs) as ligands
Although bis(N-heterocyclic imidazoliums) (NHIs) and bis(N-heterocyclic carbenes) (NHCPs) have been extensively developed as multidentate ligands, complexes supported by bis(NHOs) remain relatively rare. Two bridging methods for bis(NHOs) (281 and 282) (Scheme 99) have been reported to date.
 |
| | Scheme 99 Primary bis(NHOs) ligands 281–282. | |
In 2017, the first bis(NHOs) ligand (281) were reported.149 Treatment of 281 with phosphorus trichloride yielded the isophosphindolium chloride derivatives (283a and 283b) (Scheme 100). Both 283a and 283b featured a nearly planar PC4 five-membered ring with delocalized π-electrons, demonstrating aromaticity. The HOMO orbital of 283a and 283b were primarily located on the C–P–C moiety, indicating the nucleophilicity of the P atom. Further, the reaction of 283b with metal complexes was studied. The conversion with CuCl produced the copper phosphonium complex (284) (Scheme 100) where the P atom coordinated to the Cu center, demonstrating the potential application of 283 as a ligand. In the ferrocenyl-bridged bis(NHOs) ligand (282) the cyclopentadienyl rings were in an anti-staggered conformation, and the N atoms were in a trigonal planar environment. Conversion of 282 with [BH3(THF)] led to the isolation of 285a and 285b (Scheme 101),150 verifying their ability to act as bidentate ligands. The terminal C atoms of the exocyclic double bonds in 282 coordinated to the Lewis acid (BH3), changing from a three-coordinate to a four-coordinate state, resulting in the formation of rac- and meso-diastereomers.
 |
| | Scheme 100 Synthesis of isophosphindolium chloride derivatives 283a and 283b and its reaction with CuCl. | |
 |
| | Scheme 101 Reaction of bis(NHO) 282a and 282b with BH3. | |
4. Organocatalytic reactions enabled by N-heterocyclic carbenes
After the first study on N-heterocyclic carbenes (NHCs) organopolymerization was reported in 2010,151 this field has rapidly developed. In 2015, Dove group disclosed the preparation of poly(propylene oxide) (PPO) using N-heterocyclic olefins (NHOs) as organocatalysts.152 The nucleophile and base properties of NHOs was crucial to success in catalytic instance. Recently, the copolymerization of ethylene oxide (EO) and propylene oxide (PO) was successfully achieved through a dual catalytic system based on NHO organocatalysis and Mg(II) co-catalysis. The copolymerization behavior of PO using NHO organocatalysis (system A) and dual catalysis (system B) was compared through in situ 1H NMR measurements (Scheme 102). For system A, the reactivity ratios were determined to be rEO = 3.4 and rPO = 0.30; whereas for system B, the reactivity ratios were rEO = 7.9 and rPO = 0.13. This difference can be attributed to the presence of Mg(HMDS)2, which leads to zwitterionic propagation within the Lewis acid–base pair, further activating the epoxide mechanism.153
 |
| | Scheme 102 PO copolymerization using NHO organocatalysis (system A) and dual catalysis (system B). For system A: toluene-d8, 72 h, 50 °C; for system B: toluene-d8, 4 h, −36/−20/0 °C. | |
In 2023, after achieving the chiral amidation and trifluoromethyl thiolation of β-ketoesters,154 Dong group further developed the application of NHO organocatalysts in the α-functionalization of β-keto esters, achieving asymmetric formal coupling of β-ketoesters with quinones (Scheme 103).155 Under the optimized conditions (−78 °C, NHO* loading as low as 1 mol%), the reaction efficiently produced the desired asymmetric synthesis products with high yields (up to 96%) and high enantioselectivity (up to 99% ee), as well as high regioselectivity (up to 19:1 rr). Research revealed that the outer olefin moiety of the NHO organocatalyst significantly influenced the enantioselectivity of the reaction. Additionally, ketoesters with larger sterically hindered ester groups and naphthoquinones with electron-withdrawing groups exhibited higher enantioselectivity as substrates.
 |
| | Scheme 103 Asymmetric formal coupling of β-ketoesters with quinones achieved by NHOs. | |
Mandal group have demonstrated a novel method for the activation of CO2 (Scheme 104).156 Using NHCPs as organocatalysts. This method employs NHCPs as organiccatalysts in 10 mol%, using acetonitrile as the solvent, and successfully activates CO2 in the presence of silane, achieving the highest yield of formylated products (82%). The authors inferred through DFT calculations and intrinsic bond orbital analysis that the activation of CO2 occurs via a weakly bonded phosphinidene–silane intermediate. Notably, the phosphorus in NHCPs exhibits strong nucleophilicity, and the interaction between the phosphorus and silane plays a crucial role in the activation of CO2.
 |
| | Scheme 104 (a) CO2 activation achieved by NHCP; (b) activation of aldehydes and methyl acrylates to synthesize γ-butyrolactone catalyzed by NHCP. | |
Recently, the same group further expanded the application of NHCPs as metal-free organic catalysts for the activation of aldehydes and methyl acrylates to synthesize the corresponding γ-butyrolactones with exceptional yields up to 94%.157 The study demonstrated that the catalytic process involved a P(I)/P(III) redox cycle, forming benzoin, which successfully. Produced the target product, avoiding the harsh conditions required when using transition metals as catalysts.
Rare-earth organometallic complexes supported by imidazolin-2-iminato ligands demonstrate unique selectivity in C–H bond activation, with the smallest ionic radius Sc(III) complexes exhibiting optimal catalytic versatility. These complexes efficiently catalyze C–H alkylation reactions of anisoles, pyridines, and tertiary amines with olefins, showing remarkable preference for benzylic C(sp3)–H bond activation over ortho-aromatic C(sp2)–H bonds.112,158,159 Notably, Dong's group recently developed cationic imidazolin-2-iminato scandium alkyl catalysts (286–291) (Scheme 105) that enable highly selective hydroallylation of styrene derivatives with internal/terminal alkenes, as well as alkene dimerization, achieving exceptional yields up to 99%.160 Mechanistic studies reveal that the imidazolin-2-imine ligand plays a decisive role in modulating catalytic activity, further confirming the distinctive advantages of this ligand system in catalytic applications.
 |
| | Scheme 105 Cationic imidazolin-2-iminato scandium alkyl catalysts 286–291. | |
5. Conclusions and perspectives
This review systematically summarizes the recent advances in organometallic complexes supported by N-heterocyclic imines (NHIs), N-heterocyclic phosphinidenes (NHCPs) and N-heterocyclic olefins (NHOs). The combination of N-heterocyclic carbenes (NHCs) with X (X = N, P and C) offers significant potential for isolating metal-centered compounds with novel coordination environments. These ligands, through effective delocalization of cationic charge density within the five-membered ring system, act as strong electron donors, transferring substantial electron density to the metal center, thereby significantly enhancing the stability of the metal center. Additionally, these ligands exhibit broad tunability, allowing for the customization of backbone, ring size, and N-substituents to meet the stabilization needs of different elements.
Beyond their diverse interactions with metals, these ligands also demonstrate immense potential in the field of catalysis. Among them, NHOs have been proven to be highly efficient catalysts, widely applied in various organic synthesis reactions. However, research on NHIs and NHCPs remains in the preliminary exploration stage, with future prospects for further expanding their applications. Furthermore, future studies should prioritize computational modeling investigations of these ligand effects to provide theoretical guidance for designing novel complexes and high-efficiency catalysts.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22201210).
Notes and references
-
(a) A. J. Arduengo III, R. L. Harlow and M. Kline, A Stable Crystalline Carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef;
(b) A. J. Arduengo III, H. V. R. Dias, R. L. Harlow and M. Kline, Electronic Stabilization of Nucleophilic Carbenes, J. Am. Chem. Soc., 1992, 114, 5530–5534 CrossRef.
-
(a) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Recent Advances in the Chemistry and Applications of N-Heterocyclic Carbenes, Nat. Rev. Chem, 2021, 5, 711–725 CrossRef CAS;
(b) D. Munz, Pushing Electrons-Which Carbene Ligand for Which Application?, Organometallics, 2018, 37, 275–289 CrossRef CAS;
(c) F. E. Hahn, Introduction: Carbene Chemistry, Chem. Rev., 2018, 118, 9455–9456 CrossRef CAS PubMed.
- R. Jazzar, M. Soleilhavoup and G. Bertrand, Cyclic (Alkyl)- and (Aryl)-(amino)carbene Coinage Metal Complexes and Their Applications, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS.
- G. C. Fortman and S. P. Nolan, N-Heterocyclic Carbene (NHC) Ligands and Palladium in Homogeneous Cross-Coupling Catalysis: a Perfect Union, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- G. Frison and A. Sevin, Theoretical Study of the Bonding between Aminocarbene and Main Group Elements, J. Chem. Soc., Perkin Trans. 2, 2002, 113, 1692–1697 RSC.
-
(a) M. Majumdar, V. Huch, I. Bejan, A. Meltzer and D. Scheschkewitz, Reversible, Complete Cleavage of Si=Si Double Bonds by Isocyanide Insertion, Angew. Chem., Int. Ed., 2013, 52, 3516–3520 CrossRef CAS;
(b) A. G. Trambitas, J. Yang, D. Melcher, C. G Daniliuc, P. G. Jones, Z. Xie and M. Tamm, Synthesis and Structure of Rare-Earth-Metal Dicarbollide Complexes with an Imidazolin-2-Iminato Ligand Featuring very Short Metal-Nitrogen Bonds, Organometallics, 2011, 30, 1122–1129 CrossRef CAS.
-
(a) T. Ochiai, D. Franz and S. Inoue, Applications of N-Heterocyclic Imines in Main Group Chemistry, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC;
(b) A. Doddi, D. Bockfeld, T. Bannenberg, P. G. Jones and M. Tamm, N-Heterocyclic Carbene-Phosphinidyne Transition Metal Complexes, Angew. Chem., Int. Ed., 2014, 53, 13568–13572 (Angew. Chem., 2014, 126, 13786–13790) CrossRef CAS;
(c) R. Schuldt, J. Kästner and S. Naumann, Proton Affinities of N-Heterocyclic Olefins and Their Implications for Organocatalyst Design, J. Org. Chem., 2019, 84, 2209–2218 CrossRef CAS PubMed.
-
(a) N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, 2-Iminoimidazolines - Strong Nitrogen Bases als Ligands in Inorganic Chemistry, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS;
(b) A. G. Trambitas, T. K. Panda and M. Tamm, Rare Earth Metal Complexes Supported by Ancillary Imidazolin-2-Iminato Ligands, Z. Anorg. Allg. Chem., 2010, 636, 2156–2171 CrossRef CAS;
(c) Y. Wu and M. Tamm, Transition Metal Complexes Supported by Highly Basic Imidazolin-2-Iminato and Imidazolin-2-Imine N-Donor Ligands, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef;
(d) P. Raja, S. Revathi and T. Ghatak, Recent Advances of Imidazolin-2-Iminato in Transition Metal Chemistry, Eng. Sci., 2020, 12, 23–37 CAS.
-
(a) K. Dehnicke and A. Greiner, Unusual Complex Chemistry of Rare-Earth Elements: Large Ionic Radii - Small Coordination Numbers, Angew. Chem., Int. Ed., 2003, 42, 1340–1354 CrossRef CAS PubMed;
(b) A. Diefenbach and F. M. Bickelhaupt, Coordination Behaviour of the Isolobal Phosphoraneiminato and Cyclopentadienyl Ligands in TiCl3(NPH3), TiCl3Cp, ReO3(NPH3) und ReO3Cp, Z. Anorg. Allg. Chem., 1999, 625, 892–900 CrossRef CAS.
-
(a) L. Weber, Phosphaalkenes with Inverse Electron Density, Eur. J. Inorg. Chem., 2000, 2000, 2425–2441 CrossRef;
(b) P. Rosa, C. Gouverd, G. Bernardinelli, T. Berclaz and M. Geoffroy, Phosphaalkenes with Inverse Electron Density: Electrochemistry, Electron Paramagnetic Resonance Spectra, and Density Functional Theory Calculations of Aminophosphaalkene Derivatives, J. Phys. Chem. A, 2003, 107, 4883–4892 CrossRef CAS.
-
(a) O. Back, M. Henry-Ellinger, C. D. Martin and G. Bertrand, 31P NMR Chemical Shifts of Carbene-Phosphinidene Adducts as An Indicator of the π-Accepting Properties of Carbenes, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 (Angew. Chem., 2013, 125, 3011–3015) CrossRef CAS;
(b) S. Dutta, B. Maity, D. Thirumalai and D. Koley, Computational Investigation of Carbene-Phosphinidenes: Correlation between 31P Chemical Shifts and Bonding Features to Estimate the π-Backdonation of Carbenes, Inorg. Chem., 2018, 57, 3993–4008 CrossRef CAS.
- A. J. Arduengo III, C. J. Carmalt, J. A. C. Clyburne, A. H. Cowley and R. Pyati, Nature of the Bonding in a Carbene-Phosphinidene: a Main Group Analogue of a Fischer Carbene Complex? Isolation and Characterisation of a Bis(borane) Adduct, Chem. Commun., 1997, 981–982 RSC.
-
(a) R. Breslow, Rapid Deuterium Exchange in Thiazolium, J. Am. Chem. Soc., 1957, 79, 1762–1763 CrossRef CAS;
(b) R. Breslow, On the Mechanism of Thiamine Action. IV. Evidence from Studies on Model Systems, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS;
(c) V. Dhayalan, S. C. Gadekar, Z. Alassad and A. Milo, Unravelling Mechanistic Features of Organocatalysis with in Situ Modifications at the Secondary Sphere, Nat. Chem., 2019, 11, 543–551 CrossRef CAS PubMed;
(d) S. C. Gadekar, V. Dhayalan, A. Nandi, I. L. Zak, M. S. Mizrachi, S. Kozuch and A. Milo, Rerouting the Organocatalytic Benzoin Reaction toward Aldehyde Deuteration, ACS Catal., 2021, 11, 14561–14569 CrossRef CAS.
- N. Kuhn, H. Bohnen, D. Bläser and R. Boese, (C8H14N2)M(CO)5 (M = Mo, W)-Terminale Koordination Eines Olefins in Pentacarbonylmetall-Komplexen, Chem. Ber., 1994, 127, 1405–1407 CrossRef CAS.
- K.-M. Wang, S.-J. Yan and J. Lin, Heterocyclic Ketene Aminals: Scaffolds for Heterocycle Molecular Diversity, Eur. J. Org Chem., 2014, 2014, 1129–1145 CrossRef CAS.
- N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, 1,3,4,5-Tetramethyl-2-Methyleneimidazoline-an Ylidic Olefin, J. Chem. Soc., Chem. Commun., 1993, 1136–1137 RSC.
-
(a) B. Maji, M. Horn and H. Mayr, Nucleophilic Reactivities of Deoxy Breslow Intermediates: How does Aromaticity Affect the Catalytic Activities of N-Heterocyclic Carbenes?, Angew. Chem., Int. Ed., 2012, 51, 6231–6235 CrossRef CAS PubMed;
(b) Z. Li, P. Ji and J.-P. Cheng, Brönsted Basicities and Nucleophilicities of N-Heterocyclic Olefins in Solution: N-Heterocyclic Carbene versus N-Heterocyclic Olefin. Which is More Basic, and Which is More Nucleophilic?, J. Org. Chem., 2021, 86, 2974–2985 CrossRef CAS.
-
(a) S. Naumann, Synthesis, Properties & Applications of N-Heterocyclic Olefins in Catalysis, Chem. Commun., 2019, 55, 11658–11670 RSC;
(b) Z. Wang, Q.-H. Niu, X.-S. Xue and P. Ji, The Brönsted Basicities of N-Heterocyclic Olefins in DMSO: an Effective Way to Evaluate the Stability of NHO-CO2 Adducts, J. Org. Chem., 2020, 85, 13204–13210 CrossRef CAS.
-
(a) Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Multifunctional Metal–Organic Framework Catalysts: Synergistic Catalysis and Tandem Reactions, Chem. Soc. Rev., 2017, 46, 126–157 RSC;
(b) Q. Zhang, C. Huang, Y. Tao, Y. Zhang, J. Cui, D. Wang, P. Wang and Y.-Y. Zhang, Flexible Triboelectric Nanogenerators Based on Cadmium Metal-Organic Framework/Eethyl Cellulose Composites as Energy Harvesters for Selective Photoinduced Bromination Reaction, Small, 2025, 21, 2406623 CrossRef;
(c) S. Liu, H. Yang, Y. Zhang, F. Wang, Q. Qin, D. Wang, C. Huang and Y.-Y. Zhang, Robust Cooperative of Cadmium Sulfide with Highly Ordered Hollow Microstructure Coordination Polymers for Regulating the Photocatalytic Performance, J. Colloid Interface Sci., 2024, 663, 919–929 CrossRef CAS PubMed;
(d) F. Wang, Y.-Y. Zhang, S. Li, L. Zhang, Y. Tao, J. Cui, D. Wang, M. Jiao and C. Huang, Integration of Cobalt Coordination Polymer-Based Triboelectric Nanogenerators as Sustainable Power Sources for Self-Driven Selective Photocatalytic Reactions, Chem. Eng. J., 2025, 503, 158194 CrossRef CAS;
(e) Y. Zhang, C. Huang and L. Mi, Metal-Organic Frameworks as Acid- and/or Base-Functionalized Catalysts for Tandem Reactions, Dalton Trans., 2020, 49, 14723–14730 RSC;
(f) S. Liu, H. Yang, S. Li, Q. Qin, Y. Tao, J. Cui, D. Wang, C. Huang and Y.-Y. Zhang, Enhancement of Synergistic Photocatalytic Performance by Anchoring Cadmium Sulfide on Nanosphere Structured Coordination Polymers, Inorg. Chem., 2024, 63, 17116–17126 CrossRef CAS.
- T. Krachko and J. C. Slootweg, N-Heterocyclic Carbene-Phosphinidene Adducts: Synthesis, Properties, and Applications, Eur. J. Inorg. Chem., 2018, 2734–2754 CrossRef CAS.
- S. Revathi, P. Raja, S. Saha, M. S. Eisen and T. Ghatak, Recent Developments in Highly Basic N-Heterocyclic Iminato Ligands in Actinide Chemistry, Chem. Commun., 2021, 57, 5483–5502 RSC.
- M. M. D. Roy and E. Rivard, Pushing Chemical Boundaries with N-Heterocyclic Olefins (NHOs): from Catalysis to Main Group Element Chemistry, Acc. Chem. Res., 2017, 50, 2017–2025 CrossRef CAS PubMed.
- R. S. Ghadwal, Tuning the Electronic Properties of Main-Group Species by N-Heterocyclic Vinyl (NHV) Scaffolds, Acc. Chem. Res., 2022, 55, 457–470 CrossRef CAS PubMed.
- K. Schwedtmann, G. Zanoni and J. J. Weigand, Recent Advances in Imidazoliumyl-Substituted Phosphorus Compounds, Chem.–Asian J., 2018, 13, 1388–1405 CrossRef CAS PubMed.
- A. Doddi, M. Peters and M. Tamm, N-Heterocyclic Carbene Adducts of Main Group Elements and Their Use as Ligands in Transition Metal Chemistry, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
-
(a) D. Franz, E. Irran and S. Inoue, Synthesis, Characterization and Reactivity of an Imidazolin-2-Iminato Aluminium Dihydride, Dalton Trans., 2014, 43, 4451–4461 RSC;
(b) D. Franz and S. Inoue, Systematic Investigation of the Ring-Expansion Reaction of N-Heterocyclic Carbenes with an Iminoborane Dihydride, Chem.–Asian J., 2014, 9, 2083–2087 CrossRef CAS;
(c) D. Franz, E. Irran and S. Inoue, Isolation of a Three-Coordinate Boron Cation with a Boron-Sulfur Double Bond, Angew. Chem., Int. Ed., 2014, 53, 14264–14268 CrossRef CAS PubMed.
- C. Hering-Junghans, P. Andreiuk, M. J. Ferguson, R. McDonald and E. Rivard, Using N-Heterocyclic Vinyl Ligands to Access Stable Divinylgermylenes and a Germylium Cation, Angew. Chem., Int. Ed., 2017, 56, 6272–6275 CrossRef CAS.
- C. Hering-Junghans, I. C. Watson, M. J. Ferguson, R. McDonald and E. Rivard, Organocatalytic Hydroborylation Promoted by N-Heterocyclic Olefins, Dalton Trans., 2017, 46, 7150–7153 RSC.
- M. Lafage, A. Pujol, N. Saffon-Merceron and N. Mézailles, BH3 Activation by Phosphorus-Stabilized Geminal Dianions: Synthesis of Ambiphilic Organoborane, DFT Studies, and Catalytic CO2 Reduction into Methanol Derivatives, ACS Catal., 2016, 6, 3030–3035 CrossRef CAS.
- I. C. Watson, M. J. Ferguson, R. McDonald and E. Rivard, Zinc-Mediated Transmetalation as a Route to Anionic N-Heterocyclic Olefin Complexes in the p-Block, Inorg. Chem., 2021, 60, 18347–18359 CrossRef CAS PubMed.
- F. Zettler, H. D. Hausen and H. Hess, Die Kristall- und Molekülstruktur des Triphenylborans, J. Organomet. Chem., 1974, 72, 157–162 CrossRef CAS.
- S. R. Baird, E. Hupf, I. C. Watson, M. J. Ferguson, R. McDonald and E. Rivard, An Indium(I) Tetramer Bound by Anionic N-Heterocyclic Olefins: Ambiphilic Reactivity, Transmetallation and a Rare Indium-Imide, Chem. Commun., 2023, 59, 2903–2906 RSC.
- H. Dolati, L. Denker, B. Trzaskowski and R. Frank, Superseding β-Diketiminato Ligands: an Amido Imidazoline-2-Imine Ligand Stabilizes the Exhaustive Series of B = X Boranes (X = O, S, Se, Te), Angew. Chem., Int. Ed., 2021, 60, 4633–4639 CrossRef CAS PubMed.
- M. W. Lui, N. R. Paisley, R. McDonald, M. J. Ferguson and E. Rivard, Metal-Free Dehydrogenation of Amine-Boranes by Tunable N-Heterocyclic Iminoboranes, Chem.–Eur. J., 2016, 22, 2134–2145 CrossRef CAS PubMed.
- D. Franz, T. Szilvási, A. Pçthig and S. Inoue, Isolation of an N-Heterocyclic Carbene Complex of a Borasilene, Chem.–Eur. J., 2019, 25, 11036–11041 CrossRef CAS PubMed.
- J. Han, C. Hu, Q. Li, L. L. Liu, C.-H. Tung, P. Cui and L. Kong, Derivatization of an Alkylideneborane with B=C Bond Cleavage, Inorg. Chem., 2023, 62, 18820–18824 CrossRef CAS.
- D. Franz, E. Irran and S. Inoue, Synthesis, Characterization and Reactivity of an Imidazolin-2-Iminato Aluminium Dihydride, Dalton Trans., 2014, 43, 4451–4461 RSC.
- D. Franz and S. Inoue, Activation of Elemental Sulfur by Aluminum Dihydride: Isolationof Mono- and Bis(hydrogensulfide) Complexes of Aluminum, Chem.–Eur. J., 2014, 20, 10645–10649 CrossRef CAS.
- D. Franz, L. Sirtl, A. Pöthig and S. Inoue, Aluminum Hydrides Stabilized by N-Heterocyclic Imines as Catalysts for Hydroborations with Pinacolborane, Z. Anorg. Allg. Chem., 2016, 642, 1245–1250 CrossRef CAS.
-
(a) D. Franz, C. Jandl, C. Stark and S. Inoue, Catalytic CO2 Reduction with Boron- and Aluminum Hydrides, ChemCatChem, 2019, 11, 5275–5281 CrossRef CAS PubMed;
(b) C. Weetman, N. Ito, M. Unno and S. Inoue, NHI- and NHC-Supported Al(III) Hydrides for Amine-Borane Dehydrocoupling Catalysis, Inorganics, 2019, 7, 92 CrossRef CAS.
- H. Liu, M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, (Benz)Imidazolin-2-iminato Aluminum, Zinc, and Magnesium Complexes and Their Applications in Ring Opening Polymerization of ε-Caprolactone, Inorg. Chem., 2019, 58, 13426–13439 CrossRef CAS PubMed.
- S. Pahar, G. Kundu, C. P. George, R. G. Gonnade and S. S. Sen, Substitution at a Coordinatively Saturated Aluminum Center Stabilized by Imidazolidin-2-imine, Organometallics, 2023, 42, 276–282 CrossRef CAS.
- I. C. Watson, Y. Zhou, M. J. Ferguson, M. Kränzlein, B. Rieger and E. Rivard, Trialkylaluminum N-Heterocyclic Olefin (NHO) Adducts as Catalysts for the Polymerization of Michael-Type Monomers, Z. Anorg. Allg. Chem., 2020, 646, 547–551 CrossRef CAS.
- A. D. K. Todd, W. L. McClennan and J. D. Masuda, Organoaluminum(III) Complexes of the Bis-N,N’-(2,6-Diisopropylphenyl)Imidazolin-2-Imine Ligand, RSC Adv., 2016, 6, 69270–69276 RSC.
- J. W. Turley and H. W. Rinn, Crystal Structure of Aluminum Hydride. The Crystal Structure of Aluminum Hydride, Inorg. Chem., 1969, 8, 18–22 CrossRef CAS.
- J. Bhattacharjee, M. Peters, D. Bockfeld and M. Tamm, Isoselective Polymerization of rac-Lactide by Aluminum Complexes of N-Heterocyclic Carbene-Phosphinidene Adducts, Chem.–Eur. J., 2021, 27, 5913–5918 CrossRef CAS.
- O. Lemp, M. Balmer, K. Reiter, F. Weigendb and C. Hänisch, An NHC-Phosphinidenyl as a Synthon for New Group 13/15 Compounds, Chem. Commun., 2017, 53, 7620–7623 RSC.
- D. H. Mayo, Y. Peng, S. DeCarlo, X. Li, J. Lightstone, P. Zavalij, K. Bowen, H. Schnöckel and B. Eichhorn, K[Al4 (PPh2)7PPh]: An AlII Phosphanide/Phosphinidene Intermediate on the Path to AlP Formation, Z. Anorg. Allg. Chem., 2013, 639, 2558–2560 CrossRef CAS.
- M. Balmer and C. Hänisch, Synthesis of the Cyclic Group 13 Phosphinidenides [(NHC)PMCl2]2 (NHC = SIMes, SIDipp; M = Al, Ga), Z. Anorg. Allg. Chem., 2020, 646, 648–652 CrossRef CAS.
- L. Denker, B. Trzaskowski and R. Frank, “Give Me Five” - an Amino Imidazoline-2-Imine Ligand Stabilises the First Neutral Five-Membered Cyclic Triel(I) Carbenoides, Chem. Commun., 2021, 57, 2816–2819 RSC.
- B. Wang, W. Chen, J. Yang, L. Lu, J. Liu, L. Shena and D. Wu, N-Heterocyclic Imine-Based Bis-Gallium(I) Carbene Analogs Featuring a Four-Membered Ga2N2 ring, Dalton Trans., 2023, 52, 12454–12460 RSC.
- K. Powers, C. Hering-Junghans, R. McDonald, M. J. Ferguson and E. Rivard, Improved Synthesis of N-Heterocyclic Olefins and Evaluation of Their Donor Strengths, Polyhedron, 2016, 108, 8–14 CrossRef CAS.
- T. B. Ditri, B. J. Fox, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Effective Control of Ligation and Geometric Isomerism: Direct Comparison of Steric Properties Associated with Bis-Mesityl and Bis-Diisopropylphenyl m-Terphenyl Isocyanides, Inorg. Chem., 2009, 48, 8362–8375 CrossRef CAS PubMed.
-
(a) N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff, D. Bläser and R. Boese, Synthese and Structure of 2-Imino-l,3-dimethylimidazolin, Z. Naturforsch., B:J. Chem. Sci., 1995, 50, 1779–1784 CrossRef CAS;
(b) N. Kuhn, R. Fawzi, M. Steimann and J. Wiethoff, 2-Iminoimidazolin Derivatives of Magnesium and Aluminium, Z. Anorg. Allg. Chem., 1997, 623, 554–560 CrossRef CAS.
- M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, A Vinyl Silylsilylene and Its Activation of Strong Homo- and Heteroatomic Bonds, Chem. Sci., 2019, 10, 6476–6481 RSC.
-
(a) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, A Generic One-Pot Route to Acyclic Two-Coordinate Silylenes from Silicon(IV) Precursors: Synthesis and Structural Characterization of a Silylsilylene, Angew. Chem., Int. Ed., 2013, 52, 568–571 CrossRef CAS;
(b) T. J. Hadlington, J. A. B. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Stabilization of a Two-Coordinate, Acyclic Diaminosilylene (ADASi): Completion of the Series of Isolable Diaminotetrylenes, :E(NR2)2 (E = group 14 element), Chem. Commun., 2016, 52, 1717–1720 RSC;
(c) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sakar, C. Jandl, S. Inoue and B. Rieger, From Si(II) to Si(IV) and Back: Reversible Intramolecular Carbon-Carbon Bond Activation by an Acyclic Iminosilylene, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS;
(d) Y. K. Loh, L. Ying, M. Á. Fuentes, D. C. H. Do and S. Aldridge, An N-Heterocyclic Boryloxy Ligand Isoelectronic with N-HeterocyclicImines: Access to an Acyclic Dioxysilylene and Its Heavier Congeners, Angew. Chem., Int. Ed., 2019, 58, 4847–4851 CrossRef CAS.
- M. M. D. Roy, S. R. Baird, E. Dornsiepen, L. A. Paul, L. Miao, M. J. Ferguson, Y. Zhou, I. Siewert and E. Rivard, A Stable Homoleptic Divinyl Tetrelene Series, Chem.–Eur. J., 2021, 27, 8572–8579 CrossRef CAS PubMed.
- D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, Silicon and Oxygen's Bond of Affection: an Acyclic Three-Coordinate Silanone and Its Transformation to an Iminosiloxysilylene, J. Am. Chem. Soc., 2017, 139, 17193–17198 CrossRef CAS.
- T. Eisner, A. Kostenko, F. Hanusch and S. Inoue, Room-Temperature-Observable Interconversion between Si(IV) and Si(II) via Reversible Intramolecular Insertion into
an Aromatic C−C Bond, Chem.–Eur. J., 2022, 28, e202202330 CrossRef CAS PubMed.
- J. Y. Liu, S. Inoue and B. Rieger, Isolation and Reactivity of Silepins with a Sterically Demanding Silyl Ligand, Eur. J. Inorg. Chem., 2024, 27, e202400045 CrossRef.
- M. W. Lui, C. Merten, M. J. Ferguson, R. McDonald, Y. Xu and E. Rivard, Contrasting Reactivities of Silicon and Germanium Complexes Supported by an N-Heterocyclic Guanidine Ligand, Inorg. Chem., 2015, 54, 2040–2049 CrossRef CAS PubMed.
- D. Reiter, P. Frisch, T. Szilvási and S. Inoue, Heavier Carbonyl Olefination: the Sila-Wittig Reaction, J. Am. Chem. Soc., 2019, 141, 16991–16996 CrossRef CAS.
- D. Reiter, P. Frisch, D. Wendel, F. M. Hörmann and S. Inoue, Oxidation Reactions of a Versatile, Two-Coordinate, Acyclic Iminosiloxysilylene, Dalton Trans., 2020, 49, 7060–7068 RSC.
- H. Zhu, A. Kostenko, D. Franz, F. Hanusch and S. Inoue, Room Temperature Intermolecular Dearomatization of Arenes by an Acyclic Iminosilylene, J. Am. Chem. Soc., 2023, 145, 1011–1021 CrossRef CAS PubMed.
- H. Zhu, F. Hanusch and S. Inoue, Facile Bond Activation of Small Molecules by an Acyclic Imino(silyl)silylene, Isr. J. Chem., 2023, 63, e202300012 CrossRef CAS.
- H. Zhu, A. Kostenko, J. A. Kelly and S. Inoue, Substituent Exchange between an Imino(silyl)silylene and Aryl Isocyanides, Chem, 2024, 10, 1213–1224 CAS.
- M. E. Doleschal, A. Kostenko, J. Y. Liu and S. Inoue, Isolation of a NHC-Stabilized Heavier Nitrile and Its Conversion into an Isonitrile Analogue, Nat. Chem., 2024, 16, 2009–2016 CrossRef CAS PubMed.
- L. Wang, Y. Li, Z. Li and M. Kira, Isolable Silylenes and Their Diverse Reactivity, Coord. Chem. Rev., 2022, 457, 214413 CrossRef CAS.
- M. E. Doleschal, A. Kostenko, J. Y. Liu and S. Inoue, Silicon-Aryl Cooperative Activation of Ammonia, Chem. Commun., 2024, 60, 13020–13023 RSC.
- S. Du, H. Jia, H. Rong, H. Song, C. Cui and Z. Mo, Synthesis and Reactivity of N-Heterocyclic Silylene Stabilized Disilicon(0) Complexes, Angew. Chem., Int. Ed., 2022, 61, e202115570 CrossRef CAS.
- S. Du, F. Cao, X. Chen, H. Rong, H. Song and Z. Mo, A Silylene-Stabilized Ditin(0) Complex and Its Conversion to Methylditin Cation and Distannavinylidene, Nat. Commun., 2023, 14, 7474 CrossRef CAS.
- F. J. Kiefer, A. Kostenko, R. Holzner and S. Inoue, A Neutral Crystalline Imino-Substituted Silyl Radical, Chem. Commun., 2024, 60, 8577–8580 RSC.
-
(a) Y. Li, Y.-C. Chan, B.-X. Leong, Y. Li, E. Richards, I. Purushothaman, S. De, P. Parameswaran and C.-W. So, Trapping a Silicon(I) Radical with Carbenes: a Cationic CAAC–Silicon(I) Radical and an NHC–Parent-Silyliumylidene Cation, Angew. Chem., Int. Ed., 2017, 129, 7681–7686 CrossRef;
(b) S.-H. Zhang, E. Carter, H.-W. Xi, Y. Li, K. H. Lim and C.-W. So, Delocalized Hypervalent Silyl Radical Supported by Amidinate and Imino Substituents, Inorg. Chem., 2017, 56, 701–704 CrossRef CAS.
- M. Balmer, Y. J. Franzke, F. Weigend and C. Hänisch, Low-Valent Group 14 Phosphinidenide Complexes [({SIDipp}P)2M] Exhibit P−M pπ-pπ Interaction (M = Ge, Sn, Pb), Chem.–Eur. J., 2020, 26, 192–197 CrossRef CAS.
-
(a) K. Izod, P. Evans, P. G. Waddell and M. R. Probert, Remote Substituent Effects on the Structures and Stabilities of P=E π-Stabilized Diphosphatetrylenes (R2P)2E (E = Ge, Sn), Inorg. Chem., 2016, 55, 10510–10522 CrossRef CAS;
(b) M. Driess, S. Rell, H. Pritzkow and R. Janoschek, [R–PLi2(F–R)]2 [R = SiPri2(C6H2Pri3-2,4,6)] the First Structural Characterization of a Dilithium Phosphandiide in the Form of a Fluorosilane Complex, Chem. Commun., 1996, 2, 305–306 RSC.
- M. E. Doleschal, A. E. Ferao, A. Kostenko, F. J. Kiefer and S. Inoue, The Substituent-Effect in NHCP-Plumbylenes with a Phosphaplumbene Character, Angew. Chem., Int. Ed., 2025, e202422186 CAS.
- F. Goerigk, N. Birchall, C. M. Feil, M. Nieger and D. Gudat, Reactions of Imidazolio-Phosphides with Organotin Chlorides: Surprisingly Diverse, Eur. J. Inorg. Chem., 2022, e202101026 CrossRef CAS.
- V. Nesterov, R. Baierl, F. Hanusch, A. E. Ferao and S. Inoue, N-Heterocyclic Carbene-Stabilized Germanium and Tin Analogues of Heavier Nitriles: Synthesis, Reactivity, and Catalytic Application, J. Am. Chem. Soc., 2019, 141, 14576–14580 CrossRef CAS.
-
(a) M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, A Systematic Study of Structure and E−H Bond Activation Chemistry by Sterically Encumbered Germylene Complexes, Chem.–Eur. J., 2016, 22, 11685–11698 CrossRef CAS;
(b) W. A. Merrill, R. J. Wright, C. S. Stanciu, M. M. Olmstead, J. C. Fettinger and P. P. Power, Synthesis and Structural Characterization of a Series of Dimeric Metal(II) Imido Complexes {M(μ-NAr#)}2 [M = Ge, Sn, Pb; Ar# = C6H3−2,6−(C6H2−2,4,6−Me3)2] and the Related Monomeric Primary Amido Derivatives M{N(H)Ar#}2 (M = Ge, Sn, Pb): Spectroscopic Manifestations of Secondary Metal-Ligand Interactions, Inorg. Chem., 2010, 49, 7097–7105 CrossRef CAS PubMed.
- S. Brooker, J.-K. Buijink and F. T. Edelmann, Synthesis, Structure, and Reactivity of the First Stable Diaryllead(II), Organometallics, 1991, 10, 25–26 CrossRef CAS.
- T. Ochiai and S. Inoue, Synthesis of a Cyclopentadienyl(imino)stannylene and Its Direct Conversion into Halo(imino)stannylenes, RSC Adv., 2017, 7, 801–804 RSC.
- D. Stalke, M. A. Paver and D. S. Wright, Nucleophilic Substitution of Bis(cyclopentadienyl)-tin(II); Synthesis, Structure, and Solution Dynamicsof Trans-[(η3-Cp)Sn{μ2-N=C(NMe2)2}]2, Angew. Chem., Int. Ed. Engl., 1993, 32, 428–429 CrossRef.
- X.-X. Zhao, T. Szilvási, F. Hanusch, J. A. Kelly and S. Inoue, Isolation and Reactivity of Tetrylene-Tetrylone-Iron Complexes Supported by Bis(N-Heterocyclic Imine) Ligands, Angew. Chem., Int. Ed., 2022, 61, e202208930 CrossRef CAS PubMed.
- X.-X. Zhao, T. Szilvási, F. Hanusch and S. Inoue, An Isolable Three-Coordinate Germanone and Its Reactivity, Chem.–Eur. J., 2021, 27, 15914–15917 CrossRef CAS PubMed.
- D. Sarkar, L. Groll, D. Munz, D. Hanusch and S. Inoue, Ligand Assisted CO2 Activation and Catalytic Valorization by an NHI-Stabilized Stannylene, ChemCatChem, 2022, 14, e202201048 CrossRef CAS.
- X.-X. Zhao, S. Fujimori, J. A. Kelly and S. Inoue, Isolation and Reactivity of Stannylenoids Stabilized by Amido/imino Ligands, Chem.–Eur. J., 2023, 29, e202202712 CrossRef CAS.
- L. Groll, J. A. Kelly and S. Inoue, Reactivity of NHI-Stabilized Heavier Tetrylenes towards CO2 and N2O, Chem.–Asian J., 2024, 19, e202300941 CrossRef CAS.
- M. J. S. Gynane, D. H. Harris, M. F. Lappert, P. P. Power, P. Rivière and M. Rivière-Baudet, Subvalent Group 4B Metal Alkyls and Amides. Part 5. the Synthesis and Physical Properties of Thermally Stable Amides of Germanium(II). Tin(II), and Lead(II), J. Chem. Soc., Dalton Trans., 1977, 2004–2009 RSC.
- J. Schreiber, H. Maag, N. Hashimoto and A. Eschenmoser, Dimethyl(methylene)ammonium Iodidei, Angew. Chem., Int. Ed. Engl., 1971, 10, 330–331 CrossRef CAS.
- N. R. Paisley, M. W. Lui, R. McDonald, M. J. Ferguson and E. Rivard, Structurally Versatile Phosphine and Amine Donors Constructed from N-Heterocyclic Olefin Units, Dalton Trans., 2016, 45, 9860–9870 RSC.
- L. Y. M. Eymann, P. Varava, A. M. Shved, B. F. E. Curchod, Y. Liu, O. M. Planes, A. Sienkiewicz, R. Scopelliti, F. F. Tirani and K. Severin, Synthesis of Organic Super-Electron-Donors by Reaction of Nitrous Oxide with N-Heterocyclic Olefins, J. Am. Chem. Soc., 2019, 141, 17112–17116 CrossRef CAS PubMed.
- P. Varava, Z. Dong, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Isolation and Characterization of Diazoolefins, Nat. Chem., 2021, 13, 1055–1060 CrossRef CAS PubMed.
- P. Varava, T. Wong, Z. Dong, A. Y. Gitlina, A. Sienkiewicz, W. Feuerstein, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Head-to-Tail Dimerization of N-Heterocyclic Diazoolefins, Angew. Chem., Int. Ed., 2023, 62, e202303375 CrossRef CAS.
- T. Eisner, A. Kostenko, F. J. Kiefer and S. Inoue, Synthesis and Isolation of a Cyclic Bis-Vinyl Germylene via a Diazoolefin Adduct of Germylene Dichloride, Chem. Commun., 2024, 60, 558–561 RSC.
- Y. He, Y. Lyu, D. Tymann, P. W. Antoni and M. M. Hansmann, Cleavage of Carbodicarbenes with N2O for Accessing Stable Diazoalkenes: Two-Fold Ligand Exchange at a C(0)-Atom, Angew. Chem., Int. Ed., 2025, 64, e202415228 CrossRef CAS PubMed.
- L. P. Ho, M.-K. Zaretzke, T. Bannenberg and M. Tamm, Heteroleptic Diphosphenes and Arsaphosphenes Bearing Neutral and Anionic N-Heterocyclic Carbenes, Chem. Commun., 2019, 55, 10709–10712 RSC.
- J. Haberstroh, C. Taube, J. Fidelius, S. Schulz, N. Israel, E. Dmitrieva, R. M. Gomila, A. Frontera, R. Wolf, K. Schwedtmann and J. J. Weigand, A Neutral Diphosphene Radical: Synthesis, Electronic Structure and White Phosphorus Activation, Chem. Commun., 2024, 60, 8537–8540 RSC.
- M. A. Wünsche, T. Witteler and F. Dielmann, Lewis Base Free Oxophosphonium Ions: Tunable, Trigonal-Planar Lewis Acids, Angew. Chem., Int. Ed., 2018, 57, 7234–7239 CrossRef.
- P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Isolation, Characterization and Reactivity of Three Coordinate Phosphorus Dications Isoelectronic to Alanes and Silylium Cations, Nat. Chem., 2019, 11, 1139–1143 CrossRef CAS.
- P. Löwe, M. A. Wünsche, F. R. S. Purtscher, J. Gamper, T. S. Hofer, L. F. B. Wilm, M. B. Röthel and F. Dielmann, Terminal Methylene Phosphonium Ions: Precursors for Transient Monosubstituted Phosphinocarbenes, Chem. Sci., 2023, 14, 7928–7935 RSC.
- F. Buß, M. Das, D. Janssen-Müller, A. Sietmann, A. Das, L. F. B. Wilm, M. Freitag, M. Seidl, F. Glorius and F. Dielmann, Photoswitchable Electron-Rich Phosphines: Using Light to Modulate the Electron-Donating Ability of Phosphines, Chem. Commun., 2023, 59, 12019–12022 RSC.
- J. L. Lortie, M. Davies, P. D. Boyle, M. Karttunen and P. J. Ragogna, Chemoselective Staudinger Reactivity of Bis(azido)phosphines Supported with a π-Donating Imidazolin-2-iminato Ligand, Inorg. Chem., 2024, 63, 6335–6345 CrossRef CAS.
- K. Paul Lüdtke, E. Zander, F. Taube, J.-E. Siewert, B. Corzilius, C. Hering-Junghans, J. Bresien and A. Schulz, Reaction of NHOs with Bisphosphanes-Designing Diradicaloids, Zwitterions and Radicals, Angew. Chem., Int. Ed., 2025, e202423347 Search PubMed.
-
(a) A. Dumrath, X.-F. Wu, H. Neumann, A. Spannenberg, R. Jackstell and M. Beller, Recyclable Catalysts for Palladium-Catalyzed C–O Coupling Reactions, Buchwald-Hartwig Aminations, and Sonogashira Reactions, Angew. Chem., Int. Ed., 2010, 49, 8988–8992 CrossRef CAS;
(b) A. Dumrath, C. Lübbe, H. Neumann, R. Jackstell and M. Beller, Recyclable Catalysts for Palladium-Catalyzed Aminations of Aryl Halides, Chem.–Eur. J., 2011, 17, 9599–9604 CrossRef CAS.
- S. J. Berners-Price, L. A. Colquhoun, P. C. Healy, K. A. Byriel and J. V. Hanna, Copper(I) and Gold(I) Complexes with Cis-Bis(diphenyl-phosphino)ethylene. Crystal Structures and 31P Cross-Polarization Magic Angle Spinning Nuclear Magnetic Resonance Studies, J. Chem. Soc., Dalton Trans., 1992, 3357–3363 RSC.
- M. W. Lui, O. Shynkaruk, M. S. Oakley, R. Sinelnikov, R. McDonald, M. J. Ferguson, A. Meldrum, M. Klobukowski and E. Rivard, Engaging Dual Donor Sites within an N-Heterocyclic Olefin Phosphine Ligand, Dalton Trans., 2017, 46, 5946–5954 RSC.
- A. K. Sahoo, B. Das, S. J. Panda, C. S. Purohit and A. Doddi, N-Heterocyclic Olefin-Phosphines Based Cationic Ruthenium Complexes as Pre-Catalysts for Dual C–H Bond Functionalizations, Adv. Synth. Catal., 2024, 366, 2468–2476 CrossRef.
-
(a) T. Witteler, H. Darmandeh, P. Mehlmann and F. Dielmann, Dialkyl(1,3-diarylimidazolin-2-ylidenamino)phosphines: Strongly Electron-Donating, Buchwald-Type Phosphines, Organometallics, 2018, 37, 3064–3072 CrossRef CAS;
(b) L. F. B. Wilm, P. Mehlmann, F. Buß and F. Dielmann, Synthesis and Characterization of Strongly Electron-Donating Bidentate Phosphines Containing Imidazolin-2-Ylidenamino Substituents and Their Electron-Rich Nickel(0), Palladium(II) and Gold(I) Chelate Complexes, J. Organomet. Chem., 2020, 909, 121097 CrossRef CAS;
(c) T.-F. Leung, D. Jiang, M.-C. Wu, D. Xiao, W.-M. Ching, G. P. A. Yap, T. Yang, L. Zhao, T.-G. Ong and G. Frenking, Isolable Dicarbon Stabilized by a Single Phosphine Ligand, Nat. Chem., 2021, 12, 89–93 CrossRef;
(d) S. Anga, J. Acharya and V. Chandrasekhar, An Unsymmetric Imino-Phosphanamidinate Ligand and its Y(III) Complex: Synthesis, Characterization, and Catalytic Hydroboration of Carbonyl Compounds, J. Org. Chem., 2021, 86, 2224–2234 CrossRef CAS;
(e) H. Karmakar, S. Anga, T. K. Panda and V. Chandrasekhar, Aluminium Alkyl Complexes Supported by Imino Phosphanamide Ligand as Precursors for Catalytic Guanylation Reactions of Carbodiimides, RSC Adv., 2022, 12, 4501–4509 RSC.
- P. Löwe and F. Dielmann, Crystalline Phosphino(silyl)carbenes that Readily Form Transition Metal Complexes, Chem. Commun., 2022, 58, 11831–11834 RSC.
- M. D. Böhme, T. Eder, M. B. Röthel, P. D. Dutschke, L. F. B. Wilm, F. E. Hahn and F. Dielmann, Synthesis of N-Heterocyclic Carbenes and Their Complexes by Chloronium Ion Abstraction from 2-Chloroazolium Salts Using Electron-Rich Phosphines, Angew. Chem., Int. Ed., 2022, 61, e202202190 CrossRef.
-
(a) E. Despagnet, H. Gornitzka, A. B. Rozhenko, W. W. Schoeller, D. Bourissou and G. Bertrand, Stable Non Push-Pull (phosphino)carbenes: NMR Characterization of a Methylcarbene, Angew. Chem., Int. Ed., 2002, 41, 2835–2837 CrossRef CAS;
(b) C. Buron, H. Gornitzka, V. Romanenko and G. Bertrand, Stable Versions of Transient Push-Pull Carbenes: Extending Lifetimes from Nanoseconds to Weeks, Science, 2000, 288, 834–836 CrossRef CAS.
- S. Wang, C. Zhu, L. Ning, D. Li, X. Feng and S. Dong, Regioselective C–H Alkylation of Anisoles with Olefins by Cationic Imidazolin-2-Iminato Scandium(III) Alkyl Complexes, Chem. Sci., 2023, 14, 3132–3139 RSC.
- B. Feng, L.-W. Ye, N. Wang, H.-S. Hu, J. Li, M. Tamm and Y. Chen, Endeavoring Cerium(IV)-Alkyl, -Aryl and -Alkynyl Complexes by an Energy-Level Match Strategy, CCS Chem., 2025, 7, 1043–1053 CrossRef CAS.
- Y. Guan, K. Chang, Y. Su, X. Xu and X. Xu, Frustrated Lewis Pair-Type Reactivity of Intermolecular Rare-Earth Aryloxide and N-Heterocyclic Carbene/Olefin Combinations, Chem.–Asian J., 2024, 19, e202400190 CrossRef CAS PubMed.
- B. Wang, X. Wang, R. Zhuang, C. Zhang, H. Liu, T. Tang, M. S. Eisen and X. Zhang, Synthesis of Mono- and Bis- Benzimidazolin-2-Iminato Titanium Complexes and Their Catalytic Performances in Ethylene Homo- and Co- Polymerizations, Mol. Catal., 2021, 516, 111974 CrossRef CAS.
-
(a) I. Haas, C. Hübner, W. P. Kretschmer and R. Kempe, A Highly Efficient Titanium Catalyst for the Synthesis of Ultrahigh-Molecular-Weight Polyethylene (UHMWPE), Chem.–Eur. J., 2013, 19, 9132–9136 CrossRef CAS PubMed;
(b) M. Rep, J.-W. F. Kaagman, C. J. Elsevier, P. Sedmera, J. Hiller, U. Thewalt, M. Horáček and K. Mach, Reactions of Methyl-Substituted Titanocene-Bis(trimethylsilyl)acetylene Complexes with Acetone Azine: Crystal Structures of (η5:η1-C5HMe3CH2CMe2NH)2Ti and (C5Me5)2Ti(N CMe2), J. Organomet. Chem., 2000, 597, 146–156 CrossRef CAS;
(c) K. Nomura, J. Yamada, W. Wang and J. Liu, Effect of Ketimide Ligand for Ethylene Polymerization and Ethylene/Norbornene Copolymerization Catalyzed by (Cyclopentadienyl)(Ketimide)Titanium Complexes-MAO Catalyst Systems: Structural Analysis for Cp*TiCl2(N=CPh2), J. Organomet. Chem., 2007, 692, 4675–4682 CrossRef CAS.
- M. Koneczny, A. B. Erol, M. Mauduit, M. S. Eisen and M. Tamm, Titanium Complexes with Unsymmetrically Substituted Imidazolin-2-Iminato Ligands, Dalton Trans., 2022, 51, 11448–11456 RSC.
- M. Fischer, M. M. D. Roy, S. Hüller, M. Schmidtmannb and R. Beckhaus, Reaction of a Bis(Pentafulvene)Titanium Complex with an N-Heterocyclic Olefin: C–H activation Leads to Resonance between a Titanium Vinyl and Titanium Alkylidene Complex, Dalton Trans., 2022, 51, 10690–10696 RSC.
- I. C. Watson, Y. Zhou, M. J. Ferguson and E. Rivard, Group 4 Transition Metal Complexes with Anionic N-Heterocyclic Olefin Ligands, Z. Anorg. Allg. Chem., 2022, 648, e202200082 CrossRef CAS.
- M. Khononov, H. Liu, N. Fridman, M. Tamm and M. S. Eisen, Mono(imidazolin-2-iminato) Hafnium Complexes: Synthesis and Application in the Ring-Opening Polymerization of ε-Caprolactone and rac-Lactide, Catalysts, 2022, 12, 1201 CrossRef CAS.
- M. Peters, D. Baabe, M. Maekawa, D. Bockfeld, M.-K. Zaretzke, M. Tamm and M. D. Walter, Pogo-Stick Iron and Cobalt Complexes: Synthesis, Structures, and Magnetic Properties, Inorg. Chem., 2019, 58, 16475–16486 CrossRef CAS.
-
(a) M. D. Walter and P. S. White, {Cp’Fe(μ-OH)}3: The Synthesis of a Unique Organometallic Iron Hydroxide, Dalton Trans., 2012, 41, 8506–8508 RSC;
(b) M. D. Walter and P. S. White, Reactivity Studies on Cp’FeI2: Monomeric Amido, Phenoxo, and Alkyl Complexes, Inorg. Chem., 2012, 51, 11860–11872 CrossRef CAS PubMed;
(c) M. Reiners, D. Baabe, K. Harms, M. Maekawa, C. G. Daniliuc, M. Freytag, P. G. Jones and M. D. Walter, N-Heterocyclic Carbene Adducts to [Cp’FeI]2: Synthesis and Molecular and Electronic Structure, Inorg. Chem. Front., 2016, 3, 250–262 RSC;
(d) U. Siemeling, U. Vorfeld, B. Neumann and H.-G. Stammler, [Bis(trimethylsilyl)amido](η5-pentamethylcyclopentadienyl)-iron-(II): A Diamagnetic 14-Electron Complex with a “Pogo-Stick” Structure, Organometallics, 1998, 17, 483–484 CrossRef CAS;
(e) O. A. Groß, S. Lauk, C. Muller, W. Gidt, Y. Sun, S. Demeshko, F. Meyer and H. Sitzmann, Iron(II) High-Spin and Low-Spin Complexes from Pentaisopropylcyclopentadienyliron(II) Bis- (trimethylsilyl)amide, Eur. J. Inorg. Chem., 2017, 3635–3643 CrossRef;
(f) M. Wallasch, G. Wolmershauser and H. Sitzmann, Phenolate Complexes of Iron(II) in Different Spin States, Angew. Chem., Int. Ed., 2005, 44, 2597–2599 CrossRef CAS.
- R. Weller, A. Gonzalez, H. Gottschling, C. Hänisch and C. G. Werncke, NHC-stabilized Parent Phosphinidene Adducts of Metal(II) Hexamethyldisilazanides of Manganese - Cobalt and Their Lability in Solution, Z. Anorg. Allg. Chem., 2022, 648, e202100338 CrossRef CAS.
- L. L. Liu, D. A. Ruiz, F. Dahcheh and G. Bertrand, Isolation of a Lewis Base Stabilized Parent Phosphenium (PH2+) and Related Species, Chem. Commun., 2015, 51, 12732–12735 RSC.
-
(a) D. Bockfeld, A. Doddi, P. G. Jones and M. Tamm, Transition-Metal Carbonyl Complexes and Electron-Donating Properties of N-Heterocyclic-Carbene-Phosphinidene Adducts, Eur. J. Inorg. Chem., 2016, 3704–3712 CrossRef CAS;
(b) A. Doddi, D. Bockfeld, A. Nasr, T. Bannenberg, P. G. Jones and M. Tamm, N-Heterocyclic Carbene–Phosphinidene Complexes of the Coinage Metals, Chem.–Eur. J., 2015, 21, 16178–16189 CrossRef CAS PubMed;
(c) T. G. Larocque and G. G. Lavoie, Reactivity Study of Low-Coordinate Phosphaalkene IMes=PPh with Grubbs First-Generation Ruthenium Benzylidene Complexes, New J. Chem., 2014, 38, 499–502 RSC;
(d) M. Peters, A. Doddi, T. Bannenberg, M. Freytag, P. G. Jones and M. Tamm, N-Heterocyclic Carbene-Phosphinidene and Carbene-Phosphinidenide Transition Metal Complexes, Inorg. Chem., 2017, 56, 10785–10793 CrossRef CAS PubMed.
- A. Doddi, D. Bockfeld and M. Tamm, Synthesis and Structures of RhI and IrI Complexes Supported by N-Heterocyclic Carbene-Phosphinidene Adducts, Z. Anorg. Allg. Chem., 2019, 645, 44–49 CrossRef CAS.
- G.-W. Chen, Y.-H. Chen, H.-K. Liu and C.-Y. Lin, Three-Coordinate Halide and Methyl Complexes of Chromium, Manganese, Iron, Cobalt, and Nickel Enabled by an Imidazolin-2-iminato Ligand, Organometallics, 2023, 42, 2395–2404 CrossRef CAS.
- D. Becker, D. Bockfeld and M. Tamm, An Update on Phosphine-Imidazolin-2-Imine Iridium(I) Catalysts for Hydrogen Isotope Exchange, Adv. Synth. Catal., 2023, 365, 367–372 CrossRef CAS.
- I. C. Watson, A. Schumann, H. Yu, E. C. Davy, R. McDonald, M. J. Ferguson, C. Hering-Junghans and E. Rivard, N-Heterocyclic Olefin-Ligated Palladium(II) Complexes as Pre-Catalysts for Buchwald-Hartwig Aminations, Chem.–Eur. J., 2019, 25, 9678–9690 CrossRef CAS PubMed.
- I. Antsiburov, J. Stephan, R. J. Weininger, C. Gemel and R. A. Fischer, Copper Imidazolin-Imine Coordination Compounds as Precursors for a Cu/Al Complex, Inorg. Chem., 2024, 63, 17331–17339 CrossRef CAS PubMed.
- A. Doddi, M. Peters, D. Bockfeld and M. Tamm, Copper and Silver Complexes of N-Heterocyclic Carbene-Parent Phosphinidene Adducts, Z. Anorg. Allg. Chem., 2023, 649, e202200364 CrossRef CAS.
- H. Deka, N. Fridman, M. Koneczny, M. Tamm and M. S. Eisen, Base-Free Transfer Hydrogenation of Aldehydes and Ketones Catalyzed by Imidazolin-2-Iminato Actinide Complexes, Catal. Sci. Technol., 2023, 13, 352–361 Search PubMed.
-
(a) O. Bienemann, A. Hoffmann and S. Herres-Pawlis, (Guanidine)Copper Complexes: Structural Variety and Application in Bioinorganic Chemistry and Catalysis, Rev. Inorg. Chem., 2011, 31, 83–108 Search PubMed;
(b) I. dos Santos Vieira and S. Herres-Pawlis, Lactide Polymerisation with Complexes of Neutral N-Donors-New Strategies for Robust Catalysts, Eur. J. Inorg. Chem., 2012, 765–774 CrossRef CAS;
(c) H.-J. Himmel, Guanidinyl-Functionalized Aromatic Compounds (GFAs)-Charge and Spin Density Studies as Starting Points for the Development of a New Class of Redox-Active Ligands, Z. Anorg. Allg. Chem., 2013, 639, 1940–1952 CrossRef CAS;
(d) H.-J. Himmel, Valence Tautomerism in Copper Coordination Chemistry, Inorg. Chim. Acta, 2018, 481, 56–68 CrossRef CAS.
-
(a) A. Maronna, E. Bindewald, E. Kaifer, H. Wadepohl and H.-J. Himmel, Synthesis and Characterization of Novel Guanidine Ligands Featuring Biphenylor Binaphthyl Backbones, Eur. J. Inorg. Chem., 2011, 1302–1314 CrossRef CAS;
(b) P. Roquette, A. Maronna, M. Reinmuth, E. Kaifer, M. Enders and H.-J. Himmel, Combining NMR of Dynamic and Paramagnetic Molecules: Fluxional High-Spin Nickel(II) Complexes Bearing Bisguanidine Ligands, Inorg. Chem., 2011, 50, 1942–1955 CrossRef CAS PubMed;
(c) P. Roquette, A. Maronna, A. Peters, E. Kaifer, H.-J. Himmel, C. Hauf, V. Herz, E.-W. Scheidt and W. Scherer, On the Electronic Structure of NiII Complexes that Feature Chelating Bisguanidine Ligands, Chem.–Eur. J., 2010, 16, 1336–1350 CrossRef CAS;
(d) P. Roquette, C. Konig, O. Hubner, A. Wagner, E. Kaifer, M. Enders and H.-J. Himmel, Eur. J. Inorg. Chem., 2010, 4770–4782 CrossRef CAS;
(e) M. Reinmuth, U. Wild, D. Rudolf, E. Kaifer, M. Enders, H. Wadepohl and H.-J. Himmel, Stabilization and Activation: New Alkyl Complexes of Zinc, Magnesium and Cationic Aluminium Featuring Chelating Bisguanidine Ligands, Eur. J. Inorg. Chem., 2009, 4795–4808 Search PubMed;
(f) U. Wild, O. Hubner, A. Maronna, M. Enders, E. Kaifer, H. Wadepohl and H.-J. Himmel, The First Metal Complexes of the Proton Sponge 1,8-Bis(N,N,N/,N/-Tetramethylguanidino)naphthalene: Syntheses and Properties, Eur. J. Inorg. Chem., 2008, 4440–4447 CrossRef CAS;
(g) V. Raab, K. Harms, J. Sundermeyer, B. Kovacevic and Z. B. Maksic, 1,8-Bis(dimethylethyleneguanidino)naphthalene: Tailoring the Basicity of Bisguanidine “Proton Sponges” by Experiment and Theory, J. Org. Chem., 2003, 68, 8790–8797 CrossRef CAS PubMed;
(h) V. Raab, J. Kipke, R. M. Gschwind and J. Sundermeyer, 1,8-Bis(tetramethylguanidino)naphthalene (TMGN): a New, Superbasic and Kinetically Active “Proton Sponge”, Chem.–Eur. J., 2002, 8, 1682–1693 CrossRef CAS.
- D. Franz, T. Szilvási, A. Pçthig, F. Deiser and S. Inoue, Three-Coordinate Boron (III) and Diboron (II) Dications, Chem.–Eur. J., 2018, 24, 4283–4288 CrossRef CAS PubMed.
- F. Hanusch, D. Munz, J. Sutter, K. Meyer and S. Inoue, A Zwitterionic Heterobimetallic Gold–Iron Complex Supported by Bis(N-Heterocyclic Imine)Silyliumylidene, Angew. Chem., Int. Ed., 2021, 60, 23274–23280 CrossRef CAS PubMed.
- F. S. Tschernuth, F. Hanusch, T. Szilvási and S. Inoue, Isolation and Reactivity of Chlorotetryliumylidenes Using a Bidentate Bis(N-Heterocyclic Imine) Ligand, Organometallics, 2020, 39, 4265–4272 CrossRef CAS.
- S. V. Hirmer, F. S. Tschernuth, F. Hanusch, R. Baierl, M. Muhrb and S. Inoue, S. Modification of Bidentate Bis(N-Heterocyclic Imine) Ligands for Low-Valent Main Group Complexes, Mendeleev Commun., 2022, 32, 16–18 CrossRef CAS.
- M. Park, C. Schmidt, S. Türck, F. Hanusch, S. V. Hirmer, I. Ott, A. Casini and S. Inoue, Potent Anticancer Activity of a Dinuclear Gold(I) Bis-N-Heterocyclic Imine Complex Related to Thioredoxin Reductase Inhibition in Vitro, ChemPlusChem, 2024, 89, e202300557 CrossRef CAS.
- T. Glöge, D. Petrovic, C. G. Hrib, C. Daniliuc, E. Herdtweck, P. G. Jones and M. Tamm, Synthesis and Structural Characterisation of an Isomorphous Series of Bis(imidazolin-2-imine) Metal Dichlorides Containing the First Row Transition Metals Mn, Fe, Co, Ni, Cu and Zn, Z. Anorg. Allg. Chem., 2010, 636, 2303–2308 CrossRef.
- K. Jess, D. Baabe, T. Bannenberg, K. Brandhorst, M. Freytag, P. G. Jones and M. Tamm, Ni−Fe and Pd−Fe Interactions in Nickel(II) and Palladium(II) Complexes of a Ferrocene-Bridged Bis(imidazolin-2-imine) Ligand, Inorg. Chem., 2015, 54, 12032–12045 CrossRef CAS.
- K. Jess, D. Baabe, M. Freytag, P. G. Jones and M. Tamm, Transition-Metal Complexes with Ferrocene-Bridged Bis(imidazolin-2-imine) and Bis(diaminocyclopropenimine) Ligands, Eur. J. Inorg. Chem., 2017, 412–423 Search PubMed.
-
(a) C. Metallinos, D. Tremblay, F. B. Barrett and N. J. Taylor, 1,1’-Bis(phosphoranylidenamino)ferrocene Palladium(II) Complexes: an Unusual Case of Dative Fe → Pd Bonding, J. Organomet. Chem., 2006, 691, 2044–2047 CrossRef CAS;
(b) L. R. R. Klapp, C. Bruhn, M. Leibold and U. Siemeling, Ferrocene-Based Bis(guanidines): Superbases for Tridentate N, Fe, N-Coordination, Organometallics, 2013, 32, 5862–5872 Search PubMed.
- T. J. Hadlington, A. Kostenko and M. Driess, Synthesis and Coordination Ability of a Donor-Stabilised Bis-Phosphinidene, Chem.–Eur. J., 2021, 27, 2476–2482 CrossRef CAS PubMed.
- R. Baierl, A. Kostenko, F. Hanusch and S. Inoue, Application of Ferrocene-Bridged N-Heterocyclic Carbene Stabilised Bis-Phosphinidenes in Sn(II) Complexation, Dalton Trans., 2021, 50, 14842–14848 RSC.
- J.-K. Siewert, B. M. P. Lombardi, N. Jannsen, R. Roesler and C. Hering-Junghans, Synthesis and Ligand Properties of Chelating Bis(N-Heterocyclic Carbene)-Stabilized Bis(phosphinidenes), Inorg. Chem., 2023, 62, 16832–16841 CrossRef CAS.
- J.-K. Siewert, A. Schumann, T. Wellnitz, F. Dankert and C. Hering-Junghans, Triphosphiranes as Phosphinidene-Transfer Agents - Synthesis of Regular and Chelating NHC Phosphinidene Adducts, Dalton Trans., 2023, 52, 15747–15756 RSC.
- R. Baierl, A. Kostenko, F. Hanusch, A. D. Beck and S. Inoue, Synthesis and Reactivity of Bidentate N-Heterocyclic Carbene-Phosphinidene Supported Si(IV) Dicationic Complexes, Eur. J. Org Chem., 2022, e202201072 Search PubMed.
- C. C. Chong, B. Rao, R. Ganguly, Y. Li and R. Kinjo, Bis(N-heterocyclic olefin) Derivative: an Efficient Precursor for Isophosphindolylium Species, Inorg. Chem., 2017, 56, 8608–8614 CrossRef CAS.
- R. Guthardt, J. Mellin, C. Bruhn and U. Siemeling, 1,1’-Ferrocenylene-Bridged Bis(N-Heterocyclic Olefin) Derivatives, Eur. J. Inorg. Chem., 2022, e202100903 CrossRef CAS.
- J. Raynaud, W. N. Ottou, Y. Gnanou and D. Taton, Metal-Free and Solvent-Free Access to α,ω-Heterodifunctionalized Poly(propylene oxide)s by N-Heterocyclic Carbene-Induced Ring Opening Polymerization, Chem. Commun., 2010, 46, 3203–3205 RSC.
- S. Naumann, A. W. Thomas and A. P. Dove, N-Heterocyclic Olefins as Organocatalysts for Polymerization: Preparation of Well-Defined Poly(propylene oxide), Angew. Chem., Int. Ed., 2015, 54, 9550–9554 CrossRef CAS PubMed.
- E. Kersten, O. Linker, J. Blankenburg, M. Wagner, P. Walther, S. Naumann and H. Frey, Revealing the Monomer Gradient of Polyether Copolymers Prepared Using N-Heterocyclic Olefins: Metal-Free Anionic versus Zwitterionic Lewis Pair Polymerization, Macromol, Chem. Phys., 2023, 224, 2300097 CAS.
- S. Wang, C. Zhang, D. Li, Y. Zhou, Z. Su, X. Feng and S. Dong, New Chiral N-Heterocyclic Olefin Bifunctional Organocatalysis in α-Functionalization of β-Ketoesters, Sci. China:Chem., 2023, 66, 147–154 CrossRef CAS.
- S. Wang, J. Yang, H. Zeng, Y. Zhou, F. Wang, X. Feng and S. Dong, Asymmetric Formal Coupling of β-Ketoesters with Quinones Promoted by a Chiral Bifunctional N-Heterocyclic Olefin, Org. Lett., 2023, 25, 7247–7251 CrossRef CAS PubMed.
- P. Sreejyothi, K. Bhattacharyya, S. Kumar, P. K. Hota, A. Datta and S. K. Mandal, An NHC-Stabilised Phosphinidene for Catalytic Formylation: A DFT-Guided Approach, Chem.–Eur. J., 2021, 27, 11656–11662 CrossRef CAS.
- S. P, N. Gautam, S. Maji, K. Bhattacharyya and S. K. Mandal, Transition Metal-Mimicking Relay Catalysis by a Low-Valent Phosphorus Compound, J. Am. Chem. Soc., 2024, 146, 16743–16752 CrossRef CAS PubMed.
- D. Li, L. Ning, Q. Luo, S. Wang, X. Feng and S. Dong, C-H Alkylation of Pyridines with Olefins Catalyzed by Imidazolin-2-Iminato-Ligated Rare-Earth Alkyl Complexes, Sci. China:Chem., 2023, 66, 1804–1813 CrossRef CAS.
- D. Li, Y. Wang, S. Wang and S. Dong, The Effect of Basic Ligands and Alkenes on the Regioselectivity of C-H Additions of Tertiary Amines to Alkenes, Chem.–Eur. J., 2024, 30, e202401014 CrossRef CAS.
- S. Wang, L. Ning, T. Mao, Y. Zhou, M. Pu, X. Feng and S. Dong, Anti-Markovnikov hydroallylation reaction of alkenes via scandium-catalyzed allylic C-H activation, Nat. Commun., 2025, 16, 1423 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Minyi Yuan
* and
Minyi Yuan
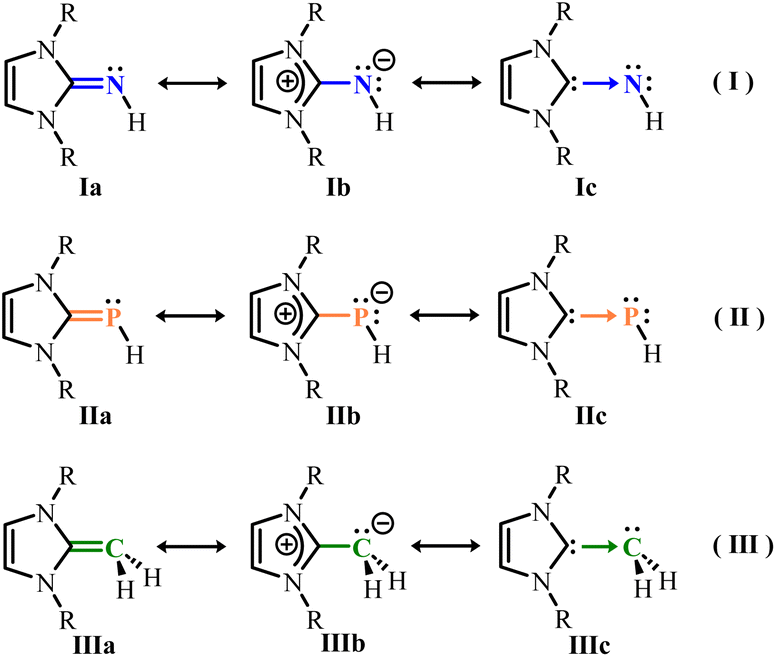











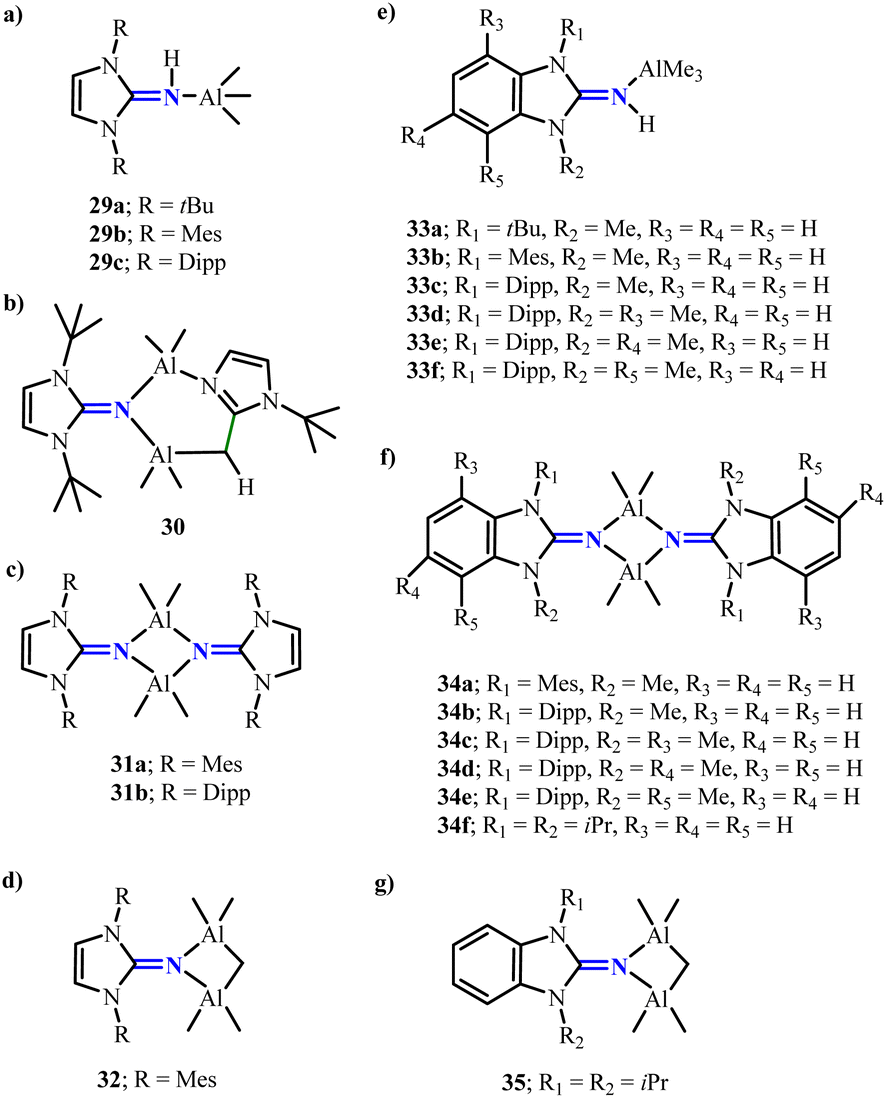
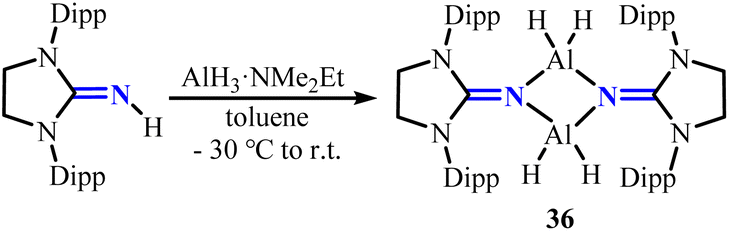



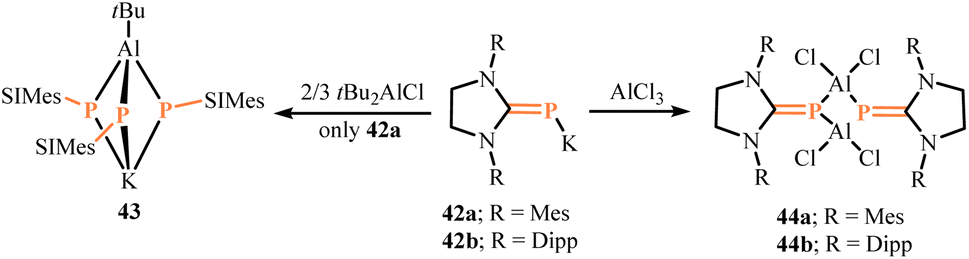









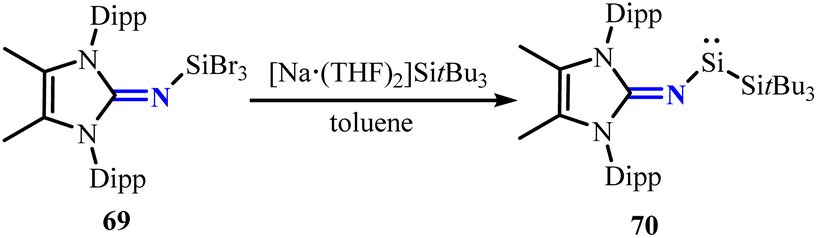


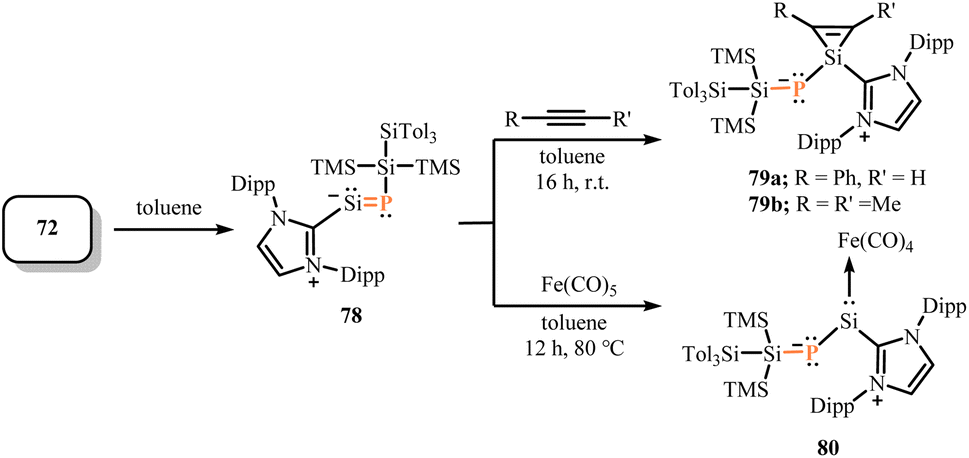
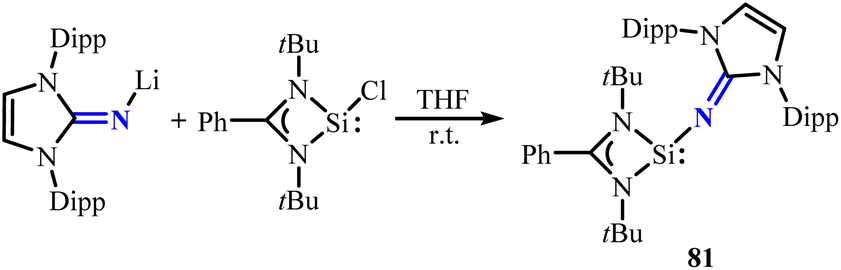







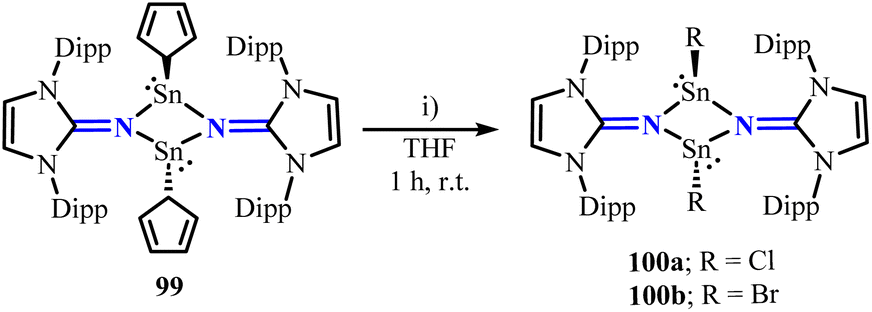


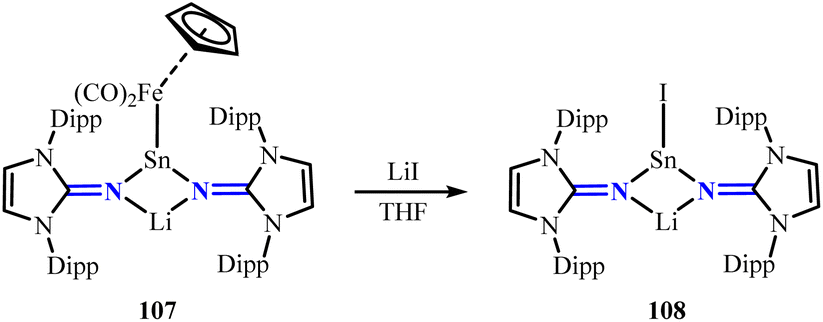












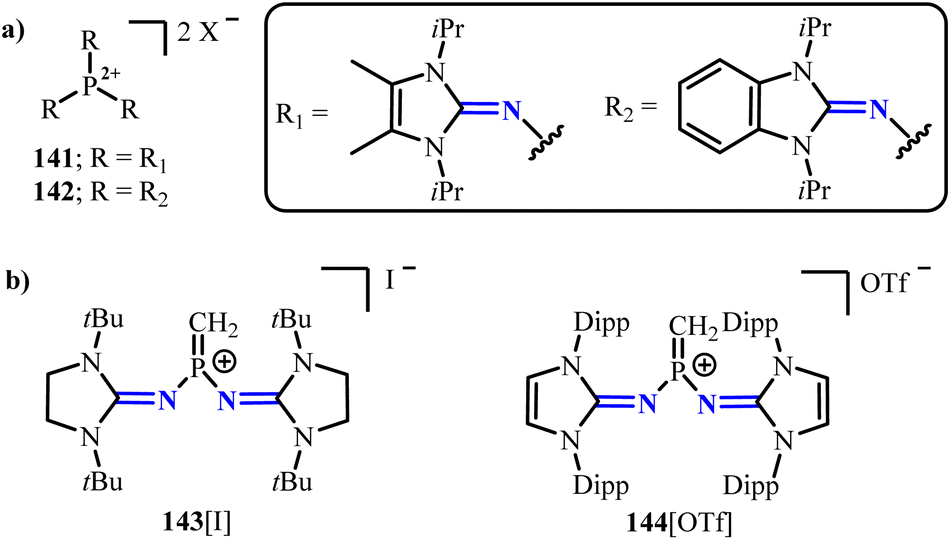



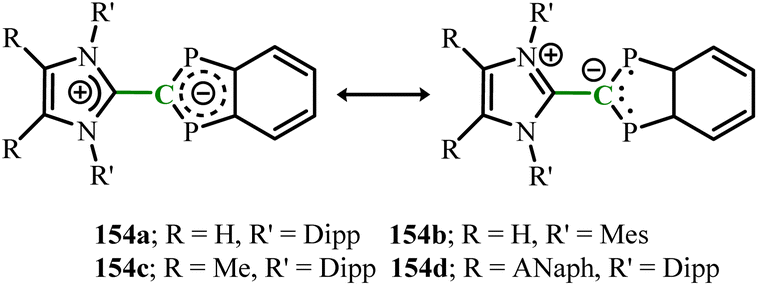







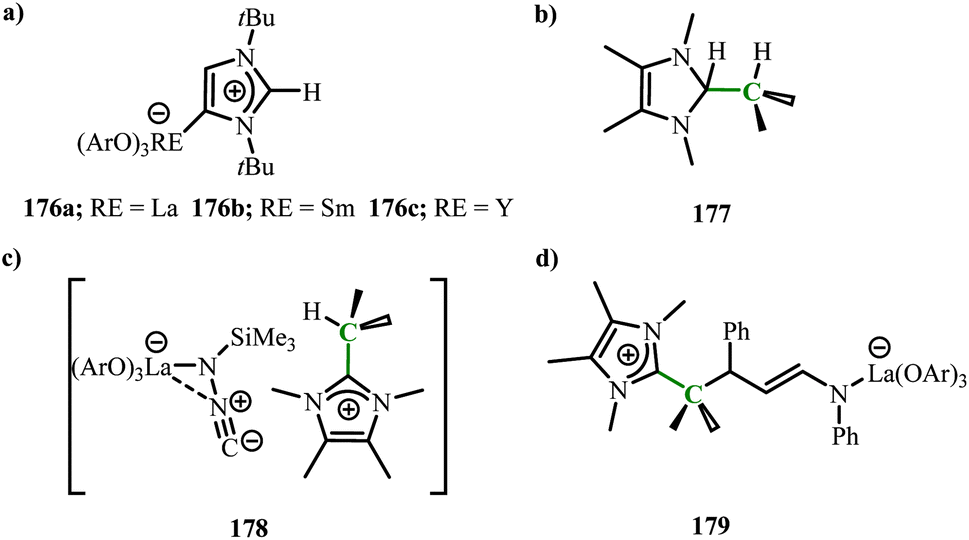


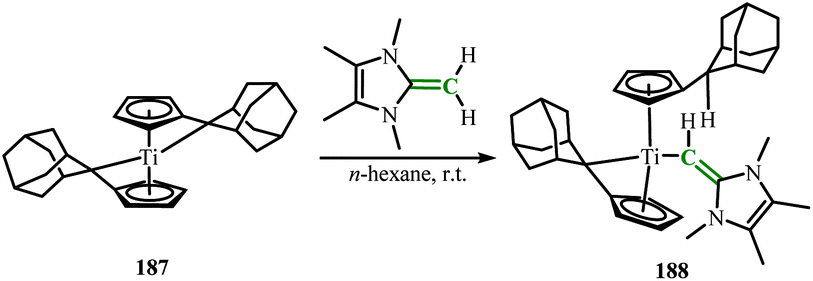
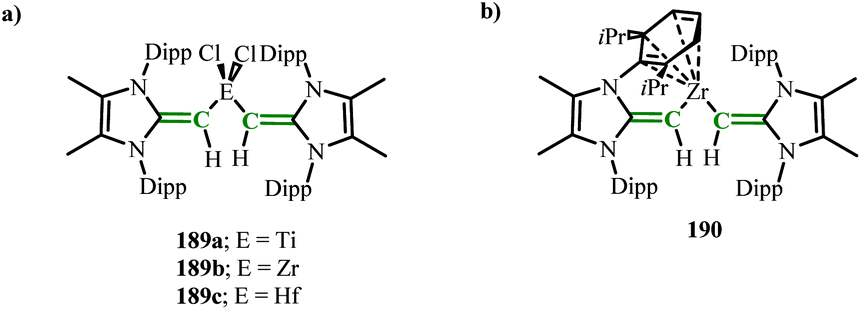
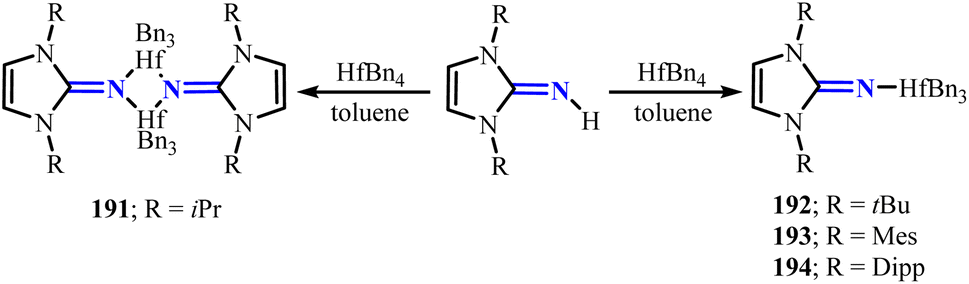

 distance of the “pogo stick” structure iron complex [Cp′Fe(NImDipp)] (199a) is 1.90 Å, which was comparable to the values of related Fe(II) high-spin species (1.88–2.05 Å) and consistent with the high-spin system of 4e−.122 The high-spin state of the Fe center was initially revealed, which was further confirmed by intermediate paramagnetic relaxation in the Fe Mössbauer spectrum. Similar cobalt complex (199b) represent a rare single-legged piano stool species. The spin state of the cobalt center in 199b exhibited temperature-dependent spin crossover. These half-sandwich imidazolin-2-imine metal complexes all showed short M–N bond lengths, which can be attributed to the multiple bond character induced by the strong 2σ, 4π electron-donating effect of the imidazolin-2-imine ligand.
distance of the “pogo stick” structure iron complex [Cp′Fe(NImDipp)] (199a) is 1.90 Å, which was comparable to the values of related Fe(II) high-spin species (1.88–2.05 Å) and consistent with the high-spin system of 4e−.122 The high-spin state of the Fe center was initially revealed, which was further confirmed by intermediate paramagnetic relaxation in the Fe Mössbauer spectrum. Similar cobalt complex (199b) represent a rare single-legged piano stool species. The spin state of the cobalt center in 199b exhibited temperature-dependent spin crossover. These half-sandwich imidazolin-2-imine metal complexes all showed short M–N bond lengths, which can be attributed to the multiple bond character induced by the strong 2σ, 4π electron-donating effect of the imidazolin-2-imine ligand.