
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVariations of heterometallic cores and magnetic properties in Co(III)–Y/Ln isobutyrate clusters with N-butyldiethanolamine: from tetranuclear to octanuclear clusters†
Dumitru Statia,
Jan van Leusen b,
Victor Ch. Kravtsova,
Yurii Chumakova,
Paul Kögerler*b and
Svetlana G. Baca*a
b,
Victor Ch. Kravtsova,
Yurii Chumakova,
Paul Kögerler*b and
Svetlana G. Baca*a
aInstitute of Applied Physics, Moldova State University, Academiei 5, 2028 Chisinau, R. Moldova. E-mail: svetlana.baca@ifa.usm.md; sbaca_md@yahoo.com
bInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
First published on 2nd June 2025
Abstract
Under similar synthetic conditions, i.e. by the interaction of Co(ib)2 (Hib = isobutyric acid) and M(NO3)3·nH2O with a tridentate organic ligand N-butyldiethanolamine (H2bdea) in acetonitrile, a series of heterometallic {Co(III)–M(III)} complexes with different cores {Co2M2}, {Co2M4}, {Co3M2}, {Co3M3}, and {Co4M4} have been isolated. The series includes tetranuclear [Co2M2(OH)2(ib)4(bdea)2(NO3)2]·2MeCN (M(III) = Y (1), Ho (2), Er (3)), pentanuclear [Co3M2(OH)(ib)5(bdea)3(NO3)3]·nMeCN (M(III) = Pr (n = 1.5, 4), Nd (n = 1.5, 5), Sm (n = 2, 6)), hexanuclear [Co2Eu4(OH)2(ib)8(bdea)2(NO3)4(H2O)2]·(7) and [Co3Yb3(OH)4(ib)6(bdea)3(H2O)3](NO3)2·3MeCN·1.5H2O (8), and octanuclear [Co4M4(OH)4(ib)8(bdea)4(NO3)4] (M(III) = Gd (9), Tb (10)) clusters. The structures of all clusters have been determined by single crystal X-ray diffraction and reveal novel structural features in the series. The tetranuclear (1–3), pentanuclear (4–6), and octanuclear (9–10) clusters are isostructural and isomorphic in their own group. Clusters 1–3 have a rhombic strictly planar tetranuclear central {Co2M2(μ3-OH)2} core resembling a common “butterfly” motif. Clusters 4–6 possess a robust and unusual topology, previously unseen in Co/Ln chemistry, of three {CoM2(μ3-OH)}, {CoM2(μ3-Obdea)}, and {Co2M(μ3-Obdea)} noncoplanar triangles sharing a common M(III) site. The arrangement of metal atoms in 9 and 10 is reminiscent of a smaller square defined by four M(III) sites inscribed in a larger square formed by four Co(III) sites, and can also be viewed as a core composed of four {CoM2(μ3-OH)} nearly coplanar triangles sharing two M(III) vertices with two neighbouring triangles. Cluster 7 can be viewed as a hexanuclear central core of two parallel {CoM2(μ3-OH)} triangles, while cluster 8 is composed of three {CoM2(μ3-OH)} triangles sharing the common M(III) vertices. DC magnetic data show non-interacting or very weakly interacting Ln(III) centers and low spin Co(III) centers. AC magnetic data show relevant out-of-phase signals only for 3 below 4 K and by application of static magnetic bias fields. First principles calculations have been performed to estimate the magnetic anisotropy energies of the prepared compounds and to calculate the exchange coupling between the lanthanides in them.
Introduction
Heterometallic Co–Ln clusters have attracted much interest because they represent prospective magnetic materials for diverse applications.1–7 In addition, such clusters have intriguing structural features, including high nuclear species and the existence of unprecedented metal topologies.7–9 Among them, 52-metal {Co10Ln42} clusters5 with acetate ligands exhibit the largest magnetocaloric effect among all known 3d–4f complexes for a {Co10Gd42} cluster (−ΔS = 41.3 J kg−1 K−1 at 2 K), surpassed only by {Mn2Gd3}8 (50.1 J kg−1 K−1), and slow relaxation of the magnetization for a {Co10Dy42} cluster. Tong et al. prepared two unprecedented wheel-shaped nanoscale clusters {Co16Ln24} (Ln = Dy, Gd) in which the dysprosium species exhibited single-molecule magnetic (SMM) behavior and the gadolinium species exhibited a relatively large magnetocaloric effect (MCE) with a maximum magnetic entropy change of 26.0 J kg−1 K−1 at 3.8 K.9 The families of {CoxLny} (x = 4, 6, 8; y = 2, 4, 6, 8) grids and cages with phosphonate ligands reported by Winpenny et al.10 showed a potential capability for magnetic cooling (the observed −ΔS value ranges from 11.8 to 28.6 J kg−1 K−1) at low temperatures. An unusual structural core and impressive magnetocaloric effects are demonstrated by {Co4Ln10} (Ln = Dy, Gd) phosphonate clusters.11 Kostakis and Escuer et al. reported a family of Co–Dy coordination clusters possessing different cores such as {Co2Dy2}, {CoDy3}, {Co4Dy4}, {Co3Dy4}, and {Co2Dy5} with robust and disk-like topologies, previously unseen in Co/Ln chemistry and exhibiting interesting magnetic properties.12,13 The combined use of carboxylate-containing ligands and alkylol amines to successfully construct heterometallic 3d–4f compounds is a widely used strategy in coordination chemistry, as they have multiple donor atoms and can reliably coordinate to 3d and 4f metal ions, exhibiting a variety of coordination modes with both chelating and bridging capabilities.4,14,15 Recently, we reported the coordination cluster compound [CoIII2DyIII4(OH)2(ib)8(bdea)2(NO3)4(H2O)2]·2MeCN, which was prepared from a one-pot reaction of cobalt(II) isobutyrate with Dy(NO3)3·6H2O and N-butyldiethanolamine (H2bdea) in MeCN.16 The cluster has a flat zig-zag {CoIII2DyIII4} core fragment with two {CoDy2(μ3-OH)} triangles connected by one of their Dy sites. Such an arrangement of metal atoms reduces the quantum tunneling of the magnetization and thus pushes up the onset of pronounced out-of-phase signals at zero bias field to 14 K. As part of our ongoing research, we report here the synthesis under similar conditions and characterization of a series of heterometallic Co–Y/Ln complexes in which the Y/Ln ion has been varied and the effect of these changes on the structural and physicochemical properties of the corresponding clusters has been studied in detail. Five different structural types of Co–Y/Ln complexes have been synthesized: tetranuclear [Co2M2(OH)2(ib)4(bdea)2(NO3)2]·2MeCN (MIII = Y (1), Ho (2), Er (3)), pentanuclear [Co3M2(OH)(ib)5(bdea)3(NO3)3]·nMeCN (MIII = Pr (n = 1.5, 4), Nd (n = 1.5, 5), Sm (n = 2, 6)), hexanuclear [Co2Eu4(OH)2(ib)8(bdea)2(NO3)4(H2O)2] (7) and [Co3Yb3(OH)4(ib)6(bdea)3(H2O)3](NO3)2·3MeCN·1.5H2O (8), and octanuclear [Co4M4(OH)4(ib)8(bdea)4(NO3)4] (MIII = Gd (9), Tb (10)) clusters (Scheme 1). | ||
| Scheme 1 Synthetic procedures and resulting structures 1–10. Color codes: C, black sticks; N, yellow, O, red balls; Co, violet, Y/Ln, green spheres. H atoms and solvent molecules are omitted for clarity. | ||
Experimental
Materials and methods
All manipulations were performed under aerobic conditions using chemicals and solvents as received without further purification. Cobalt(II) isobutyrate was prepared as reported elsewhere.17 IR transmission spectra were recorded on a PerkinElmer Spectrum 100 FT-IR in the range 650–4000 cm−1. Thermogravimetric analysis/differential scanning calorimetry (TGA/DSC) measurements were performed with a Linseis STA PT 1600 in air at a heating rate of 5 K min−1 from 25 to 600 °C for 1–11. Electrospray ionization mass spectrometry (ESI-MS) was performed using a ThermoFisher Scientific LTQ-Orbitrap XL mass spectrometer in positive and negative ion detection mode. Samples were prepared by dissolving clusters in MeOH (1–3, 5, 7–10) or MeOH + dmf (4), both of which contained 0.1% acetic acid. Powder XRD measurements were performed on freshly prepared samples of 1–10 on a RIGAKU MiniFlex 600 diffractometer, while Energy Dispersive X-ray Analysis (EDX) was performed on a Xenemetrix's X-Calibur Energy Dispersive X-ray Fluorescence spectrometer. Magnetic properties were determined using a Quantum Design MPMS-5XL SQUID magnetometer. Microcrystalline samples of 1–5, 8 were compacted and immobilized into cylindrical PTFE sample holders. The data were recorded at 0.1 and 1.0 T in the temperature range 2.0–290 K, and at 2.0 K in the field range 0.1–5.0 T. Data of 1 were used to determine the temperature independent paramagnetic contributions of the low spin Co(III) centers (resulting in χm,TIP = +1.30 × 10−4 cm3 mol−1 per Co(III) center, Fig. S25†). Additional ac magnetic susceptibility measurements were performed on samples of 2–5, 8 in the frequency range 3–937 Hz and temperature range 1.9–50 K at zero, at 500 Oe and at 1000 Oe static bias magnetic field, however, relevant out-of-phase signals were only detected only for 3. All data were corrected for χm,TIP as well as the diamagnetic contributions of the sample holder and the intrinsic contribution of the compounds: χm,dia/10−4 cm3 mol−1 = −6.01 (1), −6.77 (2), −6.80 (3), −8.18 (4), −8.22 (5), −10.46 (8).X-ray crystallography
X-ray data sets for 2, 4, 5, 8, 9, and 10 were collected on a Bruker APEX CCD diffractometer, for 1, 6 and 7 on a STOE STADIVARI diffractometer, and for 3 on an Oxford Xcalibur diffractometer equipped with a CCD detector and graphite-monochromatized MoKα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least-squares on weighted F2 values for all reflections using the SHELX suite of programs.18 All non-H atoms in the compounds were refined with anisotropic displacement parameters. Some of the solvent molecules, iso-propanol and N-butyldiethanolamine groups, and NO3 anions in the compounds 3, 4, 5, 6, and 8 were found to be disordered over two positions. Therefore, SIMU, DELU, SADI, and ISOR restraints were used to deal with the disordered moieties in the structures and to obtain reasonable geometric parameters and thermal displacement coefficients. The positions of the hydrogen atoms in the structures were located on difference Fourier maps or calculated geometrically and refined isotropically in the “rigid body” model with U = 1.2Ueq or 1.5Ueq of the corresponding O and C atoms. Some H atoms of disordered solvent molecules (H2O in 8) have not been localized. Crystallographic data and structure refinements of 1–10 are summarized in Table S1,† selected bond distances are given in Table S2.† The figures were drawn using the Mercury program. CCDC reference numbers for the structures are 2239014 (1), 2239016 (2), 2239017 (3), 2239018 (4), 2239019 (5), 2239020 (6), 2239021 (7), 2239022 (8), 2239023 (9), and 2239013 (10).Hirshfeld surface analysis
Hirshfeld surface analysis was performed and fingerprint plots were generated to quantify and decipher the intermolecular interactions in the crystal packing of compounds 1–10 using the CrystalExplorer 21.5 software.19 Atomic coordinates were imported from the CIF files. Colour coding was used in order to indicate intermolecular contacts: red, white, and blue spots show intermolecular contacts with distances less than, equal to, and larger than van der Waals radii, respectively (Fig. S19–S21†). The 2D fingerprint plots were displayed using the expanded 0.6–2.8 Å view, with the scales for the de (outside) and di (inside) distances shown on the axes of the plot (Fig. S22–S24†). Surface area, molecular volume, globularity, and asphericity for 1–10 are summarized in Table S6.† Hirshfeld surface analysis has also been used to evaluate differences that may have arisen in the coordination environment of the metal centers by comparing the corresponding local functions on the metal surface, allowing more accurate conclusions to be drawn about the nature and strength of the coordination bonds.20,21 The Hirshfeld surfaces for the Co and Y/Ln centers in 1–10 are presented in Tables S7–S9† with values for the metal surface volume, metal surface area, globularity, and asphericity.Syntheses of clusters
Compounds 2–10 were obtained in an analogous manner to that of 1.
Results and discussion
Synthesis and preliminary characterization
We have previously reported16 the structural characterization and peculiarities of magnetic behavior of the coordination cluster compound [CoIII2DyIII4(OH)2(ib)8(bdea)2(NO3)4 (H2O)2]·2MeCN, which self-assembles in a high-yield reaction of cobalt(II) isobutyrate with Dy(NO3)3·6H2O and N-butyldiethanolamine (H2bdea) in air. Due to a flat zig-zag metal skeleton, partially suppressed quantum tunneling of the magnetization was observed in this cluster, leading to a significant out-of-phase signal at zero static bias field up to 14 K. The combination of Co with other Y/Ln (Pr, Nd, Sm, Eu, Gd, Tb, Ho, Er, and Yb) cations in the presence of N-butyldiethanolamine (H2bdea) ligand was tested by the similar reaction of Co(ib)2 and yttrium/lanthanide nitrate in acetonitrile solution. The mixtures were refluxed for 30 min, during which time all ingredients dissolved and the color of the reaction mixture changed from pink to deep violet. It was then filtered and the filtrate left undisturbed at room temperature for two-three weeks to slowly concentrate by evaporation. Thus, the treatment of Co(ib)2 with Y/Ln(NO3)3 nH2O and H2bdea yielded crystals of 1–10 in 21–82% yield that were suitable for single crystal X-ray diffraction analysis. Interestingly, the similar synthetic conditions lead to the precipitation of coordination clusters with different structural topologies and nuclearities (Scheme 1). The crystals are unstable and lose crystallinity after filtration due to the loss of some solvent molecules. In these reactions, the initial CoII ions are oxidized to CoIII ions, resulting in homovalence {CoIII–MIII} clusters. Clusters 1–10 were further characterized by IR spectroscopy, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), and ESI mass spectrometry to confirm their identity.The infrared spectra of 1–10 showed strong νas(COO) and νs(COO) bands of the coordinated isobutyrates in the regions of 1569–1557 and 1418–1413 cm−1, respectively. In the range of 2966–2862 cm−1, the asymmetric and symmetric C–H stretching vibrations for the –CH3 and methylene –(CH2)– groups of the ib− and bdea2− ligands are observed, while the bands at 1504–1465 and 1373–1367 cm−1 are attributed to the asymmetric and symmetric C–H bending vibrations, respectively. The strong vibration of the coordinated NO3− anions appears at 1310–1289 cm−1.
The TGA/DSC experiments were performed to determine the thermal stability of clusters 1–10 (Fig. S1–S9†). Tetranuclear {Co2M2} (1–3) compounds show similar thermal behavior and release the remaining solvent acetonitrile molecules, with the desolvated product being thermally stable as a plateau is observed in the temperature range of 60–200 °C (Fig. S1–S3†). On further heating, the decomposition of two coordinated NO3− groups and organic ligands is initiated in several weakly resolved steps with a total weight loss of 59.1% (1), 53.8% (2) and 55.3% (3) up to ca. 410 °C. These processes are accompanied by exothermic peaks at 215, 274, 318 and 357 °C (1), 228, 277, 315 and 361 °C (2), and 214, 275 and 374 °C (3) and an endothermic peak at 80 (1) and 77 °C (3). The pentanuclear {Co2M3} clusters 4 and 5 (Fig. S4 and S5†) and the octanuclear {Co4M4} clusters 9 and 10 (Fig. S8 and S9†) remain stable up to ca. 210 °C, above which they decompose rapidly in several unresolved steps, showing a total weight loss of 58.9% (4), 60.26% (5), 58.8% (9) and 56.9% (10) in the temperature range 210–410 °C. The hexanuclear {Co2Eu4} cluster 7 exhibits thermal stability until ca. 180 °C and upon further heating decomposes in several steps with a total weight loss of 58.7% accompanied by exothermic peaks at 275 and 350 °C (Fig. S6†). Similar to the tetranuclear clusters 1–3, the hexanuclear {Co3Yb3} cluster 8 releases the remaining solvent molecules at the beginning of heating and then starts to decompose in several weakly resolved steps until ca. 450 °C with a total weight loss of 55.81%, accompanied by the endothermic (at 167 and 253 °C) and exothermic (at 207 and 358 °C) processes (Fig. S7†).
ESI-MS in positive and negative modes was used to investigate whether the structural integrity of these coordination clusters was retained in solution and to deduce information about their fragmentation characteristics.16 A summary of the fragments detected in the ESI mass spectra of 1–10 is given in Table S3.† Due to the multi-isotopic nature of naturally occurring lanthanides, the Nd-, Sm-, Gd-, Er-, and Yb-containing species displayed a characteristic isotope distribution pattern, which greatly helps in the identification of their polynuclear species. All species of 100% relative intensity also occur as singly charged cations (except 10) and anions. The mass spectrum of a tetranuclear compound {Co2Y2} (1) shows a base peak corresponding to [Co2Y2(OH)2(ib)4(bdea)2(NO3)]+ cations. A peak of 25% relative intensity was well matched with the dimeric species of type {[Co2Y2(OH)2(ib)4(bdea)2(NO3)]2(NO3)}+, suggesting that dimeric fragments do exist in the MeOH solutions and that for the growth of the assemblies a tetranuclear {Co2Y2} species could attach another tetranuclear species to form octanuclear {Co4Y4} fragments. Similar to 1, in the positive ESI-MS spectrum of 3, the fragment [Co2Er2(OH)2(ib)4(bdea)2(NO3)]+ (100%) and its dimeric fragment {[Co2Er2(OH)2(ib)4(bdea)2(NO3)]2(NO3)}+ (51%) were detected (see Fig. S10c and d† for isotope distribution). In the ESI-MS− of 1 and 3, the peaks with 100% relative intensity correspond to [Co2Y2(OH)2(ib)4(bdea)2(NO3)3]− and [Co2Er2(OH)2(ib)4(bdea)2(NO3)3]− fragments, respectively (Table S3†). The ESI mass pattern of 2, recorded in positive/negative modes, tuned out to be quite complex compared to 1 and 3 and reveals several series of peaks. A difference between adjacent peaks in each series corresponds to Δm/z = 28, which can be identified as the gradual replacement of a negatively charged ib− moiety by an acetate anion. In the main series, the peak of 100% relative intensity is attributed to the [Co2Ho2(OH)2(ib)3(bdea)2(NO3)(MeCO2)]+ fragment ion (Fig. S10a†); additional peaks result from the replacement of ib− by MeCO2− in the fragment with the general formula [Co2Ho2(OH)2(ib)4−x(bdea)2(NO3)(MeCO2)x]+ (x = 0–3), (Fig. S10b and Table S3†). The negative ESI-MS spectrum of 2 shows a series of peaks assignable to [Co2Ho2(OH)2(ib)4−x(bdea)2(NO3)3(MeCO2)x]− (x = 0–3) fragments.
In the ESI-MS+ of the{Co3Pr2} (4) cluster, pentanuclear [Co3Pr2O(ib)4(bdea)3(NO3)2]+ (100%) species were identified (Fig. S10e†). The gradual replacement of a NO3− anion by a negatively charged ib− moiety (Δm/z = 25) results in the appearance of additional peaks in the mass spectrum of 4 (see Table S3†). The presence of a peaks in the high mass region of the spectrum attributable to {[Co3Pr2(OH)(ib)5(bdea)3(NO3)2]2 + (NO3)}+ (22%, Fig. S10f†) and {[Co3Pr2(OH)(ib)6(bdea)3(NO3)2]2 + (ib)}+ fragments, respectively, reveals the assembly process from the pentanuclear {Co3Pr2} to the decanuclear {Co6Pr4} clusters in solution. Two dominant peaks of 100 and 92% relative intensity were observed in ESI-MS− in 4, corresponding to single charged pentanuclear [Co3Pr2O(ib)5(bdea)3(NO3)3]− and [Co3Pr2(OH)(ib)5(bdea)3(NO3)4]− anions, respectively. The pentanuclear {Co3Nd2} fragments {[Co3Nd2(OH)(ib)4(bdea)3(NO3)2] + (MeCO2)}+ and {[Co3Nd2(OH)(ib)5(bdea)3(NO3)(MeO)] + (MeOH)}+ for cluster 5 are observed with low intensity, due to a pure solubility of this cluster. In 6, the molecular peaks for the pentanuclear [Co3Sm2(OH)(ib)5(bdea)3(NO3)2]+ (4%), [Co3Sm2O(ib)4(bdea)3(NO3)2]+ (60%), and [Co3Sm2O3(ib)6(bdea)(MeCN)2]+ (100%) fragments were detected (Table S3†).
The ESI-MS+ spectra of the hexanuclear {Co2Eu4} (7) and {Co3Yb3} (8) clusters showed the appearance of dominant peaks due to {[Co2Eu4(OH)2(ib)9(bdea)2(NO3)3(H2O)2] + 4H2O + (MeCO2H) + H}+ (7) and {[Co3Yb3(OH)4(ib)6(bdea)3(H2O)3] + (MeO)}+ (8) species (see Fig. S11† for isotopic distribution). This indicates that the neutral [Co2Eu4(OH)2(ib)8(bdea)2(NO3)4(H2O)2] and cationic [Co3Yb3(OH)4(ib)6(bdea)3(H2O)3]2+ clusters appear to be sufficiently stable in solution. Similar to the pentanuclear cluster 4, the gradual replacement of a NO3− anion by a negatively charged ib− moiety (Δm/z = 25) results in the appearance of additional peaks in 7 (for more details see Table S3†). In case 8, the abovementioned cationic fragment eliminates an ib− moiety and additionally adds a NO3− ion, as indicated by a peak with m/z at 1826 assignable to {[Co3Yb3(OH)4(ib)5(bdea)3(NO3)(H2O)3] + (MeO)}+ (39%), and further the latter fragment loses one H2O to give {[Co3Yb3(OH)4(ib)5(bdea)3(NO3)(H2O)2] + (MeO)}+ (34%). In the higher mass region, the dodecanuclear species {[Co3Yb3O2(OH)2(ib)5(bdea)2.5(Hbdea)(NO3)(H2O)]2 + K}+ (15%) were found (see Fig. S11d† for isotopic distribution). The negative ion mode spectrum of 8 shows the presence of anions corresponding to hexanuclear {[Co3Yb3O3(ib)6(bdea)3(NO3)(MeO)(H2O)4] + K}− (100%) and dodecanuclear {[Co6Yb6O6(ib)12(bdea)6(MeO)2(H2O)8] + 2K + (NO3)}− (29%) species contaminated with potassium cations, respectively. The positive ESI-MS spectrum of 10 shows the protonated doubly charged species {[Co4Tb4O2(OH)2(ib)7(bdea)4(NO3)(MeO)(H2O)3] + H}2+, and the protonated singly charged species {[Co4Tb4O2(ib)8(bdea)4((NO3)2(MeO)2] + H}+ (see Fig. S11e and f†).
The experimental powder X-ray diffractograms for compounds 1–10 were compared with the modeling derived from the X-ray data set for single crystals (see Fig. S12†). The results indicate that the single crystals examined are representative of the microcrystalline bulk materials. Finally, the purity of the crystalline products was further tested by energy dispersive X-ray fluorescence spectroscopy, which yielded Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M ratio of 49.5:50.5 (1), 48.9:51.1 (2), 52.6:47.4 (3), 50.1:49.9 (8) in accordance with the theoretical value of 50:50; and 56.2:43.8 (4); 63.6:36.4 (5), 59.5:40.5 (6) in agreement with the theoretical value of 60:40, as expected for the crystal structures.
:M ratio of 49.5:50.5 (1), 48.9:51.1 (2), 52.6:47.4 (3), 50.1:49.9 (8) in accordance with the theoretical value of 50:50; and 56.2:43.8 (4); 63.6:36.4 (5), 59.5:40.5 (6) in agreement with the theoretical value of 60:40, as expected for the crystal structures.
Crystal structures
We have divided the heterometallic Co–Y/Ln families of the clusters into four groups according to the total number of metals in the core: tetranuclear clusters involving structures 1, 2, and 3 with a {CoIII2MIII2} core, pentanuclear clusters comprising structures 4, 5, and 6 with a {CoIII3MIII2} core, hexanuclear clusters with {CoIII2MIII4} (7) and {CoIII3MIII3} (8) cores, and octanuclear clusters with a {CoIII4MIII4} core, structures 9 and 10. Charge balance considerations, as well as the coordination geometry of the cobalt atoms in all reported structures, establish their oxidation state as +3. This is also confirmed by bond valence sum calculations (Table S4†).22 | ||
| Fig. 1 (a) Structure of [Co2Y2(OH)2(ib)4(bdea)2(NO3)2] cluster and view of {Co2Y2(OH)2} core in 1. Hydrogen atoms connected to carbon atoms and solvent molecules are omitted for clarity. (b) View of the packing clusters in 1. Metal atoms are shown as polyhedra and solvent acetonitrile molecules are shown in a space-filling mode. | ||
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group of the triclinic system (Fig. 2 and S14†). The asymmetric unit contains a heterometallic pentanuclear [Co3M2(OH)(ib)5(bdea)3(NO3)3] cluster with a novel {CoIII3MIII2} core topology (Fig. 2b) and solvent MeCN molecules. The structure of compound 6 is shown in Fig. 2 as a representative example. The cluster core in 4–6 can be described as a heterometallic {CoIIILnIII2(OH)} unit decorated with an additional {CoIII2} unit. The metal atoms in the {CoIIILnIII2} triangle are additionally linked by two ib− and three oxygen atoms of two doubly deprotonated bridging bdea2− ligands with Co1⋯Sm1, Co1⋯Sm2, and Sm1⋯Sm2 separations of 3.324(2), 5.008(1) and 4.221(1) Å, respectively. The {Co2} moiety binds to the trinuclear {CoSm2(OH)} moiety through two bridging ib− ligands and two O-bridges from two bdea2− units. The Sm1⋯Co3 and Sm2⋯Co2 distances are 3.351(2) and 3.791(2) Å, respectively. The ib− and O atoms of two bdea2− additionally bridge two Co atoms in the dimeric unit with a Co1⋯Co2 distance of 2.829(2) Å. The corresponding distances in 4 and 5 are given in Table S2.† The core {Co3Ln2} can also be viewed as three {CoLn2(μ3-OH)}, {CoLn2(μ3-Obdea)} and {Co2Ln(μ3-Obdea)} noncoplanar triangles capped by OH− anion and Obdea atoms, which in turn share the two common LnIII vertices and the common LnIII and CoIII vertices, respectively, thus all having a common LnIII atom (Fig. 2a). The dihedral angles between the planes of neighbouring triangles are 20.91 and 67.20° in 5, 21.22 and 67.02° in 6, and 21.56 and 66.85° in 7. The displacements of the OH− anion and Obdea atoms from the plane of the corresponding triangle are 0.875, 0.793, and 0.963 Å in 4, 0.871, 0.801, and 0.978 Å in 5, and 0.849, 0.805, and 0.966 Å in 6.
space group of the triclinic system (Fig. 2 and S14†). The asymmetric unit contains a heterometallic pentanuclear [Co3M2(OH)(ib)5(bdea)3(NO3)3] cluster with a novel {CoIII3MIII2} core topology (Fig. 2b) and solvent MeCN molecules. The structure of compound 6 is shown in Fig. 2 as a representative example. The cluster core in 4–6 can be described as a heterometallic {CoIIILnIII2(OH)} unit decorated with an additional {CoIII2} unit. The metal atoms in the {CoIIILnIII2} triangle are additionally linked by two ib− and three oxygen atoms of two doubly deprotonated bridging bdea2− ligands with Co1⋯Sm1, Co1⋯Sm2, and Sm1⋯Sm2 separations of 3.324(2), 5.008(1) and 4.221(1) Å, respectively. The {Co2} moiety binds to the trinuclear {CoSm2(OH)} moiety through two bridging ib− ligands and two O-bridges from two bdea2− units. The Sm1⋯Co3 and Sm2⋯Co2 distances are 3.351(2) and 3.791(2) Å, respectively. The ib− and O atoms of two bdea2− additionally bridge two Co atoms in the dimeric unit with a Co1⋯Co2 distance of 2.829(2) Å. The corresponding distances in 4 and 5 are given in Table S2.† The core {Co3Ln2} can also be viewed as three {CoLn2(μ3-OH)}, {CoLn2(μ3-Obdea)} and {Co2Ln(μ3-Obdea)} noncoplanar triangles capped by OH− anion and Obdea atoms, which in turn share the two common LnIII vertices and the common LnIII and CoIII vertices, respectively, thus all having a common LnIII atom (Fig. 2a). The dihedral angles between the planes of neighbouring triangles are 20.91 and 67.20° in 5, 21.22 and 67.02° in 6, and 21.56 and 66.85° in 7. The displacements of the OH− anion and Obdea atoms from the plane of the corresponding triangle are 0.875, 0.793, and 0.963 Å in 4, 0.871, 0.801, and 0.978 Å in 5, and 0.849, 0.805, and 0.966 Å in 6.
 | ||
| Fig. 2 (a) Structure of the [Co3Sm2(OH)(ib)5(bdea)3(NO3)3] cluster and view of a core {Co3Sm2(OH)(Obdea)2} in 6. Hydrogen atoms on carbon atoms and solvent molecules are omitted for clarity. (b) View of packing clusters illustrating that aliphatic fragments dominate in inter-cluster space. Metal atoms are shown as polyhedra and solvent acetonitrile molecules in a space-filling mode. | ||
All Co atoms have a distorted octahedral NO5 geometry (OC-6), which is confirmed by SHAPE analysis23 with a deviation value of 0.458 for Co1, 0.402 for Co2 and 0.472 for Co3 (Table S5†). Co3 is surrounded by μ3-OH, two O atoms from different ib− and two O and one N atoms from a doubly deprotonated bdea2− ligand, whereas both Co1 and Co2 are coordinated by two O atoms from different ib−, two O and one N atoms from bdea2− ligand and one O atom from another bdea2− ligand. The Co–O bond distances vary from 1.867(3) to 1.961(3) Å and the Co–N bond distances are all equal within two standard deviations [1.965(4)–1.979(4) Å]. In 4 and 5, all Co atoms also have octahedral geometries, with the values for these atoms listed in Table S5.† The Sm1 and Sm2 atoms are both nine-coordinated with an O9 surrounding formed by a μ3-OH group, two O atoms from two carboxylates, four (Sm1) or two (Sm2) O atoms from bdea2− ligands, and two (Sm1) or four (Sm2) O atoms from nitrate anions. According to SHAPE analysis,23 the geometry of the Sm1 atom adopts a spherical capped square antiprism polyhedron (CSAPR-9) with a deviation value of 2.234, and the geometry of the Sm2 atom can best be described as intermediate between a muffin pyramid (MFF-9) with a deviation value of 2.725 and a spherical capped square antiprism (CSAPR-9) with a deviation value of 2.832 (Table S5†). The Sm–O bond distances are in the range of 2.282(3)–2.820(3) Å and they are in good agreement with those found in the literature. Similar to 6, the Ln metal centers in clusters 4 (Pr) and 5 (Nd) are also nine-coordinated with one lanthanide site possessing a muffin (MFF-9) or spherical capped square antiprism (CSAPR-9) geometry and another one occupying a spherical capped square antiprism (CSAPR-9) geometry with the values listed in Table S5.† The Pr–O bond distances are in the range of 2.305(8)–2.816(8) Å, and Nd–O, 2.327(6)–2.828(6) Å (Table S2†).
space group of the triclinic system. The structure of the hexanuclear cluster 7 is similar and isomorphic to the [Co2Dy4(OH)2(ib)8(bdea)2(NO3)4(H2O)2] cluster reported in ref. 16 as an acetonitrile adduct. The asymmetric unit of the Ci-symmetric cluster 7 contains Co1, Eu1, and Eu2 metal centers, one μ3-OH− group, four ib−, one bdea2− moiety, two NO3− anions, and one coordinated H2O molecule (Fig. 3 and S15†). The cluster core of 7 can be viewed as two parallel metal atoms triangular of {CoIIIEuIII2(μ3-OH)} units, with the distance between their planes only 0.320 Å, bridged by four ib− ligands, two of which use a μ2-η1:η1 coordination mode and the other two a μ3-η1:η2:η1 coordination mode. Peripheral ligation of metal atoms is provided by a doubly deprotonated bdea2− ligand and three ib− residues. The O9 coordination environment in the form of a triangular dodecahedron (TDD-8, the deviation value of 1.564, Table S5†) around the Eu1 center contains one OH− anion, two O atoms belonging to two ib− moieties, one O atom of a doubly protonated bdea2− ligand, four O atoms of two NO3− ions and one H2O molecule. The Eu2 atom is represented in a spherical capped square antiprismatic O8 environment (CSAPR-9, the deviation value of 1.559, Table S5†) of an O atom from the OH− group, O atom from bdea2− and six O atoms belonging to five ib− groups. The Eu–O bond distances are in the range of 2.271(5)–2.643(5) Å. In 7, the polyhedra of Eu atoms share common O⋯O edges, resulting in an elongated Z-shape of the Eu4 core (Fig. S15b†) and metal separations of 4.051(1) Å for Eu1⋯Eu2 and 3.887(1) Å for Eu2⋯Eu2 (−x,−y + 2,−z). Four Eu ions in 7 are coplanar, but the two flanked Co ions are displaced from their common plane in opposite directions by 0.237(1) Å. Co1 has an octahedral NO5 environment comprising one hydroxy O atom, two O atoms from different ib− ligands, and one N atom and two O atoms from the bdea2− residue. The Co–O [1.870(5)–1.919(5) Å] and Co–N [1.965(6) Å] bond distances are in good agreement with those reported for the related {Co2Dy4} cluster.14 In the triangular {CoIIIEuIII2(μ3-OH)} unit, the OH− anion is displaced from the plane of the metal atoms at 0.921 Å and the Co1⋯Eu1 and Co1⋯Eu2 distances equal 3.331(2) and 3.285(2) Å, respectively (Table S2†). The coordinated water molecules (O18) are involved in intramolecular H-bonds (2.728(7) Å) with oxygen atoms (O7 [−x, −y + 2, −z]) of the isobutyrate and in intermolecular H-bonds with oxygen atoms of the isobutyrate (O3 [−x + 1, −y + 2, −z], 2.785(7) Å) belonging to the neighboring cluster. Finally, the OH− group (O1) participates in weak inter-molecular H-bonds (2.995(8) Å) with oxygen atoms (O17 [−x + 1, −y + 2, −z]) of the NO3− ions forming a supramolecular zig-zag chain as shown in Fig. 3c, which are packing in parallel mode via lipophilic interactions, Fig. 3b.
 | ||
| Fig. 3 (a) Structure of the [Co2Eu4(OH)2(ib)8(bdea)2(NO3)4(H2O)2] cluster in 7. Hydrogen atoms and on carbon atoms are omitted for clarity. (b) Packing of chains in 7, viewed along the a axis. (c) H-bonded chain running along the a axis in 7. Hydrogen bonds are shown as dotted blue lines. The coordinated water molecules are highlighted as red balls. | ||
Single crystal X-ray analysis reveals that [Co3Yb3(OH)4(ib)6(bdea)3(H2O)3](NO3)2·3MeCN·1.5H2O (8) crystallizes in the P21/c space group of the monoclinic system. The asymmetric unit of 8 contains a cationic [Co3Yb3(OH)4(ib)6(bdea)3(H2O)3]2+ cluster, two NO3− outer sphere anions, and three MeCN and one and half H2O solvent molecules (Fig. 4 and S16†). The cluster core of 8 is composed of three {CoYb2(μ3-OH)} noncoplanar triangles capped by OH− anions and sharing the common Yb vertices. The dihedral angles between the planes of the neighbouring triangles are 51.01, 34.99, and 50.50°. The displacements of O2, O3, and O4 atoms of OH− anions from the plane of the corresponding CoYb2 triangle are 0.900, 0.932 and 0.920 Å. Three Yb atoms in the centre of the core also form triangles capped by μ3-hydroxy group, which is displaced from the plane of this triangle by 0.777 Å. The atom Co1 and the atoms Co2 and Co3 are located on opposite sides of the plane of three Yb atoms and are displaced from it by 2.255, 0.870 and 0.954 Å, respectively. Corresponding CoYb2 triangles form dihedral angles of 58.93, 19.25, and 21.35° with the Yb3 plane. Three CoIII and three YbIII atoms bridged by four μ3-OH groups and six ib− moieties, and additionally linked by three fully deprotonated bdea2− ligands. Thus, the arrangement of metal atoms in 8 can also be viewed as two nearly equilateral triangles: an intersection of a smaller triangle defined by three Yb sites with Yb⋯Yb distances of 3.817(2)–3.828(9) Å and a larger triangle formed by Co sites [Co⋯Co, 6.126(2)–6.175(2) Å].
 | ||
| Fig. 4 (a) Structure of a cationic [Co3Yb3(OH)4(ib)6(bdea)3(H2O)3]2+ cluster in 8. Color codes: C, grey; O, red; N, blue sticks; Co, violet; Yb, green spheres. The disordered and hydrogen atoms, NO3− anions, and solvent molecules are omitted for clarity. (b) Schematic top and side views of a {Co3Yb3} core in 8. (c) Intersection of metal core triangles. (d) Fragment of packing illustrating the formation of H-bonded chain along the c axis between alternating charged components in 8. Metal atoms are shown as polyhedra and solvent MeCN molecules in a space-filling mode. | ||
The dihedral angle between the Yb3 and Co3 triangular planes is equal to 36.7°. The “triangle-in-triangle” structure of 8 resembles the pivalate clusters with a similar {Co3Dy3} or {Co3Gd3} core.27–29 All of CoIII atoms in this structure are six-coordinated and adopt a distorted octahedral NO5 geometry (OC-6) formed by an O atom of μ3-OH group, two O atoms from two izobutyrates, two O atoms and a N atom of the bdea2− ligand. The deviation values from the ideal polyhedron equal to 0.252 (Co1), 0.263 (Co2), and 0.224 (Co3) as listed in Table S5.† The Co–O and Co–N bond distances are in the range of 1.868(6)–1.929(7) and 1.964(9)–1.975(8) Å, respectively (Table S2†), which are in good agreement with the values reported for the related {Co3Ln3} (Ln = Gd, Dy) clusters.27–29 Each of the YbIII atoms is eight-coordinated, and its O8 environment is completed by three OH groups, two O atoms from different ib− moieties and two O atoms from two bdea2− ligands. A H2O molecule completes the coordination sphere of the YbIII atoms. The coordination geometry around the Yb1 and Yb2 atoms is best described as a distorted square-antiprismatic (SARP-8) with a deviation value of 1.027 and 0.657, respectively, whereas the Yb3 atom adopts a triangular dodecahedron (TDD-8) with a deviation value of 0.971 from the ideal geometry (Table S5†). The Yb–O bond distances range from 2.255(6) to 2.380(7) Å. The coordinated water molecules (O23, O24, O25) and hydroxyl groups (O1, O2, O3, O4) are involved in intermolecular H-bonds with NO3− ions and solvent molecules. One of the symmetry independent NO3− ions bridges the cationic clusters by H-bonds, resulting in a supramolecular linear rod along the c axis. The crystal packing in 8 is provided by lipophilic interactions of parallel rods (Fig. 4c).
 | ||
| Fig. 5 (a) Structure of the [Co4Tb4(OH)4(ib)8(bdea)4(NO3)4] cluster in 10. Color codes: C, grey; O, red; N, blue sticks; Co, violet; Tb, cyan spheres. The disordered atoms are omitted for clarity. (b) Schematic view of a metal core build-up from four {CoTb2(μ3-OH)} units. (c) Planar “square-in-square” topology of the {Co4Tb4} core. (d) The packing of the clusters in 10. | ||
Hirshfeld surface analysis
Intermolecular interactions between neighboring clusters units in the solid state can significantly affect their magnetic properties.31 The Hirshfeld surface (HS) analysis helps to identify all the intermolecular interactions of a cluster unit with its neighbors present in the crystal and can help in other studies such as theoretical calculations.32 The intermolecular interactions in 1–10 were further investigated and visualized by HS analysis. To view characteristic red sports indicating specific contact points in crystals 1–10, the Hirshfeld surfaces mapped over dnorm were calculated (Fig. S19–S21†) and representative examples are shown in Fig. 6. The two-dimensional (2D) fingerprint plots (Fig. S22–S24†) quantitatively revealed the contribution of these interactions in the crystal structures, and Fig. 7 shows the relative contributions to the Hirshfeld surface area for each type of intermolecular contact in these compounds. The most prominent type of contacts in 1–10 corresponds to H⋯H contacts, which account from 65.1% to 82.4% of the total surface contacts. The second most frequent interactions on the surface with a percentage from 13.6% to 31.5%, correspond to O⋯H/H⋯O contacts, which refer to O–H⋯N, O–H⋯O and C–H⋯O interactions. The percentage contributions of particular interatomic contacts to the HSs indicated that H⋯H and O⋯H contacts play a crucial role in the stabilization of crystal systems of 1–10. The other types of interactions for the whole structures of 1–10 are C⋯H/H⋯C (with a contribution from 0.6% to 2.3%) contact points to the whole crystal packing. For clusters 1–6 and 8, where there are lattice solvents in the structures, the distribution of contact points N⋯H/H⋯N (3.0–6.2%) is asymmetric with respect to the main diagonal in the 2D fingerprint plots (Fig. S22–S24†), and the contact points are mainly between the clusters and the acetonitrile molecules. For cluster 7 without solvent molecules, the contribution of N⋯H/H⋯N is 0.9% and corresponds to the interactions between the coordinated NO3− and OH− groups. | ||
| Fig. 6 View of the Hirshfeld surface mapped over dnorm for 1, 5, 7, 8, and 9, showing the points of contacts derived from different interactions. | ||
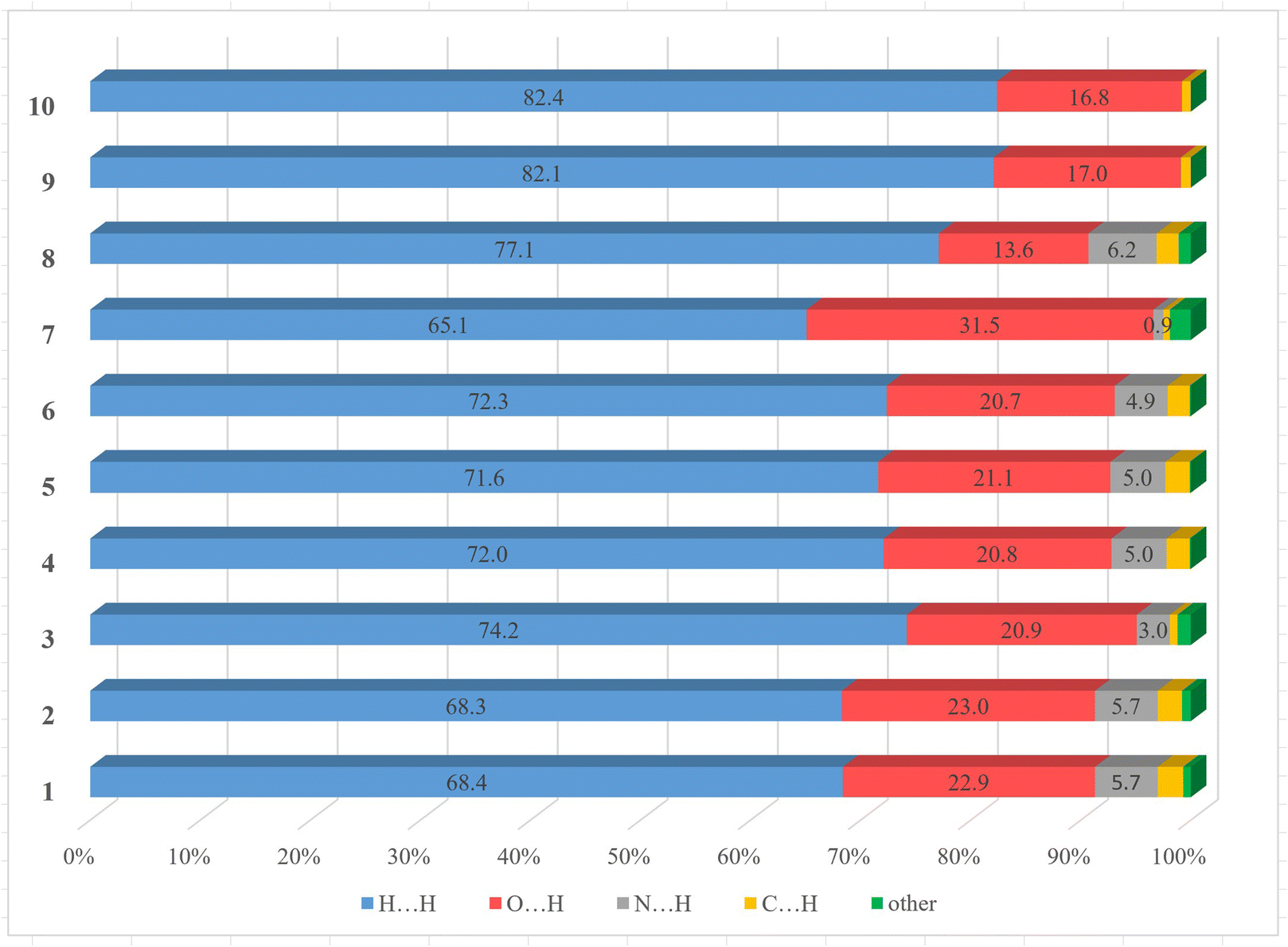 | ||
| Fig. 7 Relative contributions to the Hirshfeld surface for the major intermolecular contacts in 1–10. | ||
In order to evaluate differences that may have arisen in the coordination environment of the metal centers by comparing the corresponding local functions on the metal surface, the Hirshfeld surfaces for the Co and Y/Ln centers in 1–10 were calculated. The Hirshfeld surfaces for metal centers shown in Tables S7–S8† with the shape index function reflect the direction and relatively strong metal–ligand coordination bonds as red regions perpendicular to the bonds. In particular, the metal surfaces of CoIII with the shortest coordination bonds are characterized by an almost cubic shape in 1–10 (Table S7†), as the coordination bonds through the series do not significantly differ in nature and strength, in all cases there is a NO5 environment around CoIII (Co–O, 1.864(7)–1.978(6) Å; Co–N, 1.964(9)–1.999(6) Å). This is also accompanied by a similarity in globularity (0.854–0.882), indicating a degree of deviation from the sphere (<1), while the asphericity, which is a measure of anisotropy, is found to be 0.001–0.009. Y/LnIII centers have more distorted metal surfaces than CoIII centers, with globularity ranging from 0.718 to 0.839 and asphericity ranging from 0.046 to 0.257 (Table S8†). The Hirshfeld surfaces for Y/Ln also have the largest volume and area, resulting from the generally longer coordination distances (Y/Ln–O, 2.238(3)–2.828(8) Å) compared to CoIII.
Magnetic properties
Due to the presence of low spin CoIII centers in all compounds, which are well known for small temperature independent paramagnetic (TIP) contributions of the Van-Vleck type, i.e. caused by localized electrons, we analyzed the magnetic data of 1 to determine their magnitude. The result is χm,TIP = +1.30 × 10−4 cm3 mol−1 per CoIII center (Fig. S25†). Taking into account the TIP besides the usual diamagnetic contributions, the remaining data characteristic for each compound (2–5 and 8) with paramagnetic lanthanide centers are shown in Fig. 8. At 290 K and at 0.1 as well as 1.0 T, the χmT values are 27.4 (2), 21.6 (3), 3.05 (4), 3.06 (5) and 7.29 cm3 K mol−1 (8). These values are in or just below the expected ranges33 for the two or three, respectively, non-interacting Ln3+ centers in each compound: 26.5–27.6 (2), 22.1–22.6 (3), 2.9–3.2 (4), 2.9–3.1 (5) and ca. 7.6 cm3 K mol−1 (8). Upon cooling the compounds, the χmT values continuously decrease, and drop of at 100 K (2), 50 K (3) and 25 K (4), respectively. At 2.0 K, the χmT values are 9.36 (2), 8.71 (3), 0.16 (4), 1.42 (5) and 4.17 cm3 K mol−1 (8) at 0.1 T, and 6.64 (2), 5.98 (3), 0.15 (4), 1.38 (5) and 3.45 cm3 K mol−1 (8) at 1.0 T. The characteristic decrease of χmT for lanthanides (all but 4f7 centers) are due to the thermal depopulation of the mJ energy states that are basically split due to spin–orbit coupling, electron–electron interrepulsion and the ligand field. The difference between the data sets at 0.1 T and 1.0 T are due to the Zeeman effect that is almost negligible at 0.1 T. The molar magnetizations Mm vs. the applied magnetic field B at 2.0 K are shown in Fig. 8b. For 2, 3, 5 and 8, Mm linearly increases for magnetic fields up to 1 T, and increases at higher field with distinctly lesser slope without reaching saturation, while for 4 Mm is almost linear up to 5.0 T. At 2.0 K and 5.0 T the values of Mm are 8.9 (2), 8.3 (3), 0.7 (4), 2.8 (5) and 5.3 NAμB (8), which are well below the saturation values33 of 20 (2), 18 (3), 6.4 (4), 6.5 (5) and 12 NAμB (8). The observed deviations from the saturation values at 5.0 T are due to the magnetically anisotropic nature of the Ln3+ centers and that the data were taken from a powder sample, i.e. the data represent the mean values of statistically oriented, anisotropic magnetic moments resulting in smaller values. In addition, note that very weak Heisenberg exchange interactions between the lanthanide centers, particularly of antiferromagnetic nature, cannot be excluded from the χmT vs. T or Mm vs. B data without further analyses.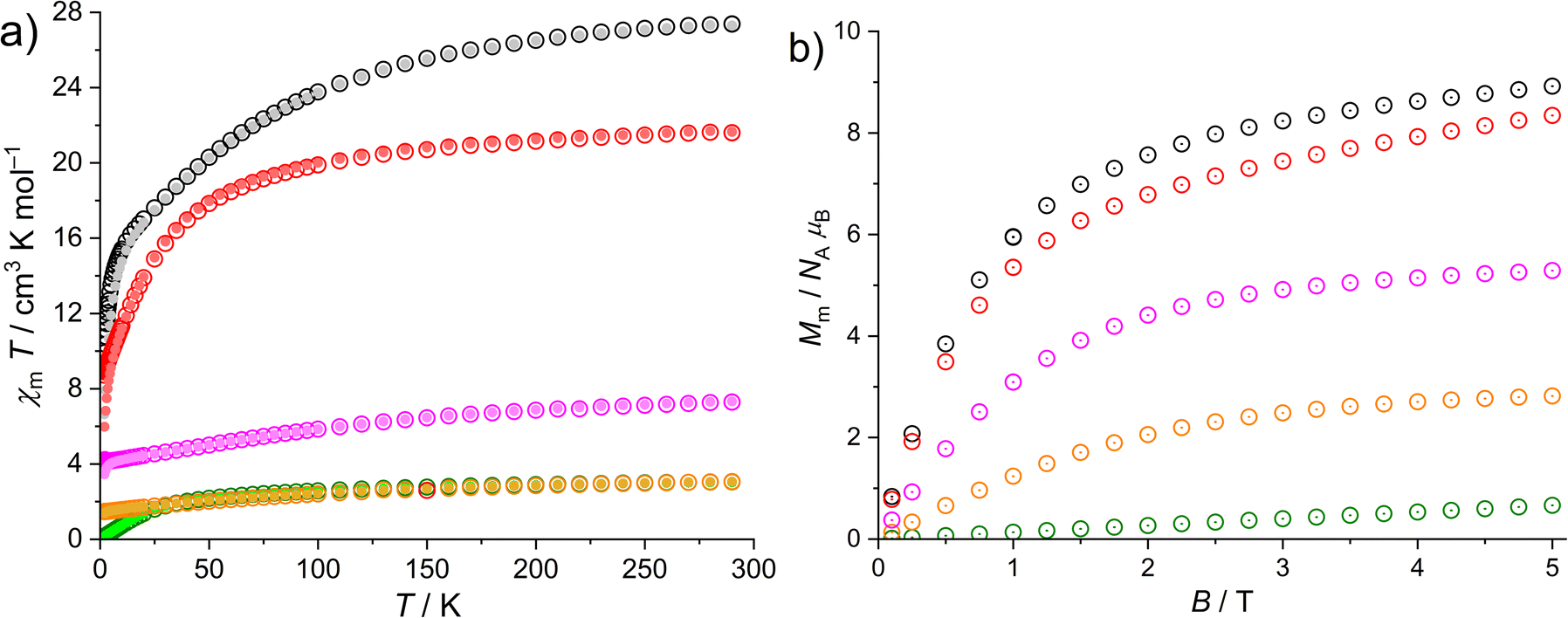 | ||
| Fig. 8 Temperature dependence of χmT at 0.1 T (open symbols) and 1.0 T (full circles) of 2–5 and 8 (a); molar magnetization Mm vs. applied field B at 2.0 K (b); color codes: black (2), red (3), green (4), orange (5), magenta (8). | ||
Regarding ac magnetic susceptibility measurements, only compound 3 showed relevant out-of-phase signals although only below 4 K and with application of a static magnetic bias field. The best results were observed at a bias field of 1000 Oe (Fig. S26†). A generalized Debye expression34 was simultaneously fitted to the in-phase magnetic susceptibility  vs. frequency f and out-of-phase magnetic susceptibility
vs. frequency f and out-of-phase magnetic susceptibility  vs. f data. The fits yield the solid lines shown in Fig. S26a, c and d† and the relaxation times τ shown in Fig. S26b.† The corresponding distribution of relaxation times α = 0.172 ± 0.022 suggests the presence of few relaxation pathways. Therefore, the best reproduction of the τ vs. 1/T data was found by consideration of a direct relaxation process induced by applying the static bias field and a Raman relaxation. Such relaxation processes are described by the formula τ−1 = AT + CTn. Fitting this formula to the data yields the constant A = (4.63 ± 0.35) × 103 s−1 K−1 for the direct process as well as C = (1.23 ± 2.39) s−1 K−n and n = 9.61 ± 1.92 for the Raman process. Since the error margins of the Raman parameters are rather large, they could indicate either the well-known T7 dependency for Kramers ions in a Raman process by spin–two-phonon interaction or a T9 dependency in a process by spin-one-phonon interaction in second order.35
vs. f data. The fits yield the solid lines shown in Fig. S26a, c and d† and the relaxation times τ shown in Fig. S26b.† The corresponding distribution of relaxation times α = 0.172 ± 0.022 suggests the presence of few relaxation pathways. Therefore, the best reproduction of the τ vs. 1/T data was found by consideration of a direct relaxation process induced by applying the static bias field and a Raman relaxation. Such relaxation processes are described by the formula τ−1 = AT + CTn. Fitting this formula to the data yields the constant A = (4.63 ± 0.35) × 103 s−1 K−1 for the direct process as well as C = (1.23 ± 2.39) s−1 K−n and n = 9.61 ± 1.92 for the Raman process. Since the error margins of the Raman parameters are rather large, they could indicate either the well-known T7 dependency for Kramers ions in a Raman process by spin–two-phonon interaction or a T9 dependency in a process by spin-one-phonon interaction in second order.35
The TB2J Python package, based on the Green's function method, was used to estimate the magnetic interactions in the prepared compounds, as described in the ESI.† As input, the package used the output of the Siesta code, which provided the calculation of the magnetic anisotropy energy of compounds 2–10.
Conclusions
The synthesis, crystal structures and an investigation of the magnetic behavior of a series of Co–Y/Ln polynuclear clusters afforded by isobutyrate and N-butyldiethanolamine ligands and Y (1), Ho (2), Er (3), Pr (4), Nd (5), Sm (6), Eu (7), Yb (8), Gd (9), and Tb (10) as the yttrium/lanthanide ions are presented. The preference from a lower to a higher nuclear structure was only partly observed on going across the yttrium/lanthanide series brought about by the variation in the size of the YIII/LnIII ions. Despite the large structural diversity and different topologies of the metal cores in 1–10, all these compounds contain {CoIIIYIII/LnIII2(μ3-OH)} building units. In addition to the μ3-OH bridge, the Co and YIII/LnIII atoms in this building unit are linked by the carboxylic group of the ib− ligands coordinated in a μ2-η1:η1 mode and by an oxygen atom of the bdea2− ligand coordinated in a μ2-η1 fashion. The {CoIIIYIII/LnIII2(μ3-OH)} building unit shares common Y/Ln atoms with a neighbouring one in 1–3, two neighbouring units in 8–10, and two such units are separated in 7. In clusters 4–6, this unit shares two Ln atoms with a {CoIIILnIII2(μ3-Obdea)} fragment, and the latter shares Co and Ln atoms with {CoIII2LnIII(μ3-Obdea)} one. The latter two fragments differ in their connectivity from the common to all compound building units.Note, the CSD survey revealed eight clusters with a tetranuclear {CoIII2LnIII2} (Ln = Tb, Dy, Ho, Er, and Yb) metal core containing the {CoIIILnIII2(μ3-OH)} building units with the same connectivity of Co and Ln atoms as in 1–10, and no one with {CoIIIYIII2(μ3-OH)} building units. Clusters with a pentanuclear {CoIII3LnIII2} metal core and the above-mentioned building unit have not been found. For hexanuclear {CoIII2LnIII4}-containing clusters, only one compound with a {CoIII2DyIII4} core and the {CoIIIDyIII2(μ3-OH)} building units has recently been published by us.14 Four compounds have been found for hexanuclear {Co3IIILnIII3} clusters with Ln = Gd and Dy.27–29 Only one octanuclear cluster with a {CoIII4DyIII4} core comprising {CoIIILnIII2(μ3-OH)} building units has been found in ref. 36. However, in this compound the metal core is essentially non-planar as in the above-mentioned compounds with a {Mn4Ln4} core30 in contrast to the reported herein compounds 9 and 10 with a planar metal core.
The dc magnetic data that were taken of 1, 2–5 and 8 are in agreement with the expectation of non-interacting or very weakly interacting lanthanide centers. AC magnetic measurements revealed relevant out-of-phase signals only for 3 indicating direct and Raman relaxation processes below 4 K at a static magnetic bias field of 1000 Oe.
The new DFT-TB2J approach has been applied to estimate the magnetic anisotropy energy (MAE) of compounds 2–10 and to calculate the exchange coupling between lanthanides in these compounds. Large values of MAE have been found for the majority of the compounds studied, the magnitudes of which, as well as the magnetic exchange interactions, depend significantly on the nature of the lanthanides.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1–10 has been deposited at the CCDC under 2239014 (1), 2239016 (2), 2239017 (3), 2239018 (4), 2239019 (5), 2239020 (6), 2239021 (7), 2239022 (8), 2239023 (9), and 2239013 (10) accessing numbers, and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Author contributions
DS: chemical synthesis and growth of crystals, writing of the paper, analysis of the data; JvL: analysis of the data, generation of images, writing of the paper; VChK: data collection and refinement, analysis of the data, generation of images, writing of the paper; YCh: formal analysis of the data; PK: conceptualization, analysis of the data, generation of images, writing of the paper; SGB: conceptualization, analysis of the data, generation of images, writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the subprogram 011202 financed by the Ministry of Education and Research of R. Moldova and the bilateral project “MultiClust” (01DK25013 and 25.80013.5007.07GER) supported by the German Federal Ministry of Education and Research (BMBF) and the Moldovan National Agency for Research and Development (NARD). The authors thank the National Initiatives for Open Science in Europe for granting ‘MAGNANO’. We acknowledge the provided access to the e-infrastructure purchased under the National Roadmap for RI, financially coordinated by the MES of the Republic of Bulgaria (grant no. D01-325/01.12.2023). D. S. thanks the World Federation of Scientists for the excellence scholarship.Notes and references
- G. E. Kostakis, S. P. Perlepes, V. A. Blatov, D. M. Proserpio and A. K. Powell, Coord. Chem. Rev., 2012, 256, 1246 CrossRef CAS.
- M. Murrie, Chem. Soc. Rev., 2010, 39, 1986 RSC.
- E. C. Yang, D. N. Hendrickson, W. Wernsdorfer, M. Nakano, L. N. Zakharov, R. D. Sommer, A. L. Rheingold, M. Ledezma-Gairaud and G. Christou, J. Appl. Phys., 2002, 91, 7382 CrossRef CAS.
- K. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2015, 289–290, 74 Search PubMed.
- J.-B. Peng, Q.-C. Zhang, X.-L. Kong, Y.-Z. Zeng, Y.-P. Ren, L.-S. Long, R. B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2012, 134, 3314 CrossRef CAS PubMed.
- J.-L. Liu, Y.-C. Chen, F.-S. Guo and M.-L. Tong, Coord. Chem. Rev., 2014, 281, 26 Search PubMed.
- C. Papatriantafyllopoulou, E. E. Moushi, G. Christou and A. J. Tasiopoulos, Chem. Soc. Rev., 2016, 45, 1597 Search PubMed.
- F.-S. Guo, Y.-C. Chen, J.-L. Liu, J.-D. Leng, Z.-S. Meng, P. Vrabel, M. Orendac and M.-L. Tong, Chem. Commun., 2012, 48, 12219 Search PubMed.
- Z.-M. Zhang, L.-Y. Pan, W.-Q. Lin, J.-D. Leng, F.-S. Guo, Y.-C. Chen, J.-L. Liu and M.-L. Tong, Chem. Commun., 2013, 49, 8081 Search PubMed.
- Y.-Z. Zheng, M. Evangelisti, F. Tuna and R. E. P. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057 CrossRef CAS PubMed.
- E. M. Pineda, F. Tuna, R. C. Pritchard, A. C. Regan, R. E. P. Winpenny and E. J. McInnes, Chem. Commun., 2013, 49, 3522 RSC.
- B. Berkoff, K. Griffiths, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, A. Escuer and G. E. Kostakis, Dalton Trans., 2015, 44, 12788 RSC.
- E. Loukopoulos, B. Berkoff, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, A. Escuer and G. E. Kostakis, Eur. J. Inorg. Chem., 2015, 2646 CrossRef CAS.
- J. W. Sharples and D. Collison, Coord. Chem. Rev., 2014, 260, 1 CrossRef CAS PubMed.
- S. G. Baca, S. Herringer, Sh.-X. Liu and S. Decurtins, Polynuclear Clusters Based on Fe/Fe-Ln Carboxylates with Selected Magnetic Properties, in Comprehensive Coordination Chemistry, ed. E. C. Constable, G. Parkin and L. Que Jr, 3rd edn, 2021, pp. 29–65 Search PubMed.
- D. Stati, J. van Leusen, N. Ahmed, V. Ch. Kravtsov, P. Kögerler and S. G. Baca, Cryst. Growth Des., 2023, 23, 395 CrossRef CAS.
- I. L. Malaestean, M. Speldrich, A. Ellern, S. G. Baca and P. Kogerler, Polyhedron, 2010, 29, 1990 CrossRef CAS.
- M. A. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef CAS.
- C. B. Pinto, L. H. R. Dos Santos and B. L. Rodriues, Acta Crystallogr., 2019, C75, 707 Search PubMed.
- C. B. Pinto, L. H. R. Dos Santos and B. L. Rodriues, Cryst. Growth Des., 2020, 20, 4827 CrossRef CAS.
- R. M. Wood and H. J. Palenik, Inorg. Chem., 1998, 16, 4149 CrossRef PubMed.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE 2.0, Universitat de Barcelona, Barcelona, 2010 Search PubMed.
- S. K. Langley, N. F. Chilton, L. Ungur, B. Moubaraki, L. F. Chibotaru and K. S. Murray, Inorg. Chem., 2012, 51, 11873 Search PubMed.
- S. K. Langley, N. F. Chilton, B. Moubaraki and K. S. Murray, Chem. Commun., 2013, 49, 6965 RSC.
- S. K. Langley, N. F. Chilton, B. Moubaraki and K. S. Murray, Inorg. Chem., 2013, 52, 7183 CrossRef CAS PubMed.
- I. Radu, V. Ch. Kravtsov, S. M. Ostrovsky, O. S. Reu, K. Krämer, S. Decurtins, S.-X. Liu, S. I. Klokishner and S. G. Baca, Inorg. Chem., 2017, 56, 2662 CrossRef CAS.
- J. A. Sheikh, H. S. Jena and S. Konar, Molecules, 2022, 27, 1130 CrossRef CAS PubMed.
- J. A. Sheikh, S. Goswami and S. Konar, Dalton Trans., 2014, 43, 14577 RSC.
- V. Mereacre, M. N. Akhtar, Y. Lan, A. M. Ako, R. Clerac, Ch. E. Anson and A. K. Powell, Dalton Trans., 2010, 39, 4918 RSC.
- R. Tiron, W. Wernsdorfer, N. Aliaga-Alcalde and G. Christou, Phys. Rev. B, 2003, 68, 140407 CrossRef.
- V. Psycharis, D. Dermitzaki and C. P. Raptopoulou, Crystals, 2021, 11, 1246 CrossRef CAS.
- H. Lueken, Magnetochemie, Teubner Verlag, Stuttgart, 1999 Search PubMed.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS.
- K. N. Shrivastava, Phys. Status Solidi B, 1983, 117, 437 CrossRef CAS.
- K. R. Vignesh, S. K. Langley, K. S. Murray and G. Rajaraman, Chem.–Eur. J., 2017, 23, 1654 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: [TGA-DSC curves (Fig. S1–S9); ESI-MS isotope patterns (Fig. S10 and S11); PXRD spectra (Fig. 12); asymmetric units and additional structure plots (Fig. S13–S18); fingerprint plots and Hirshfeld surfaces (Fig. S19–S24); dc magnetic data of 1 (Fig. S25); ac magnetic data of 3 (Fig. S26); calculation details (Fig. S27); additional crystallographic and structural details (Tables S1 and S2); ESI mass spectrometry data (Table S3); BVS calculations for Co atoms (Table S4); continuous shape measures (CShM) of the coordination geometries of metal centers in 1–10 using SHAPE v2.0 (Table S5); patterns on Hirshfeld surfaces (Table S6) and quantitative data from Hirshfeld surface of the metal centers in 1–10 (Tables S7–S9); theoretical calculations (Table S10)]. CCDC 2239013, 2239014, 2239016–2239023. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra02513c |
| This journal is © The Royal Society of Chemistry 2025 |