
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable carbon-based catalysts for oxygen reduction-enhanced FDCA production at ultra-low cell voltage†
Phongphot Sakulaueab,
Sarinya Woraphutthapornc,
Takafumi Ishii d,
Kanokwan Kongpatpaniche,
Pongkarn Chakthranont*c and
Khanin Nueangnoraj*a
d,
Kanokwan Kongpatpaniche,
Pongkarn Chakthranont*c and
Khanin Nueangnoraj*a
aSchool of Bio-Chemical Engineering and Technology, Sirindhorn International Institute of Technology, Thammasat University, Pathum Thani 12120, Thailand. E-mail: khanin@siit.tu.ac.th
bDivision of Chemical Engineering, Faculty of Engineering, Rajamangala University of Technology Krungthep, Bangkok 10120, Thailand
cNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani 12120, Thailand. E-mail: pongkarn.cha@nanotec.or.th
dInternational Research and Education Center for Element Science, Faculty of Science and Technology, Gunma University, Japan
eDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong, 21210, Thailand
First published on 30th June 2025
Abstract
The electrochemical oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) offers a sustainable pathway for bio-based polyester production. While extensive studies have focused on electrocatalyst development for this reaction, the integration of the oxygen reduction reaction (ORR) with HMF oxidation (HMFOR) remains largely underexplored. This study presents a novel electrochemical system that synergistically combines ORR with HMFOR, utilizing a tea leaf-derived carbon cathode and a nickel foam anode. The carbonization temperature of tea leaf waste was systematically optimized to finely tune the microstructure, electronic properties, and chemical compositions of the carbon electrodes. The optimal temperature of 800 °C produced a metal-free carbon catalyst with high activity and selectivity for the 2-electron ORR to H2O2, enabling efficient gas diffusion electrode fabrication. In a membrane-free flow cell, coupling ORR with HMFOR significantly enhanced FDCA production, achieving an 82% yield at a cell voltage as low as 1 V, compared to 72% at 2.75 V under N2. Interestingly, H2O2 did not significantly enhance HMFOR, suggesting that O2 primarily lowers the reaction potential rather than directly contributing to oxidation. Notably, the tea leaf-derived carbon outperformed commercial Pt/C as a cathodic catalyst for HMFOR application, delivering a higher FDCA yield at similarly low cell voltages. Our reaction design concept demonstrates a sustainable and cost-effective approach to FDCA production by utilizing biomass-derived catalysts, reducing material costs and energy requirements while enhancing scalability and efficiency.
1. Introduction
The depletion of fossil fuels and the urgent need to reduce greenhouse gas emissions have intensified the search for renewable, sustainable, and green feedstocks for the industrial processes and product use (IPPU) sector.1–4 Biomass, particularly lignocellulose, is a highly promising resource, serving as a versatile feedstock for biofuels and chemicals through biorefinery technologies, analogous to petroleum refineries.5–8 A key chemical derived from lignocellulose is 5-hydroxymethylfurfural (HMF), a platform molecule for synthesizing various renewable-based chemicals.9,10 Hydrogenating HMF yields 2,5-dimethylfuran (DMF), which serves as a biofuel.11,12 Oxidation of HMF, on the other hand, produces valuable compounds such as 2,5-diformylfuran (DFF), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 5-formyl-2-furancarboxylic acid (FFCA), and, most notably, 2,5-furandicarboxylic acid (FDCA).13,14 FDCA has garnered significant attention15–17 as a precursor for producing poly(ethylene-2,5-furandicarboxylate) (PEF), a sustainable bioplastic alternative to polyethylene terephthalate (PET).18,19 PEF offers superior properties compared to PET, including higher thermal stability, enhanced mechanical strength, and improved gas barrier performance. These attributes make PEF an ideal material for lightweight applications, durable packaging, textiles, and electronic components.20,21 Furthermore, PEF products are estimated to emit 33% fewer greenhouse gases throughout their lifecycle compared to PET products, solidifying their environmental benefits.22Significant efforts have been devoted to developing commercially viable routes for FDCA production, with the electrochemical HMF oxidation reaction (HMFOR) emerging as a promising alternative.23–25 Life cycle assessments highlight several advantages of this approach, including efficient process control under mild conditions (low temperature and pressure), higher yields, and reduced environmental impact, especially when powered by renewable electricity.26 The success of HMFOR systems hinges on the selection of oxidation catalysts and the design of the electrochemical process. Early studies demonstrated that precious metal catalysts, such as Pt, Pd, Au, and Au–Pd, significantly influence the reaction pathways for HMF conversion.27–29 For instance, Chadderdon et al. showed that Au/C preferentially oxidizes aldehyde groups over alcohol groups at low overpotentials, leading to HMFCA production. In contrast, Pd/C catalyzes the oxidation of both alcohol and aldehyde groups at moderate rates. Notably, bimetallic Au–Pd catalysts outperform monometallic ones, effectively producing FDCA at low potentials.29
Despite their high efficiency, the practicality of noble metal catalysts is limited by their high cost. As a result, non-precious metal catalysts, particularly Ni-based materials, have emerged as promising alternatives due to their ability to utilize high oxidation states (Ni3+ and Ni4+).30–32 These states serve as electron acceptors, producing Ni2+ while oxidizing HMF to intermediates such as HMFCA and FFCA, ultimately yielding FDCA. The higher oxidation states are then electrochemically regenerated under the applied potential. This mechanism improves selectivity, minimizes side reactions, and enhances the overall efficiency of Ni-based systems, making them a viable and cost-effective option for FDCA production. The efficacy of Ni-based catalysts for scalable FDCA production has been demonstrated in several studies. Most notably, Latsuzbaia et al. reported a continuous electrochemical process using a NiOOH catalyst, achieving 90% FDCA production with a faradaic efficiency of ∼80% in an alkaline medium (pH > 12).33 Their system delivered an impressive production rate of ∼30 g h−1 through a two-step reaction in bench-top electrolyzers. While promising for large-scale applications, the process faced challenges such as low product purity and high device costs.
Building on this, our recent work scaling up FDCA production to kilogram-scale daily output demonstrated that cost-effective production rates exceeding 80 g h−1 could be achieved in a continuous stirred tank reactor (CSTR).34 This system utilized NiFeOOH and Pt as the anodic and cathodic catalysts, respectively. Additionally, we discovered the pivotal role of oxygen (O2) sparging in increasing both the yield and purity of the FDCA product. Under O2-rich conditions, the oxygen reduction reaction (ORR) becomes thermodynamically more favorable than the hydrogen evolution reaction (HER) and HMF reduction. Hence, the presence of O2 improves overall performance by acting as an electron scavenger and mitigating parasitic reduction of HMF, allowing the cell to be operated membrane-free. This enhanced energy efficiency, stability, and FDCA yield, moving the process further towards industrial feasibility. This discovery opens new possibilities for designing ORR electrodes that work synergistically with anodic HMF oxidation, enabling a more sustainable and industrially feasible FDCA production process.
Generally, electrochemical ORR can proceed via two primary pathways: a 4-electron pathway that produces water (H2O) or a 2-electron pathway that generates hydrogen peroxide (H2O2).35–37 The 4-electron process, requiring a higher reduction potential of 1.23 V vs. RHE compared to 0.7 V vs. RHE for the 2-electron pathway, is better suited for energy applications like fuel cells and air batteries due to its higher cell voltage and limiting current density.38 However, it typically relies on costly noble metal and transition metal catalysts, particularly platinum (Pt).39–42 In contrast, the 2-electron pathway is more facile, particularly under alkaline conditions, making it an ideal counter-reaction for organic oxidation processes.43 Crucially, it can be catalyzed by non-metal catalysts such as carbon-based materials. These materials offer a large surface area and sustainability advantages over metal-based catalysts.44–48 Incorporating heteroatoms into the carbon structure has been shown to significantly enhance ORR performance.48–52 For instance, nitrogen-doped carbon nanofibers accelerate ORR by up to 100 times compared to undoped fibers,53 and nitrogen doping of graphene edges has demonstrated catalytic activity comparable to platinum-on-carbon in both acidic and basic environments.54
Studies on paired electrolysis systems have demonstrated the feasibility of co-producing H2O2 and oxidized organic compounds, such as furfural55 and HMF,56 in membrane flow cells that separate catholyte and anolyte for efficient product collection. Notably, H2O2 is a versatile and environmentally friendly oxidizing agent that can facilitate the conversion of HMF into various valuable products,57 including β-formylacrylic acid,58 5-hydroxy-2(5H)-furanone,59 maleic acid (MA),60 fumaric acid (FA),61 and FDCA.62,63 Hence, it is both fundamentally intriguing and practically relevant to investigate whether in situ generated H2O2 at the cathode can enhance the rate of HMFOR at the anode in a membrane-free system.
In this work, we developed a highly efficient HMFOR system using nitrogen-doped carbon derived from tea leaves as the cathodic catalyst and nickel foam as the anodic catalyst. By optimizing the carbonization process, the natural nitrogen compounds in tea leaves were converted into pyrrolic N-rich carbon, which exhibited high activity for the 2-electron ORR. Additionally, the high-surface-area carbon catalyst functioned as a microporous layer (MPL), enabling the cost-effective fabrication of a gas diffusion cathode that enhanced O2 mass transport to active sites. This approach offers an environmentally friendly solution for HMFOR, utilizing waste biomass and eliminating the need for precious metal catalysts. When paired with ORR, our HMFOR system operates at significantly lower potentials compared to systems utilizing HER as the cathodic reaction, achieving a 60% reduction in cell voltage while enhancing FDCA yield. This study demonstrates a cost-effective, scalable, and sustainable method for FDCA production, paving the way for bio-based plastic development with reduced environmental impact.
2. Experimental methods
2.1 Synthesis of tea leaf-derived carbon catalyst
The nitrogen-doped carbon derived from tea leaves was synthesized as in an earlier report.64 The tea leaves waste was obtained from Ichitan Co. Ltd, Thailand. Briefly, the carbon was prepared by carbonizing and activating tea leaves pretreated with potassium carbonate (K2CO3) at a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5. The treated tea leaves were placed in an alumina boat and heated in a tube furnace. The activation process was carried out at varying temperatures from 600 and 900 °C with a heating rate of 5 °C per minute and held for 1 hour under a N2 flow of 200 mL min−1. The activated carbon was then ground to a powder and thoroughly washed with RO water to remove impurities and neutralize the pH. Then the carbon was dried at 120 °C for 24 hours. The resulting nitrogen-doped carbons were labeled TL600, TL700, TL800, and TL900 according to their respective activation temperatures.
:1.5. The treated tea leaves were placed in an alumina boat and heated in a tube furnace. The activation process was carried out at varying temperatures from 600 and 900 °C with a heating rate of 5 °C per minute and held for 1 hour under a N2 flow of 200 mL min−1. The activated carbon was then ground to a powder and thoroughly washed with RO water to remove impurities and neutralize the pH. Then the carbon was dried at 120 °C for 24 hours. The resulting nitrogen-doped carbons were labeled TL600, TL700, TL800, and TL900 according to their respective activation temperatures.
2.2 Structural characterization of tea leaf-derived carbon catalyst
The porous properties of the carbons were analyzed by nitrogen adsorption–desorption measurements at −196 °C (BELsorp miniX, MicrotracBEL, Corp.). The specific surface area was determined using the Brunauer–Emmett–Teller (BET) method. The total pore volume (Vtotal) was estimated from the nitrogen adsorption data at a relative pressure of 0.99. The micropore volume (Vmicro) was calculated using the Dubinin–Radushkevich (DR) equation and the mesopore volume (Vmeso) was determined by subtracting the micropore volume from the total pore volume. In addition, the pore size distribution was evaluated using non-local density functional theory (NLDFT). Raman spectra were acquired using a Horiba LabRam HR Evolution spectrometer using a 532 nm excitation. The intensity ratio of the disordered (D) and graphitic (G) peaks (ID/IG) was determined by calculating the ratio of the peak areas fitted with Gaussian functions. The crystal structure was determined by X-ray powder diffraction (XRD, Bruker-AXS D8 Advance) in transmission geometry with a Cu Kα1,2 source (λ1 = 1.5406 Å and λ2 = 1.54443 Å) and a LYNEXE XE-T detector. The spectra were measured in a 2θ range of 10–80°, 0.02° per step. The graphitic interlayer space, d-spacing (d002), was calculated from the (002) diffraction peak of graphite. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos AXIS NOVA instrument (Shimadzu). Deconvolution was performed by CasaXPS and the C 1s binding energy was set to 284.5 eV to correct for charge shifts.2.3 Preparation of the tea leaf-derived carbon gas diffusion electrode
The carbon slurry was prepared using a combination of carbon catalyst (TL800), carbon black (CB), and polyvinylidene fluoride (PVDF) at a weight ratio of 75:5:20 in an N-methyl-2-pyrrolidone (NMP) solution. The addition of carbon black was intended to increase electrical conductivity and improve electron transport across the electrode.65 NMP was carefully added to adjust the solids content to 10–20% by weight. The resulting mixture was uniformly mixed with a magnetic stirrer and then cast onto carbon fiber paper (Toray, TGP-H-120) with a doctor blade thickness of 250 μm. The cast film was dried in a vacuum oven at 60 °C for 6 hours and this casting step was repeated three times to obtain a dense electrode. The electrode was then cut to a specific size (3.5 × 3.5 cm2). The carbon loading was calculated based on the total weight of carbon on the carbon fiber paper. For comparison, a similar method was used to make the electrode with the commercially available Pt/C powder (20 wt%, Sigma Aldrich). It should be noted that the PVDF binder was specifically chosen for the gas diffusion electrode (GDE) experiment due to its excellent oxygen reduction reaction (ORR) performance.66 PVDF provides good gas permeability and hydrophobicity, which improves ORR kinetics and overall electrochemical performance.
2.4 Electrochemical characterizations
The selectivity of H2O2 was calculated using eqn (1) below:
 | (1) |
| ERHE = EAg/AgCl + 0.059 × pH + 0.197 V | (2) |
Cyclic voltammetry of the cathode was performed at a scan rate of 5 mV s−1 over a potential range of 0.1 to −1.3 V vs. RHE. Chronopotentiometry (CP) was performed at a current density of 10 mA cm−2 for 6 hours. The total cell voltage was monitored using a digital multimeter. Electrochemical impedance spectroscopy (EIS) was conducted at open-circuit potential over a frequency range of 100 kHz to 1 Hz with an amplitude of 10 mV to assess ohmic loss of the cell.
2.5 Products quantification
| 2Ce4+ + H2O2 → 2Ce3+ + 2H+ + O2 | (3) |
This method utilizes the yellow color of Ce4+ and the colorless nature of Ce3+. A stock solution of 0.5 mM Ce(SO4)2 (97%, Sigma-Aldrich) was prepared in 0.1 M H2SO4 (96%, Sigma-Aldrich). During ORR device testing, electrolyte samples were mixed with Ce4+ solution at a 1:300 volume ratio and allowed to react at room temperature for 30 minutes. The residual Ce4+ concentration was measured using UV-vis spectroscopy (Thermo Scientific, Evolution 350) at 317 nm and compared to a standard curve (0.05–0.5 mM Ce4+).
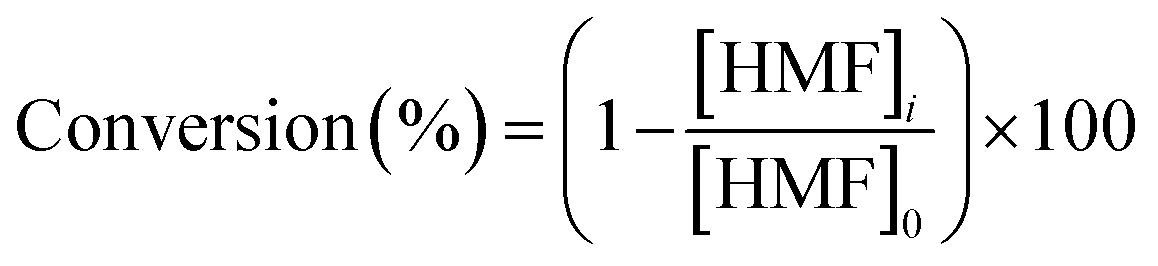 | (4) |
 | (5) |
3. Results and discussion
3.1 Structural and chemical characterizations of the tea leaf-derived carbons
N2 adsorption–desorption measurements were used to investigate the porous structure of the obtained carbons. The specific surface area (SBET), total pore volume (Vtotal), micropore volume (Vmicro), and mesopore volume (Vmeso) were calculated and listed in Table S1.† The results show that both the surface area and the pore volume increase with increasing activation temperature. All tea leaf-derived carbons show a combination of type I and IV isotherms (Fig. 1a), which can be attributed to the presence of both micro- and mesoporosity. Fig. 1b shows that the pore size distribution is mainly dominated by micropores with a pore diameter of about 1 nm (Fig. S1†). Increasing the activation temperature slightly increases mesopores but significantly enhances micropores, suggesting that the majority of the increased surface area is primarily attributed to micropores. | ||
| Fig. 1 (a) N2 adsorption–desorption isotherms, (b) micro–mesopore volume and SBET, (c) Raman spectra, and (d) XRD patterns of the tea leaf-derived carbons obtained from different activation temperatures. | ||
The Raman spectra reveal the structural evolution of the carbon derived from tea leaf at different activation temperatures (Fig. 1c). At 1350 and 1590 cm−1, two prominent peaks corresponding to the D and G bands are observed, reflecting the degree of graphitization of the carbon structure. The D-band is associated with defects in the sp2 structure, while the G-band represents the vibrational mode of the graphitic structure. In order to better quantify the relative intensities, the area ratio of the D and G bands (ID/IG) was calculated and presented in Table 1. The results show that the ID/IG ratio decreases with increasing activation temperature, indicating an increase in the graphitic domain of the carbon structure.
| Sample | ID/IG | d002 (Å) | Elemental composition (at%) | ||
|---|---|---|---|---|---|
| C 1s | N 1s | O 1s | |||
| TL600 | 3.33 | 3.59 | 69.6 | 2.9 | 27.5 |
| TL700 | 3.11 | 3.53 | 73.0 | 2.1 | 24.9 |
| TL800 | 2.99 | 3.50 | 74.9 | 1.4 | 23.7 |
| TL900 | 2.86 | 3.43 | 79.8 | 0.5 | 19.6 |
Fig. 1d shows the X-ray diffraction (XRD) patterns of all carbon samples, which provide information about the degree of graphitization, the interlayer spacing, and the structural arrangement. The two main peaks observed at about 26° and 44° correspond to the (002) and (100) diffraction planes of graphite (COD 9011577). The (002) peak originates from the ordered stacking of the graphene layers, while the (100) peak represents diffraction in the plane of the graphene sheets. The broad nature of these peaks indicates a high degree of disorder and a low degree of graphitization, especially at lower activation temperatures. In particular, the degree of graphitization improves slightly as the activation temperature increases from 600 °C to 900 °C, as evidenced by a slight decrease in the interlayer spacing (d002, listed in Table 1). The shift of the (002) peak in TL900 indicates a reduction in interlayer spacing (d002), attributed to increased graphitization at higher activation temperatures. Additionally, a small peak observed at 30° likely corresponds to K2CO3 (PDF 70-0292), a residue from the activation process that persisted despite washing with RO water.67 This suggests that some K2CO3 may be physically trapped in the porous structure, especially at higher activation temperatures. However, its presence is minimal and should not affect the electrochemical effect as K2CO3 is not thermodynamically reactive with carbonaceous materials under typical carbonization conditions (ΔG > 0).68
The surface chemistry of catalysts plays a crucial role in ORR activity; therefore, the surface chemical compositions of tea leaf-derived carbons were analyzed by X-ray photoelectron spectroscopy (XPS). All carbons prepared in this study contain three main elements, which are C, N, and O. The atomic concentrations of these elements are listed in Table 1. It is evident that the carbon content (C 1s) increases with activation temperature, while both nitrogen (N 1s) and oxygen (O 1s) contents decrease, a trend typically observed in the carbonization/activation process. The increase in carbon content is primarily due to the decomposition of volatile components at higher carbonization temperatures. Oxygen, which is mainly present in the form of carboxyl, hydroxyl, carbonyl, and ether groups, is released during the carbonization process as volatile compounds such as CO2 and H2O. Nitrogen, which is present in smaller quantities in the form of amine, imine, and heterocyclic groups, is also removed as part of the volatile by-products. This result aligns with the findings from Raman spectroscopy and XRD analysis, confirming that higher activation temperatures promote graphitization of the carbon structure while reducing both structural and chemical defects.
The chemical states of the tea leaf-derived carbons were determined by deconvoluting the XPS peaks. The C 1s spectra reveal the presence of C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds at 284, 285.8, and 288.0 eV, respectively (Fig. S2†). The N 1s spectra, shown in Fig. 2a, display the peaks at 396.0, 398.0, 400.0, and 401.0 eV, corresponding to metal nitride, pyridinic-N, pyrrolic-N, and graphitic-N, respectively. The percentages of each functional group, calculated from the peak areas after deconvolution, are summarized in Fig. 2c. The results show that while the overall nitrogen content decreased with increasing activation temperature, pyrrolic-N remains dominant, accounting for more than half of the total nitrogen species across all activation temperatures. Conversely, the fraction of pyridinic-N decreases, while that of graphitic-N increases with activation temperature.
O bonds at 284, 285.8, and 288.0 eV, respectively (Fig. S2†). The N 1s spectra, shown in Fig. 2a, display the peaks at 396.0, 398.0, 400.0, and 401.0 eV, corresponding to metal nitride, pyridinic-N, pyrrolic-N, and graphitic-N, respectively. The percentages of each functional group, calculated from the peak areas after deconvolution, are summarized in Fig. 2c. The results show that while the overall nitrogen content decreased with increasing activation temperature, pyrrolic-N remains dominant, accounting for more than half of the total nitrogen species across all activation temperatures. Conversely, the fraction of pyridinic-N decreases, while that of graphitic-N increases with activation temperature.
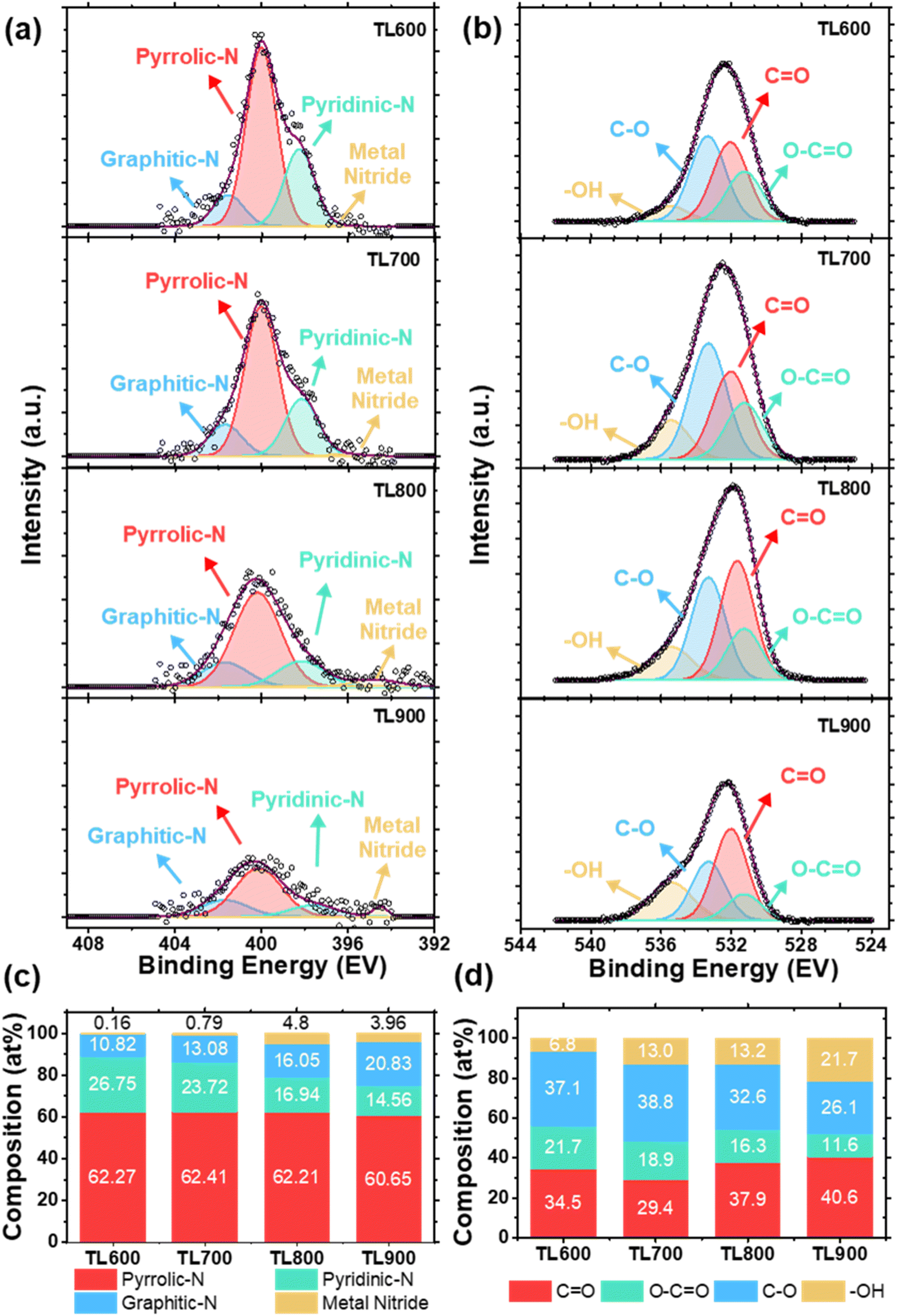 | ||
| Fig. 2 (a) N 1s spectra and (b) O 1s spectra; (c) the relative composition of each nitrogen functional group and (d) the relative composition of each oxygen functional group of tea leaf-derived carbons under different activation temperatures. | ||
The high-resolution O 1s spectra (Fig. 2b) reveal four distinct peaks corresponding to carboxyl groups (O–CO) at 531.3 eV, carbonyl groups (CO) at 532.0 eV, ether groups (C–O) at 533.4 eV, and hydroxyl groups (–OH) at 535.5 eV. The relative percentages of each functional group, estimated from the peak areas, are shown in Fig. 2d. It can be observed that the fraction of C–O and CO groups remain relatively stable with increasing activation temperature, indicating that these functional groups are more thermally stable under the carbonization conditions. On the other hand, hydroxyl groups (–OH) increase, while carboxyl groups (O–CO) decrease significantly with activation temperature.
3.2 Evaluation of the ORR performances
To evaluate the ORR performance of the obtained carbon catalysts, RRDE testing was performed at 1600 rpm in an O2-saturated 0.1 M KOH electrolyte. After baseline correction in an Ar-saturated electrolyte, the current densities measured on the disk and the ring (accounted for ring collection efficiency) are shown in Fig. 3a. The disk current reflects the overall ORR activity on the catalyst surface, while the ring current corresponds to the oxidation of H2O2 produced during the reaction, enabling the calculation of instantaneous H2O2 selectivity and electron transfer number (n). Key activity parameters, including onset potential (Eonset) and limiting current density, are summarized in Table 2. The Eonset, which indicates the potential at which ORR begins, is more anodic for TL800 and TL900 (0.80 V vs. RHE), indicating superior catalytic activity compared to TL600 and TL700 (Eonset values of 0.68 and 0.75 V vs. RHE, respectively). Additionally, TL800 (4.66 mA cm−2) and TL900 (4.68 mA cm−2) exhibited higher limiting current densities than TL600 (3.01 mA cm−2) and TL700 (3.16 mA cm−2), suggesting more efficient O2 mass transfer, which is crucial for overall activity. These results demonstrate that higher carbonization temperatures enhance ORR catalytic performance, likely due to increased conductivity and improved graphitization of TL800 and TL900. Furthermore, RRDE measurements under O2 and Ar (Fig. S3†) confirm the role of O2 in the cathodic reaction. A significant cathodic current was only observed under O2, indicating an active ORR, while a negligible current under Ar indicates a shift towards HER, which occurs at higher overpotentials and leads to an increased cell voltage.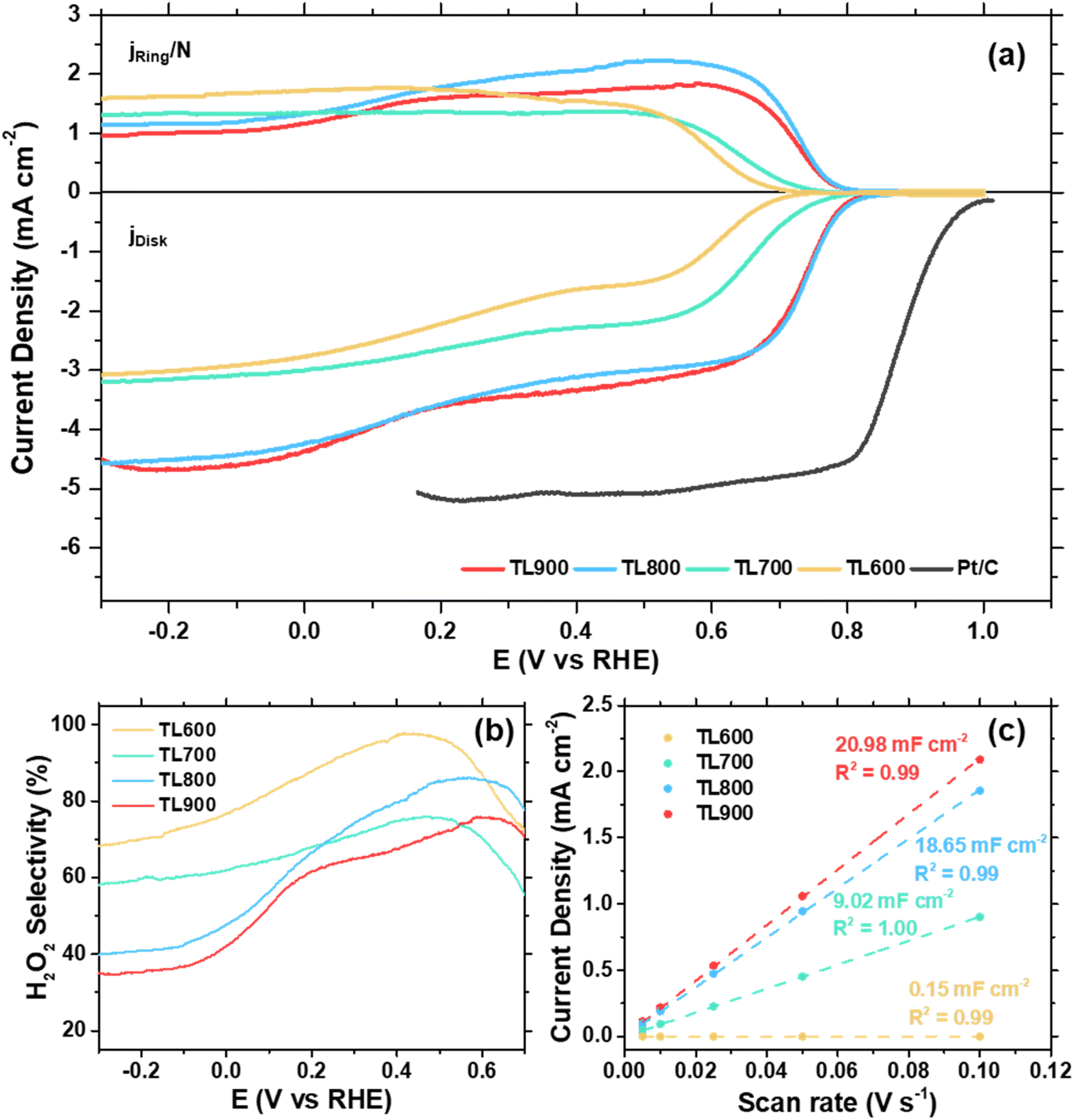 | ||
| Fig. 3 (a) ORR polarization curves obtained from RRDE experiments, (b) H2O2 selectivity, and (c) double-layer capacitances (Cdl) of the tea leaf-derived carbons of different activation temperatures. | ||
| Sample | Eonset at −0.2 mA cm−2 (V vs. RHE) | Limiting current density (mA cm−2) | Number of electron transfer (n) at 0.5 V vs. RHE | H2O2 selectivity at 0.5 V vs. RHE (%) |
|---|---|---|---|---|
| TL600 | 0.68 | 3.01 | 2.07 | 96.3 |
| TL700 | 0.75 | 3.16 | 2.49 | 75.6 |
| TL800 | 0.80 | 4.66 | 2.30 | 85.2 |
| TL900 | 0.80 | 4.68 | 2.57 | 71.5 |
The H2O2 selectivity (Fig. 3b) indicates that all tea leaf-derived carbons predominantly catalyze ORR via the 2-electron pathway. The number of electron transfers (n) at 0.5 V vs. RHE, shown in Table 2, ranges from 2 to 2.6. Among all, TL600 has the highest H2O2 selectivity (96.3%). However, as the activation temperature increases, TL700, TL800, and TL900 show slightly lower selectivities (75.6%, 85.2%, and 71.5%, respectively). This effect may arise from the content of pyrrolic-N and CO groups in the carbon, which are more abundant at lower activation temperatures. Pyrrolic-N and carbonyl functional groups are known to be key active sites for the 2-electron ORR pathway.50,69 As the activation temperature increases, the content of these functional groups decreases, resulting in slightly reduced H2O2 selectivity.
Despite the slightly lower H2O2 selectivity, tea leaf-derived carbons activated at higher temperatures benefit from improved onset potentials and higher limiting current densities. In addition to the higher degree of graphitization, the differences in electrochemically active surface area (ECSA) contribute to this enhancement. ECSA is typically correlated to the double-layer capacitance (Cdl) of the catalyst, which can be measured by cyclic voltammetry at varying scan rates in the non-faradaic region. As shown in Fig. 3c, Cdl increases with activation temperature, following a trend similar to that of SBET. TL600 and TL700 exhibit lower Cdl (0.15 and 9.02 mF cm−2, respectively), corresponding to their limited surface area and higher ID/IG, indicating poor conductivity and significant structural disorder. These factors contribute to their lower Eonset and limiting current densities. In contrast, TL800 and TL900 show comparable Cdl (18.65 and 20.98 mF cm−2), despite differing surface areas (SBET = 1236 and 1639 m2 g−1). This can be attributed to the dominance of micropores in their total surface area, as micropores are less electrochemically accessible mesopores.
Overall, the results demonstrate that the carbonization temperature significantly influences the catalytic performance of tea leaf-derived carbons by altering their surface area, pore structure, conductivity, and nitrogen and oxygen species distribution. TL800 stands out as the optimal catalyst, achieving a balance of high Eonset, efficient electron transfer, and moderate H2O2 selectivity, making it an excellent candidate for use in ORR electrodes paired with the HMFOR process.
To incorporate the tea leaf-derived catalysts into a practical device, TL800 was cast three times onto carbon fiber to form a dense electrode. Under an optical microscope, the cross-sectional area of TL800 on the fiber is clearly visible (Fig. 4a). The carbon paper displays a reflective, fibrous structure, while the carbon ink forms an opaque black layer. The layer thickness, measured using ImageJ software, was approximately 126 μm. The carbon loading was calculated to be 4.9 mg cm−2 based on the weight of carbon in a defined area of 3.5 × 3.5 cm2. The electrochemically active surface area (ECSA), as indicated by the Cdl, was 83 mF cm−2.
 | ||
| Fig. 4 (a) Optical microscopic image of TL800 electrode. (b) Schematic illustration of the GDE cell system for ORR coupled HMFOR. (c) Cyclic voltammetry of TL800 electrode and Pt/C electrode under O2 and N2 feeds. | ||
Fig. 4b illustrates the GDE cell configuration used for ORR-coupled HMFOR electrolysis. In addition to TL800, a Pt/C catalyst was included as a control due to its high efficiency in the 4-electron ORR pathway. Fig. 4c presents the cyclic voltammograms of the two GDEs under O2 and N2 environments. Both TL800 and Pt/C exhibited significantly higher currents in the O2 environment than in the N2, confirming their ORR activity. Notably, the ORR activity of TL800 closely matches that of Pt/C in terms of current density, highlighting the effectiveness of the TL800 electrode. These results establish TL800 as a highly promising cathode candidate for ORR applications.
3.3 Electrochemical HMFOR to FDCA and the overall cell voltage
The oxidation of HMF to FDCA was conducted under various conditions in the GDE cell (Fig. 4b) with bare Ni foam serving as the anode. On the cathodic side, GDEs based on TL800 and Pt/C catalysts were employed with either O2 or N2 gas feeds. N2 gas was used as a control to assess the FDCA electrosynthesis performance when HER was used as counter reaction instead of ORR. In addition, the effects of H2O2 was also investigated by introducing 10 mM H2O2 into the electrolyte. This concentration was chosen based on the H2O2 levels produced by the TL800 electrode during ORR at 50 mA cm−2 over 30 minutes, as shown in Fig. S4.†As shown in Fig. 5a, the concentration of HMF decreased over time, while the yield of FDCA increased. Small amounts of intermediates, including HMFCA, FFCA, and DFF, were observed. Notably, HMFCA was detected in greater quantities than DFF, indicating that the oxidation pathway predominantly proceeds via HMFCA. For the system employing the TL800 cathode, the FDCA yields after 5 hours were markedly different: 82% with O2 and 72% with N2. These findings highlight that the HMFOR process is more efficient under O2 conditions, resulting in higher FDCA yield and improved carbon balance. Conversely, in the N2 system, HMF appears to undergo alternative reaction pathways rather than the desired HMFOR, likely due to reductive degradation at the cathode under a highly reducing potential.70 This results in lower product yields and significant discrepancies in carbon balance. Evidence of HMF degradation in the N2 environment is seen in the yellow coloration of the electrolyte after electrolysis (Fig. S5†), suggesting the formation of by-products unlikely to convert into FDCA, thereby reducing the overall yield. When the system is purged with O2, this parasitic reduction is suppressed, as O2 reduction is thermodynamically more favorable. Thus, O2 acts as an effective electron scavenger, suppressing undesirable side reactions at the cathode and lowering its reductive potential, ultimately enhancing FDCA yield.
 | ||
| Fig. 5 (a) HMF conversion and yields of FDCA, HMFCA, FFCA, and DFF, under different electrolysis conditions: O2, N2, without and with 10 mM H2O2 addition in the electrolyte. (b) Cathode potential and (c) cell voltage obtained during a chronopotentiometry measurement at 10 mA cm−2 over 6 hours. | ||
When comparing the FDCA yields of TL800 and Pt/C, TL800 achieved a superior yield of 82% compared to 75% for Pt/C. Interestingly, the addition of 10 mM H2O2 to the TL800 system led to a decrease in FDCA yield under both O2 and N2 conditions, with yields dropping to 74% and 55%, respectively. These results suggest that in situ generated H2O2 does not significantly aid the oxidation process and that excessive H2O2 concentrations may accelerate HMF degradation, reducing the efficiency of FDCA production. Considering that all systems utilize the same Ni foam anode, the superior HMFOR performance in the TL800 system likely arises from the ability to effectively reduce O2 without interfering with the HMF reactant of TL800 catalyst. This contributes to a higher carbon balance compared to the Pt/C system. These findings highlight the advantages of the N- and O-doped carbon structure of TL800, which combines high ORR activity with moderate H2O2 selectivity. In addition, the stability test after HMFOR activity over 6 hours, the LSV curves show only minimal changes (Fig. S6†), indicating that the TL800 electrode exhibits excellent electrochemical stability even under prolonged ORR operation. As an effective and cost-efficient cathode material, TL800 is a promising candidate for enabling more efficient and sustainable HMFOR processes.
Apart from improved yield, the key benefits of our reaction configuration of synergistically combining ORR with HMFOR is the significantly reduced power consumption and thus the operating costs of the electrochemical process. Fig. 5b shows the catalytic stability of TL800 and Pt/C in O2 and N2 gas systems evaluated by chronopotentiometry (CP) at 10 mA cm−2 over a duration of 6 hours in the GDE cell. All CP profiles show stable cathodic potential with no significant fluctuations, indicating minimal degradation of the electrode and excellent electrochemical stability over the entire reaction period. Remarkably, TL800 shows comparable ORR potential compared to Pt/C. This demonstrates the superior catalytic efficiency of TL800 in facilitating ORR at the cathode.
Fig. 5c shows the operating cell voltage for ORR coupled with HMFOR at a current density of 10 mA cm−2. The cell voltages stabilized after 30 minutes and remained constant during the entire 6-hour operation: 2.75 V for TL800 under N2, 1.09 V for TL800 under O2, and 1.02 V for Pt/C under O2. Comparing the TL800 electrode under O2 and N2 systems, the voltage reduction reached up to 60%, underscoring the pivotal role of ORR in minimizing energy consumption. Additionally, this system achieved a significant reduction in cell voltage compared to previous work using a small CSTR reactor with a highly optimized NiFeOOH/Ni foam anode and Pt/Ni foam cathode, which reported a total cell voltage of approximately 2.2 V.70 This is even better than other electrochemical reaction systems for FDCA production reported in literature (as listed in Table S2†), further highlighting and benchmarking the performance of electrocatalysts in this study. The presented design achieved up to a 55% reduction in cell voltage, primarily due to the introduction of a carbon-based catalyst. This carbon material not only served as a catalyst but also acted as a microporous layer in the GDE, maintaining the balance between gas and liquid phases and enhancing O2 mass transport, thereby improving overall cell efficiency. These findings demonstrate that the integration of biomass-derived carbon-based catalysts and an innovative cell design can enable more selective and energy-efficient FDCA electrosynthesis. Further optimization, including the incorporation of advanced HMFOR catalysts reported in the literature, could further enhance the performance of this system.
4. Conclusion
In this study, we have designed and successfully developed a selective and cost-effective electrochemical process for the production of FDCA by pairing HMFOR with ORR in a membrane-free GDE flow cell. The cathode, fabricated using a tea leaf-derived N-doped carbon catalyst (TL800), demonstrated exceptional ORR activity and moderate selectivity to H2O2 (∼85%) due to its high surface area, favorable electronic properties, and abundance of pyrrolic-N and carbonyl functional groups. When incorporated into a GDE and operated under O2 feed, TL800 enabled an 82% FDCA yield at a significantly reduced cell voltage of 1.09 V, representing a 60% energy reduction compared to the operation under N2 feed and a substantial improvement over prior systems. In comparison to Pt/C, TL800 delivered a similar cell voltage while achieving superior FDCA yield, highlighting its promise as a cost-effective and scalable alternative for HMFOR applications. This work demonstrates a sustainable and economically viable pathway for bio-based FDCA production, leveraging biomass-derived catalysts and innovative reaction design. By integrating high ORR activity with efficient GDE architecture of TL800, the process offers both operational and economic benefits that pave the way for scalability and industrial implementation. Future advancements in cathode and anode optimization could further enhance the system's performance, solidifying its potential as a transformative approach to sustainable chemical production.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science, Research and Innovation Fund, Thailand Science Research and Innovation (TSRI) (Grant No. FFB670076/0337) and Thammasat University Research Fund (TUFT 44/2567). P. S. acknowledges the support from Thammasat Post Doctoral Fellowship (Contract No. TUPD 4/2567). The authors would like to acknowledge Sakvarit Nitrathorn, Naphat Ormsapsin, Warith Powichien, and Patcharapol Intrasorn for their help with collecting preliminary results that led to this research.References
- K. Sudesh and T. Iwata, Sustainability of Biobased and Biodegradable Plastics, Clean:Soil, Air, Water, 2008, 36(5–6), 433–442, DOI:10.1002/clen.200700183.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, A roadmap towards green packaging: the current status and future outlook for polyesters in the packaging industry, Green Chem., 2017, 19(20), 4737–4753, 10.1039/c7gc02521a.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr, J. P. Hallett, D. J. Leak and C. L. Liotta, et al., The path forward for biofuels and biomaterials, Science, 2006, 311(5760), 484–489, DOI:10.1126/science.1114736.
- B. Kamm, M. Kamm, P. R. Gruber and S. Kromus, Biorefinery Systems – An Overview, in Biorefineries-Industrial Processes and Products, 2005, pp. 1–40 Search PubMed.
- X. Tong, Y. Ma and Y. Li, Biomass into chemicals: Conversion of sugars to furan derivatives by catalytic processes, Appl. Catal., A, 2010, 385(1–2), 1–13, DOI:10.1016/j.apcata.2010.06.049.
- X. Tang, M. Zuo, Z. Li, H. Liu, C. Xiong, X. Zeng, Y. Sun, L. Hu, S. Liu and T. Lei, et al., Green Processing of Lignocellulosic Biomass and Its Derivatives in Deep Eutectic Solvents, ChemSusChem, 2017, 10(13), 2696–2706, DOI:10.1002/cssc.201700457.
- M. Brodin, M. Vallejos, M. T. Opedal, M. C. Area and G. Chinga-Carrasco, Lignocellulosics as sustainable resources for production of bioplastics – A review, J. Cleaner Prod., 2017, 162, 646–664, DOI:10.1016/j.jclepro.2017.05.209.
- I. Delidovich, P. J. Hausoul, L. Deng, R. Pfutzenreuter, M. Rose and R. Palkovits, Alternative Monomers Based on Lignocellulose and Their Use for Polymer Production, Chem. Rev., 2016, 116(3), 1540–1599, DOI:10.1021/acs.chemrev.5b00354.
- B. Zhu, C. Chen, L. Huai, Z. Zhou, L. Wang and J. Zhang, 2,5-Bis(hydroxymethyl)furan: A new alternative to HMF for simultaneously electrocatalytic production of FDCA and H2 over CoOOH/Ni electrodes, Appl. Catal., B, 2021, 297, 120396, DOI:10.1016/j.apcatb.2021.120396.
- C. Yang, X. Li, Z. Zhang, B. Lv, J. Li, Z. Liu, W. Zhu, F. Tao, G. Lv and Y. Yang, High efficient catalytic oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid under benign conditions with nitrogen-doped graphene encapsulated Cu nanoparticles, J. Energy Chem., 2020, 50, 96–105, DOI:10.1016/j.jechem.2020.03.003.
- N. Ma, Y. Song, F. Han, G. I. N. Waterhouse, Y. Li and S. Ai, Highly selective hydrogenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran at low temperature over a Co–N–C/NiAl-MMO catalyst, Catal. Sci. Technol., 2020, 10(12), 4010–4018, 10.1039/d0cy00363h.
- H. Xia, S. Xu, H. Hu, J. An and C. Li, Efficient conversion of 5-hydroxymethylfurfural to high-value chemicals by chemo- and bio-catalysis, RSC Adv., 2018, 8(54), 30875–30886, 10.1039/c8ra05308a.
- M. A. Lilga, R. T. Hallen and M. Gray, Production of Oxidized Derivatives of 5-Hydroxymethylfurfural (HMF), Top. Catal., 2010, 53(15–18), 1264–1269, DOI:10.1007/s11244-010-9579-4.
- Z. Lin, X. Chen, L. Lu, X. Yao, C. Zhai and H. Tao, Recent advances in electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid: Mechanism, catalyst, coupling system, Nanotechnol. Rev., 2023, 12(1), 1–20, DOI:10.1515/ntrev-2022-0518.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Toward biomass-derived renewable plastics: Production of 2,5-furandicarboxylic acid from fructose, Sci. Adv., 2018, 4(1), eaap9722, DOI:10.1126/sciadv.aap9722.
- S. Prasad, A. J. Khalid, V. Narishetty, V. Kumar, S. Dutta and E. Ahmad, Recent advances in the production of 2,5-furandicarboxylic acid from biorenewable resources, Mater. Sci. Energy Technol., 2023, 6, 502–521, DOI:10.1016/j.mset.2023.04.005.
- R. O. Rajesh, T. K. Godan, R. Sindhu, A. Pandey and P. Binod, Bioengineering advancements, innovations and challenges on green synthesis of 2, 5-furan dicarboxylic acid, Bioengineered, 2020, 11(1), 19–38, DOI:10.1080/21655979.2019.1700093.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, M. Majdoub, G. Z. Papageorgiou and D. N. Bikiaris, Effect of catalyst type on recyclability and decomposition mechanism of poly(ethylene furanoate) biobased polyester, J. Anal. Appl. Pyrolysis, 2017, 126, 357–370, DOI:10.1016/j.jaap.2017.05.010.
- A. Marshall, B. Jiang, R. M. Gauvin and C. M. Thomas, 2,5-Furandicarboxylic Acid: An Intriguing Precursor for Monomer and Polymer Synthesis, Molecules, 2022, 27(13), 4071, DOI:10.3390/molecules27134071.
- J. G. van Berkel, N. Guigo, H. A. Visser and N. Sbirrazzuoli, Chain Structure and Molecular Weight Dependent Mechanics of Poly(ethylene 2,5-furandicarboxylate) Compared to Poly(ethylene terephthalate), Macromolecules, 2018, 51(21), 8539–8549, DOI:10.1021/acs.macromol.8b01831.
- J. G. van Berkel, N. Guigo, J. J. Kolstad and N. Sbirrazzuoli, Biaxial Orientation of Poly(ethylene 2,5-furandicarboxylate): An Explorative Study, Macromol. Mater. Eng., 2018, 303(3), 1700507, DOI:10.1002/mame.201700507.
- M. G. Davidson, S. Elgie, S. Parsons and T. J. Young, Production of HMF, FDCA and their derived products: a review of life cycle assessment (LCA) and techno-economic analysis (TEA) studies, Green Chem., 2021, 23(9), 3154–3171, 10.1039/d1gc00721a.
- M. T. Bender, X. Yuan, M. K. Goetz and K.-S. Choi, Electrochemical Hydrogenation, Hydrogenolysis, and Dehydrogenation for Reductive and Oxidative Biomass Upgrading Using 5-Hydroxymethylfurfural as a Model System, ACS Catal., 2022, 12(19), 12349–12368, DOI:10.1021/acscatal.2c03606.
- M. J. Kang, H. Park, J. Jegal, S. Y. Hwang, Y. S. Kang and H. G. Cha, Electrocatalysis of 5-hydroxymethylfurfural at cobalt based spinel catalysts with filamentous nanoarchitecture in alkaline media, Appl. Catal., B, 2019, 242, 85–91, DOI:10.1016/j.apcatb.2018.09.087.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, Electrochemical Oxidation of 5-Hydroxymethylfurfural with NiFe Layered Double Hydroxide (LDH) Nanosheet Catalysts, ACS Catal., 2018, 8(6), 5533–5541, DOI:10.1021/acscatal.8b01017.
- P. Patel, D. Schwartz, X. Wang, R. Lin, O. Ajao and A. Seifitokaldani, Technoeconomic and Life-Cycle Assessment for Electrocatalytic Production of Furandicarboxylic Acid, ACS Sustain. Chem. Eng., 2022, 10(13), 4206–4217, DOI:10.1021/acssuschemeng.1c08602.
- K. R. Vuyyuru and P. Strasser, Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis, Catal. Today, 2012, 195(1), 144–154, DOI:10.1016/j.cattod.2012.05.008.
- H. G. Cha and K. S. Choi, Combined biomass valorization and hydrogen production in a photoelectrochemical cell, Nat. Chem., 2015, 7(4), 328–333, DOI:10.1038/nchem.2194.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on supported Au and Pd bimetallic nanoparticles, Green Chem., 2014, 16(8), 3778–3786, 10.1039/c4gc00401a.
- B. J. Taitt, D.-H. Nam and K.-S. Choi, A Comparative Study of Nickel, Cobalt, and Iron Oxyhydroxide Anodes for the Electrochemical Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid, ACS Catal., 2018, 9(1), 660–670, DOI:10.1021/acscatal.8b04003.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang and X. Li, et al., Biomass Valorization via Paired Electrosynthesis Over Vanadium Nitride-Based Electrocatalysts, Adv. Funct. Mater., 2019, 29(42), 1904780, DOI:10.1002/adfm.201904780.
- Z. Zhou, Y.-n. Xie, L. Sun, Z. Wang, W. Wang, L. Jiang, X. Tao, L. Li, X.-H. Li and G. Zhao, Strain-induced in situ formation of NiOOH species on CoCo bond for selective electrooxidation of 5-hydroxymethylfurfural and efficient hydrogen production, Appl. Catal., B, 2022, 305, 121072, DOI:10.1016/j.apcatb.2022.121072.
- R. Latsuzbaia, R. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt and E. Goetheer, et al., Continuous electrochemical oxidation of biomass derived 5-(hydroxymethyl)furfural into 2,5-furandicarboxylic acid, J. Appl. Electrochem., 2018, 48(6), 611–626, DOI:10.1007/s10800-018-1157-7.
- P. Chakthranont, S. Woraphutthaporn, C. Sanpitakseree, K. Srisawad and K. Faungnawakij, Kilogram-scale production of high purity 2,5-furandicarboxylic acid via sustainable leap in continuous electrochemical oxidation of 5-hydroxymethylfurfural, Chem. Eng. J., 2023, 476, 146478, DOI:10.1016/j.cej.2023.146478.
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts, Nat. Catal., 2018, 1(4), 282–290, DOI:10.1038/s41929-018-0044-2.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin and Y. Liu, et al., High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials, Nat. Catal., 2018, 1(2), 156–162, DOI:10.1038/s41929-017-0017-x.
- E. Jung, H. Shin, W. Hooch Antink, Y.-E. Sung and T. Hyeon, Recent Advances in Electrochemical Oxygen Reduction to H2O2: Catalyst and Cell Design, ACS Energy Lett., 2020, 5(6), 1881–1892, DOI:10.1021/acsenergylett.0c00812.
- S. Boonlha, P. Chakthranont and S. Kityakarn, 3DOM Cerium Doped LaCoO3 Bifunctional Electrocatalysts for the Oxygen Evolution and Reduction Reactions, ChemCatChem, 2022, 14(3), e202101398, DOI:10.1002/cctc.202101398.
- A. R. Khataee, M. Safarpour, M. Zarei and S. Aber, Electrochemical generation of H2O2 using immobilized carbon nanotubes on graphite electrode fed with air: Investigation of operational parameters, J. Electroanal. Chem., 2011, 659(1), 63–68, DOI:10.1016/j.jelechem.2011.05.002.
- A. Moraes, M. H. M. T. Assumpção, R. Papai, I. Gaubeur, R. S. Rocha, R. M. Reis, M. L. Calegaro, M. R. V. Lanza and M. C. Santos, Use of a vanadium nanostructured material for hydrogen peroxide electrogeneration, J. Electroanal. Chem., 2014, 719, 127–132, DOI:10.1016/j.jelechem.2014.02.009.
- J. Zhang, H. Zhang, M. J. Cheng and Q. Lu, Tailoring the Electrochemical Production of H(2) O(2) : Strategies for the Rational Design of High-Performance Electrocatalysts, Small, 2020, 16(15), e1902845, DOI:10.1002/smll.201902845.
- A. Morozan, B. Jousselme and S. Palacin, Low-platinum and platinum-free catalysts for the oxygen reduction reaction at fuel cell cathodes, Energy Environ. Sci., 2011, 4, 1238–1254, 10.1039/c0ee00601g.
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Recent Progress of Electrochemical Production of Hydrogen Peroxide by Two-Electron Oxygen Reduction Reaction, Adv. Sci., 2021, 8(15), 2100076, DOI:10.1002/advs.202100076.
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S. Z. Qiao, Tailoring Selectivity of Electrochemical Hydrogen Peroxide Generation by Tunable Pyrrolic-Nitrogen-Carbon, Adv. Energy Mater., 2020, 10(21), 2000789, DOI:10.1002/aenm.202000789.
- Z. Guo, Z. Xiao, G. Ren, G. Xiao, Y. Zhu, L. Dai and L. Jiang, Natural tea-leaf-derived, ternary-doped 3D porous carbon as a high-performance electrocatalyst for the oxygen reduction reaction, Nano Res., 2016, 9(5), 1244–1255, DOI:10.1007/s12274-016-1020-2.
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins and R. Sinclair, et al., Defective Carbon-Based Materials for the Electrochemical Synthesis of Hydrogen Peroxide, ACS Sustain. Chem. Eng., 2017, 6(1), 311–317, DOI:10.1021/acssuschemeng.7b02517.
- G. F. Han, F. Li, W. Zou, M. Karamad, J. P. Jeon, S. W. Kim, S. J. Kim, Y. Bu, Z. Fu and Y. Lu, et al., Building and identifying highly active oxygenated groups in carbon materials for oxygen reduction to H(2)O(2), Nat. Commun., 2020, 11(1), 2209, DOI:10.1038/s41467-020-15782-z.
- Y. Sun, S. Li, Z. P. Jovanov, D. Bernsmeier, H. Wang, B. Paul, X. Wang, S. Kuhl and P. Strasser, Structure, Activity, and Faradaic Efficiency of Nitrogen-Doped Porous Carbon Catalysts for Direct Electrochemical Hydrogen Peroxide Production, ChemSusChem, 2018, 11(19), 3388–3395, DOI:10.1002/cssc.201801583.
- D. W. Kim, O. L. Li and N. Saito, Enhancement of ORR catalytic activity by multiple heteroatom-doped carbon materials, Phys. Chem. Chem. Phys., 2015, 17(1), 407–413, 10.1039/c4cp03868a.
- P. Chakthranont, S. Nitrathorn, S. Thongratkaew, P. Khemthong, H. Nakajima, R. Supruangnet, T. Butburee, N. Sano and K. Faungnawakij, Rational Design of Metal-free Doped Carbon Nanohorn Catalysts for Efficient Electrosynthesis of H2O2from O2Reduction, ACS Appl. Energy Mater., 2021, 4(11), 12436–12447, DOI:10.1021/acsaem.1c02260.
- G. P. Kharabe, S. Barik, S. K. Veeranmaril, A. Nair, R. Illathvalappil, A. Yoyakki, K. Joshi, C. P. Vinod and S. A. Kurungot, Nitrogen-Dual-Doped Reduced Graphene Oxide Co-Existing with Cobalt-Encapsulated Graphitic Carbon Nanotube as an Activity Modulated Electrocatalyst for Oxygen Electrocatalyst for Oxygen Electrochemistry Applications, Small, 2024, 20(35), e2400012, DOI:10.1002/smll.202400012.
- G. P. Kharabe, N. Manna, A. Nadeema, S. K. Singh, S. Mehta, A. Nair, K. Joshi and S. Kurungot, A pseudo-boehmite AlOOH supported NGr composite-based air electrode for mechanically rechargeable Zn-air battery applications, J. Mater. Chem. A, 2022, 10(18), 10014–10025, 10.1039/d2ta00546h.
- S. Maldonado and K. J. Stevenson, Influence of Nitrogen Doping on Oxygen Reduction Electrocatalysis at Carbon Nanofiber Electrodes, J. Phys. Chem. B, 2005, 109(10), 4707–4716, DOI:10.1021/jp044442z.
- X. Fu, J. Jin, Y. Liu, Z. Wei, F. Pan and J. Zhang, Efficient oxygen reduction electrocatalyst based on edge-nitrogen-rich graphene nanoplatelets: toward a large-scale synthesis, ACS Appl. Mater. Interfaces, 2014, 6(6), 3930–3936, DOI:10.1021/am405130w.
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S.-Z. Qiao, Tailoring Selectivity of Electrochemical Hydrogen Peroxide Generation by Tunable Pyrrolic-Nitrogen-Carbon, Adv. Energy Mater., 2020, 10(21), 2000789, DOI:10.1002/aenm.202000789.
- J. Wang, X. Liu, C. Ma, H. Fu, S. Chen, N. Li, Y. Li, X. Fan and W. Peng, Simulation-guided atomic Ni catalyst with oxygen-enriched coordination environment for H2O2 electrosynthesis coupled with 5-HMF oxidation, Chem Catal., 2024, 4(9), 101090, DOI:10.1016/j.checat.2024.101090.
- S. P. Teong, X. Li and Y. Zhang, Hydrogen peroxide as an oxidant in biomass-to-chemical processes of industrial interest, Green Chem., 2019, 21(21), 5753–5780, 10.1039/C9GC02445J.
- L. A. Badovskaya and V. V. Poskonin, Rearrangements and Tautomeric Transformations of Heterocyclic Compounds in Homogeneous Reaction Systems Furfural–H2O2–Solvent, Russ. J. Gen. Chem., 2018, 88(8), 1568–1579, DOI:10.1134/s1070363218080030.
- L. A. Badovskaya, V. M. Latashko, V. V. Poskonin, E. P. Grunskaya, Z. I. Tyukhteneva, S. G. Rudakova, S. A. Pestunova and A. V. Sarkisyan, Catalytic Oxidation of Furan and Hydrofuran Compounds. 7. Production of 2(5H)-Furanone by Oxidation of Furfural with Hydrogen Peroxide and Some of Its Transformations in Aqueous Solutions, Chem. Heterocycl. Compd., 2002, 38(9), 1040–1048, DOI:10.1023/a:1021288711593.
- Y. Lou, S. Marinkovic, B. Estrine, W. Qiang and G. Enderlin, Oxidation of Furfural and Furan Derivatives to Maleic Acid in the Presence of a Simple Catalyst System Based on Acetic Acid and TS-1 and Hydrogen Peroxide, ACS Omega, 2020, 5(6), 2561–2568, DOI:10.1021/acsomega.9b02141.
- N. Araji, D. D. Madjinza, G. Chatel, A. Moores, F. Jérôme and K. De Oliveira Vigier, Synthesis of maleic and fumaric acids from furfural in the presence of betaine hydrochloride and hydrogen peroxide, Green Chem., 2017, 19(1), 98–101, 10.1039/c6gc02620f.
- S. Li, K. Su, Z. Li and B. Cheng, Selective oxidation of 5-hydroxymethylfurfural with H2O2 catalyzed by a molybdenum complex, Green Chem., 2016, 18(7), 2122–2128, 10.1039/c5gc01991e.
- S. Zhang and L. Zhang, A facile and effective method for preparation of 2.5-furandicarboxylic acid via hydrogen peroxide direct oxidation of 5-hydroxymethylfurfural, Pol. J. Chem. Technol., 2017, 19(1), 11–16, DOI:10.1515/pjct-2017-0002.
- S. S. Desa and K. Nueangnoraj, Enhanced Redox Activity of Anthraquinone on N-Doped Porous Carbon Electrode, ChemistrySelect, 2024, 9(21), e202303953, DOI:10.1002/slct.202303953.
- X. Zhang, X. Xia, I. Ivanov, X. Huang and B. E. Logan, Enhanced activated carbon cathode performance for microbial fuel cell by blending carbon black, Environ. Sci. Technol., 2014, 48(3), 2075–2081, DOI:10.1021/es405029y.
- H. Su, S. Pasupathi, B. Bladergroen, V. Linkov and B. G. Pollet, Optimization of gas diffusion electrode for polybenzimidazole-based high temperature proton exchange membrane fuel cell: Evaluation of polymer binders in catalyst layer, Int. J. Hydrogen Energy, 2013, 38(26), 11370–11378, DOI:10.1016/j.ijhydene.2013.06.107.
- C. Nita, B. Zhang, J. Dentzer and C. Matei Ghimbeu, Hard carbon derived from coconut shells, walnut shells, and corn silk biomass waste exhibiting high capacity for Na-ion batteries, J. Energy Chem., 2021, 58, 207–218, DOI:10.1016/j.jechem.2020.08.065.
- C. Lu, S. Xu and C. Liu, The role of K2CO3 during the chemical activation of petroleum coke with KOH, J. Anal. Appl. Pyrolysis, 2010, 87(2), 282–287, DOI:10.1016/j.jaap.2010.02.001.
- H. Zhang, K. Lv, B. Fang, M. C. Forster, R. Dervişoğlu, L. B. Andreas, K. Zhang and S. Chen, Crucial role for oxygen functional groups in the oxygen reduction reaction electrocatalytic activity of nitrogen-doped carbons, Electrochim. Acta, 2018, 292, 942–950, DOI:10.1016/j.electacta.2018.09.175.
- P. Chakthranont, S. Woraphutthaporn, C. Sanpitakseree, K. Srisawad and K. Faungnawakij, Kilogram-scale production of high purity 2,5-furandicarboxylic acid via sustainable leap in continuous electrochemical oxidation of 5-hydroxymethylfurfural, Chem. Eng. J., 2023, 476, 146478, DOI:10.1016/j.cej.2023.146478.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02265g |
| This journal is © The Royal Society of Chemistry 2025 |