
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted synthesis of a trimethoxyphenyltetrahydropyrimidine analogue designed as a DNA intercalator: in silico, multi-spectroscopic, thermodynamic, and in vitro approaches†
Ahmed A. Al-Karmalawy *ab,
Ayman Abo Elmaatycd,
Galal Magdyef,
Aya Saad Radwanb,
Radwan Alnajjarg,
Moataz A. Shaldamh,
Arwa Omar Al Khatibi,
Salem Salman Almujrij,
Abdullah Yahya Abdullah Alzahranik and
Haytham O. Tawfik*l
*ab,
Ayman Abo Elmaatycd,
Galal Magdyef,
Aya Saad Radwanb,
Radwan Alnajjarg,
Moataz A. Shaldamh,
Arwa Omar Al Khatibi,
Salem Salman Almujrij,
Abdullah Yahya Abdullah Alzahranik and
Haytham O. Tawfik*l
aDepartment of Pharmaceutical Chemistry, College of Pharmacy, The University of Mashreq, Baghdad 10023, Iraq. E-mail: akarmalawy@horus.edu.eg
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Horus University-Egypt, New Damietta 34518, Egypt
cMedicinal Chemistry Department, Faculty of Pharmacy, Port Said University, Port Said 42526, Egypt
dMedicinal Chemistry Department, Clinical Pharmacy Program, East Port Said National University, Port Said 42526, Egypt
ePharmaceutical Analytical Chemistry Department, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, 33511, Egypt
fDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Mansoura National University, Gamasa, 7731168, Egypt
gCADD Unit, Faculty of Pharmacy, Libyan International Medical University, Benghazi 16063, Libya
hDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh 33516, Egypt
iFaculty of Pharmacy, Hourani Center for Applied Scientific Research, Al-Ahliyya Amman University, Amman, Jordan
jDepartment of Pharmacology, College of Pharmacy, King Khalid University, Asir-Abha 61421, Saudi Arabia
kDepartment of Chemistry, Faculty of Science, King Khalid University, Abha 61413, Saudi Arabia
lDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Tanta University, Tanta, 31527, Egypt. E-mail: haytham.omar.mahmoud@pharm.tanta.edu.eg
First published on 8th May 2025
Abstract
Based on the rational design of DNA intercalators and Topo-II inhibitors and taking into consideration the main pharmacophoric features of doxorubicin (Dox) as a reference standard, we theoretically designed novel substituted tetrahydropyrimidine analogues (T1–35). The designed analogues (T1–35) were investigated for their inhibitory potential towards the hybrid DNA and Topo-II target receptor using molecular docking. Interestingly, the theoretically designed analogue T30 with a 3,4,5-trimethoxy phenyl side chain was found to be the superior candidate, achieving a binding score of −7.06 kcal mol−1, compared with two reference standards, doxorubicin (Dox) and a co-crystal ligand (EVP). Moreover, the docked candidates (T30, Dox, and EVP) were further subjected to molecular dynamics simulations for 500 ns. Furthermore, MM-GBSA calculations showed that the target candidate (T30) achieved superior ΔG binding energy (−33.86 kcal mol−1) compared with Dox and EVP. Moreover, T30 was found to be the most promising candidate that could be conveniently synthesized based on its order in the chemical synthesis scheme. In addition, to evaluate the antiproliferative activity and scope of compound T30, we requested the National Cancer Institute (NCI) to test it against nine cancer cell types. Interestingly, compound T30 exhibited very strong antiproliferative activity with a mean GI% of 122% and a mean GI50 of 4.10 μM. It exhibited the highest anticancer activity towards all 59 cell lines. Moreover, the in vitro binding interaction of compound T30 with calf thymus DNA (ctDNA) was examined using various techniques, such as spectrofluorimetry, UV-vis spectrophotometry, viscosity measurements, ionic strength measurements, and thermodynamics to confirm its mechanism of action. Investigating the intermolecular binding interaction between small compounds and DNA can provide valuable insights for designing drugs with enhanced effectiveness and improved targeted activities.
1. Introduction
Cancer remains a leading global health challenge, claiming approximately 8 million lives annually. Alarmingly, projections suggest a rise of over 50% in new cancer cases in the coming years.1 By 2030, the annual global burden of cancer is expected to reach a staggering 22 million cases.2 This represents a significant increase from the current 8 million cases per year, underlining the urgent need for advancements in cancer prevention, diagnosis, and treatment. A growing concern in cancer treatment is the emergence of acquired resistance to chemotherapy in various cancer types.3,4 Unfortunately, many chemotherapeutic drugs can also affect healthy dividing cells, causing side effects such as nausea, a weakened immune system, anemia, and hair loss. This is because these drugs are not specific in targeting only cancer cells.5,6This critical need for improved cancer treatments underscores the urgency of developing new anticancer agents with a superior therapeutic index. Many successful anticancer drugs work by causing DNA damage in cancer cells, ultimately leading to programmed cell death (apoptosis).7,8 Some anticancer drugs, called DNA intercalators, work by embedding themselves between the strands of DNA. This disrupts the normal function of DNA, leading to cell cycle disruptions and eventually cell death.9,10 By imagining the DNA molecule as a twisted ladder, DNA intercalators are similar to tiny wedges that can squeeze between the rungs of the ladder (base pairs). These wedges disrupt the normal functioning of the DNA, ultimately leading to cell death. Intercalation is stabilized by a combination of forces, including π-electron interactions, electrostatic attractions, and hydrophobic/polar interactions.11 Acridine derivatives (e.g. amsacrine),12 anthracyclines (e.g., mitoxantrone, doxorubicin (Dox), and nogalamycin),13,14 are examples of FDA-approved DNA intercalators, as shown in Fig. 1.
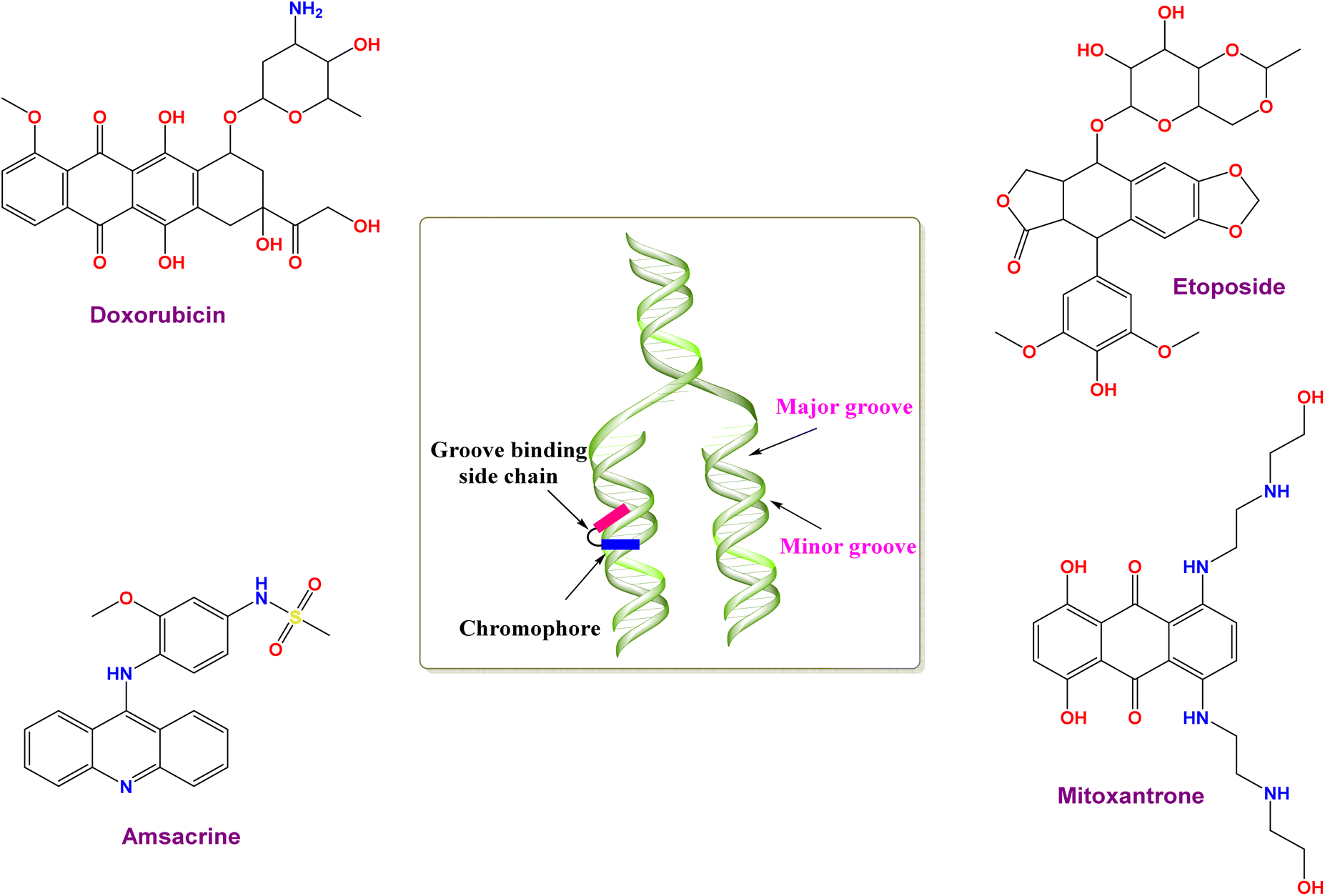 | ||
| Fig. 1 Illustration of some approved topoisomerase II (Topo-II) inhibitors and DNA intercalators. | ||
Compounds can interact with DNA through either non-covalent or covalent interactions, with the non-covalent pattern typically being more prevalent. Non-covalent binding can be categorized into three types: intercalation, electrostatic, and groove binding.15 The molecule can undergo binding at several sites, including the minor groove and major groove, between two base pairs (full intercalation), on the exterior of the helix, and through electrostatic binding.15,16 Typically, there is no singular method that can offer a definitive understanding of the interaction between drugs and DNA. Therefore, it is crucial to provide effective, rapid, and economical experimental and computational methods to evaluate the interaction between DNA and other substances, with the purpose of assisting in drug design and discovery.
DNA topoisomerases are like tiny cellular machines (enzymes) that help manage the complex structure of DNA. DNA topoisomerases use energy from ATP to manipulate the molecular motion of DNA, preventing it from getting tangled.17 There are two main types of DNA topoisomerases: Type I and Type II. Notably, Type I enzymes (topoisomerase I) can make changes to the structure of a single strand of DNA, while Type II (topoisomerase II) enzymes can modify the structure involving both strands of DNA.17
Topoisomerase II (Topo-II) is regarded as a prominent enzyme in our cells that helps manage the complex structure of DNA. It temporarily aids in cutting both strands of the DNA molecule and then sealing the cuts back up. This allows DNA to unwind and be used in important cellular processes like copying chromosomes, cell division, and making proteins.18 Topoisomerase II inhibitors can be categorized into two major classes: Topo-II poisons and Topo-II catalytic inhibitors. Topo-II poisons are characterized by their ability to enhance the rate of Topo-II/DNA complex cleavage, ultimately leading to the formation of DNA strand lesions.19 Examples of Topo-II poisons are anthracyclines (e.g., Dox13 and mitoxantrone14). However, Topo-II catalytic inhibitors work by specifically inhibiting the catalytic activity of Topo-II and do not change the rate of Topo-II/DNA complex cleavage.20 An example of a Topo-II catalytic inhibitor is Etoposide.21 Hence, DNA intercalation and Topo-II inhibition are emerging as attractive and efficient tools in the fight against cancer. Their ability to target DNA makes them valuable approaches for cancer therapy.
Additionally, the pyrimidine derivatives are privileged scaffolds in medicinal chemistry with various biological activities, such as anti-microbial,22–24 anti-hypertensive,25 anti-diabetic,26–28 and anticancer agents.29–32 Hence, in this current work, our main aim is to design and synthesize novel ethyl 6-(chloromethyl)-1-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate derivatives as DNA intercalators and Topo-II inhibitors.
1.1. Rationale of the design
Herein, many DNA intercalators and Topo-II inhibitors, such as Dox, share key pharmacophores that are essential for their function. These pharmacophores include a planar polyaromatic chromophore, a groove binder (a component that fits into a specific area of the DNA molecule), and a positively charged region (cationic center).10,33 These structural features underpin the efficacy of DNA intercalators and Topo-II inhibitors by facilitating their interaction with designated regions of the DNA molecule. This interaction disrupts the enzyme's activity and consequently interferes with the maintenance of proper DNA topology, as the rational design is based on maintaining the main essential pharmacophores with the possibility of slight molecular modifications to pursue activity change. In this current work, we retained the main essential pharmacophores of Topo-II inhibitors and DNA intercalators with some molecular modifications for SAR studies. Moreover, we have considered the pharmacophoric nature of the parent compounds, not their exact chemical entities. Replacing the anthracene nucleus of Doxorubicin (Dox) with a tetrahydropyrimidine scaffold is a strategy rooted in medicinal chemistry and drug design, aiming to improve certain pharmacological properties, while retaining or modifying the therapeutic activity. For example, the quinone and aromatic rings in the anthracene nucleus are involved in redox cycling, generating reactive oxygen species (ROS) that damage cardiac tissue.34 Replacing this core with a less redox-active scaffold-like tetrahydropyrimidine could help minimize ROS generation and reduce cardiotoxic effects. So, the anthracene nucleus of Dox was replaced by a tetrahydropyrimidine scaffold (to be inserted in between the DNA base pairs, where both act as intercalators between the DNA nucleobases regardless of the difference in size between pyrimidine and anthracene scaffolds), the –NH2 group of Dox was kept in our designed compounds (to act as a cationic center that can be protonated at physiological pH to interact with the phosphate group of the DNA sugar moiety), and the groove binder of Dox was replaced by similarly acting diverse substituted phenyl motifs to be directed towards the minor groove of DNA (Fig. 2). It is worth noting that the 3,4,5-trimethoxy phenyl side chain as a groove binder can contribute to superior stability, lower RMSD, and better ΔG binding energy compared to Dox. Regarding stability, the 3,4,5-trimethoxyphenyl group can fit particularly well into the DNA minor groove, owing to the 1-steric compatibility (the narrow groove accommodates small, hydrophobic, and electron-rich aromatic rings), 2-hydrogen bonding potential (the methoxy groups can act as hydrogen bond acceptors), and the 3-hydrophobic interactions (groove interiors are relatively hydrophobic and the 3,4,5-trimethoxyphenyl non-polar surface matches this environment). Regarding RMSD, the 3,4,5-trimethoxyphenyl group side chain contributes to low RMSD because it locks into the groove via different interactions. Moreover, unlike Dox's intercalation, which can disturb the DNA geometry or flip out the bases, the groove binding of 3,4,5-trimethoxyphenyl is less invasive, so the ligand can stay more stable with lower RMSD values. The binding free energy (ΔG) may be more favorable in 3,4,5-trimethoxyphenyl-based groove binders since methoxy groups at positions 3, 4, and 5 can interact with DNA bases and sugar-phosphate backbone oxygens via hydrogen bonds. The phenyl ring can make tight van der Waals contacts with the groove wall, in addition to groove environments that favor non-polar interactions. The 3,4,5-trimethoxyphenyl group can achieve this type of interaction, affording favorable binding free energy (ΔG). In conclusion, our designed compounds retained the main pharmacophores of Dox required for DNA intercalation and Topo-II inhibition. | ||
| Fig. 2 The design rationale of the investigated tetrahydropyrimidine derivatives illustrates the main pharmacophores of doxorubicin as a DNA intercalator and Topo-II inhibitor. | ||
2. Results and discussion
2.1. In silico studies
 | ||
| Scheme 1 Cyclocondensation of different aromatic aldehydes 1 with ethyl 4-chloroacetoacetate 2 and N-methylurea 3 to obtain the target analogues (T1–35). | ||
These analogues were designed based on their novelty in synthesis and diversity. Afterward, all of the designed analogues (T1–35) were investigated for their inhibitory potential towards the hybrid DNA and Topo-II target receptor (PDB ID: 3QX3) using molecular docking. This was done to select the most promising analogue to be synthesized, and accordingly save effort, time, and cost in the drug discovery process. First, the active site residues were identified to be Met782, Gln778, Asp479, Lys456, Glu477, Gly478, Gly504, Leu502, Arg503, Ala817, DA12, DC11, DC14, and DG13. Moreover, both Dox and the co-crystallized inhibitor of the target receptor (EVP) were utilized as two reference standards.
Interestingly, the theoretically designed tetrahydropyrimidine analogue (T30) with a 3,4,5-trimethoxy phenyl side chain was found to be the superior candidate, achieving a binding score of −7.06 kcal mol−1 (RMSD = 1.56 Å). This binding score was comparable to both reference standards (Dox and EVP), which exhibited binding scores of −7.44 kcal mol−1 (RMSD = 1.58 Å) and −7.45 kcal mol−1 (RMSD = 1.57 Å), respectively. Additionally, the docking scores, RMSD, and binding interactions of the target novel substituted tetrahydropyrimidine analogues (T1–35) are summarized in the ESI data (Table S1†).
The binding mode of the designed analogue (T30) was highly similar to both reference standards (Dox and EVP), as shown in Fig. 3, illustrating the formation of one hydrogen bond with Met782 and one pi-hydrogen bond with DA12 through its trimethoxy phenyl side chain. Moreover, it formed two hydrogen bonds with DC11 and Ala817 through its tetrahydropyrimidine moiety extensions at C2 and C6. Accordingly, both hydrogen and pi-hydrogen bonds were clarified to be crucial types of interactions to stabilize analogue (T30) inside the binding pocket of DNA and the Topo-II target receptor. Moreover, Dox showed the formation of two hydrogen bonds with DA12 and Gln778. However, the docked co-crystallized EVP bound both DA12 and Met782 with two hydrogen bonds. These findings indicate the antagonistic potential of the designed analogue (T30) towards the hybrid DNA and Topo-II target receptor (PDB ID: 3QX3).
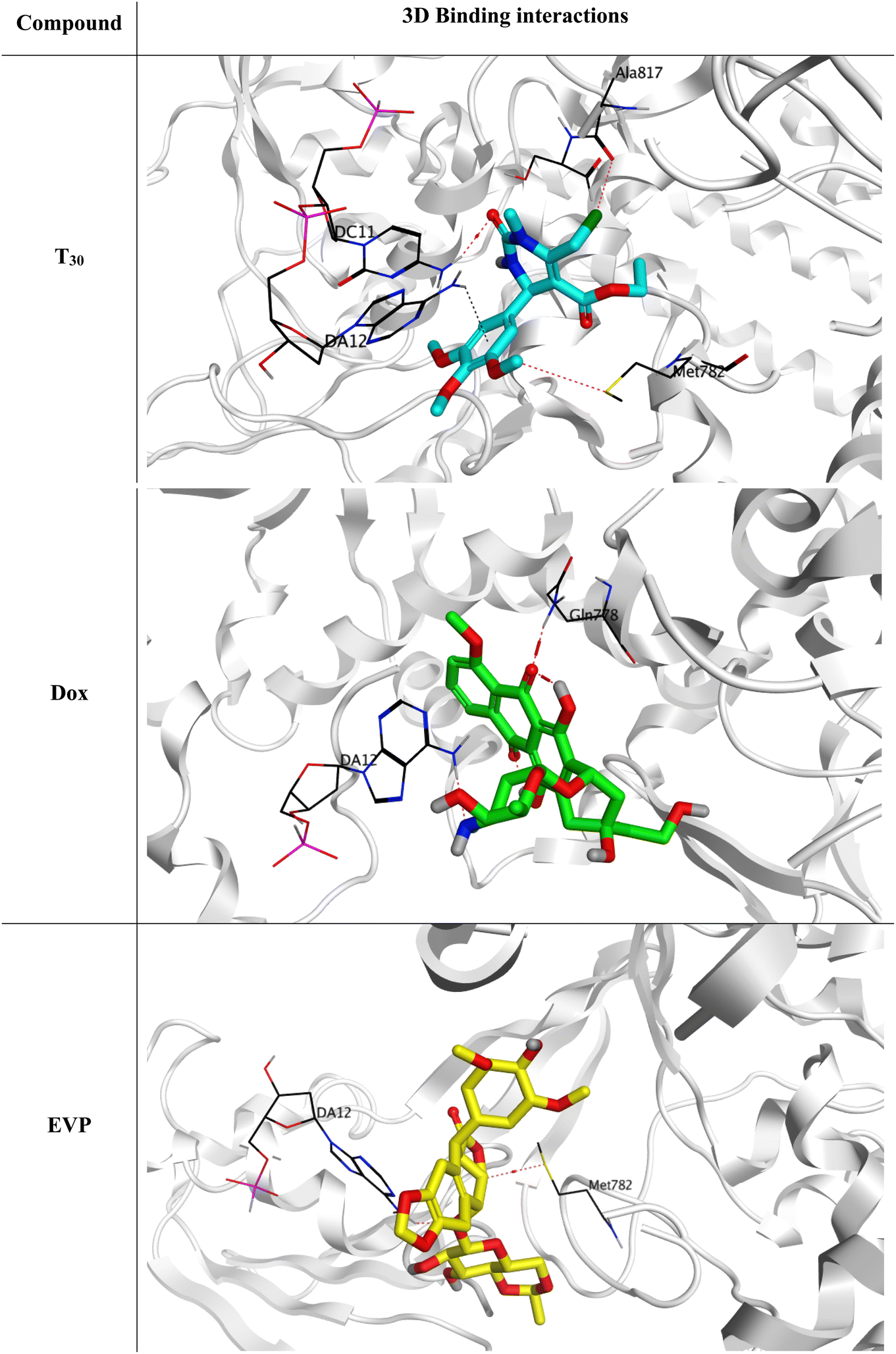 | ||
| Fig. 3 3D Binding interactions of the superior theoretically designed tetrahydropyrimidine analogue (T30), doxorubicin, and the docked co-crystallized EVP towards the hybrid DNA and Topo-II target receptor (PDB ID: 3QX3). | ||
2.1.2.1. RMSD analysis. RMSD was performed to judge the system's stability. The RMSD describes the deviation degree of the complex protein with respect to its initial position in a quantitative way. The three examined complexes showed moderate stability with RMSD values <5.4 Å (Fig. 4A). Moreover, the ligand RMSD (L-RMSD) was studied to evaluate the exact behaviour of each ligand within the hybrid DNA and Topo-II target receptor (PDB ID: 3QX3) binding domain (Fig. 4B). Interestingly, compound T30 showed the most stable behaviour with an RMSD value of 6.3 Å as the maximum fluctuation level. However, Dox and EVP fluctuated to 8 and 9 Å, respectively, indicating their less stable behaviours compared to T30.
 | ||
| Fig. 4 (A) Protein RMSD and (B) ligand RMSD for the complexes (T30, Dox, and EVP) of the hybrid DNA and Topo-II target receptor (PDB ID: 3QX3) binding domain as a function of simulation time (500 ns). | ||
2.1.2.2. Binding interactions histogram and heat map analysis. To completely describe the hybrid DNA and Topo-II target receptor (PDB ID: 3QX3)-ligand interactions, both histograms and heat maps were analyzed in detail (Fig. 5 and ESI Fig. S1†), respectively.
 | ||
| Fig. 5 Histogram describing the binding interactions between the hybrid DNA and Topo-II target receptor (PDB ID: 3QX3) and its ligands: (A) T30, (B) Dox, and (C) EVP. | ||
The histogram of the hybrid DNA and Topo-II target receptor in complex with compound T30 showed that Gln778 contributed the most to the interactions with about 58%. The types of interactions included hydrogen bonds and water bridges (Fig. 5A). However, Dox, Ala817, Gln789, and Asn786 contributed the most to the interactions at 80%, 70%, and 65%, respectively, with hydrogen bonds and water bridge interactions for the three amino acids (Fig. 5B). Moreover, the hybrid DNA and Topo-II receptor-EVP histogram showed that Asn520 and Gln778 were the superior amino acids with 52% and 45% contributions, respectively, through hydrogen bonds and water bridge interactions (Fig. 5C).
Briefly, it can be concluded that T30 shared the co-crystal (EVP) interactions with Gln778 amino acid, which may be considered the most crucial residue to produce the antagonistic activity towards the hybrid DNA and Topo-II receptor.
Furthermore, the exact time of the binding interactions for the hybrid DNA and Topo-II target receptor residues with T30, Dox, and EVP candidates were represented by heat map output (ESI Fig. S1†).
The heat map of the hybrid DNA and Topo-II target receptor-T30 complex showed that the interactions of Gln778 were more prominent after 100 ns of the simulation time, and were preserved until the end of the simulation (Fig. S1,† A). Conversely, more intense behaviours were observed for Ala817, Gln789, and Asn786 interactions with Dox (Fig. S1B†). The interactions of Ala817 were more intense in the first half of the simulation time. In contrast, the interactions of Gln789 and Asn786 were more intense in the second half of the simulation time. Additionally, the hybrid DNA and Topo-II target receptor-EVP complex heat map showed that Asn520 and Gln778 continually contributed to the binding interactions throughout most of the simulation time (Fig. S1C†).
| Complex | ΔG binding | Coulomb | Covalent | H-bond | Lipo | Bind packing | Solv_GB | VdW |
|---|---|---|---|---|---|---|---|---|
| a Coulomb: Coulomb energy; covalent: covalent binding energy; H-bond: hydrogen-bonding energy; lipo: lipophilic energy; Solv_GB: generalized born electrostatic solvation energy; VdW: van der Waals energy; and St. Dev.: standard deviation. | ||||||||
| T30 | −33.86 | −963.07 | 6.41 | −0.43 | −10.79 | −0.39 | 1024.80 | −90.38 |
| Dox | −33.80 | −1046.22 | 5.18 | −0.50 | −13.04 | −0.51 | 1133.57 | −112.27 |
| EVP | −27.94 | −930.40 | 6.02 | −0.26 | −10.52 | −0.06 | 998.07 | −90.79 |
2.2. Chemistry
As presented in Scheme 2, the most promising candidate (T30) was conveniently synthesized through Biginelli multicomponent reaction (MCR).36,37 It involves the cyclocondensation of 3,4,5-trimethoxybenzaldehyde 1 with ethyl 4-chloroacetoacetate 2 and N-methylurea 3 in reflux with absolute ethanol, in addition to citric acid and triethylorthoformate (TEOF) to remove the water molecule and the formation of the tetrahydropyrimidine (THPM) analogue T30 with high yield (93%). | ||
| Scheme 2 Synthesis of the THPM derivative T30. Reagents and conditions: (i) citric acid, TEOF, absolute ethanol, and reflux 10 h. | ||
Elemental analysis and spectroscopic data (1H NMR, 13C NMR, DEPTQ, HMQC, mass spectroscopy, and HPLC) of the newly synthesized compound were in agreement with the expected chemical structure, as reported in the experimental section. Generally, the 1H NMR spectral data of the target compound showed a pair of doublet signals at δ 5.08 and 8.06 ppm, representing 4-H and 3-NH, respectively, at the THPM ring, and the disappearance of the singlet signals of proton of the starting aldehyde and the two protons of the active methylene (–CH2) of ethyl 4-chloroacetoacetate 2 at around δ 9.82 and 3.41 ppm, respectively. 13C NMR spectra showed the presence of 4-C at the THPM ring at δ 52.59 ppm, the disappearance of the carbonyl group of the starting aldehyde, and one of the two carbonyl groups of ethyl 4-chloroacetoacetate 2 in the expected range of δ 190–210 ppm. The conducted HRMS further assured the chemical structure of compound T30 (cald./found 399.13174/399.13194 m/z).
The 13C NMR-DEPTQ approach was used to discriminate between carbons that carried an odd number of protons (CH and CH3) and those bearing an even number of protons or without protons (CH2 and CQ) by revealing signals upfield (positive) and downfield (negative), respectively, to the baseline. Compound T30 showed two aliphatic peaks downfield to the baseline at 38.50 and 60.91 ppm, respectively, for the CH2Cl and CH2O groups, in addition to all quaternary carbons. In addition, it showed peaks above the baselines related to all CH3 groups (three OCH3, NCH3, and OCH2CH3) and all CH groups (two benzene CH at positions 2/6 and one THPM CH at position 4).
Furthermore, the two-dimensional 1H–13C HMQC (Heteronuclear Multiple Quantum Coherence) technique was used to correct and complete the 13C NMR peak assignments. From the 1H–13C HMQC spectrum shown in Fig. 6, two separate cross-peaks can easily be seen by correlation of proton HA at 4.99 ppm with the carbon signal at 38.49 ppm, and that of proton HB at 5.11 ppm with the carbon signal at the same chemical shift (38.49 ppm). Based on this, the signal at 38.49 ppm could easily be assigned to the carbon atom attached to both protons (HA and HB) of compound T30. The previous conclusion was supported by the same coupling constant of both doublet signals at 4.99 and 5.11 ppm (J = 10.0 Hz). Although there is a third doublet signal for a proton in that region (5.08 ppm), it was possible to conclude that it couples with the doublet signal of the NH group (8.06 ppm) because of the same coupling constant value (J = 5.0 Hz). Thus, we can conclude that it is the proton belonging to the carbon number four at the THPM ring.
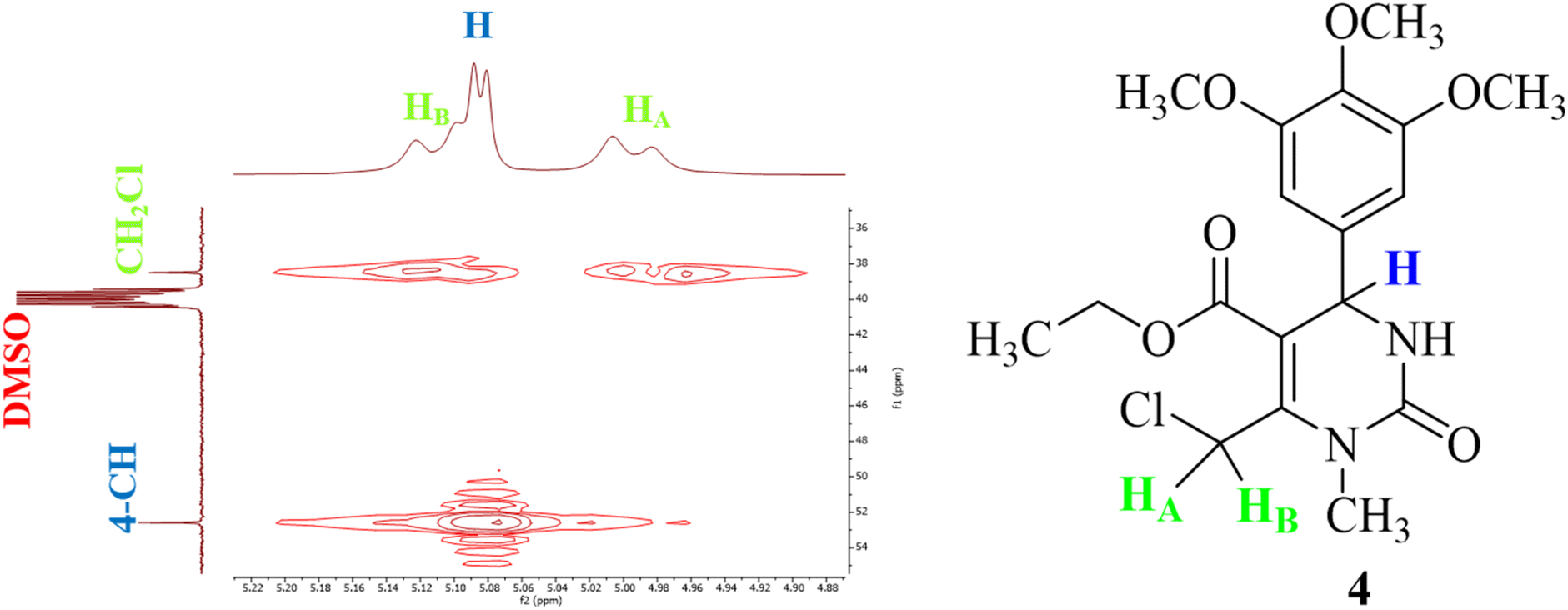 | ||
| Fig. 6 1H–13C HMQC NMR spectrum of compound T30 in the regions (1H) 4.87–5.23 ppm and (13C) 35–55 ppm. | ||
The mass spectra of the synthesized compound showed a peak corresponding to the molecular ion and its isotope. According to HPLC analysis, compound T30 has a purity level greater than 98.9% (see the purity value listed for the lead compound in the experimental section and Fig. S7†).
2.3. Biological evaluation
Compound T30 exhibited significant antiproliferative activity, with mean GI% = 122% and mean GI50 = 4.10 μM. It had the highest level of effectiveness against all 59 cell lines in terms of inhibiting cancer growth. Compound T30 demonstrated deadly effects in 39 instances of cancer cells, with growth inhibition percentages ranging from 101% to 196% (Fig. S8†). In addition, it exhibited remarkable activity with GI50 values ranging from 1.43 to 13.5 μM (6 of them being less than 2.00 μM) and LC50 values ranging from 7.16 to greater than 100.0 μM (Fig. S9†).
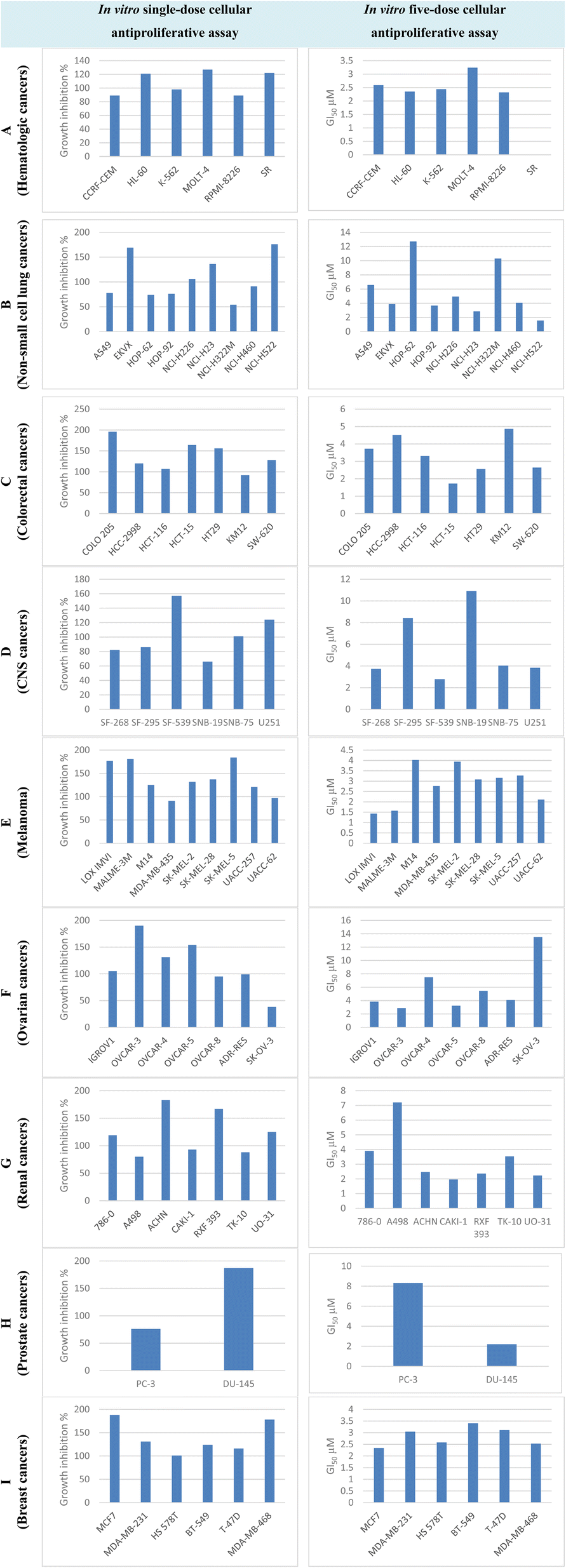 | ||
| Fig. 7 GI% and GI50 of cell lines representing diverse cancer subtypes by compound T30. | ||
2.4. Spectroscopic approaches
2.4.2.1. UV spectrophotometric study. This study relies on the addition of increasing concentrations of compound T30 to ctDNA and observing the alterations in the intensity and position of the DNA's distinctive peak at 260 nm, which is associated with the π–π* transition of the base pairs of DNA.54 The study revealed that the intensity of the ctDNA peak increased as the concentration of compound T30 increased, as depicted in Fig. 8. Furthermore, the peak position exhibited a red shift towards a shorter wavelength. Consequently, based on the shift rule of the DNA distinctive absorption peak, the binding process is anticipated to occur through intercalation mode rather than groove.55
 | ||
| Fig. 8 UV spectrophotometric spectra of ctDNA (97.0 μM) with increasing concentrations of compound T30 from 0 to 5 (0, 5.0, 10.0, 20.0, 30.0 and 40.0) μM. | ||
2.4.2.2. Viscosity studies. Viscosity studies are crucial for investigating alterations in the DNA length and how it binds.56 The traditional intercalative binding process significantly affects the viscosity of the DNA solution due to its requirement for ample space between adjacent base pairs to elongate the double helix and accommodate micro-molecules.57 Conversely, the impact of the electrostatic and groove binding processes on the DNA viscosity is minimal or nonexistent, as supported by many studies.58,59 The data presented in Fig. 9 demonstrates that the relative specific viscosity of ctDNA (η/η0)1/3 varies on subsequent addition of compound T30, providing evidence for the intercalation binding mode of compound T30 with ctDNA. This finding is in line with the results obtained from the spectrophotometric investigation.
 | ||
| Fig. 9 Effect of different concentrations of compound T30 (0–50.0 μM) on the ctDNA viscosity (97.0 μM) in Tris–HCl buffer at 298 K. | ||
2.4.2.3. Spectrofluorimetric competitive binding interactions. Competitive binding studies were carried out utilizing Rhodamine B (RB) and Ethidium bromide (EB) fluorescent probes to investigate the binding of compound T30 to ctDNA. According to reports, RB binds to the DNA's minor groove, particularly in areas rich in AT base pairs. In contrast, EB binds to DNA by intercalation.60,61 The fluorescence intensities of EB and RB increase when they attach to DNA.62 The analysis revealed that the fluorescence intensity of the EB-DNA complex decreased as the concentration of compound T30 increased. However, there was no impact on the fluorescence intensity of the RB-DNA complex, which further supports the intercalation binding process (Fig. 10).
 | ||
| Fig. 10 (A) Fluorescence emission spectra of the EB-ctDNA complex in the presence and absence of compound T30 at 298 K. C (ctDNA): 97.0 μM; C (EB): 7.0 μM; C (compound T30): 0, 5.0, 10.0, 20.0, 30.0, 40.0 μM, (λex/λem = 525/603 nm). (B) Fluorescence emission spectra of the RB-ctDNA complex in the presence and absence of compound T30 at 298 K. C(ctDNA): 97.0 μM; C(RB): 1.27 μM; C (compound T30) (0 → 5): 0, 5.0, 10.0, 20.0, 30.0, 40.0 μM, (λex/λem = 465/576 nm). | ||
2.4.2.4. Ionic strength evaluation. The degree of electrostatic contact is substantially influenced by the ionic strength of the reaction media.63 Thus, in order to assess the potential for electrostatic interaction between ctDNA and compound T30, the impact of different NaCl concentrations on their binding relationship was examined. The data presented in Fig. 11 demonstrate that the absorbance of the complex with ctDNA remains constant as the concentration of NaCl increases from 0 to 0.07 M. This suggests the absence of any electrostatic interaction. Briefly, the previously revealed experimental data demonstrate an intercalation binding interaction between compound T30 and ct-DNA.
 | ||
| Fig. 11 Influence of the NaCl ionic strength on the absorbance of the ctDNA-compound T30 complex. The conc. of compound T30 and ctDNA were 2.0 μM and 97.0 μM, respectively. The conc. of NaCl was 0–0.07 M. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex formed between compound T30 and ctDNA:64
:1 complex formed between compound T30 and ctDNA:64
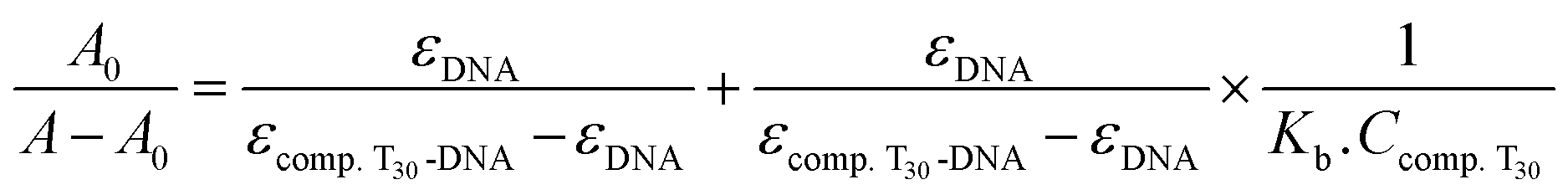 | (1) |
A linear relationship was observed when plotting 1/Ccompound 4 versus A0/(A − A0) at various temperatures (298, 303, 308 K), as shown in Fig. 12. This outcome indicates that the resultant complex exhibits a stoichiometry of 1:1. Table 2 displays the various Kb values acquired for the resultant complex utilizing eqn (1). The Kb values were in the order of 106 M−1, indicating that compound T30 has a significant affinity for binding to ctDNA. In addition, the values seem to be comparable to those of the intercalators, with a Kb of 1.4 × 106 M−1 (ref. 65) and higher than groove binders,61 demonstrating the involvement of the intercalation binding process in the binding of ctDNA-compound T30. Furthermore, when the temperature increases, the binding affinity between ctDNA and compound T30 increases, suggesting that the interaction between compound T30 and ctDNA is an endothermic process.54
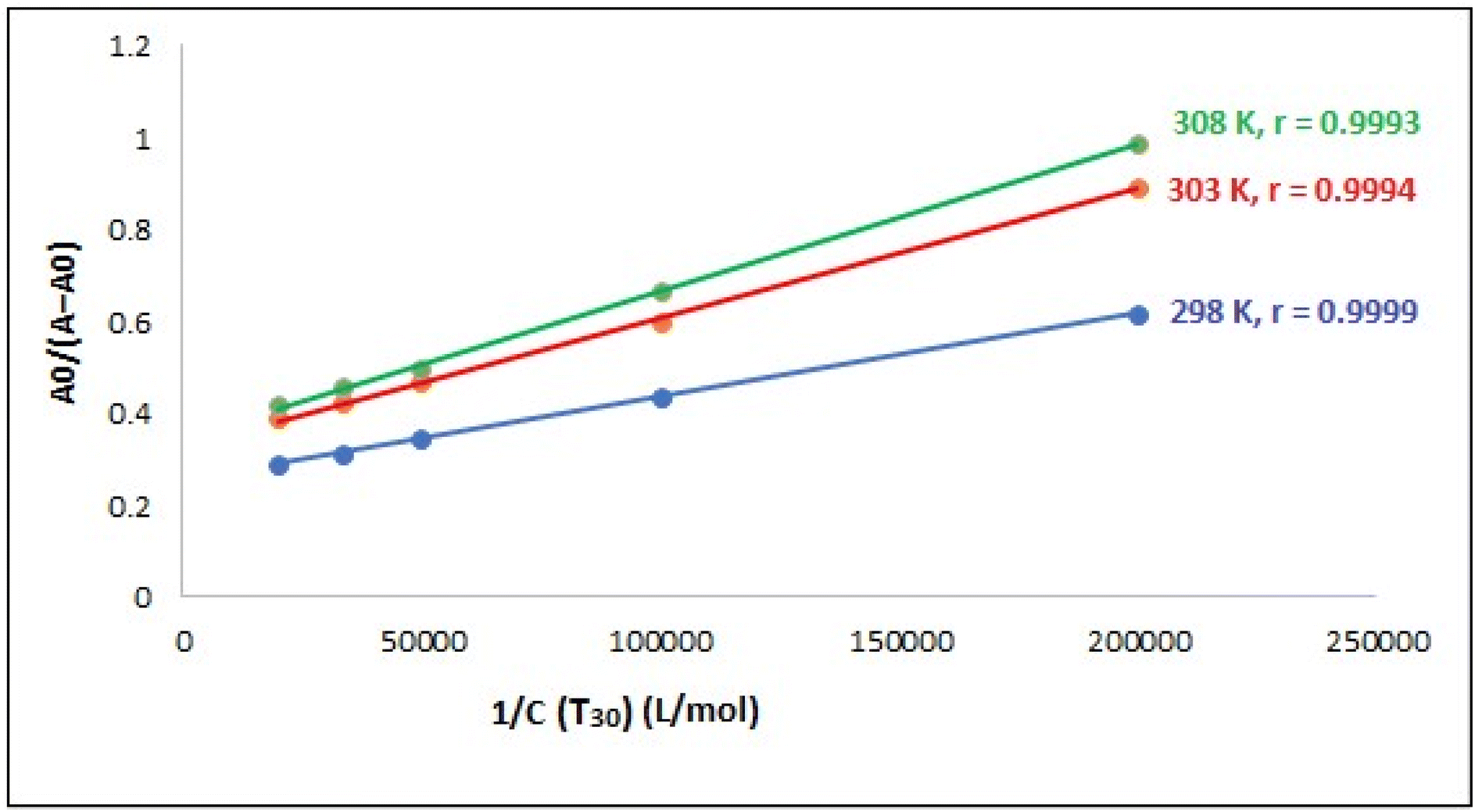 | ||
| Fig. 12 A plot of A0/(A − A0) versus 1/Ccompound T30 at different temperature settings (CDNA = 97.0 μM), where r is the correlation coefficient. | ||
 | (2) |
| ΔG° = ΔH° − TΔS° | (3) |
The graph depicted in Fig. 13 illustrates the relationship between ln Kb and 1/T, according to Van't Hoff's equation. The plot's slope and intercept were utilized to estimate ΔH° and ΔS°, correspondingly, which are shown in Table 2. The presence of hydrophobic contacts typically leads to positive values of ΔH° and ΔS°. Since the result of ΔH° was greater than zero, it provided further confirmation that the binding process was endothermic. Furthermore, it was found that ΔG° is negative, indicating a spontaneous binding association. Therefore, the process of compound T30 binding with ctDNA is both endothermic and spontaneous. The main binding forces in this interaction appear to be hydrophobic interactions.
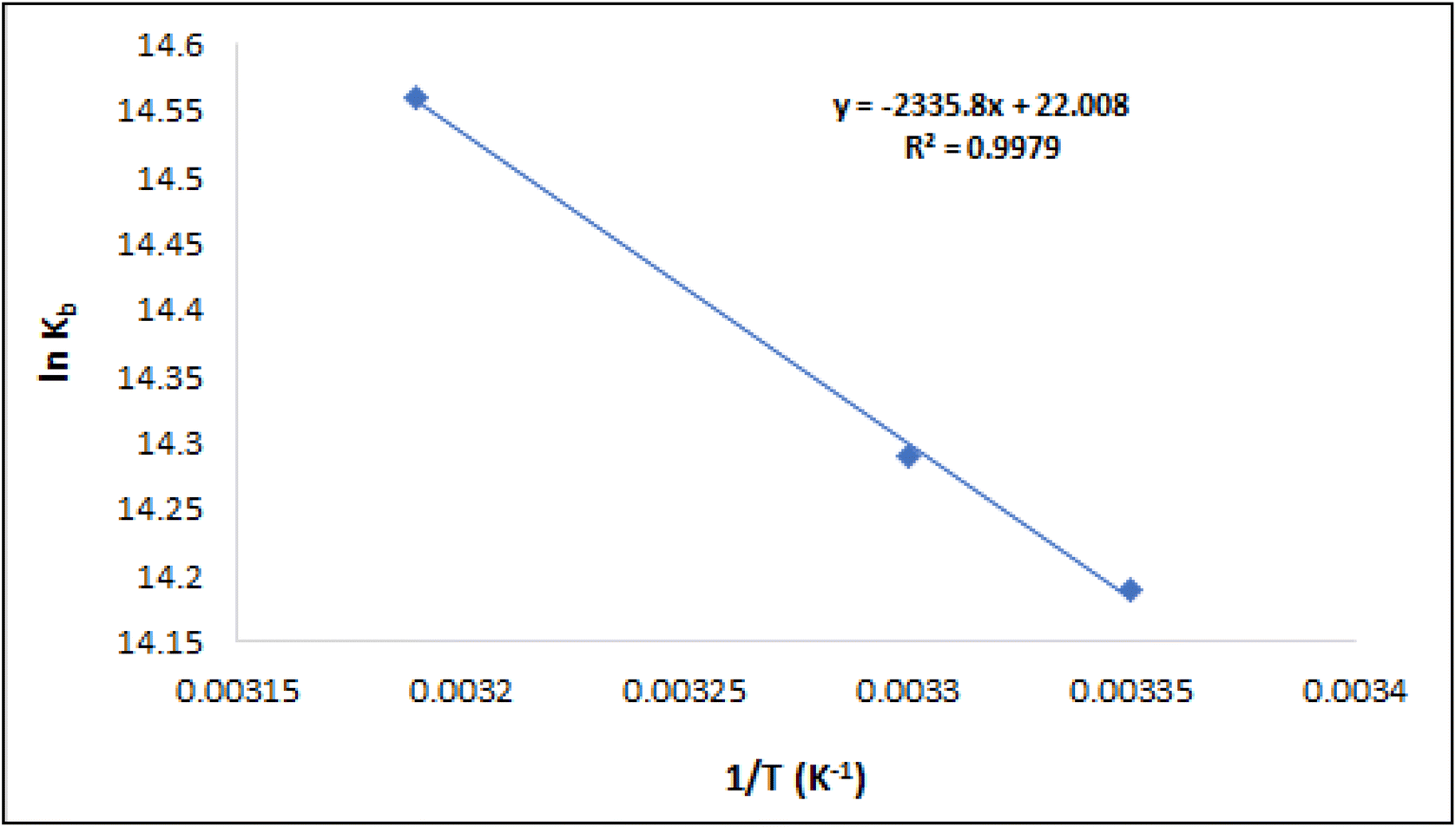 | ||
| Fig. 13 Vanʼt Hoff graph for the ctDNA-compound T30 complex. | ||
3. Conclusion
In this study, the theoretically designed novel substituted tetrahydropyrimidine analogues (T1–35) were designed as promising DNA intercalators and Topo-II inhibitors. Notably, the theoretically designed analogue (T30) with a 3,4,5-trimethoxy phenyl side chain was found to be the superior candidate among the investigated compounds in comparison to both reference standards, Dox and EVP. Molecular dynamics simulation revealed that compound T30 showed the most stable behaviour with an L-RMSD value of 6.3 Å, as a maximum fluctuation level. However, Dox and EVP fluctuated to 8 and 9 Å, respectively, indicating their less stable behaviours compared to T30. Moreover, the target candidate (T30) achieved superior ΔG binding energy (−33.86 kcal mol−1), which was better than that of both Dox and EVP. Furthermore, the most promising candidate (T30) was synthesized and denoted as compound T30. Compound T30 revealed very strong antiproliferative activity with mean growth = −22% and mean GI% = 122%, and showed the greatest anticancer activity towards all 59 cell lines. We conclude that compound T30 is remarkably effective against non-small cell lung cancer (NCI–H522) and melanoma (LOX IMVI). Moreover, the binding interaction was investigated using several spectroscopic techniques, ionic strength studies, viscosity tests, and in silico investigations. The data demonstrated a strong affinity between compound T30 and ctDNA, indicating an intercalation binding relationship. Furthermore, the binding between compound T30 and ctDNA was spontaneous, and the resulting complex was mostly held together by hydrophobic interactions. In summary, this study presented comprehensive data on the characteristics of this interaction, including how the molecules connect, the strength of the binding, the specific location of the binding site, and the forces involved in the interaction. This information is highly significant for rationally designing anticancer candidates, intending to enhance their activity and selectivity.4. Experimental
4.1. In silico studies
4.2. Chemistry
The initial chemicals and reagents were acquired from Sigma-Aldrich and Merck Millipore, and were utilized without additional purification. The progression of the reactions was tracked using a pre-coated sheet of thin-layer chromatography obtained from Eastman Kodak Co. The mobile phase was a mixture of n-hexane and ethyl acetate at a ratio of 2:3. The melting point was determined using a Stuart SMP10 instrument and was not adjusted for any corrections. The PerkinElmer 2400 CHNS analyzer was used to conduct elemental analysis, and the results were found to be within ±0.40 of the theoretical values. 1D and 2D NMR spectra were recorded on a JEOL spectrometer using DMSO-d6. The HRMS were recorded on LC/Q-TOF, 6530 (Agilent Technologies, Santa Clara, CA, USA) at the Faculty of Pharmacy, Fayoum University. Compound T30 is 98.96% pure by HPLC analysis. The column used in HPLC was ZORBAX Eclipse plus C18 (150 × 4.6 mm, 3.5 μm). The mobile phase consisted of acetonitrile (ACN)/0.05 M KH2PO4 buffer pH 3.7 (30:70), with a flow rate of 1.5 mL min−1, and the run time was set as double the elution time. The ESI file† contains the spectrum data, as well as their interpretation.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OOCH2). 13C NMR-DEPTQ spectrum, CH and CH3 [positive (up)], CQ and CH2 [negative (down)], revealed the following signals at δ: 14.49 (OCH2CH3 ↑), 29.39 (NCH3 ↑), 38.50 (CH2Cl ↓), 52.59 (THPM 4-CH ↑), 56.24 (3,5-diOCH3 ↑), 60.48 (4-OCH3 ↑), 60.91 (OCH2CH3 ↓), 103.59 (2-CH and 6-CH of benzene ring ↑), 105.70 (5-CQ of THPM ↓), 137.32 (1-CQ of benzene ring ↓), 138.95 (4-CQ of benzene ring ↓), 148.15 (6-CQ of THPM ↓), 153.38 (3-CQ and 5-CQ of benzene ring ↓), 153.55 (CO of THPM ↓), 164.99 (COOCH2 ↓). HRMS (ESI): m/z: Cald./Found 399.13174/399.13194 (C18H2435ClN2O6, [M + H]+), 401.12879/401.12784 (C18H2437ClN2O6, [M + H+2]+). Anal. Calcd. for C18H23ClN2O6: C, 54.21; H, 5.81; N, 7.02. Found: C, 53.99; H, 5.80; N, 6.97.
OOCH2). 13C NMR-DEPTQ spectrum, CH and CH3 [positive (up)], CQ and CH2 [negative (down)], revealed the following signals at δ: 14.49 (OCH2CH3 ↑), 29.39 (NCH3 ↑), 38.50 (CH2Cl ↓), 52.59 (THPM 4-CH ↑), 56.24 (3,5-diOCH3 ↑), 60.48 (4-OCH3 ↑), 60.91 (OCH2CH3 ↓), 103.59 (2-CH and 6-CH of benzene ring ↑), 105.70 (5-CQ of THPM ↓), 137.32 (1-CQ of benzene ring ↓), 138.95 (4-CQ of benzene ring ↓), 148.15 (6-CQ of THPM ↓), 153.38 (3-CQ and 5-CQ of benzene ring ↓), 153.55 (CO of THPM ↓), 164.99 (COOCH2 ↓). HRMS (ESI): m/z: Cald./Found 399.13174/399.13194 (C18H2435ClN2O6, [M + H]+), 401.12879/401.12784 (C18H2437ClN2O6, [M + H+2]+). Anal. Calcd. for C18H23ClN2O6: C, 54.21; H, 5.81; N, 7.02. Found: C, 53.99; H, 5.80; N, 6.97.4.3. Biological evaluation
4.4. DNA-binding study
A stock solution of compound T30 with a conc. of 1.0 × 10−3 M was obtained by dissolving it in a solution of DMSO/methanol. Additionally, RB with a concentration of 2.0 × 10−3 M and EB with a concentration of 1.2 × 10−3 M were prepared in ethanol, and stored in a dark environment at a temperature of 4 °C.
Data availability
All data generated or analyzed during this study are included in this published article and its ESI material.†Author contributions
Conceptualization and supervision: Ahmed A. Al-Karmalawy; data curation and visualization: Haytham O. Tawfik, Ayman Abo Elmaaty, Galal Magdy, Aya Saad Radwan, Radwan Alnajjar, Moataz A. Shaldam, and Ahmed A. Al-Karmalawy; methodology: Haytham O. Tawfik, Galal Magdy, Aya Saad Radwan, Radwan Alnajjar, Moataz A. Shaldam, Salem Salman Almujri, Abdullah Yahya Abdullah Alzahrani, and Ahmed A. Al-Karmalawy; Writing–review & editing: Haytham O. Tawfik, Ayman Abo Elmaaty, Galal Magdy, Aya Saad Radwan, Radwan Alnajjar, Moataz A. Shaldam, Arwa Omar Al Khatib, Salem Salman Almujri, Abdullah Yahya Abdullah Alzahrani, Ahmed A. Al-Karmalawy. All authors revised and approved the final submitted version of the manuscript.Conflicts of interest
The authors declared no conflict of interest.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through the large Group Research Project under grant number (RGP2/630/46).References
- F. Bray, et al., The ever-increasing importance of cancer as a leading cause of premature death worldwide, Cancer, 2021, 127(16), 3029–3030 CrossRef PubMed.
- F. Bray, et al., Global cancer transitions according to the Human Development Index (2008–2030): a population-based study, Lancet Oncol., 2012, 13(8), 790–801 CrossRef PubMed.
- H. O. Tawfik, et al., Rationale design of novel substituted 1,3,5-triazine candidates as dual IDH1(R132H)/IDH2(R140Q) inhibitors with high selectivity against acute myeloid leukemia: In vitro and in vivo preclinical investigations, Bioorg. Chem., 2024, 149, 107483 CrossRef CAS PubMed.
- R. S. Al Taher, et al., Correlation Between ImageJ and Conventional Manual Scoring Methods for Programmed Death-Ligand 1 Immuno-Histochemically Stained Sections, Technol. Cancer Res. Treat., 2024, 23, 15330338241242635 CrossRef CAS PubMed.
- S. Shaaban, et al., Repurposed organoselenium tethered amidic acids as apoptosis inducers in melanoma cancer via P53, BAX, caspases-3, 6, 8, 9, BCL-2, MMP2, and MMP9 modulations, RSC Adv., 2024, 14(26), 18576–18587 RSC.
- A. A. Gaber, et al., Multi-target rational design and synthesis of novel diphenyl-tethered pyrazolopyrimidines targeting EGFR and topoisomerase II with potential DNA intercalation and apoptosis induction, Bioorg. Chem., 2024, 145, 107223 CrossRef CAS.
- T. Helleday, et al., DNA repair pathways as targets for cancer therapy, Nat. Rev. Cancer, 2008, 8(3), 193–204 CrossRef CAS PubMed.
- T. A. El-Masry, et al., Therapeutic efficiency of Tamoxifen/Orlistat nanocrystals against solid ehrlich carcinoma via targeting TXNIP/HIF1-α/MMP-9/P27 and BAX/Bcl2/P53 signaling pathways, Biomed. Pharmacother., 2024, 180, 117429 CrossRef CAS.
- R. Ducray, et al., Novel 3-alkoxy-1H-pyrazolo [3, 4-d] pyrimidines as EGFR and erbB2 receptor tyrosine kinase inhibitors, Bioorg. Med. Chem. Lett., 2008, 18(3), 959–962 CrossRef CAS PubMed.
- M. M. Khalifa, et al., Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: design, synthesis, anti-proliferative, docking, and in vivo studies, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 299–314 CrossRef CAS PubMed.
- N. J. Wheate, et al., DNA intercalators in cancer therapy: organic and inorganic drugs and their spectroscopic tools of analysis, Mini-Rev. Med. Chem., 2007, 7(6), 627–648 CrossRef CAS.
- I. H. Eissa, et al., Diphenylurea derivatives for combating methicillin-and vancomycin-resistant Staphylococcus aureus, Eur. J. Med. Chem., 2017, 130, 73–85 CrossRef CAS.
- L. F. Liu, DNA topoisomerase poisons as antitumor drugs, Annu. Rev. Biochem., 1989, 58(1), 351–375 CrossRef CAS PubMed.
- T. D. Shenkenberg and D. D. Von Hoff, Mitoxantrone: a new anticancer drug with significant clinical activity, Ann. Intern. Med., 1986, 105(1), 67–81 CrossRef CAS.
- H. Nawaz, et al., Electrochemical DNA biosensor for the study of ciprofloxacin–DNA interaction, Anal. Biochem., 2006, 354(1), 28–34 CrossRef CAS.
- M. Alqaraleha, et al., Evaluation of the antiproliferative activity and molecular docking of selected branched fatty acids, J. Med. Pharm. Chem. Res., 2024, 6, 1340–1353 Search PubMed.
- K. B. Loboda, et al., Design and synthesis of 3, 5-substituted 1, 2, 4-oxadiazoles as catalytic inhibitors of human DNA topoisomerase IIα, Bioorg. Chem., 2020, 99, 103828 CrossRef CAS.
- E. M. Abbass, et al., Design, efficient synthesis, docking studies, and anticancer evaluation of new quinoxalines as potential intercalative Topo II inhibitors and apoptosis inducers, Bioorg. Chem., 2020, 104, 104255 CrossRef CAS PubMed.
- I. H. Eissa, A. M. El-Naggar and M. A. El-Hashash, Design, synthesis, molecular modeling and biological evaluation of novel 1H-pyrazolo [3, 4-b] pyridine derivatives as potential anticancer agents, Bioorg. Chem., 2016, 67, 43–56 CrossRef CAS.
- M. S. Alesawy, et al., Design and discovery of new 1, 2, 4-triazolo [4, 3-c] quinazolines as potential DNA intercalators and topoisomerase II inhibitors, Arch. Pharm., 2021, 354(3), 2000237 CrossRef CAS.
- E. Baldwin and N. Osheroff, Etoposide, topoisomerase II and cancer, Curr. Med. Chem.:Anti-Cancer Agents, 2005, 5(4), 363–372 CrossRef CAS PubMed.
- M. M. Mohamed, et al., Design, synthesis of new pyrimidine derivatives as anticancer and antimicrobial agents, Synth. Commun., 2017, 47(16), 1441–1457 CrossRef CAS.
- S. Suryawanshi, et al., Design, synthesis and biological evaluation of aryl pyrimidine derivatives as potential leishmanicidal agents, Bioorg. Med. Chem. Lett., 2013, 23(18), 5235–5238 CrossRef CAS.
- P. Liu, et al., Design and synthesis of novel pyrimidine derivatives as potent antitubercular agents, Eur. J. Med. Chem., 2019, 163, 169–182 CrossRef CAS.
- A. M. Farghaly, et al., Design, synthesis, and antihypertensive activity of new pyrimidine derivatives endowing new pharmacophores, Med. Chem. Res., 2019, 28, 360–379 CrossRef CAS.
- H. W. Lee, et al., Molecular design, synthesis, and hypoglycemic and hypolipidemic activities of novel pyrimidine derivatives having thiazolidinedione, Eur. J. Med. Chem., 2005, 40(9), 862–874 CrossRef CAS PubMed.
- Y. Fang, et al., Design and synthesis of novel pyrimido [5, 4-d] pyrimidine derivatives as GPR119 agonist for treatment of type 2 diabetes, Bioorg. Med. Chem., 2018, 26(14), 4080–4087 CrossRef CAS.
- A. Mobinikhaledi, B. Asghari and M. Jabbarpour, Design and synthesis of new benzimidazole and pyrimidine derivatives as α-glucosidase inhibitor, J. Pharm. Res., 2015, 14(3), 723 Search PubMed.
- V. N. Madia, et al., Design, synthesis and biological evaluation of new pyrimidine derivatives as anticancer agents, Molecules, 2021, 26(3), 771 CrossRef CAS.
- D. A. Ibrahim and A. M. El-Metwally, Design, synthesis, and biological evaluation of novel pyrimidine derivatives as CDK2 inhibitors, Eur. J. Med. Chem., 2010, 45(3), 1158–1166 CrossRef CAS PubMed.
- L. H. Amin, et al., Design, synthesis, anticancer evaluation and docking studies of new pyrimidine derivatives as potent thymidylate synthase inhibitors, Bioorg. Chem., 2019, 91, 103159 CrossRef.
- E. M. Mohi El-Deen, et al., Novel pyridothienopyrimidine derivatives: design, synthesis and biological evaluation as antimicrobial and anticancer agents, Molecules, 2022, 27(3), 803 CrossRef CAS PubMed.
- M. M. Hammoud, et al., Design, synthesis, biological evaluation, and SAR studies of novel cyclopentaquinoline derivatives as DNA intercalators, topoisomerase II inhibitors, and apoptotic inducers, New J. Chem., 2022, 46(23), 11422–11436 RSC.
- A. Narezkina, H. K. Narayan and A. E. Zemljic-Harpf, Molecular mechanisms of anthracycline cardiovascular toxicity, Clin. Sci., 2021, 135(10), 1311–1332 CrossRef.
- M.-D. I. Tools, Schrödinger, New York, NY, USA, 2017 Search PubMed.
- M. H. El-Hamamsy, et al., Design, synthesis, and molecular docking study of new monastrol analogues as kinesin spindle protein inhibitors, Arch. Pharm., 2020, 353(8), 2000060 CrossRef CAS.
- H. O. Tawfik, et al., Design, synthesis, and bioactivity of dihydropyrimidine derivatives as kinesin spindle protein inhibitors, Bioorg. Med. Chem., 2019, 27(23), 115126 CrossRef CAS.
- H. A. Elsebaie, et al., Novel 4-(2-arylidenehydrazineyl)thienopyrimidine derivatives as anticancer EGFR inhibitors: design, synthesis, biological evaluation, kinome selectivity and in silico insights, Bioorg. Chem., 2023, 140, 106799 CrossRef CAS.
- H. O. Tawfik, et al., Discovery of new carbonic anhydrase IX inhibitors as anticancer agents by toning the hydrophobic and hydrophilic rims of the active site to encounter the dual-tail approach, Eur. J. Med. Chem., 2022, 232, 114190 CrossRef CAS PubMed.
- P. Allart-Vorelli, et al., Haematological cancer and quality of life: a systematic literature review, Blood cancer J., 2015, 5(4), e305 CrossRef CAS.
- A. V. Kudryavtseva, et al., Important molecular genetic markers of colorectal cancer, Oncotarget, 2016, 7(33), 53959 CrossRef.
- A. Shergalis, et al., Current challenges and opportunities in treating glioblastoma, Pharmacol. Rev., 2018, 70(3), 412–445 CrossRef CAS PubMed.
- H. Tsao, et al., Melanoma: from mutations to medicine, Genes Dev., 2012, 26(11), 1131–1155 CrossRef CAS.
- T. M. Grzywa, W. Paskal and P. K. Włodarski, Intratumor and intertumor heterogeneity in melanoma, Transl. Oncol., 2017, 10(6), 956–975 CrossRef PubMed.
- B. A. Goff, Advanced ovarian cancer: what should be the standard of care?, J. Gynecol. Oncol., 2013, 24(1), 83–91 CrossRef CAS PubMed.
- A. Desai, et al., Epithelial ovarian cancer: An overview, World J. Transl. Med., 2014, 3(1), 1 CrossRef PubMed.
- H. J. Lim and W. Ledger, Targeted therapy in ovarian cancer, Women’s Health, 2016, 12(3), 363–378 Search PubMed.
- T. Grunewald and J. A. Ledermann, Targeted therapies for ovarian cancer, Best Pract. Res. Clin. Obstet. Gynaecol., 2017, 41, 139–152 CrossRef.
- L. Dirven and M. J. Taphoorn, Epidemiology of Central Nervous System Metastases, Central nervous system metastases, 2020: pp. 3–14 Search PubMed.
- A. T. Beksac, et al., Heterogeneity in renal cell carcinoma, in Urologic Oncology: Seminars and Original Investigations, Elsevier, 2017 Search PubMed.
- V. Tzelepi, et al., Modeling a lethal prostate cancer variant with small-cell carcinoma features, Clin. Cancer Res., 2012, 18(3), 666–677 CrossRef CAS PubMed.
- T. J. Key, P. K. Verkasalo and E. Banks, Epidemiology of breast cancer, Lancet Oncol., 2001, 2(3), 133–140 CrossRef CAS PubMed.
- K. J. Chavez, S. V. Garimella and S. Lipkowitz, Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer, Breast Dis., 2010, 32(1–2), 35 Search PubMed.
- K.-Y. Chen, et al., Exploring the binding interaction of calf thymus DNA with lapatinib, a tyrosine kinase inhibitor: multi-spectroscopic techniques combined with molecular docking, J. Biomol. Struct. Dyn., 2019, 37(3), 576–583 CrossRef CAS.
- M. Sirajuddin, S. Ali and A. Badshah, Drug–DNA interactions and their study by UV-Visible, fluorescence spectroscopies and cyclic voltametry, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- R. Palchaudhuri and P. J. Hergenrother, DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action, Curr. Opin. Biotechnol., 2007, 18(6), 497–503 CrossRef CAS.
- J. Saucier, B. Festy and J.-B. Le Pecq, The change of the torsion of the DNA helix caused by intercalation: II—measurement of the relative change of torsion induced by various intercalating drugs, Biochimie, 1971, 53(9), 973–980 CrossRef CAS PubMed.
- E. Grueso, et al., Thermodynamic and structural study of phenanthroline derivative ruthenium complex/DNA interactions: probing partial intercalation and binding properties, J. Inorg. Biochem., 2012, 106(1), 1–9 CrossRef CAS PubMed.
- S. Mahadevan and M. Palaniandavar, Spectroscopic and voltammetric studies on copper complexes of 2, 9-dimethyl-1, 10-phenanthrolines bound to calf thymus DNA, Inorg. Chem., 1998, 37(4), 693–700 CrossRef CAS.
- A. H. Hegde, S. Prashanth and J. Seetharamappa, Interaction of antioxidant flavonoids with calf thymus DNA analyzed by spectroscopic and electrochemical methods, J. Pharm. Biomed. Anal., 2012, 63, 40–46 CrossRef CAS PubMed.
- M. M. Islam, et al., Binding of DNA with Rhodamine B: Spectroscopic and molecular modeling studies, Dyes Pigm., 2013, 99(2), 412–422 CrossRef CAS.
- J. Li and C. Dong, Study on the interaction of morphine chloride with deoxyribonucleic acid by fluorescence method, Spectrochim. Acta, Part A, 2009, 71(5), 1938–1943 CrossRef CAS.
- D. Sahoo, P. Bhattacharya and S. Chakravorti, Quest for mode of binding of 2-(4-(dimethylamino) styryl)-1-methylpyridinium iodide with calf thymus DNA, J. Phys. Chem. B, 2010, 114(5), 2044–2050 CrossRef CAS.
- J.-H. Shi, et al., Characterization of interaction of calf thymus DNA with gefitinib: spectroscopic methods and molecular docking, J. Photochem. Photobiol., B, 2015, 147, 47–55 CrossRef CAS PubMed.
- J. Olmsted III and D. R. Kearns, Mechanism of ethidium bromide fluorescence enhancement on binding to nucleic acids, Biochemistry, 1977, 16(16), 3647–3654 CrossRef.
- P. D. Ross and S. Subramanian, Thermodynamics of protein association reactions: forces contributing to stability, Biochemistry, 1981, 20(11), 3096–3102 CrossRef CAS PubMed.
- J.-H. Shi, et al., Binding interaction between sorafenib and calf thymus DNA: spectroscopic methodology, viscosity measurement and molecular docking, Spectrochim. Acta, Part A, 2015, 136, 443–450 CrossRef CAS.
- M. A. Aziz, et al., Design, Synthesis, Biological Evaluation, 2D-QSAR Modeling, and Molecular Docking Studies of Novel 1H-3-Indolyl Derivatives as Significant Antioxidants, Int. J. Mol. Sci., 2021, 22(19), 10396 CrossRef CAS PubMed.
- C. Inc, Molecular Operating Environment (MOE), Chemical Computing Group Inc, 2016, p. 1010 Search PubMed.
- M. M. Hammoud, et al., Synthesis, structural characterization, DFT calculations, molecular docking, and molecular dynamics simulations of a novel ferrocene derivative to unravel its potential antitumor activity, J. Biomol. Struct. Dyn., 2022, 1–18 CrossRef.
- A. Belal, et al., Design, synthesis and molecular docking of new fused 1H-pyrroles, pyrrolo[3,2-d]pyrimidines and pyrrolo[3,2-e][1, 4]diazepine derivatives as potent EGFR/CDK2 inhibitors, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 1884–1902 CrossRef CAS PubMed.
- M. A. Salem, et al., Potential Valorization of Edible Nuts By-Products: Exploring the Immune-Modulatory and Antioxidants Effects of Selected Nut Shells Extracts in Relation to Their Metabolic Profiles, Antioxidants, 2022, 11(3), 462 CrossRef CAS PubMed.
- K. I. Eissa, et al., Design, synthesis, and biological evaluation of thienopyrimidine derivatives as multifunctional agents against Alzheimer’s disease, Drug Dev. Res., 2023, 84(5), 937–961 CrossRef CAS PubMed.
- D. Elebeedy, et al., In vitro and computational insights revealing the potential inhibitory effect of Tanshinone IIA against influenza A virus, Comput. Biol. Med., 2022, 141, 105149 CrossRef CAS.
- Y. Rahman, et al., Unravelling the interaction of pirenzepine, a gastrointestinal disorder drug, with calf thymus DNA: An in vitro and molecular modelling study, Arch. Biochem. Biophys., 2017, 625, 1–12 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02179k |
| This journal is © The Royal Society of Chemistry 2025 |