
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrain effects on catalytic activity and stability of PdM nanoalloys with grain boundaries†
Junpeng Wang‡
ab,
Tao Jin‡c,
Longfei Guoab,
Zhen Liab,
Chongyang Wangab,
Shuang Shanab,
Quan Tangab,
Bowei Panab and
Fuyi Chen *ab
*ab
aState Key Laboratory of Solidification Processing, Northwestern Polytechnical University, Xi'an 710072, China. E-mail: fuyichen@nwpu.edu.cn
bSchool of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an, 710072, China
cLongmen Laboratory, Luoyang, 471000, China
First published on 23rd May 2025
Abstract
Formate has emerged as a promising liquid hydrogen carrier for fuel cell applications, yet the kinetic limitations and stability issues of catalysts for formate dehydrogenation (FDH) and oxidation (FOR) remain challenging. Through systematic density functional theory (DFT) calculations, we computationally investigated how strain engineering modulates the electronic structure and catalytic behavior of PdM38 and PdM79 nanoalloys (M = Ir/Ag). Our theoretical models revealed that Ir atoms exhibit surface segregation driven by hydrogen/oxygen adsorption, effectively alleviating core lattice strain. Compressive strain was computationally observed to induce a negative shift in the d-band center of surface Pd sites. First-principles calculations identified core–shell PdIr and Janus-type PdAg configurations as optimal candidates, demonstrating enhanced theoretical activity for both FDH and FOR. This improvement was attributed to the elevated hydrogen adsorption free energy at Ir-enriched surfaces. By establishing a correlation between atomic strain, electronic structure, and catalytic descriptors, this computational study provides a theoretical framework for designing strain-engineered Pd-based catalysts, highlighting the critical role of element-specific segregation patterns in optimizing formate-based hydrogen storage systems as a hydrogen carrier and fuel.
1. Introduction
Hydrogen is a promising clean energy carrier, capable of storing excess energy from renewable sources through water electrolysis.1,2 However, its widespread adoption is hindered by inefficient storage technologies.3 Storing hydrogen in chemical bonds (e.g., formate salts, MHCO2, M = Na+, K+, and NH4+)2 offers a hydrogen storage capacity of 1.2–1.6 wt% (20–28 g H2 per L) and ambient reversibility.4–6 Formate further enables a closed carbon cycle through CO2 electroreduction,7 making it a sustainable energy carrier. Since Sasson et al. (1986) pioneered the sodium formate hydrogen storage system, research on FDH catalysts has been continuously advancing.8 Although the homogeneous Ru/Ir systems have reversible hydrogen storage capabilities, they face the bottleneck of homogeneous separation.9,10 Heterogeneous Pd-based catalysts (such as Pd/C and PdAg core–shell structures) achieve high activity (with a TOF of up to 311 h−1) through morphology/carrier regulation.7,9,11,12 Current research mostly focuses on physical dimensions (morphology, composition, carrier), but there is still a lack of systematic first-principles analysis of the in-depth mechanisms for performance improvement, such as atomic scale theories including electron transfer pathways, the regulation of the d-band center by strain effects, and the interfacial dynamic adsorption mechanism. This has restricted the targeted design of catalysts and breakthroughs in their performance.Formate shows potential as an energy-conversion device. In direct formate fuel cells (DFFC), it oxidizes easily to generate electricity, with a theoretical voltage of 1.45 V.12,13 Formate for DFFC can be produced via CO2 electroreduction, promoting carbon-cycle sustainability.13 However, DFFC's commercialization is hindered by the lack of efficient, stable catalysts for the anodic formate oxidation reaction (FOR).14 Pd-based catalysts avoid CO poisoning but suffer from slow FOR kinetics due to strong hydrogen intermediate (H*) binding, which blocks reactant adsorption. PdAg and PdCeO2 catalysts show excellent FOR activity.11,15–20 Adding Ir to PdAgIr nanoflower catalysts lowers the FOR onset potential. Doping AgPd core–shell nanoalloys with Ni creates AgPd Janus nanoalloys, but atomic-scale mechanisms remain unclear.21 Computational analysis suggests alloying Pd with Ag/Ir weakens H* adsorption, crucial for FOR kinetics.
Simulations of small clusters (38/79 atoms) reveal how truncated octahedral geometry links structural symmetry to catalytic activity, similar to high-index crystal plane behavior. Yudanov et al. simulated large particles (1000 atoms) using smaller clusters (∼300 atoms), optimizing the outer shell while keeping the core as bulk structure.22 Standard DFT calculations scale as O(N3) with valence electrons, limiting nanoparticle modeling to diameters of about 2–3 nm, smaller than the typical 4–10 nm range studied experimentally.23 Cao et al. found optimal specific activity for Pt–Ni octahedral catalysts at edge lengths >5.5 nm and Pt0.85Ni0.15 composition, while mass activity peaks at 3.3–3.8 nm and Pt0.8Ni0.2 composition. For nanoparticles of 10 nm (ref. 24) and 50 nm,21 compressive or tensile strain can be regulated to alter the d-band center and intermediate adsorption, enhancing FOR electrocatalytic activity or FDH catalytic performance.
In polymetallic nanomaterials, grain boundaries, as critical structural features, have a significant impact on catalytic performance.25 Firstly, the presence of grain boundaries increases the number of active sites, providing more adsorption positions for reactants.26 Secondly, the local strain at grain boundaries optimizes the electronic structure of the catalyst, adjusts the position of the d-band center, thereby enhancing the adsorption capacity for reaction intermediates.25 Moreover, the existence of grain boundaries stabilizes the catalyst structure, reduces the migration of defects such as dislocations and dissolution, thereby improving the durability of the catalyst.24 Taking the core–shell and sandwich structures of PdIr and AgPd as examples, the grain boundaries in these structures play a crucial role in enhancing catalytic activity and stability. Research shows that the presence of grain boundaries not only increases the number of active sites but also enhances the strength of the material by hindering dislocation movement, thereby improving the stability of the catalyst.
Structural distortions at grain boundaries can induce localized strain fields, which in turn cooperatively regulate the position of the d-band center. Strain effects can enhance catalytic performance.27 Atomic strain is controlled by adjusting metal composition and structure.28 Size effects are significant in catalysis, as size changes alter atomic strain and electronic properties.29 A 1% atomic strain shifts the Pt catalyst's d-band center by 0.1 eV, altering intermediate adsorption energy.30 Strain engineering optimizes intermediate adsorption energy through d-band center tuning, combined with elemental segregation dynamics, providing a theoretical framework for catalyst design. Jin et al. reported that compressive strain in AgPd bulk alloys shifts the d-band center downward, weakening intermediate adsorption and enhancing FOR catalytic efficiency.19
The precise synthesis of PdM38 and PdM79 (M = Ir, Ag) nanoalloys remains a significant challenge. However, recent advancements in the spatially controlled growth of bimetallic nanoparticles, such as PdAu Janus and PtNi core-frame structures, provide promising pathways.21,31 In PdAg systems, the high miscibility of Pd and Ag may facilitate interfacial alloying, yet the thermodynamic preference for Janus configurations can be strategically controlled through kinetic regulation during reduction.32,33 Notably, Zhang et al. successfully synthesized Au@PdAg and Au@PtRh sandwich structures using a seed-assisted reduction method, demonstrating the experimental feasibility of multi-layer metal deposition.34 This achievement offers experimental validation for the PdIr and PdAg sandwich structures predicted by DFT in this study. Although the synthesis of PdIr and PdAg sandwich structures still faces challenges, including metal compatibility, interface control, and precise control over size and morphology, existing experimental methods suggest their feasibility. Future research should prioritize optimizing synthesis techniques, such as ligand-assisted approaches, and validating their catalytic performance to substantiate the theoretically predicted structures and properties. No systematic theoretical study has explored PdIr and PdAg nanoalloys' catalytic activities for FDH and FOR. FDH and FOR occur in liquid phase, where catalysts are embedded in solvents, and adsorbates accumulate on the surface.
Surface segregation in alloy nanoparticles, induced by CO, H, and adsorbed oxygen, raises the question of whether H (an FDH/FOR intermediate) and O in oxidative settings can induce segregation.35,36 In this computational study, we employ density functional theory (DFT) and molecular dynamics simulations to systematically investigate PdIr and PdAg nanoalloys (Janus, core–shell, sandwich) for FDH/FOR, along with investigating the electronic structure and atomic strain of PdM38 and PdM79 (M = Ir, Ag) nanoalloys. We also explore segregation, anti-segregation phenomena and the structural stability of Janus and core–shell nanoalloys. Results show Pd32Ir6, Pd60Ir19 core–shell and Pd19Ag19, Pd34Ag45 Janus nanoalloys have the highest FDH and FOR catalytic activities in relevant groups. In vacuum, Ag segregates from core to shell; with H and O adsorption, Pd in PdAg and Ir in PdIr do so, relieving internal stress. The component redistribution induced by segregation behavior alters the strain field distribution near grain boundaries, thereby enabling dynamic regulation of active sites.
Additionally, the analysis of atomic strain shows tensile strain in the Ag region and compressive strain in the Pd region of PdAg Janus and sandwich nanoalloys. In PdAg core–shell ones, cores have tensile strain and shells have compressive strain. Compressive strain exists at the interface of PdIr sandwich nanoalloys. This research deepens understanding of PdIr and PdAg nanoalloys' effects on formate reactions, guiding catalyst development and providing new views on formate-related power sources for portable devices. Importantly, the proposed core–shell and Janus configurations build upon experimentally realized analogs: PdAg core–shell structures10 and PdAu Janus nanoparticles32 synthesized via kinetically controlled reduction, demonstrating the feasibility of our design principles.
2. Methods
2.1 Nanoalloy structure generation
The Birmingham Nanoalloy Genetic Algorithm (BCGA) software,37 in combination with a many-body Gupta potential,38 was utilized to create PdM38 and PdM79 (M = Ir, Ag) nanoalloys. These nanoalloys include pure monometallic nanoalloys of Ir, Ag, and Pd, as well as bimetallic nanoalloys with Janus, sandwich, and core–shell structures. The choice of nanoalloys with 38 and 79 atoms was based on their magical numbers, resulting in complete truncated octahedral (TO) structures.39 Both (100) and (111) surfaces enclose these nanoalloys. Henceforth, the PdM38 and PdM79 (M = Ir, Ag) nanoalloys were donated as Ir38, Ag38, Pd38, Pd19M19 Janus, Pd20M18 sandwich, Pd32M6 core–shell, Pd34M45 Janus, Pd19M60 sandwich, and Pd60M19 core–shell (M = Ir, Ag) nanoalloys, respectively. The Gupta potential35 accurately describes the surface energy of a metal compared to other empirical potential, such as the embedded atom method (EAM) potential.40 It can be applied at the appropriate time corresponding to each structure, ensuring the structure converges directly to its stable conformation regardless of the initial configuration.2.2 Molecular dynamics (MD) simulations
The MD simulations were used to determine the atomic strain fields and stress distributions of the PdIr and PdAg nanoalloys. The simulations employed the neural network (NN) interatomic potential41 with the LAMMPS software42 due to the absence of an empirical potential for PdIr. To achieve the equilibrium configuration, conjugate gradient (CG) minimization was initially applied to the entire nanoalloy, following by MD simulations using the canonical ensemble (NVT) at 50 K with a Nose–Hoover thermostat43 for 100 picoseconds. The atomic strain was calculated by comparing the final configuration with the initial configuration and then mapped atom-wise onto the final configuration using the OVITO software.44 The ESI† contains detailed instructions for calculating and visualizing the atomic strain using the OVITO software. This methodology is consistent with previously reported studies.45–472.3 Density functional theory (DFT) calculations
The DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) employing the periodic plane-wave method.48–50 The electron exchange and correlation interaction were described using the projected augmented wave (PAW) scheme51 and the Perdew–Burke–Ernzerhof (PBE)52 functional, employing the generalized gradient approximation (GGA). To ensure complete structural relaxation, all model atomic configurations were relaxed. The plane wave cutoff was set to 450 eV, and the self-consistent field and atomic force convergence criteria were set to 1 × 10−7 eV and 0.02 eV Å−1, respectively, for both structural relaxation and other calculations. The nanoalloys were positioned in the centre of a suitably large supercell (30 Å) to ensure a sufficient separation between periodic images. The Brillouin zone was sampled using the Γ point. Additionally, ab initio molecular dynamics simulations (AIMD) were performed in the canonical ensemble (NVT)53 with Nose–Hoover thermostate54 at 300, 400, 500 and 600 K for a time period of 20 ps.2.4 Thermodynamic treatment
The thermodynamics of the formate oxidation and formate dehydrogenation reaction mechanisms were evaluated by computing the Gibbs free energy of the various reaction steps as follows:ΔG = ΔH − TΔSwhere ΔH is the enthalpy change (including total energy of electrons, vibrational contribution, zero-point energy (ZPE) and neglecting pΔV), T is set to the room temperature of 298.15 K, and ΔS is the vibrational entropy change (obtained from the partition function as the reactant molecules are adsorbed on the catalyst surface and the translational and rotational entropy is restricted). The total energy of the adsorbent was calculated by VASP, and the thermodynamic corrections including the ZPE, the vibrational internal energy, and the vibrational entropy obtained from the partition function are obtained from the VASPKIT script.55 The other details of the calculations are provided in the ESI.†3. Results and discussion
Initially, the PdM38 and PdM79 (M = Ir, Ag) nanoalloys obtained through genetic algorithm were subjected to DFT structural relaxation. Fig. S1† illustrates the atomic structures of PdM38 and PdM79 (M = Ir, Ag) nanoalloys with truncated octahedral (TO) structures following DFT relaxation. Fig. S1(a) and (c)† demonstrate that the PdIr38 and PdAg38 nanoalloys maintained complete TO structures after DFT relaxtion, while Fig. S1(b) and (d)† reveal that the Pd34Ir45 Janus and Pd34Ag45 Janus nanoalloys displayed minor deformations. These nanoalloys have a core–shell structure, with Pd atoms forming the shell and Ir or Ag atoms occupying the core.The DFT calculations were conducted on PdM38 and PdM79 (M = Ir, Ag) nanoalloys to investigate their catalytic activity for FDH and electrocatalytic activity for FOR. At room temperature, formate can release hydrogen through the dehydrogenation reaction, and the elementary steps of FDH56 are as follows:
| HCOO− + * → HCOO* | (3.1) |
| HCOO* → H* + CO2 | (3.2) |
| H* → 1/2H2 + * | (3.3) |
The format oxidation reaction (FOR) plays a critical role as the anode reaction in the DFFC, ultimately determining its performance. According to prior research,15,17 the electrochemical oxidation of HCOO− to CO2 and H2O in an alkaline environment follows a direct associative pathway, typically involving of the following elementary steps:
| HCOO− + * → HCOO*− | (3.4) |
| HCOO*− → H* + CO2 + e− | (3.5) |
| H* + OH− → H2O + * + e− | (3.6) |
The adsorption of reactants onto the catalyst surface is a fundamental process in catalytic reactions, and accordingly, the adsorption energy serves as a crucial determinant of reaction catalytic activity. Excessive or insufficient adsorption energy can hinder the catalytic reaction. Therefore, it is imperative to optimize the adsorption energy of the catalyst towards the reactants and their intermediate products to achieve optimal catalytic activity. The adsorption energies of H, OH, and HCOO on PdM38 and PdM79 (M = Ir, Ag) nanoalloys are presented in Fig. 1.
 | ||
| Fig. 1 The absorption energy of H, OH, and HCOO on (a) PdIr38, (b) PdIr79, (c) PdAg38 and (d) PdAg79 nanoalloys. | ||
Fig. 1(a) and (b) shows that the core–shell PdIr nanoalloy displays moderate adsorption energy for H, OH, and HCOO, whereas the sandwich and Janus PdIr nanoalloys exhibit strong adsorption energy for these species. This difference is attributed to the Ir element's robust adsorption energy on its surface for H, OH, and HCOO. As Fig. 1(c) and (d) demonstrates, the Janus structure of PdAg nanoalloy exhibits moderate adsorption energy for H, OH, and HCOO, whereas the sandwich and core–shell structures exhibit stronger adsorption energy.
Fig. 2 summarizes the FDH catalytic properties of PdM38 and PdM79 (M = Ir, Ag) nanoalloys. The results reveal that the primary limitation to FDH activity stems from the hindered desorption of hydrogen intermediates (denoted as ΔG3), which results from excessively strong hydrogen adsorption free energy, as illustrated in Fig. 2(a–d). Consequently, ΔG3 emerges as the crucial evaluation parameter for assessing FDH reaction performance.
 | ||
| Fig. 2 Catalytic activity of the formate dehydrogenation reaction (FDH) on PdM38 and PdM79 (M = Ir, Ag) nanoalloys. The free energy diagram of FDH on (a) PdIr38, (b) PdIr79, (c) PdAg38 and (d) PdAg79 nanoalloys. (e) The ΔG3 of FDH of the PdM38 and PdM79 (M = Ir, Ag) nanoalloys. (f) A plot of the ΔG3 of FDH versus the hydrogen adsorption free energy. | ||
Fig. 2(e) illustrates the ΔG3 for the FDH of PdM38 and PdM79 (M = Ir, Ag) nanoalloys. The Ir38, Ag38, Ir79 and Ag79 nanoalloys exhibit inferior FDH catalytic activity, with ΔG3 values of 0.536, 0.568, 0.563 and 0.613 eV, respectively. In contrast, the Pd38 and Pd79 nanoalloys demonstrate ΔG3 values of 0.362 and 0.369 eV, respectively, indicating Pd as the active element for FDH. Moreover, the ΔG3 values for FDH of Pd32Ir6 core–shell, Pd60Ir19 core–shell, Pd19Ag19 Janus and Pd34Ag45 Janus nanoalloys are 0.263, 0.275, 0.194 and 0.206 eV, respectively. These findings suggest that synthesizing catalysts with specific shapes can enhance the catalytic activity of FDH. Additionally, we computed the catalytic activity of FDH in bulk Ir, Ag, and Pd, comparing it to that of nanoalloys. As illustrated in Fig. S2,† the FDH catalytic activity of the nanoalloys surpasses that of the bulk Ir, Ag, and Pd.
Fig. 2(f) provides a summary of the relationship between hydrogen adsorption free energy and ΔG3 in FDH. The free energy of hydrogen adsorption exhibits a volcano-type relationship with FDH activity, with an optimum free energy value yielding the highest activity. This is due to the stronger hydrogen adsorption free energy hindering the desorption of H intermediates, while the weaker hydrogen adsorption free energy is not conducive to the reaction. Among the PdM38 and PdM79 (M = Ir, Ag) nanoalloys, the Pd19Ag19 Janus nanoalloy exhibits the highest FDH catalytic activity, primarily attributed to its moderate H adsorption free energy.
Furthermore, a previous study57 has indicated that the selectivity of PdAu nanoalloys in HCOOH dehydrogenation relies on the surface Pd atoms. In particular, the Pd atom positioned at the Pd–Au interface demonstrates a higher inclination for HCOOH dehydrogenation, while the Pd atoms located at the Pd(111) surface, lacking coordination with Au atom, preferentially facilitate the dehydration of HCOOH. Consequently, the Janus-structured PdAu nanoalloy proves favourable for HCOOH dehydrogenation, consistent with the current calculations demonstrating that the Pd19Ag19 Janus nanoalloy exhibits the best FDH catalytic activity. The Janus structure also serves as a miniature model of grain boundaries. Grain boundaries have a significant impact on the adsorption energy. The local strain at grain boundaries can optimize the electronic structure of the catalyst, thereby modulating the adsorption energy of reactants. For example, the Pd19Ag19 Janus nanoalloy exhibits moderate H adsorption energy at the grain boundaries, contributing to its superior FDH catalytic activity. This electronic structure optimization induced by grain boundaries offers new insights for catalyst design.
The catalytic activities of PdM38 and PdM79 (M = Ir, Ag) nanoalloys for the FOR were evaluated by calculating their overpotentials, as depicted in Fig. 3. Fig. 3(a–d) presents the free energy diagram of PdM38 and PdM79 (M = Ir, Ag) nanoalloys at equilibrium potential, revealing that the obstacle step for Ir38, Ag38, Pd38, Ir79, Ag79, Pd79, Pd19Ir60 sandwich, Pd34Ag45 Janus and Pd60Ag19 core–shell nanoalloys is the breaking of the C–H bond in the formate molecule. Conversely, for Pd19Ir19 Janus, Pd20Ir18 sandwich, Pd32Ir6 core–shell, Pd34Ir45 Janus, Pd60Ir19 core–shell, Pd20Ag18 sandwich, Pd32Ag6 core–shell and Pd19Ag60 sandwich nanoalloys, the obstacle step is the oxidation of the H intermediate to H2O. The Pd32Ir6 core–shell, Pd60Ir19 core–shell, Pd19Ag19 Janus and Pd34Ag45 Janus nanoalloys exhibit the highest FOR catalytic activity among the PdM38 and PdM79 (M = Ir, Ag) nanoalloys. The core–shell structure of the PdIr38 and PdIr79 nanoalloys demonstrates the best FOR catalytic activity due to the excessive hydrogen adsorption free energy of Ir elements on the surface of Janus and sandwich structures.
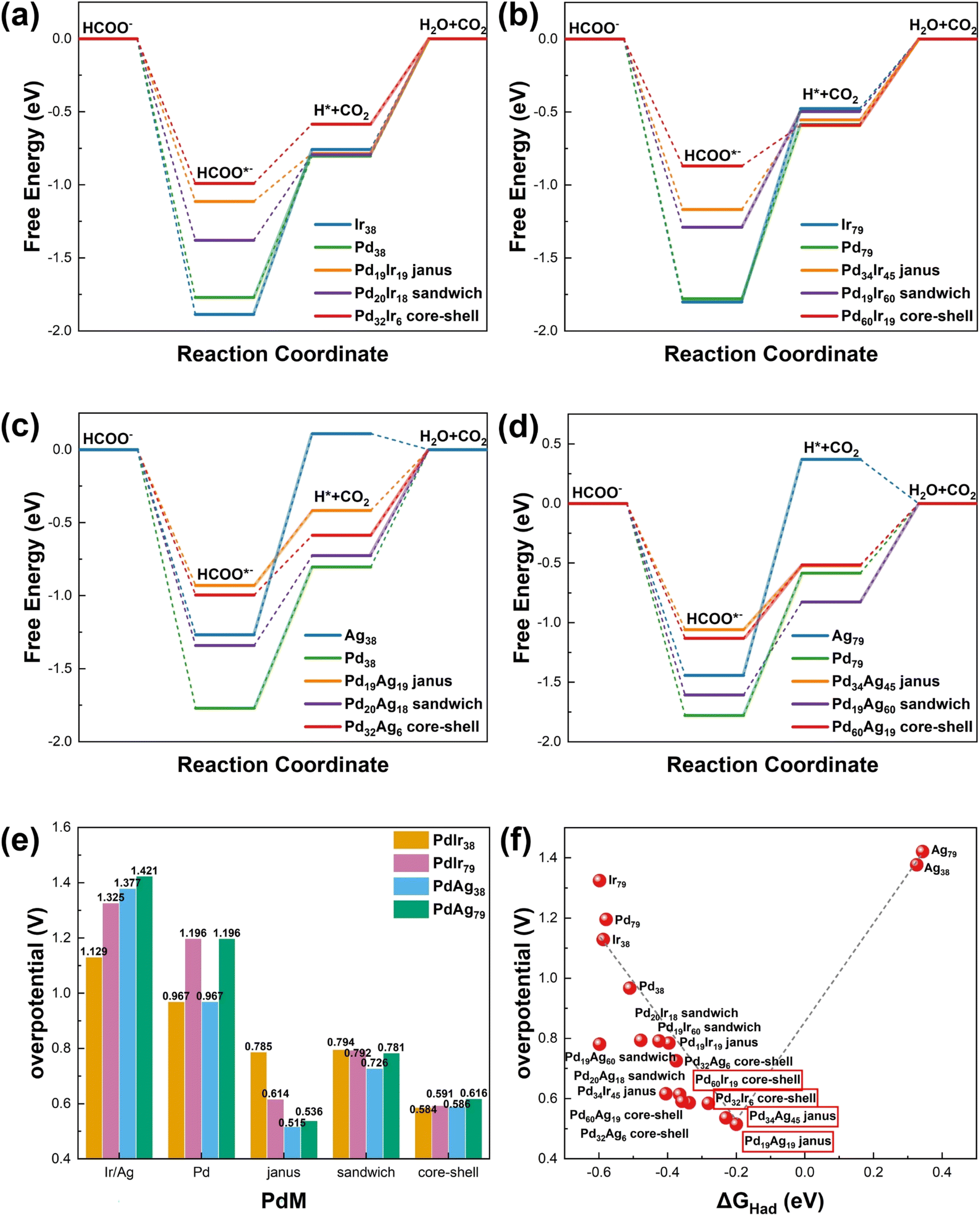 | ||
| Fig. 3 Catalytic activity of the formate oxidation reaction (FOR) on PdM38 and PdM79 (M = Ir, Ag) nanoalloys. The free energy diagram of FOR on (a) PdIr38, (b) PdIr79, (c) PdAg38 and (d) PdAg79 nanoalloys. (e) The overpotential of FOR of the PdM38 and PdM79 (M = Ir, Ag) nanoalloys. (f) A plot of the overpotential of FOR versus the hydrogen adsorption free energy. | ||
Fig. 3(e) presents the FOR overpotentials of PdM38 and PdM79 (M = Ir, Ag) nanoalloys. The FOR overpotentials for Ir38, Ag38, Pd38, Ir79, Ag79, and Pd79 nanoalloys are 1.129, 1.377, 0.967, 1.325, 1.421 and 1.196 V, respectively. However, the inclusion of the alloying effect of Pd with Ir or Ag significantly reduced the overpotentials of Pd32Ir6 core–shell, Pd60Ir19 core–shell, Pd19Ag19 Janus and Pd34Ag45 Janus nanoalloys to 0.584, 0.591, 0.515 and 0.536 V, respectively. Fig. 3(f) illustrates the relationship between the H adsorption free energy and FOR overpotential, indicating the presence of an optimum free energy value that results in the highest activity. This relationship is consistent with the relationship between H adsorption free energy and FDH catalytic activity. Fig. S3–S6† illustrates the FOR free energy of PdM38, PdM79 (M = Ir, Ag) nanoalloys at 0 V, equilibrium potential and overpotential, as well as the overpotential of FOR. Additionally, we calculated the free energy and overpotential of the FOR for bulk Ir, Ag, and Pd. This data is illustrated in Fig. S7 and S8.† The FOR catalytic activity of the PdM38 and PdM79 (M = Ir, Ag) nanoalloys surpasses that of the bulk Ir, Ag, and Pd. Table 1 summarizes the catalytic and electrocatalytic activities of nanoalloy PdM38 and PdM79 (M = Ir, Ag) for the FDH and FOR, as well as the adsorption free energy of hydrogen and formate, in comparison to their bulk counterparts, Ir, Ag, and Pd.
| ΔGHad/eV | ΔGHCOOad/eV | ΔG3/eV | η/V | ΔGHad/eV | ΔGHCOOad/eV | ΔG3/eV | η/V | ||
|---|---|---|---|---|---|---|---|---|---|
| Ir(100) | −0.505 | −2.471 | 0.657 | 1.281 | Ir(111) | −0.539 | −2.082 | 0.628 | 1.079 |
| Ag(100) | 0.425 | −1.909 | 0.987 | 1.338 | Ag(111) | 0.461 | −1.682 | 1.052 | 1.203 |
| Pd(100) | −0.491 | −1.863 | 0.437 | 0.659 | Pd(111) | −0.513 | −1.756 | 0.385 | 0.607 |
| Ir38 | −0.587 | −3.060 | 0.536 | 1.129 | Ir79 | −0.598 | −2.974 | 0.563 | 1.325 |
| Ag38 | 0.326 | −2.440 | 0.568 | 1.377 | Ag79 | 0.343 | −2.593 | 0.613 | 1.421 |
| Pd38 | −0.510 | −2.164 | 0.362 | 0.967 | Pd79 | −0.579 | −2.043 | 0.369 | 1.196 |
| Pd19Ir19 Janus | −0.396 | −2.944 | 0.324 | 0.785 | Pd34Ir45 Janus | −0.365 | −2.938 | 0.317 | 0.614 |
| Pd20Ir18 sandwich | −0.478 | −2.286 | 0.334 | 0.794 | Pd19Ir60 sandwich | −0.425 | −2.952 | 0.322 | 0.792 |
| Pd32Ir6 core–shell | −0.281 | −2.254 | 0.263 | 0.584 | Pd60Ir19 core–shell | −0.358 | −2.462 | 0.275 | 0.591 |
| Pd19Ag19 Janus | −0.200 | −2.104 | 0.194 | 0.515 | Pd34Ag45 Janus | −0.230 | −2.523 | 0.206 | 0.536 |
| Pd20Ag18 sandwich | −0.375 | −2.515 | 0.504 | 0.726 | Pd19Ag60 sandwich | −0.598 | −2.386 | 0.619 | 0.781 |
| Pd32Ag6 core–shell | −0.337 | −2.168 | 0.364 | 0.586 | Pd60Ag19 core–shell | −0.405 | −2.491 | 0.397 | 0.616 |
For a catalyst, a lower relative d-band center to the Fermi level results in lower adsorption energy for the adsorbed intermediates. As the d-band center shifts upward, a distinctive antibonding state emerges above the Fermi level.58 The empty antibonding states above the Fermi level correspond to stronger bonds, which becomes increasingly stronger as their numbers increase.59 Consequently, strong bonding occurs when the antibonding states are shifted upward though the Fermi level, resulting in their emptiness, whereas weak bonding occurs when the antibonding states are shifted downward through the Fermi level, leading to their filling.60
We calculated the projected density of states (PDOS) and d-band center for PdM38 and PdM79 (M = Ir, Ag) nanoalloys. As shown in Fig. S9 and S10,† the Pd32Ir6 core–shell, Pd60Ir19 core–shell, Pd19Ag19 Janus and Pd34Ag45 Janus nanoalloys have a d-band center of −2.320, −2.819, −2.855 and −2.598 eV, located between the Ir38, Ag38, Pd38, Ir79, Ag79, and Pd79 nanoalloys. These findings suggest that the Pd32Ir6 core–shell, Pd60Ir19 core–shell, Pd19Ag19 Janus and Pd34Ag45 Janus nanoalloys have moderate d-band center, indicating moderate adsorption free energy of H intermediates. The d-band center of PdM38 and PdM79 (M = Ir, Ag) nanoalloys is shown in Fig. S10.† This d-band modulation mechanism provides a unified framework for understanding both FDH and FOR enhancements. While FDH activity is directly governed by H desorption energetics (ΔG3), the improved FOR performance in core–shell/Janus structures originates from dual effects: (1) strain-induced d-band downshifting weakens HCOO adsorption to facilitate C–H cleavage in simple structures, and (2) optimized H adsorption energy enables efficient H oxidation kinetics in complex architectures. The structural design flexibility allows decoupling of adsorption energetics for different intermediates, circumventing the traditional scaling relationship limitations.
Given that a large library of atomically precise metal nanocatalysts have been structurally resolved, it is important to probe the segregation or anti-segregation during the reactions from ab initio calculations to advance the fundamental understanding of the catalytic stability.61 Fig. S11† illustrates the vacuum Eseg of core–shell PdIr and PdAg nanoalloys, Ag@Pd and Ir@Pd nanoalloys as −0.100 and 0.488 eV, respectively, indicating an Ag surface segregation trend for Ag@Pd, while Ir shows no tendency to segregate. This behavior is due to the higher surface energy of Ir (3.048 J m−2) compared to that of Pd (2.003 J m−2) and Ag (1.246 J m−2).62 Conversely, under H and O adsorption, the segregation energy is above zero for Ag@Pd and below zero for Ir@Pd, signifying a propensity for Pd atom surface segregation in Ag@Pd and Ir atom surface segregation in Ir@Pd during the reaction. This occurrence arises from the stronger binding energy of H and O on Ir in comparison to Pd and Ag.63
The stability of Janus and core–shell nanoalloys exhibiting higher FDH and FOR catalytic activities was investigated using AIMD simulations. The illustration in Fig. S12† demonstrates that Ir atoms consistently occupy the core, aligning with the segregation energy calculations. Conversely, Ag atoms segregate from the core to the shell at a temperature of 600 K under vacuum conditions. This observation is consistent with prior research,64 indicating that the segregation processes can be stopped at any time by lowering temperature below 573 K. Consequently, subsequent AIMD simulations were conducted at a temperature of 600 K. Fig. S13† displays the atomic structures of Ir6@Pd32, Ag6@Pd32, Pd6@Ag32 core–shell nanoalloys, and Pd19M19 Janus (M = Ir, Ag) nanoalloys after DFT optimization and AIMD simulation. After AIMD simulation, Ag atoms initially situated in the core of the Ag6@Pd32 nanoalloy undergo segregated into the shell. Conversely, the remaining core–shell nanoalloys did not experience segregation, and the Janus nanoalloys preserved their structural stability after the AIMD simulation.
The atomic structure of PdM38 Janus (M = Ir, Ag) nanoalloys under various H and O coverages is depicted in Fig. 4, following DFT optimization and AIMD simulation. After DFT optimization and AIMD simulations, the Pd19M19 Janus (M = Ir, Ag) nanoalloys demonstrate sustained structural stability. This stability is further reinforced by grain boundaries, which play a pivotal role in enhancing durability by inhibiting dislocation motion, as evidenced in PdIr/PdAg core–shell and sandwich configurations. Additionally, grain boundaries significantly contribute to the structural stability of catalysts, mitigating deactivation during reactions. For example, in the core–shell and sandwich structures of PdIr and PdAg, grain boundaries stabilize the catalyst structure and enhance its durability. This stability is essential for the long-term performance of catalysts in practical applications.
 | ||
| Fig. 4 Atomic structure evolution of PdIr Janus (a), PdAg Janus (b) nanoalloys after adsorption of hydrogen and oxygen atoms with varying coverage, investigated using DFT optimization and AIMD simulations at 600 K. The PdM Janus (M = Ir, Ag) nanoalloys maintain structural stability after AIMD simulations. | ||
The atomic structure of PdM38 core–shell (M = Ir, Ag) nanoalloys is illustrated in Fig. S14,† following DFT optimization and AIMD simulation at different H and O atomic coverage. Upon the adsorption of a single H and O atom on the surface of the Pd32Ir6 core–shell nanoalloy, Ir atoms remain confined within the core and do not segregate to the shell. Conversely, when eight or more H and O atoms are adsorbed on the surface, AIMD simulations reveal the segregation of Ir atoms from the core to the shell, as depicted in Fig. S14(a).† In the case of the Pd32Ag6 core–shell nanoalloy, the adsorption of H and O atoms onto the surface does not prompt the segregation of Ag atoms from the core to the shell. This outcome is attributed to the stronger binding energy of Pd with H and O, as visually depicted in Fig. S14(b).† To establish a comparison, we conducted additional investigations into the segregation behavior of the Pd6@Ag32 core–shell nanoalloy under different H and O coverage. As shown in Fig. S15,† the adsorption of 8 or more H and O atoms onto the surface led to the segregation of Pd atoms from the core to the shell. This phenomenon can be attributed to the higher binding energy between Pd and H or O. The total energy versus the time for AIMD simulations is depicted in Fig. S16 and S17.†
Several critical factors contribute to segregation. Firstly, differences in cohesive energy play a significant role. Secondly, disparities in surface energy drive the tendency for elements with lower surface energy to segregate to the surface. Thirdly, variations in atomic size, with larger atomic radii, promote surface segregation, aiding the release of internally accumulated stress. Lastly, variations in binding energy are also influential.65
The relationship between atomic strain and the activity of formate dehydrogenation and oxidation was investigated through theoretical calculations. To assess the impact of size on atomic strain, we focused on comparing two isomers of PdM (M = Ir, Ag) nanoalloys containing 38 and 79 atoms. Fig. S1† demonstrates that both PdM38 and PdM79 (M = Ir, Ag) nanoalloys have a substantial density of edge and corner sites on their surfaces. During the minimization stage of the MD simulation, surface atoms have a tendency to minimize their energy by deviating from their original positions, which is termed as atomic strain.42
Fig. 5 depict the stress distribution in the surface and cross-section of PdIr38 nanoalloy along the x-, y-, and z-directions. The results show that the subsurface of the nanoalloy experienced compressive stress, which is consistent with previous research.66 In addition, the outer surface atoms, which lack coordinating atoms, were found to be under tensile stress. The Ir38 and Pd38 nanoalloy exhibits tensile strain at the face center and compressive strain at the corners and edges. For the Pd19Ir19 Janush and Pd20Ir18 sandwich nanoalloys, compressive strain was observed at the Pd–Ir interface. The Pd32Ir6 core–shell nanoalloy has a slight tensile strain. This similarity in atomic radii, with Ir and Pd measuring 1.27 and 1.28 Å,64 respectively, could account for this observation.
 | ||
| Fig. 5 The stress distribution and atomic strain field maps of Ir38, Pd38, Pd19Ir19 Janus, Pd20Ag18 sandwich, and Pd32Ir6 core–shell nanoalloys. (a) and (b) The x-direction, (c) and (d) y-direction, and (e) and (f) z-direction for surface and cross-sectional stress. (g) and (h) The atomic strain at the surface and in the cross-section. The Pd19Ir19 Janus and Pd20Ag18 sandwich nanoalloys exhibit compressive strain at the Ir–Pd interface. The unit of measurement for stress is bar. | ||
Fig. 6 illustrates the stress distribution and atomic strain field of the PdAg38 nanoalloy. The subsurface of the nanoalloy experienced compressive stress, which is consistent with previous research.63 In addition, the outer surface atoms, which lack coordinating atoms, were found to be under tensile stress. The Ag38 cluster exhibits compressive strain at the face center and tensile strain at the corners and edges. In contrast, the Pd38 cluster has the opposite distribution of atomic strain, with tensile strain at the face center and compressive strain at the corners and edges. For the Pd19Ag19 Janus and Pd20Ag18 sandwich clusters, tensile strain was observed in the Ag atoms region, and compressive strain in Pd atoms region. The Pd32Ag6 core–shell cluster has the same distribution of atomic strain as the Pd38 cluster, with tensile strain at the face center and compressive strain at the corners and edges. The atomic strain distribution in the cross-section of the PdAg38 nanoalloy is consistent with the atomic strain at the surface, as shown in Fig. 6(h). The compressive strain on the surfaces of Ag38, Pd38, Pd19Ag19 Janus, Pd20Ag18 sandwich and Pd32Ag6 core–shell clusters is −0.77%, −0.79%, −0.75%, −0.87% and −0.70%, respectively. This is due to the larger atomic radius of Ag than that of Pd, which are 1.34 and 1.28 Å,67 respectively.
 | ||
| Fig. 6 The stress distribution and atomic strain field maps of Ag38, Pd38, Pd19Ag19 Janus, Pd20Ag18 sandwich, and Pd32Ag6 core–shell nanoalloys. (a) and (b) The x-direction, (c) and (d) y-direction, and (e) and (f) z-direction for surface and cross-sectional stress. (g) and (h) The atomic strain at the surface and in the cross-section. In the Pd19Ag19 Janus and Pd20Ag18 sandwich nanoalloys, the Ag atoms experience tensile strain while the Pd atoms experience compressive strain. The unit of measurement for stress is bar. | ||
To investigate the impact of size on atomic strain in PdM (M = Ir, Ag) nanoalloys, we calculated the atomic strain field of PdIr79 and PdAg79 nanoalloys, as shown in Fig. S18 and S19.† The PdIr79 and PdAg79 nanoalloy indicates compressive stress in the subsurface, consistent with the results of the PdIr38 and PdAg38 nanoalloy. Additionally, the surface of the PdIr79 and PdAg79 nanoalloy experiences slightly tensile stressed due to the lack of coordination atoms. The compressive strain of Ag79, Pd79, Pd34Ag45 Janus, Pd19Ag60 sandwich and Pd60Ag19 core–shell nanoalloys is −1.01%, −1.12%, −1.27%, −0.88% and −0.89%, respectively. These results indicate that increasing the size of the nanoalloys leads to higher compressive strain, while the tensile strain remains constant. The displacements of PdM38 and PdM79 (M = Ir, Ag) nanoalloys along the x, y, and z direction after MD simulations are shown in Fig. S20–S23.†
In this study, the PdIr core–shell structure (e.g., Pd32Ir6) shifts the d-band center of surface Pd downward (−2.32 eV) through interfacial compressive strain (∼−0.79%), reducing hydrogen adsorption strength (ΔG_H = 0.263 eV) and enhancing FDH/FOR activity. Under H/O adsorption, Ir migrates to the surface to relieve internal stress, while high surface energy inhibits segregation. In the PdAg Janus structure (e.g., Pd19Ag19), tensile strain in the Ag region and compressive strain in the Pd region synergistically regulate the d-band center (−2.86 eV), balancing adsorption/desorption kinetics (ΔG3 = 0.194 eV) to meet “volcano-type” activity requirements, while maintaining dynamic stability at 600 K.
Based on the presented calculation results, it can be inferred that Pd30Ir6 core–shell, Pd60Ir19 core–shell, Pd19Ag19 Janus and Pd34Ag45 Janus nanoalloys exhibit superior catalytic activities for FDH and FOR. This can be attributed to the strain present on the surface of nanoalloy, which directly modulates the d-band center (Fig. S10†) as evidenced by: (1) −0.75% compressive strain in Pd19Ag19 Janus lowering the d-band center to −2.855 eV, (2) corresponding ΔGH reduction to 0.194 eV (Fig. 2(e)), and (3) simultaneous FOR overpotential minimization to 0.515 V (Fig. 3(e)). The complete strain → d-band → ΔGH → activity pathway is quantitatively validated through cross-referencing Fig. 5, 6, S10 with 2e, 3e. The Pd30Ir6 core–shell and Pd60Ir19 core–shell nanoalloys is the optimal catalyst candidate for FDH and FOR due to its maximum tensile strain on its surface. The Pd19Ag19 Janus and Pd34Ag45 Janus nanoalloys is the optimal catalyst candidate for FDH and FOR due to its maximum compressive strain on its surface. These findings are consistent with a previous theoretical study54 on the formic acid dehydrogenation activity of PdAu nanoalloys, which showed that Janus-shaped PdAu nanoalloys have better formic acid dehydrogenation selectivity and activity. In practical FDH and FOR conditions, Ir atoms originally positioned in the core may undergo segregation towards the shell, while PdAg Janus nanoalloys exhibit the capability to sustain their structural stability. Segregation in nanoalloys can be attributed to the interplay between variations in binding energy for H and O, differences in surface energy, and the alleviation of internal stress.
4. Conclusions
In this work, we utilized the Birmingham Nanoalloy Genetic Algorithm software to obtained atomic structures of PdM38 and PdM79 (M = Ir, Ag) nanoalloys. Three representative atomic arrangements, including Janus, sandwich, and core–shell structures, were considered. The results show that compressive stress is present inside both PdM38 and PdM79 (M = Ir, Ag) nanoalloys. The atomic strain distribution in Janus and sandwich structures is element dependent, with tensile strain present in the Ag atoms region and compressive strain present in the Pd atoms region. In addition, our calculations reveal that as the size of the nanoalloy increases, the compressive atomic strain on the surface becomes more increased, while the tensile strain remains constant. Density functional theory (DFT) calculations suggest that PdIr nanoalloys with core–shell structures exhibit optimal formate dehydrogenation (FDH) and oxidation reactions (FOR) catalytic activity, while PdAg nanoalloys with Janus structures exhibit optimal FDH and FOR catalytic activity due to the excessive H adsorption free energy of Ir elements on the Janus and sandwich surfaces. Our calculations demonstrate that decreasing the adsorption free energy of H intermediates can improve FDH and FOR catalytic activity. Based on the segregation energy and ab initio molecular dynamics (AIMD) simulations, it is observed that Ag segregates to the shell under vacuum conditions, whereas Ir segregates to the shell under H and O conditions. Overall, this work provides valuable theoretical insights for the design and development of efficient formate dehydrogenation catalysts and anode catalysts for direct formate fuel cells.Data availability
The data underlying this study are available in the published article and its online ESI.†Author contribution
J. W. and T. J. contributed equally to this work. J. W.: data curation, project administration, validation, writing manuscript. T. J.: figures prepared, data curation, project administration, validation. L. G.: project administration. Z. L. and C. W.: validation, experimental design. S. S. and Q. T.: validation. B. P.: figures prepared. F. C.: project administration, funding acquisition, writing manuscript. All the authors have approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 51874243, 51271148 and 50971100), the Research Fund of State Key Laboratory of Solidification Processing (NPU), China (Grant no. 2020-TS-02), the Project of Transformation of Scientific and Technological Achievements of NWPU (grant no. 19-2017), the Funding for Innovation and Venture Capital of Student Work Department of Party Committee of NWPU (grant no. 2021-CXCY-018), the Open Fund of State Key Laboratory of Advanced Technology for Material Synthesis and Processing (Wuhan University of Technology grant no. 2018-KF-18), the Frontier Exploration Projects of Longmen Laboratory (no. LMQYQN202408), and the Natural Science Foundation of Henan Province (252300420790). We would like to thank the Analytical & Testing Center of Northwestern Polytechnical University for TEM characterizations.References
- S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC.
- K. Grubel, H. Jeong, C. W. Yoon and T. Autrey, J. Energy Chem., 2020, 41, 216–224 CrossRef.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- Q.-Y. Bi, J.-D. Lin, Y.-M. Liu, X.-L. Du, J.-Q. Wang, H.-Y. He and Y. Cao, Angew. Chem., Int. Ed., 2014, 53, 13583–13587 CrossRef CAS PubMed.
- K. Müller, K. Brooks and T. Autrey, Energy Fuels, 2018, 32, 10008–10015 CrossRef.
- Z. Dong, A. Mukhtar, T. Ludwig, S. A. Akhade, S. Kang, B. Wood, K. Grubel, M. Engelhard, T. Autrey and H. Lin, Appl. Catal., B, 2023, 321, 122015 CrossRef CAS.
- H. Wiener, Y. Sasson and J. Blum, J. Mol. Catal., 1986, 35, 277–284 CrossRef CAS.
- K. Nakajima, M. Tominaga, M. Waseda, H. Miura and T. Shishido, ACS Sustain. Chem. Eng., 2019, 7, 6522–6530 CrossRef CAS.
- B. S. Choi, J. Song, M. Song, B. S. Goo, Y. W. Lee, Y. Kim, H. Yang and S. W. Han, ACS Catal., 2019, 9, 819–826 CrossRef CAS.
- B. Pan, S. Shan, J. Wang, Q. Tang, L. Guo, T. Jin, Q. Wang, Z. Li, M. Usman and F. Chen, Nanoscale, 2023, 15, 7032–7043 RSC.
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef PubMed.
- T. Jin, F. Chen, L. Guo, Q. Tang, J. Wang, B. Pan, Y. Wu and S. Yu, J. Phys. Chem. C, 2021, 125, 19497–19508 CrossRef CAS.
- L. F. Guo, T. Jin, Q. Tang, J. P. Wang, B. W. Pan, Q. Wang, Z. Li, C. Y. Wang, J. W. Liu and F. Y. Chen, J. Mater. Chem. A, 2022, 10, 13998–14010 RSC.
- Q. Tang, F. Chen, T. Jin, L. Guo, Q. Wang and H. Liu, J. Mater. Chem. A, 2019, 7, 22996–23007 RSC.
- Q. Wang, F. Chen, L. Guo, T. Jin, H. Liu, X. Wang, X. Gong and Y. Liu, J. Mater. Chem. A, 2019, 7, 16122–16135 RSC.
- J. L. Wang, F. Y. Chen, Y. C. Jin, L. F. Guo, X. F. Gong, X. L. Wang and R. L. Johnston, Nanoscale, 2019, 11, 14174–14185 RSC.
- T. Jin, L. Guo, Q. Tang, J. Wang, B. Pan, C. Wang, Z. Li and F. Chen, J. Phys. Chem. C, 2022, 126, 9683–9695 CrossRef CAS.
- Y. Jin, F. Chen, L. Guo, J. Wang, B. Kou, T. Jin and H. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 26694–26703 CrossRef CAS PubMed.
- Q. Wang, F. Chen, Q. Tang, L. Guo, T. T. Gebremariam, T. Jin, H. Liu, B. Kou, Z. Li and W. Bian, Appl. Catal., B, 2020, 270, 118861 CrossRef CAS.
- I. V. Yudanov, M. Metzner, A. Genest and N. Rösch, J. Phys. Chem. C, 2008, 112, 20269–20275 CrossRef CAS.
- L. Cao and T. Mueller, J. Am. Chem. Soc., 2023, 145, 7352–7360 CrossRef CAS PubMed.
- J. Wang, Z. Li, C. Wang, S. Shan, L. Guo, Q. Tang, Q. Wang, X. Wu and F. Chen, Appl. Surf. Sci., 2025, 688, 162454 CrossRef CAS.
- Z. H. Xia and S. J. Guo, Chem. Soc. Rev., 2019, 48, 3265–3278 RSC.
- L. Hou, Z. Li, H. Jang, M. G. Kim, J. Cho, S. Liu and X. Liu, Angew. Chem., Int. Ed., 2024, 63, e202315633 CrossRef CAS PubMed.
- H. Wang, S. Xu, C. Tsai, Y. Li, C. Liu, J. Zhao, Y. Liu, H. Yuan, F. Abild-Pedersen and F. B. J. S. Prinz, Mater. Sci., 2016, 354, 1031–1036 CAS.
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Nature, 2021, 598, 76–81 CrossRef CAS PubMed.
- D. Nafday, S. Sarkar, P. Ayyub and T. Saha-Dasgupta, ACS Nano, 2018, 12, 7246–7252 CrossRef CAS PubMed.
- J. Wu, L. Qi, H. You, A. Gross, J. Li and H. Yang, J. Am. Chem. Soc., 2012, 134, 11880–11883 CrossRef CAS PubMed.
- Y. Xia, K. D. Gilroy, H.-C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- J. S. Santana, K. M. Koczkur and S. E. Skrabalak, React. Chem. Eng., 2018, 3, 437–441 RSC.
- Z. Lyu, S. Zhu, L. Xu, Z. Chen, Y. Zhang, M. Xie, T. Li, S. Zhou, J. Liu, M. Chi, M. Shao, M. Mavrikakis and Y. Xia, J. Am. Chem. Soc., 2021, 143, 149–162 CrossRef CAS PubMed.
- J.-X. Zhang, C.-L. Lv, C. Tang, A.-J. Wang, L.-P. Mei, P. Song and J.-J. Feng, Sens. Actuators, B, 2023, 382, 133497 CrossRef CAS.
- H. Liao, A. Fisher and Z. J. Xu, Small, 2015, 11, 3221–3246 CrossRef CAS PubMed.
- C. Li, D. Raciti, T. Pu, L. Cao, C. He, C. Wang and T. Mueller, J. Phys. Chem. C, 2018, 122, 18040–18047 CrossRef CAS.
- R. L. Johnston, Dalton Trans., 2003, 22, 4193–4207 RSC.
- R. P. Gupta, Phys. Rev. B, 1981, 23, 6265–6270 CrossRef CAS.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, Nanoscale, 2014, 6, 1153–1165 RSC.
- M. J. Mehl and D. A. Papaconstantopoulos, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 4519–4530 CrossRef CAS PubMed.
- S.-D. Huang, C. Shang, P.-L. Kang, X.-J. Zhang and Z.-P. Liu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1415 CAS.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in 't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2010, 18, 015012 CrossRef.
- T. Wu, M. Sun and B. Huang, Angew. Chem., Int. Ed., 2021, 60, 22996–23001 CrossRef CAS PubMed.
- H. W. Huang, H. H. Jia, Z. Liu, P. F. Gao, J. T. Zhao, Z. L. Luo, J. L. Yang and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 3594–3598 CrossRef CAS PubMed.
- T. Jin, L. Guo, Q. Tang, J. Wang, B. Pan, Z. Li, C. Wang, S. Shan and F. Chen, Nanoscale, 2023, 15, 11131–11140 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- Q. Luo, G. Feng, M. Beller and H. Jiao, J. Phys. Chem. C, 2012, 116, 4149–4156 CrossRef CAS.
- W.-Y. Yu, G. M. Mullen, D. W. Flaherty and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 11070–11078 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, J. Chem. Phys., 2004, 120, 10240–10246 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- B. Hammer and J. K. Norskov, in Advances in Catalysis; Impact of Surface Science on Catalysis, ed. B. C. Gates and H. Knozinger, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- X. Zhang, S. Han, B. Zhu, G. Zhang, X. Li, Y. Gao, Z. Wu, B. Yang, Y. Liu, W. Baaziz, O. Ersen, M. Gu, J. T. Miller and W. Liu, Nat. Catal., 2020, 3, 411–417 CrossRef CAS.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- H. Lv, Y. A. Zheng, Y. Z. Wang, J. L. Wang, B. Liu and Z. A. Qiao, Angew. Chem., Int. Ed., 2023, 62, 202304420 CrossRef PubMed.
- Z. Kaszkur, W. Juszczyk and D. Łomot, Phys. Chem. Chem. Phys., 2015, 17, 28250–28255 RSC.
- A. Aguado, in Nanoalloys, ed. F. Calvo, Elsevier, Oxford, 2013, pp. 75–111, doi: DOI:10.1016/B978-0-12-394401-6.00003-5.
- E. Panizon and R. Ferrando, Nanoscale, 2016, 8, 15911–15919 RSC.
- J. C. Slater, J. Chem. Phys., 1964, 41, 3199–3204 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02127h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |