
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical pretreatment of a glassy carbon electrode for sensitive determination of epinephrine†
Tong Menga,
Yanshu Zhua,
Hangyu Guob and
Juan Zhang *a
*a
aSchool of Basic Medical Sciences, Ningxia Medical University, Yinchuan 750004, China. E-mail: zhangjuano13@126.com
bSchool of Inspection, Ningxia Medical University, Yinchuan 750004, China
First published on 29th May 2025
Abstract
In this study, a novel pretreatment of a glassy carbon electrode was conducted via cyclic voltammetry (CV), successfully fabricating an activated glassy carbon electrode (AGCE). A simple two-step electrochemical pretreatment method was established. During the first stage, anodic oxidation pretreatment was achieved by potentiometric scanning across a wide potential range and at a high anodic potential. The reduction stage was subsequently executed through potentiometric scanning within a narrow potential window at a reduced anodic potential. Field atomic force microscopy (AFM), electrochemical impedance spectroscopy (EIS), and CV were utilized to characterize the morphology and properties of the pretreated electrodes. The results indicate that the AGCE possesses a rough surface and exhibits increased oxidation peak currents and decreased overpotential during epinephrine (EP) oxidation. Quantitative determination of EP was performed via linear sweep voltammetry (LSV), revealing that the AGCE can effectively recognize EP in a wide range of interferences, spanning a broad linear range of 0.1–8, 8–100, and 100–700 μM. The detection limit (DL) was 0.032 μM. The AGCE exhibited favorable selectivity, stability, and reproducibility in the detection of EP.
1 Introduction
Epinephrine (EP), an essential catecholamine neurotransmitter produced in chromaffin cells of the adrenal medulla and certain central nervous system neurons, plays a pivotal role in nerve conduction and cardiovascular regulation. Alterations in adrenaline levels have been associated with the onset of various neurological disorders, including schizophrenia, Alzheimer's disease, and Parkinson's disease.1 EP has various medical applications, including the treatment of conditions such as cardiac arrest, bronchitis, allergic reactions, and emphysema. Additionally, it is utilized as a vasoconstrictor and coagulation promoter for skin and mucous membranes.2 However, EP has been misused by athletes in competitions as a stimulant and is included on the list of substances prohibited by the World Anti-Doping Agency (WADA).3 Therefore, detecting concentrations of EP is essential for clinical diagnosis and doping screening. Currently, a variety of analytical techniques are employed for the quantitative determination of EP, including high-performance liquid chromatography,4 chemiluminescence,5 capillary electrophoresis,6 mass spectrometry,7 fluorescence,8 and electrochemical methods.9 Among these detection techniques, electrochemical measurements have attracted significant interest because of their ease of use, rapidity, high sensitivity, and good controllability. However, electrochemical analysis of EP in biological fluids faces two major challenges: (1) extremely low concentration of EP; and (2) high concentration of interfering substances that are highly compatible with the redox properties of EP. Among them, endogenous ascorbic acid (AA) leads to severe electrochemical cross-reactivity due to the almost complete overlap of its oxidation potential with EP, and uric acid (UA), as a common co-existing electroactive substance, has oxidized signals that are prone to introduce background interference. In addition, the structural analog phenylephrine (PE) is widely present in biological samples as a drug, further exacerbating the difficulty of selective detection. Therefore, the development of novel electrochemical sensors with higher selectivity and sensitivity to detect EP is of great theoretical and applied value.The glassy carbon electrode (GCE) is an electrode material that possesses excellent electrochemical properties and is favored by researchers for electrochemical detection because of its low cost, outstanding electrical conductivity, electrochemical inertness, chemical stability under a wide potential window, and ease of surface modification and functionalization,10 making it a popular choice. However, bare electrodes often exhibit poor sensitivity and selectivity, which has led to significant research exploring electrode modification. Various nanomaterials, such as conducting polymers,11 metal nanostructures,12 carbon quantum dots,13 carbon nanotubes,14 graphene and its derivatives, as well as several novel composites15–17 have been successfully applied to electrochemical sensors for detecting EPs. Although these electrochemical sensing methods for detecting EP have yielded good experimental results, however, the synthesis of the modifying materials and their modification on the electrode surfaces are usually cumbersome processes that limit further applications.18
Carbon electrodes undergo an electrochemical pretreatment process, resulting in the formation of a rough and porous surface. Moreover, many oxygen-containing functional groups such as phenol, aldehyde, quinone, and carboxyl groups are generated. These groups greatly enhance the electrochemical activity of the electrodes, demonstrated by numerous studies.19–26 The most common methods of electrochemical oxidation used for pretreating GCEs include constant potential oxidation, constant current oxidation, and cyclic voltammetry (CV). It has been shown that constant potential pretreatment can form deep microporous structures on the electrode surface through continuous anodic polarization, while CV, by virtue of its dynamic polarization characteristics, can precisely regulate the generation of a uniform mesoporous structure on the surface layer of the GCE and significantly enhance the density of active sites on the surface.27 During electrochemical pretreatment, improper selection of parameters (e.g. potential window, scan rate, or treatment time deviating from the optimized range) may lead to surface corrosion and loss of structural integrity of glassy carbon electrodes.28 To minimize the risk of electrode damage, the electrode was pretreated by the CV method with higher controllability. The CV activation process can be divided into three characteristic potential intervals: anodic region, middle potential region, and cathodic region.29 Anodic oxidation induces a dense oxide passivation layer, which has excellent chemical stability but limited sensing sensitivity due to the low density of active sites; cathodic reduction significantly improves the sensitivity by localized carbon lattice reconfiguration to form a porous active layer, but overreduction is prone to structural collapse; anodic oxidation followed by cathodic reduction achieves better stability and activity.30 To improve the stability and electrochemical activity of the pretreated electrodes, and at the same time to mitigate the damage caused by the electrochemical pretreatment to the electrode itself, the two-step potentiodynamic CV method, which separates the anodic oxidation and cathodic reduction, was used for the first time in this study for the pretreatment of GCE, and the pretreatment conditions were optimized. In the first stage, anodic oxidation pretreatment was performed by scanning with a CV method at a large potential range and high anodic potential. In the second stage, reduction pretreatment was carried out by scanning with the CV method in a small potential range and at a low anodic potential. The activated glassy carbon electrode (AGCE) prepared under optimal parameter conditions has a wide linear range, low detection limit, and high stability, and can be used for the electrochemical detection of EP, which is a simple, low-cost, and eco-friendly electrode.
2 Experiments
2.1 Reagents
(±)-Epinephrine hydrochloride, (R)-(−)-phenylephrine hydrochloride, AA, UA, hexamine ruthenium chloride ([Ru(NH3)6]Cl3), potassium ferricyanide (K3[Fe(CN)6]), and potassium hexacyanoferrate (K4[Fe(CN)6]) were purchased from Aladdin (Shanghai, China). Potassium dihydrogen phosphate (KH2PO4), disodium hydrogen phosphate (Na2HPO4), potassium chloride (KCl), and sodium chloride (NaCl) were obtained from Sinopharm Group Chemical Reagent Co. Ltd (Shanghai, China). D(+)-glucose was purchased from Damao Chemical Reagent Factory (Tianjin, China). L-glycine and anhydrous ethanol were purchased from Guangnuo Chemical Technology Co. Ltd (Shanghai, China). Phosphate buffer (PB) solutions of different pH values were prepared by adjusting the ratio of disodium hydrogen phosphate to potassium dihydrogen phosphate. All reagents were of analytical grade and could be used directly without further purification. All aqueous solutions for the experiments were prepared with ultrapure water (UPR-II-40L, 18.25 MΩ cm, Ulupure Ultrapure Technology Co. Ltd, Sichuan, China).2.2 Instruments
CHI660E electrochemical workstation (CH Instrument Inc., Shanghai, China); a conventional three-electrode system consisting of a glassy carbon electrode (GCE, 0.07 cm2 geometric area) as the working electrode and a platinum foil (2 cm2 geometric area) as the counter electrode, sourced from Gaossunion Technology Ltd (Wuhan, China) and a commercially available saturated calomel electrode (SCE, CH Instrument Inc., Shanghai, China) as the reference electrode.KQ-100B ultrasonic cleaner (Ultrasonic Instrument Co. Ltd, Kunshan, China); Leici PHS-25 pH meter (Yidian Scientific Instrument Co. Ltd, Shanghai, China); MFP-3D-Origin (AFM, Oxford Instruments Co. Ltd, Britain) were used to characterize the surface morphology of the electrodes, infrared spectroscopy was carried out in the attenuated total reflection (ATR) mode (Thermo Scientific® Nicolet iS50) using a DTGS detector and a diamond crystal.
2.3 Preparation of activated pretreated GCE
The GCE was polished in a 0.05 μm Al2O3 suspension,31 then sequentially sonicated in ultrapure water, anhydrous ethanol, and ultrapure water for 15 s. To obtain AGCE, the cleaned GCE was immersed in 0.2 M PB (pH 5.0) with a CV between 0.5 and 2.0 V and a scan rate of 50 mV s−1 for 10 cycles, and then the potentiometric window was set to −0.5 to 1.0 V for 6 cycles at the same scan rate.2.4 Electrochemical measurements
EP was detected in PB at pH 7.4 via CV and linear sweep voltammetry (LSV) over a potential range of −0.2 to 0.6 V at a scan rate of 100 mV s−1. Control CV scans of PB without analytes were used as blank controls. After each measurement, the AGCE was immersed in 0.5 M H2SO4 aqueous solution for 2 min and then rinsed with deionized water to remove the residual EP and possible oxidation product from the electrode surface.The electrochemical impedance spectroscopy (EIS) measurements were performed via a specified solution containing 5.0 mM [Fe(CN)6]3−/4− in a 0.1 M NaCl solution at the open circuit potential. The signal amplitude was set to 5 mV, ranging from 105 Hz to 10−2 Hz. The data analysis was conducted via ZSimpWin software. All the electrochemical experiments were conducted at room temperature (25 ± 2 °C) and atmospheric pressure.
3 Results and discussion
3.1 Optimization of conditions of electrode pretreatment
The effects of potential window, pH, scanning rate, number of scanning cycles in the oxidation stage, and number of scanning cycles in the reduction stage in electrode pretreatment were investigated by detection of the CV oxidation peak current of 50 μM EP. | ||
| Fig. 1 Effect of (A and B) potential window, (C) pH, (D) scanning rate, (E) number of scanning cycles in the first stage, and (F) number of scanning cycles in the second stage in electrode pretreatment and each data point is based on 3 samples. | ||
The duration of the electrochemical reduction process is indirectly regulated by manipulating the number of scanning cycles during the reduction stage. The effect of different scanning cycles on AGCE was investigated during the electrochemical pretreatment of the reduction stage, as shown in Fig. 1F. The results show that the peak oxidation current of EP increases and then decreases with the change of the number of scanning cycles during the pretreatment process in the reduction stage, and the maximum value of the peak oxidation current of EP occurs when the number of scanning cycles is 6 cycles. After several repetitive detections, the oxidation peak current of EP was relatively stable, as shown in Fig. S2.† Therefore, six cycles are selected as the number of scanning cycles in the reduction stage.
3.2 Characterization of pretreated electrodes
The morphology of GCE and AGCE was characterized using AFM, and the AFM data were analyzed using Gwyddion software. Fig. 2(A and B) shows the two-dimensional (2D) and three-dimensional (3D) images of GCE, respectively, and the results show that there are a large number of polishing scratches on the newly polished GCE surface resulting in root-mean-square roughness (RMSR) and average roughness (Ra) of 2.87 nm and 2.28 nm. Fig. 2(C and D) shows the 2D and 3D images of AGCE, respectively, which have a rougher surface with RMSR and Ra of 5.16 nm and 4.07 nm, respectively, compared to GCE, and deeper and larger voids can be observed, which is attributed to the pretreatment. X-ray energy dispersive spectroscopy (EDS) reveals the distribution of carbon and oxygen on the surfaces of the GCE and AGCE in Fig. S3 and Table S1.† As shown in Fig. S3 and Table S1,† compared with that of the GCE, the oxygen content on the surface of the AGCE was 3.10% greater, which demonstrates that AGCE introduces active oxygen-containing functional groups, as aligns with the literature.40–43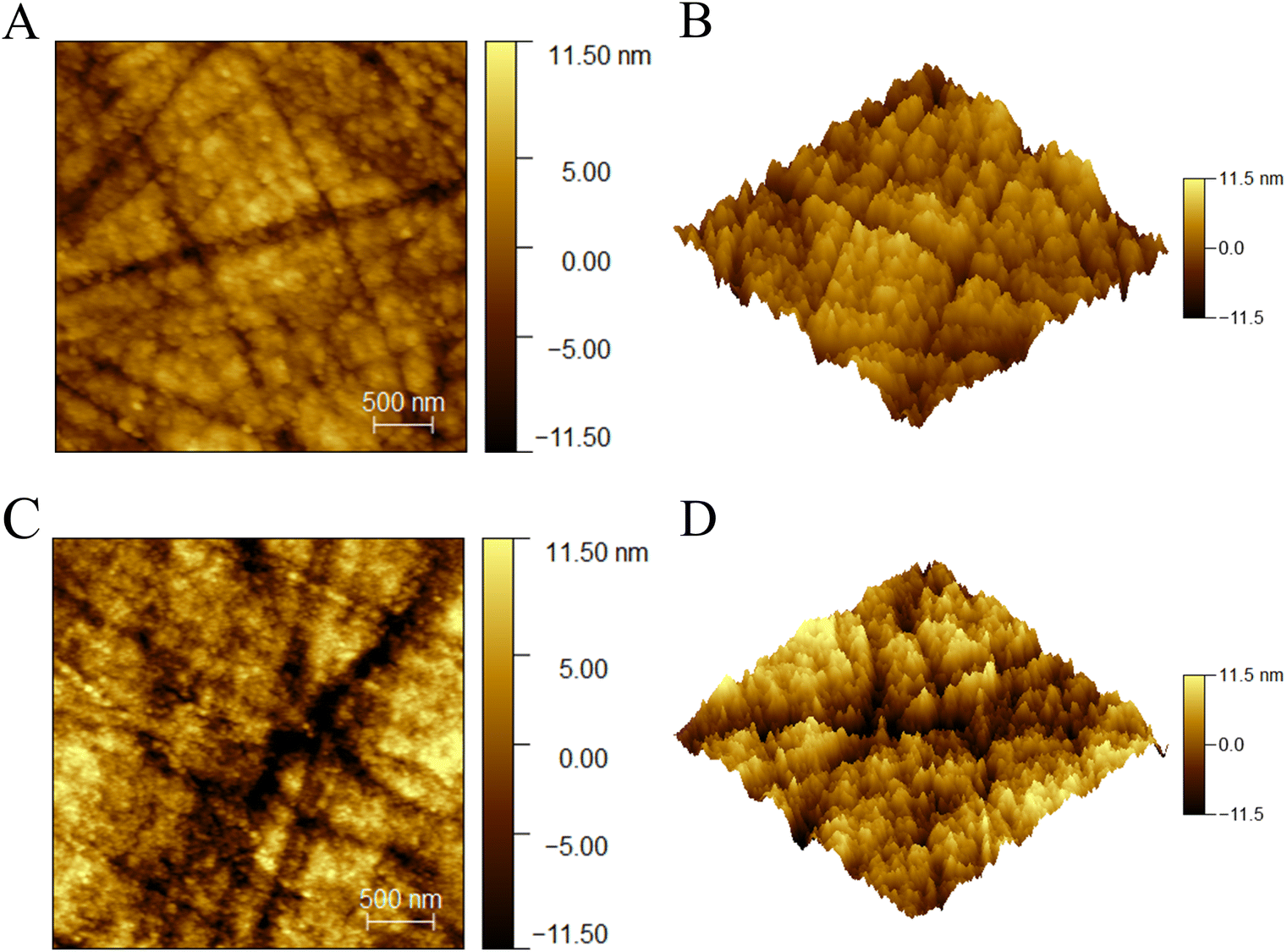 | ||
| Fig. 2 Atomic force microscopy images of the (A and B) GCE and (C and D) AGCE with a scanning range of 3 μm × 3 μm. | ||
The characterization of AGCE by using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) is shown in Fig. S4.† Several new characteristic bands of oxygen-containing functional groups appeared in the ATR-FTIR spectra of AGCE compared to GCE, with bands in the range of 1400–1800 cm−1 attributed to the carboxylate COO− group at 1683 cm−1, the quinone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group at 1558 cm−1, and the CC stretching vibration at 1505 cm−1.24 In addition, the bands in the range of 3050–3800 cm−1 are associated with hydroxyl stretching vibrations, i.e., hydroxyl O–H stretching vibrations at 3566 cm−1. The bands at 1000–1300 cm−1 can be attributed to ether or epoxy groups, i.e., stretching vibration of C–O single bond of the ether or epoxy at 985 cm−1.42 The results indicate that oxygen-containing functional groups have been successfully introduced on the surface of the GCE by a two-step electrochemical pretreatment process.
O group at 1558 cm−1, and the CC stretching vibration at 1505 cm−1.24 In addition, the bands in the range of 3050–3800 cm−1 are associated with hydroxyl stretching vibrations, i.e., hydroxyl O–H stretching vibrations at 3566 cm−1. The bands at 1000–1300 cm−1 can be attributed to ether or epoxy groups, i.e., stretching vibration of C–O single bond of the ether or epoxy at 985 cm−1.42 The results indicate that oxygen-containing functional groups have been successfully introduced on the surface of the GCE by a two-step electrochemical pretreatment process.
The interfacial properties of the AGCE were examined via electrochemical impedance spectroscopy (EIS) and CV. To ensure a more accurate comparison, the same GCE with or without pretreatment was utilized. The Nyquist plots of the GCE, GCE-ox, and AGCE are illustrated in Fig. 3A. The Nyquist fitting diagram and fitting results for a single electrode are shown in Fig. S5 and Table S2.† In the electrochemical impedance spectrum, the semicircle in the high-frequency region corresponds to the charge transfer process, the formation of which originates from the electron transfer resistance of the bilayer charging and electrochemical reaction at the interface between the electrode and the electrolyte, and the diameter of the semicircle directly corresponds to the electron transfer resistance. Fig. 3A shows that the diameter of the semicircle of the GCE-ox in the high-frequency region is significantly larger than that of the GCE and AGCE. This is due to an oxide layer, which hinders the transfer of electrons. However, the semicircular diameter of AGCE is slightly larger than that of GCE in the high-frequency region, which may be because, after the second stage of pretreatment, part of the oxidized layer still exists inside AGCE, forming a structure with a dense interior and a rough surface. In Table S2,† Rs is the solution resistance, which is related to the ionic concentration and conductivity of the electrolyte; Rct is the electron transfer resistance, which is related to the active sites on the electrode surface and the concentration of the reactants; Cdl is the double-electric-layer capacitance, which is related to the electrode surface area and the roughness; Qdl is the constant-phase-angle component related to the double-electric-layer capacitance, which is commonly used to describe the behavior of a nonideal double-electric layer; and Zw is the Warburg impedance that commonly used to characterize the effect of diffusion processes on the kinetics of electrochemical reactions. The same electrode was used to compare the redox behavior of anion probes at different pretreatment stages, as shown in Fig. 3B. The CV curve of GCE in a 0.1 M NaCl solution containing 5.0 mM [Fe(CN)6]3−/4− as shown in Fig. 3B(a). The redox peak of [Fe(CN)6]3−/4− disappears in Fig. 3B(b), indicating that GCE-ox forms an oxidized layer on the electrode surface that hinders the electron transfer, and the redox peak of [Fe(CN)6]3−/4− in Fig. 5B(c) peak was restored, indicating that AGCE formed a rough porous structure in the oxide layer on the electrode surface and restored the electron transfer, but the peak current was still slightly smaller than that of GCE may be that part of the oxide layer still existed inside the AGCE hindering part of the electron transfer,44 which was in agreement with the results after EIS analysis.
 | ||
| Fig. 3 (A) Nyquist plots of the GCE, GCE-ox, and AGCE; (B) CV curves of (a) GCE, (b) GCE-ox, (c) AGCE. All the experiments were performed in 0.1 M NaCl electrolyte containing 5.0 mM [Fe(CN)6]3−/4−. Illustration: partial enlargement. | ||
3.3 Electrochemical behavior of pretreated electrode
The CV method was used for 5.0 mM [Fe(CN)6]3−/4− anionic probe or 1.0 mM [Ru(NH3)6]3+ cationic probe on the AGCE and GCE. As shown in Fig. 4A, the electrochemical response of [Fe(CN)6]3−/4− is affected slightly by the electrostatic repulsion of negatively charged oxygen-containing functional groups, which can be attributed to the sufficiently large pores on the AGCE surface.45 There is a small amount of oxide layer inside the AGCE to prevent electron transfer, so the redox current of [Fe(CN)6]3−/4− is reduced slightly. As shown in Fig. 4B, the AGCE enhances the redox response of [Ru(NH3)6]3+, which may be attributed to the generation of negatively charged oxygen-containing functional groups on the surface of the AGCE that attract [Ru(NH3)6]3+ cation.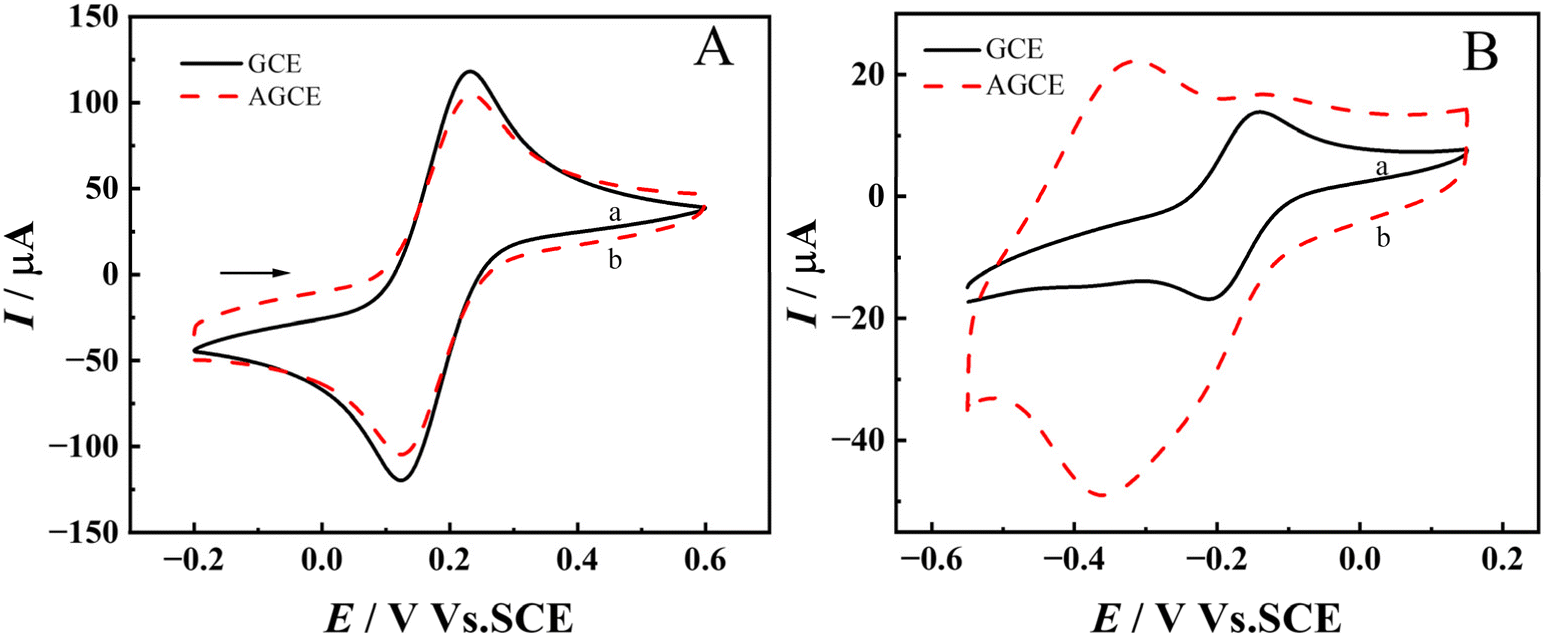 | ||
| Fig. 4 (A) CV curves of (a) GCE and (b) AGCE in 0.1 M NaCl with 5.0 mM [Fe(CN)6]3−/4−; (B) CV curves of (a) GCE and (b) AGCE in 0.1 M containing 1.0 mM [Ru(NH3)6]3+ in NaCl electrolyte. | ||
The electrochemical properties of 50 μM EP on the GCE and AGCE were investigated via CV, and the experimental results are shown in Fig. 5. EP has pKa = 8.66 and exists in its cationic form at pH 7.4.46 The negatively charged oxygen-containing groups on the surface of AGCE have a strong electrostatic attraction to the NH3+ groups of EP, so EP can be enriched in large quantities on the surface of AGCE, which greatly enhances the peak oxidation current, which is about 28 times of the peak oxidation current on GCE, and the data are shown in Table S3.† The negative shift of the oxidation peak potential of EP detected by AGCE compared to GCE suggests that the introduced oxygen-containing functional groups have a catalytic effect on the oxidation of EP, which is consistent with the literature reports.47 Then, the effect of accumulation time on the oxidation peak current of EP detected by the AGCE was investigated, and the experimental results are shown in Fig. S6.† The oxidation peak current of EP detected by the AGCE reached its maximum value when the accumulation time reached 360 s, and with increasing time, the oxidation peak current remained constant. Therefore, an accumulation time of 360 s was chosen for EP detection.
 | ||
| Fig. 5 CV curves of the (a) GCE and (b) AGCE in 0.2 M PB containing 50 μM EP (pH 7.4). | ||
3.4 Selectivity of the pretreated electrodes
Mixtures of EP measurements and interferents were tested on AGCEs with AA, UA, PE, glucose, and glycine (Gly) serving as coexisting compounds or structural analogs of EP. The selectivity of the AGCE towards EP was also investigated in a mixture containing EP and its possible interferents by comparing the peak current ratio (Is/I0) in where Is and I0 are the oxidation peak current measured in EP solution with (Is) or without (I0) interferents.48 The current of EP at AGCE with the presence of equal, ten, twenty, or one hundred-fold concentration of interferents is shown in Fig. 6A and S7.† The current ratio (Is/I0) remains relatively stable, exhibiting a maximum decrease of less than 14% in Is when compared with I0. The results showed that AA and UA did not interfere significantly with the EP assay, mainly because the pKa of AA and UA were 4.1 and 3.7, respectively, and existed in anionic form in the PB at pH 7.4,49 and the negatively charged oxygen-containing groups on AGCE had a strong electrostatic attraction to EP, whereas they had an electrostatic repulsion to AA and UA. The LSV responses of a solution containing equal concentrations (50 μM) of EP and the possible interferents such as AA, UA, and PE in pH 7.4 phosphate buffer at AGCE were presented in Fig. 6B, and the electrochemical parameters are shown in Table S4.† The oxidation peaks of AA, EP, UA, and PE appeared at −25, 145, 269, and 615 mV, respectively. The peak potential differences (ΔEp) between EP and AA, UA, and PE were 170 mV, 124 mV, and 470 mV, respectively, which were all greater than 120 mV. Additionally, the oxidation peak currents of EP were significantly greater than those of AA, UA, and PE at the same concentration. These findings indicate that these interfering substances do not interfere with the detection of EP on AGCE. | ||
| Fig. 6 (A) LSV peak current ratio (Is/I0) for EP with (Is) or without (I0) interference on AGCE and (B) LSV curves for mixtures of the same concentrations of EP, AA, UA, and PE on AGCE. All the experiments were performed in pH 7.4 phosphate buffer with 50 μM EP. | ||
3.5 Linearity, repeatability, and stability
The AGCE was used to measure various concentrations of EP by LSV in a PB solution at a pH of 7.4, and the resulting experimental data are displayed in Fig. 7. The peak current of EP oxidation increased with the increase of EP concentration, which was linear in the three concentration intervals of 0.1–8, 10–100, and 100–700 μM. The linear regression equations, linear correlation coefficients, and detection limits are listed in Table 1. | ||
| Fig. 7 LSV curves of the AGCE electrode in pH 7.4 phosphate buffer with different EP concentrations, a–u: 0.1, 0.2, 0.4, 0.6, 0.8, 2, 4, 6, 8, 10, 20, 40, 60, 80, 100, 200, 300, 400, 500, 600, and 700 μM. | ||
| Linear range (μM) | Linear regression equation (c: μM) | Linear correlation coefficient | Detection limit (μM) |
|---|---|---|---|
| 0.1–8.0 | Ip (μA) = 1.5957c − 0.0372 | 0.9998 | 0.032 |
| 10–100 | Ip (μA) = 0.42647c + 13.6953 | 0.9997 | |
| 100–700 | Ip (μA) = 0.1309c + 43.989 | 0.9993 |
The detection limit (DL) was calculated via the following equation:50
| DL = 3s/m |
The repeatability of the electrodes was assessed by conducting three separate measurements under consistent experimental conditions using the same AGCE. The resulting data are displayed in ESI Table S6,† with an RSD value of 1.23%. Repeatability tests were performed on different pretreated electrodes prepared under the same conditions, and the experimental results are shown in Table S7† with an RSD of 3.40%. The long-term stability of the pretreated electrode was assessed after eight days of storage at ambient temperature, as shown in Fig. S8.† The current response of the electrode demonstrated a retention rate of 98.54% compared with its initial value, indicating that the pretreated electrode exhibited excellent repeatability and stability.
3.6 Real sample analysis
AGCE was used to analyze EP in bovine serum samples by the standard addition method under optimal sensing conditions. Firstly, 100 μL of bovine serum was diluted with 100 mL of PBS of pH 7.4 to obtain a 1000-fold diluted serum solution, which could reduce the contamination of biomolecules on the surface of the electrode. Then, 10 mL of the diluted serum solution was added with 0 μL, 20 μL, 40 μL, 60 μL of 1 mmol per L EP standard reserve solution to produce spiked samples of 0 μM, 2 μM, 4 μM, and 6 μM in sequence. The spiked samples were measured using AGCE, and the measured values were substituted into the standard curve for the concentration range of 0.1–8 μM as described above to obtain the concentration measurement values of the spiked samples. The sample recoveries were obtained from the actual and measured concentrations of the spiked samples and are shown in Table 2. The results showed that AGCE had good recoveries for the detection of EP in real samples.4 Conclusion
In this study, we developed a new GCE pretreatment method that separates anodic oxidation and cathodic reduction through a two-step CV. By performing anodic oxidation in a large potential window and high anodic potential, followed by reduction in a small potential window and low anodic potential, a surface structure with a dense inner layer and a rough and porous outer layer was designed, which combines the advantages of a dense oxide film and a porous structure. It was shown that AGCE obtained under optimal pretreatment conditions significantly improved the electrochemical response of EP. This result can be attributed to the introduction of different oxygen-containing functional groups on the surface of AGCE and the rough surface structure that improves the electrochemical performance. The AGCE was prepared using a simple, eco-friendly, easy-to-handle, and low-cost method, and it showed good selectivity, sensitivity, stability, repeatability, and low detection limit when applied to the detection of EP.Data availability
The authors conform that the data supporting the findings are available within the main article.Author contributions
Tong Meng: conceptualization, methodology, validation, resources, data curation, formal analysis and investigation, visualization, writing – original draft. Yanshu Zhu: resources, data curation. Hangyu Guo: resources, data curation. Juan Zhang: conceptualization, methodology, formal analysis and investigation, supervision, writing – review & editing, funding acquisition, project administration.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the Ningxia Provincial Natural Science Foundation (Project No. 2021AAC03137).References
- S. Banerjee, S. McCracken, M. F. Hossain and G. Slaughter, Biosensors, 2020, 10, 101–118 CrossRef CAS PubMed.
- S. Madhurantakam, J. B. Karnam, D. Brabazon, M. Takai, I. U. Ahad, J. B. Balaguru Rayappan and U. M. Krishnan, ACS Chem. Neurosci., 2020, 11, 4024–4047 CrossRef CAS PubMed.
- T. Tavana, M. A. Khalilzadeh, H. Karimi-Maleh, A. A. Ensafi, H. Beitollahi and D. Zareyee, J. Mol. Liq., 2012, 168, 69–74 Search PubMed.
- L. Ma, T. Zhao, P. Zhang, M. Liu, H. Shi and W. Kang, Anal. Biochem., 2020, 593, 113594 Search PubMed.
- Y. Zhao, S. Zhao, J. Huang and F. Ye, Talanta, 2011, 85, 2650 CrossRef CAS PubMed.
- A. Roychoudhury, K. A. Francis, J. Patel, S. K. Jha and S. Basu, RSC Adv., 2020, 10, 25487–25495 RSC.
- K. Syslová, L. Rambousek, M. Kuzma, V. Najmanová, V. Bubeníková-Valešová, R. Šlamberová and P. Kačer, J. Chromatogr. A, 2011, 1218, 3382–3391 CrossRef PubMed.
- C. E. Cardoso, R. O. R. Martins, C. U. A. S. Telles and R. Q. Auc Lio, Mikrochim. Acta, 2004, 146, 79–84 CrossRef CAS.
- X. Yang, P. Zhao, Z. Xie, M. Ni, C. Wang, P. Yang, Y. Xie and J. Fei, Talanta, 2021, 233, 122545 CrossRef CAS PubMed.
- A. F. Craievich, Mater. Res. Bull., 1976, 11, 1249–1255 Search PubMed.
- S. E. Elugoke, O. E. Fayemi, A. S. Adekunle, E. M. Sherif and E. E. Ebenso, Heliyon, 2022, 8, e10835 CrossRef CAS PubMed.
- S. A. Leau, C. Lete, C. Matei and S. Lupu, Biosensors, 2023, 13, 781–797 Search PubMed.
- S. E. Elugoke, O. E. Fayemi, A. S. Adekunle, P. Ganesh, S. Kim and E. E. Ebenso, J. Electroanal. Chem., 2023, 929, 117120 Search PubMed.
- S. Kumar, A. Awasthi, M. D. Sharma, K. Singh and D. Singh, Mater. Chem. Phys., 2022, 290, 126656 Search PubMed.
- S. Immanuel and R. Sivasubramanian, Bull. Mater. Sci., 2020, 43, 79–89 CrossRef CAS.
- K. Sen, S. Ali, D. Singh, K. Singh and N. Gupta, FlatChem, 2021, 30, 100288 Search PubMed.
- H. Tang, M. Cui, M. Zhang and Y. Zhang, Bioelectrochemistry, 2024, 155, 108591 Search PubMed.
- Y. Xu, Z. Zhang and Y. Wang, J. Electroanal. Chem., 2023, 932, 117239 Search PubMed.
- T. Nagaoka and T. Yoshino, Anal. Chem., 1986, 58, 1037–1042 Search PubMed.
- L. J. Kepley and A. J. Bard, Anal. Chem., 1988, 60, 1459–1467 Search PubMed.
- A. H. Oghli, E. Alipour and M. Asadzadeh, RSC Adv., 2015, 5, 9674–9682 RSC.
- H. Zhang, S. Li, F. Zhang, M. Wang, X. Lin and H. Li, J. Solid State Electrochem., 2017, 21, 735–745 CrossRef CAS.
- Y. Li, J. Zhou, J. Song, X. Liang, Z. Zhang, D. Men, D. Wang and X. Zhang, Biosens. Bioelectron., 2019, 144, 111534 CrossRef CAS PubMed.
- A. M. Abdel-Aziz, H. H. Hassan and I. H. A. Badr, Anal. Chem., 2020, 92, 7947–7954 CrossRef CAS PubMed.
- R. Peng, Y. Gao and W. Chen, J. Electrochem. Soc., 2021, 168, 067509 CrossRef CAS.
- M. Wang, J. Lin, J. Gong, M. Ma, H. Tang, J. Liu and F. Yan, RSC Adv., 2021, 11, 9021 RSC.
- K. S. K. Shiu, Anal. Chem., 2002, 74, 879–885 CrossRef PubMed.
- Y. Yi, G. Weinberg, M. Prenzel, M. Greiner, S. Heumann, S. Becker and R. Schlögl, Catal. Today, 2017, 295, 32–40 Search PubMed.
- M. Noel and P. N. Anantharaman, Surf. Coat. Technol., 1986, 28, 161–179 CrossRef CAS.
- Q. Zhao, L. Bao, Q. Luo, M. Zhang, Y. Lin, D. Pang and Z. Zhang, Biosens. Bioelectron., 2009, 24, 3003–3007 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- S. H. Choudhury, Y. Ding, Y. Yi, C. Rohner, W. Frandsen, T. Lunkenbein, M. Greiner, R. Schlögl and S. Heumann, ChemElectroChem, 2022, 9, e202200637 CrossRef CAS.
- A. Dekanski, J. Stevanovic, R. Stevanovic, B. Z. Nikolic and V. M. Jovanovic, Carbon, 2001, 39, 1195–1205 CrossRef CAS.
- H. L. Hu and N. Li, Electrochemical Measurements, Chemical Industry Press, Beijing, 2020 Search PubMed.
- M. Noel and P. N. Anantharaman, Analyst, 1985, 110, 1095–1103 RSC.
- M. Santhiago, C. M. Maroneze, C. C. C. Silva, M. N. L. Camargo and L. T. Kubota, ChemElectroChem, 2015, 2, 761–767 CrossRef CAS.
- F. Li, M. Xue, X. Ma, M. Zhang and T. Cao, Anal. Chem., 2011, 83, 6426–6430 Search PubMed.
- D. Mao, P. Duan and Y. Piao, J. Electroanal. Chem., 2022, 925, 116898 Search PubMed.
- M. G. Sullivan, B. Schnyder, M. Bärtsch, D. Alliata, C. Barbero, R. Imhof and R. Kötz, J. Electrochem. Soc., 2000, 147, 2636–2643 Search PubMed.
- S. Thiagarajan, T. Tsai and S. Chen, Biosens. Bioelectron., 2009, 24, 2712–2715 Search PubMed.
- T. Bystron, E. Sramkova, F. Dvorak and K. Bouzek, Electrochim. Acta, 2019, 299, 963–970 CrossRef CAS.
- S. Uruc, O. Gorduk and Y. Sahin, Int. J. Environ. Anal. Chem., 2021, 103, 3544–3562 CrossRef.
- A. Atchabarova, S. Abdimomyn, D. Abduakhytova, K. Kishibayev, Y. Zlobina, A. Kurbatov, G. L. Turdean and T. Djenizian, J. Solid State Electrochem., 2023, 28, 2425–2436 CrossRef.
- R. C. Engstrom, Anal. Chem., 1982, 54, 2310–2314 CrossRef CAS.
- Q. Zhao, Z. Zhang, L. Bao and D. Pang, Electrochem. Commun., 2008, 10, 181–185 Search PubMed.
- R. Álvarez-Diduk and A. Galano, J. Phys. Chem. B, 2015, 119, 3479–3491 CrossRef PubMed.
- L. Xuan, W. Liao, M. Wang, H. Zhou, Y. Ding, F. Yan, J. Liu, H. Tang and F. Xi, Talanta, 2021, 225, 122066 CrossRef CAS PubMed.
- J. Zhang, X. Guo, J. Zhou, G. Liu and S. Zhang, Mater. Sci. Eng., C, 2018, 91, 696–704 Search PubMed.
- M. Zhao, Z. Li, X. Zhang, J. Yu, Y. Ding, H. Li and Y. Ma, Sens. Actuators, B, 2020, 309, 127757 CrossRef CAS.
- Y. Wu, P. Deng, Y. Tian, J. Feng, J. Xiao, J. Li, J. Liu, G. Li and Q. He, J. Nanobiotechnol., 2020, 18, 112–124 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02123e |
| This journal is © The Royal Society of Chemistry 2025 |