
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable production and antibacterial efficacy of silver nanoparticles on cellulose nanofibers from mushroom waste
Charzen Mae Kinoan ab and
Haliza Katas*a
ab and
Haliza Katas*a
aCentre for Drug Delivery Technology and Vaccine (CENTRIC), Faculty of Pharmacy, Universiti Kebangsaan Malaysia, Jalan Raja Muda Abdul Aziz, Kuala Lumpur, 50300, Malaysia. E-mail: haliza.katas@ukm.edu.my; Fax: +60-3-26983271; Tel: +60-3-9289797
bDepartment of Pharmacy, College of Pharmacy and Medical Technology, University of San Agustin, Iloilo City, Philippines
First published on 11th June 2025
Abstract
Underutilized agricultural wastes, such as spent mushroom substrate (SMS), present valuable opportunities for developing sustainable biomedical materials. In this study, cellulose nanofibers (CNFs) were successfully isolated from SMS through a chemo-mechanical process, while the water extract of SMS (WESMS) served as a green reducing agent for the simultaneous synthesis and in situ loading of silver nanoparticles (AgNPs) onto TEMPO-oxidized CNFs (AgNP/ToCNF). The chemical structure of the isolated cellulose was characterized using ATR-FTIR, while UV-vis spectroscopy confirmed the successful synthesis and AgNPs loading, showing a maximum absorbance at 424 nm. The resulting hybrid nanomaterial exhibited a nanofiber width diameter range of 273.5–318.5 nm, while the AgNPs had an average diameter of 34.04 nm. The antimicrobial efficacy of AgNP/ToCNF was evaluated against S. aureus, P. aeruginosa, and E. coli using agar well diffusion, broth microdilution, time-kill, and cell membrane leakage assays. AgNP/ToCNF exhibited MIC90 values of 250 μg mL−1 against S. aureus and 125 μg mL−1 against P. aeruginosa and E. coli, whereas free-state AgNPs showed MIC90 values of 62.5 μg mL−1 against S. aureus and 31.25 μg mL−1 against P. aeruginosa and E. coli. Both compounds demonstrated bactericidal activity against all three bacterial strains. Cytotoxicity was assessed using the LDH assay, revealing a concentration-dependent toxicity pattern. Notably, AgNP/ToCNF exhibited minimal toxicity to human dermal fibroblasts (HDFs) at concentrations ≤500 μg mL−1 after 72 hours, while free-state AgNPs induced >67% cytotoxicity. Although CNFs derived from SMS lacked intrinsic antimicrobial activity, their incorporation with AgNPs significantly enhanced antibacterial efficacy while simultaneously reducing AgNPs-induced cytotoxicity in mammalian cells. These findings underscore the potential of SMS-derived CNFs as biocompatible nanocarriers for AgNPs and other antibacterial agents, offering a sustainable and eco-friendly approach to developing antimicrobial biomaterials. This study explores the feasibility of upcycling SMS into high-value biomedical products, creating opportunities for future applications in wound healing, antimicrobial coatings, and medical nanocomposites.
Introduction
The sustainable management of agricultural waste is a growing global concern due to its environmental and economic implications.1–3 Among various types of biomass waste, spent mushroom substrate (SMS)—a byproduct generated in large quantities during mushroom cultivation that is high in organic matter and low in toxic elements—represents an underutilized resource with high potential for value-added applications.4,5 Approximately 5 kilograms are generated for every kilogram of mushrooms cultivated, creating a notable challenge in terms of disposal.6,7 Conventional disposal techniques, such as incineration and landfilling, contribute to environmental degradation through the release of harmful greenhouse gases,8–10 and the increased risk of public health issues such as respiratory problems.11 Therefore, the development of sustainable and innovative strategies to repurpose SMS is both timely and necessary.SMS is predominantly composed of lignocellulosic biomass, such as cellulose, which accounts for 25–47% of its dry weight.12–18 Cellulose and its derivatives have been extensively studied as bio-based materials for tissue engineering and drug delivery due to their biocompatibility, functionality, flexibility, and mechanical strength.19–21 Recent advances in nanotechnology have enabled the extraction of nanocellulose—a biodegradable nanomaterial with enhanced surface area, mechanical performance, and functionality—from various lignocellulosic masses. Nanocellulose has demonstrated significant potential in fields such as food packaging, pharmaceuticals, and regenerative medicine.22–25 However, its application in wound healing remains limited due to the absence of inherent antimicrobial properties.26–28 One promising strategy to overcome this limitation involves the functionalization of nanocellulose with antimicrobial metal nanoparticles, such as zinc, gold, and silver. This approach not imparts antimicrobial properties to the nanomaterial but also controls the release of the metal ions and reduces their cytotoxicity.29
SMS is not only an abundant source of nanocellulose but its water extract has also been studied as a reducing agent for metal nanoparticles.30 LC-MS analysis of the water extract of SMS (WESMS) reveals the presence of abundant fatty acid derivatives, such as vanillic and decanoic acid – compounds useful for synthesizing metal nanoparticles.30–32 This positions SMS as a promising starting material for developing nanomaterials applicable in various scientific fields.
Based on our current understanding, the integrated use of SMS both as a source of cellulose nanofibers (CNF) and as a green reducing agent for in situ synthesis of silver nanoparticles (AgNPs) has not been explored. In this study, we present a novel and sustainable method for producing AgNP-loaded TEMPO-oxidized cellulose nanofibers (AgNP/ToCNF) from SMS. The antibacterial activity of AgNP/ToCNF was investigated using agar well diffusion, microbroth dilution assay, time-kill kinetics, and cell membrane leakage assay. Furthermore, cell proliferation and nanomaterial cytotoxicity were also assessed using lactate dehydrogenase (LDH) and Alamar Blue assays. This dual-functional approach to utilizing SMS not only contributes to waste valorization but also offers a potential platform for developing low-cost, biodegradable, and antimicrobial biomaterials for medical applications.
Experimental section
Materials
Spent mushroom substrate (SMS) of Pleurotus ostreatus (oyster mushroom) was kindly provided by Nas Agro Farm, Sepang, Malaysia. Silver nitrate (AgNO3) (ACS reagent grade) and low molecular weight (LMW) chitosan (50–190 kDa, 75–85% deacetylated) were purchased from Sigma-Aldrich, MO, USA. Sodium hydroxide (NaOH) and 10% sodium hypochlorite (NaOCl), used in the isolation of cellulose nanofibers, were obtained from Bio3 Scientific Sdn. Bhd., Puchong, Malaysia. 2,2,6,6-Tetramethylpiperidine-1-oxyl (TEMPO) and sodium bromide (NaBr), used for TEMPO-mediated oxidation, were acquired from Sigma-Aldrich, MO, USA, and Bio3 Scientific Sdn. Bhd., Puchong, Malaysia, respectively.Three bacterial strains—Staphylococcus aureus ATCC 25923, Pseudomonas aeruginosa ATCC 27853, and Escherichia coli ATCC 25927—were obtained from Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia. Mueller–Hinton broth (MHB), Mueller–Hinton agar (MHA), and ciprofloxacin hydrochloride were purchased from TargetMol, MA, USA.
Primary human dermal fibroblasts (HDF, passage 0) were purchased from ATCC, VA, USA. Dulbecco's Modified Eagle Medium/Ham's F-12 (DMEM/F-12), supplemented with L-glutamine, sodium pyruvate, and HEPES, as well as trypsin–EDTA solution (2.5 g per L trypsin, 1 mmol per L EDTA), were obtained from Nacalai Tesque, Kyoto, Japan. Fetal bovine serum (FBS) and penicillin–streptomycin solutions were sourced from Tico Europe, Amstelveen, Netherlands. The Alamar Blue cell proliferation reagent was purchased from Invitrogen, MA, USA, and the LDH cytotoxicity assay kit was obtained from Canvax, Valladolid, Spain.
Water extraction of SMS (WESMS)
The starch binder from the SMS was removed, and the fibers were dried at 40 °C for 48 hours. The dried samples were then ground using a Pulverisette 14 grinder equipped with a 1.0 mm sieve ring at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and stored in a sealed container. Subsequently, 20 g of SMS fibers were soaked in 100 mL of purified water and heated at 60 °C for 30 minutes with continuous stirring. The mixture was filtered using Whatman No. 1 filter paper. The solid residue was retained for CNF isolation (see Subsection: Preparation of SMS Fibers). The resulting filtrate was centrifuged at 5000 rpm, filtered again to completely remove any remaining fibers, and stored at 4 °C until use.
000 rpm and stored in a sealed container. Subsequently, 20 g of SMS fibers were soaked in 100 mL of purified water and heated at 60 °C for 30 minutes with continuous stirring. The mixture was filtered using Whatman No. 1 filter paper. The solid residue was retained for CNF isolation (see Subsection: Preparation of SMS Fibers). The resulting filtrate was centrifuged at 5000 rpm, filtered again to completely remove any remaining fibers, and stored at 4 °C until use.
Isolation of CNF from SMS
The percentages of lignin, hemicellulose, and cellulose in the biomass were then calculated as follows:
 | (1) |
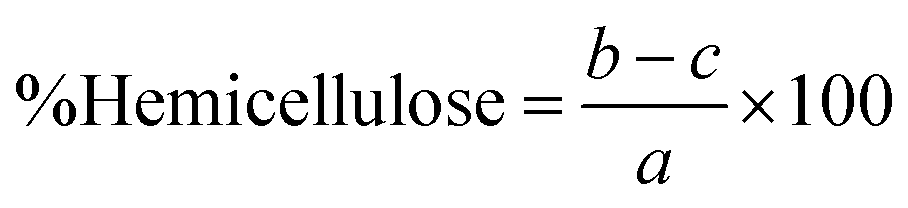 | (2) |
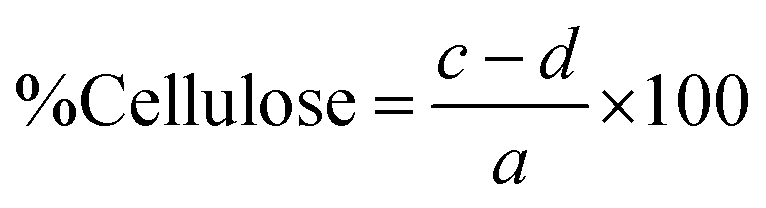 | (3) |
 | (4) |
 | (5) |
:100 (w/v) ratio. Oxidation was initiated by adding 10 mmol of NaOCl per gram of fiber to the suspension at room temperature, under continuous stirring at 500 rpm. The pH was maintained at 10 using 0.5 M NaOH throughout the reaction. After one hour, the oxidation was quenched by adding 30 mL of denatured alcohol. The oxidized cellulose nanofibers were then washed twice with distilled water via vacuum filtration. Structural modification was confirmed by ATR-FTIR analysis (PerkinElmer Spectrum 100, Waltham, MA, USA).Simultaneous green synthesis and in situ loading of AgNPs to ToCNFs
The nanocomposite was prepared following the procedure described by Shin et al. and Suleman Ismail Abdalla et al. with some modifications.30,34 Briefly, a slurry of ToCNFs and 0.01 M AgNO3 were mixed in a 1:1 ratio and heated at 37 °C for 1 hour. The fibers were then collected and washed to remove unreacted AgNO3. The ToCNFs/Ag+ were subsequently reacted with SMS water extract in a 1:5 ratio until the mixture turned reddish brown—a visual indication of AgNP formation. To confirm the synthesis of AgNPs, the mixture was analyzed by UV-vis spectroscopy (Genesys™, ThermoFisher, Waltham, MA, USA) across a scan range of 200–700 nm. A characteristic absorption peak between 400 and 500 nm confirmed the presence of AgNPs. The mixture was then sonicated for 20 minutes and stirred at room temperature for 30 minutes. It was subjected to ultracentrifugation (15000 rpm for 15 minutes) to remove unreacted components and washed three times with purified water. The resulting AgNP-loaded ToCNFs were resuspended in 5 mL of deionized water and stored at −80 °C prior to lyophilization. Freeze-drying was performed using a ScanVac Coolsafe (Labogene, Lillerød, DK) at −40 °C to −60 °C for 48 hours until a constant weight was achieved. The morphology of the lyophilized nanocomposite was examined using SEM.
Characterization of ToCNF and AgNP/ToCNF
Evaluation of antibacterial activity
:100 with MHB to achieve approximately 1 × 106 CFU mL−1. Subsequently, 100 μL of each bacterial strain suspension was added to wells of a 96-well plate containing 100 μL of the treatment samples, resulting in a final bacterial concentration of 5 × 105 CFU mL−1. The plates were then incubated at 37 °C for 18 hours. Bacterial growth was measured by optical density at 600 nm (OD600) using a microplate reader (Multiskan™ FC, ThermoFisher, Waltham, MA, USA). The percentage of growth inhibition was calculated to determine the minimum concentration that inhibits 90% of bacterial growth (MIC90).10 CFU mL−1 reduction of −1 (90% killing), −2 (99% killing), and −3 (99.9% killing). Assay readings at 3, 6, 12, and 24 hours were compared to bacterial counts at 0 hours. Bactericidal activity was defined as the lowest antimicrobial agent concentration that reduced the original inoculum by ≥3log10 CFU mL−1 (99.9%), while bacteriostatic activity was defined as a reduction of <3log10 CFU mL−1. All Petri dishes with no signs of bacterial growth were allowed to incubate for an additional 24 hours to confirm bactericidal activity.In vitro proliferative effects and cytotoxicity
 | (6) |
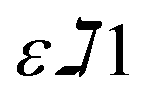 and
and 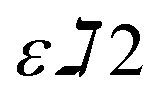 are constants representing the molar extinction coefficient of AB at 570 and 600 nm, respectively, in the oxidized (εox) and reduced (εred) forms. The constant values are 117216
are constants representing the molar extinction coefficient of AB at 570 and 600 nm, respectively, in the oxidized (εox) and reduced (εred) forms. The constant values are 117216 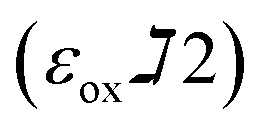 , 80586
, 80586  , 155677
, 155677  , and 14652
, and 14652 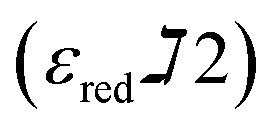 .
.  and
and  represent the absorbance of the test wells at 570 and 600 nm, respectively.
represent the absorbance of the test wells at 570 and 600 nm, respectively.  and
and  represent the absorbance of the negative control wells at 570 and 600 nm, respectively. The values of AB reduction % were corrected for background values of negative controls containing medium without cells.
represent the absorbance of the negative control wells at 570 and 600 nm, respectively. The values of AB reduction % were corrected for background values of negative controls containing medium without cells.The relative cell proliferation was then calculated to quantify the cell viability effects of the test compounds to HDF. The values obtained from treated and untreated cells were used to compute the relative cell proliferation percentage (%RCP) using the formula below (eqn (7)):
 | (7) |
 | (8) |
Statistical analysis
All statistical analyses were performed using GraphPad Prism 10. Data are presented as mean ± standard deviation (SD). For comparisons involving a single independent variable, one-way ANOVA was followed by Tukey's post hoc test. For analyses involving two independent variables, two-way ANOVA were performed followed by Tukey's post hoc test for pairwise comparisons. A p-value of <0.05 was considered statistically significant.Results and discussion
Isolation of CNF from SMS
Fig. 1 shows the diagram illustrating the isolation of CNF extracted from SMS. SMS fibers underwent alkaline hydrolysis using varying concentrations of NaOH, followed by bleaching with 10% NaOCl to remove lignin—a complex polymer in plant cell walls that can hinder the extraction of cellulose fibers. Different concentrations of NaOH were tested to determine the optimal level for effectively removing lignin from the raw material. Subsequently, the fibers were bleached with 10% NaOCl for 3 cycles to eliminate any remaining lignin and to decolorize the fibers. | ||
| Fig. 1 Schematic diagram of the chemo-mechanical process used to isolate cellulose nanofibers (CNF) from spent mushroom substrate (SMS). | ||
As shown in Fig. 2, the highest yield of 43.21% ± 0.73 was obtained using 2% NaOH under bleaching conditions. Increasing the NaOH concentration to 4% and 8% resulted in a gradual decline in yield to 40.24% ± 0.48 and 38.25% ± 0.44, respectively. At even higher concentrations (16% and 32%), the yield further dropped to approximately 32.8%, indicating a significant reduction in CNF recovery. This trend suggests that as the NaOH concentration increases, the CNF yield decreases. Apart from losses incurred during transferring, filtering, and washing, a reduction in weight typically indicates the effective degradation or removal of lignin and other non-cellulosic components from the biomass. These components—including hemicellulose, lignin, and extractives – contribute to the bulk of the raw material but are undesirable for pure cellulose extraction.36,37 Further loss in weight was observed after bleaching due to the removal of the remaining impurities, which resulted in a whiter appearance of the fibers.38 The biomass composition of the untreated and treated SMS fibers was analyzed using the Chesson–Datta method to quantify the relative amounts of lignocellulosic materials—cellulose, hemicellulose, and lignin—as well as other residual impurities remaining after pre-treatment. Fig. 3 shows the changes in the percentages of these components after the pre-treatment processes. Untreated SMS fibers derived from P. ostreatus contained 24.19% ± 0.46 cellulose. Previous studies on SMS from the same mushroom species reported cellulose contents ranging from approximately 29 to 37%.14,15,18 These variations in the cellulose content are influenced by the differences in the initial substrate materials used and cultivation practices.4 In addition to cellulose, the untreated SMS also contained 18.98% ± 0.35 hemicellulose, and 28.41% ± 0.47 lignin, which are comparable to the findings of Adi et al., who reported similar levels of structural polysaccharides in SMS.39
 | ||
| Fig. 2 Yield of cellulose nanofibers (CNF) obtained from spent mushroom substrate (SMS) following various chemical treatments, n = 9. | ||
 | ||
| Fig. 3 Biomass composition (hemicellulose, cellulose, lignin, ash, and extractives) of untreated and chemically treated SMS fibers. Treatments include NaOH at varying concentrations (2%, 4%, 8%, 16%, and 32%) followed by bleaching, where A–B denotes alkali (NaOH) treatment followed by bleaching. Data are expressed as percentage of dry weight, n = 9. | ||
After treatment with increasing NaOH concentrations, significant compositional changes were observed. Cellulose content increased progressively from 29.8% at 2% to 64.36% at 32% NaOH, indicating effective preservation of cellulose. This substantial increase in cellulose content recovery is accompanied by a pronounced reduction in hemicellulose content, from 18.46% at 2% to a minimum of 12.62% at 16% NaOH, with a slight increase to 14.54% at 32% NaOH, suggesting some variability in hemicellulose removal at higher alkali concentrations. Lignin content showed a notable reduction from 29.67% at 2% to approximately 13.26% and 14.3% at 16% and 32% NaOH, respectively, confirming effective delignification with stronger alkali treatments. Ash content similarly declined sharply from 9.15% at 2% NaOH to just above 1% at 16% and 32% NaOH, indicating effective removal of inorganic impurities. Extractives also decreased from 12.93% at 2% NaOH to 5.68% at 32% NaOH, further evidencing purification.
Although both 16% and 32% NaOH treatments effectively removed impurities, no significant difference in the final cellulose content was detected between the two. Therefore, 16% NaOH-treated fibers were selected for the isolation of CNF, given their economic advantages, including lower chemical consumption and easier handling compared to the 32% NaOH-treated fibers. Additionally, the use of 16% NaOH-treated fibers offers a cost-effective and practical solution for large-scale production.
ATR-FTIR analysis of CNF
The extracted cellulose from the selected treatment was further subjected to TEMPO-mediated oxidation—a selective chemical modification process that converts the primary hydroxyl groups (–OH) on the cellulose chains into carboxylate groups (–COOH). This transformation enhances the surface charge of the fibers and improves their dispersibility in water, which is important for producing high-quality CNF. The oxidation process was confirmed using Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) to identify organic functional groups in the sample.Fig. 4 presents the spectra of the SMS fibers after each treatment. The dominant spectral band observed around ∼3350 corresponds to O–H stretching vibrations, while the peak ∼1640 cm−1 is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carboxylate groups in the TEMPO-oxidized sample—indicating the successful conversion of the primary hydroxyl groups to carboxylates. Bands in the region of 1630–1650 cm−1 observed in the other samples represent the presence of adsorbed water, which is commonly retained within the cellulose matrix due to strong hydrogen bonding interactions and is difficult to eliminate completely.
O stretching of the carboxylate groups in the TEMPO-oxidized sample—indicating the successful conversion of the primary hydroxyl groups to carboxylates. Bands in the region of 1630–1650 cm−1 observed in the other samples represent the presence of adsorbed water, which is commonly retained within the cellulose matrix due to strong hydrogen bonding interactions and is difficult to eliminate completely.
 | ||
| Fig. 4 ATR-FTIR spectra of untreated SMS, 16% A (16% NaOH-treated), 16% A–B (16% NaOH-treated followed by bleaching), and TEMPO (TEMPO-oxidized fibers). | ||
Additional peaks at ∼1370 cm−1 and ∼1316 cm−1 correspond to C–H and O–H bending vibrations, respectively. The prominent band at ∼1040 cm−1 is assigned to C–O–C stretching vibration in pyranose ring of cellulose, a hallmark of the cellulose backbone. Furthermore, the signal at 890–898 cm−1 is characteristic of cellulosic β-glycosidic bond stretching vibration in cellulose, confirming the preservation of the structural integrity of cellulose.
Overall, the ATR-FTIR analysis verified the successful oxidation of the cellulose, as indicated by the emergence of carboxylate-specific peaks while maintaining the key structural features of cellulose. These modifications improve the nanomaterial's functional properties—particularly water affinity and surface charge—making it better suited for applications requiring well-dispersed, high-performance CNF.40,41
Zeta potential, particle size distribution, and surface morphology of ToCNF
In addition to chemical oxidation, the fibers were subjected to mechanical disintegration to further reduce their diameter. Mechanical treatments, such as ultrasonication, are commonly employed to break down the cellulose fibers into nanofibers by applying shear forces that separate the fibers and reduce their size. This process is especially important in the production of CNF, as it significantly increase the surface area, improves dispersion, and enhances their mechanical strength and functional interactions with other composite systems.42The resulting ToCNFs demonstrated a zeta potential ranging from 34.68 to −40.9 mV (Fig. 5), indicating a negatively charged surface area due to the introduction of the carboxylate groups during the oxidation process. A zeta potential of below −30 mV typically signifies strong electrostatic repulsion between the fibers, which is associated with good colloidal stability in aqueous suspensions.43 This high surface charge helps prevent aggregation and ensures stable dispersion.44
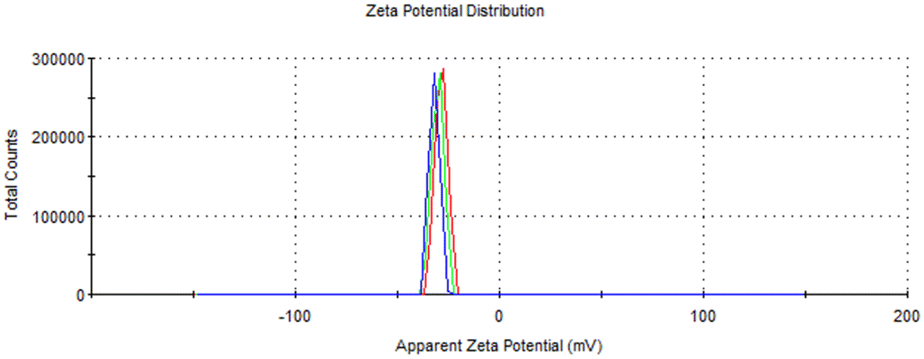 | ||
| Fig. 5 Zeta potential distribution of TEMPO-oxidized cellulose nanofibers (ToCNF). | ||
SEM micrographs (Fig. 6) reveal that the ToCNFs possess high aspect ratio, with fiber diameters ranging from 42 nm to 216 nm. The nanofibers appeared web-like, overlapping bundles, making it challenging to determine their precise length due to the bundling and entanglement. This morphology is typical of cellulose nanofibers produced by TEMPO-mediated oxidation, consistent with the observations of Isogai and Zhou, who reported the overlapping, branched fibers with extended lengths in ToCNFs.45 The observed network-like structure suggests that ToCNFs can form strong entanglements through hydrogen bonding, which is critical for reinforcing polymer matrices and improving mechanical properties in composite materials.46,47 Fiber curling was also observed, a common morphological change during fibrillation and drying processes.48 Similar curling phenomenon was reported by Wang et al., who noted that this morphology can be hypothetically attributed to the residual starch and hemicellulose creating a physical barrier.49 Levanič et al. likewise noted fiber curling and demonstrated that it can be minimized by fiber swelling from prolonged TEMPO oxidation time and thorough pulp washing.41 In this study, the observed curling may be further attributed to the lyophilization step, which commonly induces fiber shrinkage and aggregation due to the removal of bound water and structural collapse during freeze-drying.50
 | ||
| Fig. 6 SEM micrographs of ToCNFs taken at 15000× magnification with a scale bar of 1 μm and the corresponding particle size distribution histogram illustrating the diameter range of the nanofibers, n = 60. | ||
Simultaneous green synthesis and in situ loading of AgNPs to ToCNFs
Fig. 7 illustrates the synthesis route for the fabrication of AgNP/ToCNF. The process began with ion exchange between silver ions (Ag+) and sodium ions (Na+) present on the sodium salt form of the oxidized nanofibers, converting them into Ag+-ToCNFs complex. This ion exchange reaction was conducted at 37 °C for 1 hour. During the thermal treatment, the fiber color changed from white to brown, indicating successful silver ion incorporation. Thereafter, the in situ reduction of the Ag+ to AgNPs was carried out using WESMS. | ||
| Fig. 7 Pictorial diagram of the simultaneous green synthesis and in situ loading of AgNPs onto ToCNFs. | ||
In a previous study, WESMS was analyzed by LC-MS to identify its phytochemical constituents.30 The presence of vanillic acid and decanoic acid derivatives in WESMS has been reported to act as reducing agents for producing AgNPs and other metal nanoparticles.31,32 The mixture was monitored daily using UV-vis spectroscopy to track the formation of AgNPs. A characteristic absorption peak between 400 and 500 nm indicates the successful reduction to AgNPs, attributable to the surface plasmon resonance of the AgNPs, which corresponds to the observed reddish-brown color change.51 Fig. 8 presents the UV-vis spectra of Day 0 and Day 3 of the mixtures. On Day 3, the ToCNFs/Ag+ sample showed no distinct absorption peak in the 400–500 nm range, whereas the ToCNFs/Ag+/WESMS mixture exhibited clear absorption peaks within this range, confirming the role of WESMS as a reducing agent in the in situ synthesis of AgNPs on the ToCNFs. After thoroughly removing the unreacted reactants from the mixture by centrifugation at 15000 rpm, AgNP/ToCNF was successfully isolated, with the maximum absorbance detected at 424 nm (Fig. 9). Additionally, the peaks observed at 200–230 nm corresponds to the characteristic absorption bands of CNF.
 | ||
| Fig. 8 UV-vis absorption spectra of AgNO3, ToCNFs, ToCNFs/Ag+, WESMS, ToCNFs/Ag+/WESMS on Day 0 (a) and Day 3 (b). | ||
 | ||
| Fig. 9 UV-vis absorption spectrum of synthesized AgNP/ToCNF nanocomposite. | ||
A similar reduction procedure was also performed on AgNPs without ToCNFs, and the reduction using WESMS was observed on Day 5, consistent with the findings of Suleman Ismail Abdalla et al.30 This indicates that the presence of ToCNFs accelerates the nucleation and growth of AgNPs, likely due to the functional groups on the nanofiber surface acting as nucleation sites and stabilizers.52 This synergistic effect between WESMS and ToCNFs not only facilitates faster reduction kinetics but also enables stable loading of AgNPs on the cellulose nanofiber matrix, which is advantageous for applications requiring well-dispersed metal nanoparticles with antimicrobial activity.
Zeta potential, particle size distribution, and surface morphology of AgNP/ToCNF
The zeta potential of the AgNP/ToCNF composite was −48.3 ± 0.58 mV, indicating good colloidal stability. SEM analysis revealed the size and surface morphology of the nanocomposite (Fig. 10). As shown in Fig. 10b, the mean diameter of reduced AgNPs loaded on the ToCNFs surface was 34.04 nm, while the ToCNFs exhibited a mean width diameter of 296 nm (Fig. 10d). The increase in the width diameter of ToCNFs compared to neat ToCNFs is attributed to the spherical AgNP deposition on the fiber surface. Similar morphological changes were observed by Smiechowicz et al., where AgNP decoration led to increased fiber dimensions and enhanced surface roughness.53 | ||
| Fig. 10 SEM micrographs of (a) AgNPs at 40000× magnification (scale bar = 500 nm) and their corresponding size distribution (b); (c) ToCNFs at 40000× magnification (scale bar = 500 nm) with size distribution (d); and surface morphology comparison of (e) neat ToCNFs and (f) AgNP-loaded ToCNFs at 15000× magnification (scale bar = 1 μm). Particle size distributions are based on n = 60 measurements. | ||
This decoration of AgNPs resulted in a distinctly roughened nanocomposite surface (Fig. 10f) compared to the smooth surface observed on the neat ToCNFs (Fig. 10e), which is consistent with the findings of Jatoi et al., who reported similar surface topography changes in AgNP-modified cellulose nanofibers.54 Notably, the reduced AgNPs were well dispersed without significant agglomeration—an improvement over typical AgNP synthesis processes, where nanoparticle agglomeration is a common challenge.55,56 This selective loading is likely due to the preferential attachment of silver nanoparticles to the oxidized regions of the cellulose structure, as previously reported.57
While the current study demonstrates the successful synthesis of AgNP/ToCNF nanocomposite, it is important to note that long-term stability assessments were beyond the scope of this work. Future studies should focus on evaluating the structural and functional stability of the materials under various storage and physiological conditions to further validate their practical applicability.
Evaluation of antibacterial activity
Preliminary screening for antibacterial activity of ToCNFs, AgNPs, and AgNP/ToCNF. The antibacterial activity of the test compounds was initially assessed using the agar well diffusion method. A zone of inhibition measuring 4.0 mm or greater was considered indicative of effective antibacterial activity. Fig. 11 shows the inhibition zones produced by the tested samples against the three bacterial strains, S. aureus, E. coli, and P. aeruginosa. | ||
| Fig. 11 Zones of inhibition of 500 μg mL−1 (a) and 250 μg mL−1 (b) of the different test compounds against S. aureus (left), E. coli (middle), and P. aeruginosa (right) grown on Mueller–Hinton agar. (Legend: C = ciprofloxacin, B = blank, T = ToCNF, A = AgNP, AT = AgNP/ToCNF). | ||
Ciprofloxacin (20 μg mL−1) served as the positive control and exhibited the largest zones of inhibition, with mean diameters of 25.29 ± 1.19 mm, 27.85 ± 0.64 mm, and 15.02 ± 1.12 mm for S. aureus, E. coli, and P. aeruginosa, respectively (Table 1). As expected, ToCNF alone (both at 250 and 500 μg mL−1) showed no antibacterial activity against any tested strain, consistent with previous findings by Jiang et al.58. In contrast, AgNPs demonstrated significant antibacterial effects at 500 μg mL−1, producing large inhibition zones of 18.34 ± 1.47 mm, 20.61 ± 1.43 mm, and 13.38 ± 0.82 mm for S. aureus, E. coli, and P. aeruginosa, respectively. These values indicate strong antibacterial efficacy, approaching the activity of ciprofloxacin, especially against P. aeruginosa. The AgNP/ToCNF composite also exhibited antibacterial activity, though slightly reduced compared to AgNPs alone, with inhibition zones at 500 μg mL−1 measuring 13.17 ± 0.95 mm for S. aureus, 16.96 ± 0.40 mm for E. coli, and 11.23 ± 1.04 mm for P. aeruginosa. At the lower concentration of 250 μg mL−1, both AgNPs and AgNP/ToCNF showed diminished antibacterial activity but maintained measurable zones of inhibition. The lack of activity in ToCNFs alone confirms that the observed antibacterial effects are primarily attributable to the silver nanoparticles. These findings suggest that the incorporation of AgNPs into ToCNF effectively imparts antibacterial properties, making the composite a promising candidate for antimicrobial applications.
| Inhibition zone diameter in mm | |||
|---|---|---|---|
| S. aureus | E. coli | P. aeruginosa | |
| Mean ± SD | Mean ± SD | Mean ± SD | |
| a Abbreviations: Cipro20 = ciprofloxacin 20 μg mL−1; T500 = ToCNF 500 μg mL−1; A500 = AgNP 500 μg mL−1; AT500 = AgNP/ToCNF 500 μg mL−1. | |||
| Cipro20 | 25.29 ± 1.19 | 27.85 ± 0.64 | 15.02 ± 1.12 |
| dH2O | — | — | — |
| T500 | — | — | — |
| T250 | — | — | — |
| A500 | 18.34 ± 1.47 | 20.61 ± 1.43 | 13.38 ± 0.82 |
| A250 | 13.63 ± 1.32 | 15.87 ± 0.91 | 11.51 ± 0.87 |
| AT500 | 13.17 ± 0.95 | 16.96 ± 0.40 | 11.23 ± 1.04 |
| AT250 | 10.38 ± 0.68 | 11.62 ± 0.41 | 8.67 ± 1.21 |
 | ||
| Fig. 12 MIC90 against S. aureus of AgNP (a) and AgNP/ToCNF (b) and photograph of the wells (c), against E. coli (d–f), and P. aeruginosa (gi); bar graphs data present the percentage of growth inhibition after 24 hours of treatment exposure measured by OD600. MIC90 against S. aureus of AgNP (a) and AgNP/ToCNF (b) and photograph of the wells (c), against E. coli (d–f), and P. aeruginosa (g–i); bar graphs data present the percentage of growth inhibition after 24 hours of treatment exposure measured by OD600. The experiment was done in triplicate and performed in three trials. | ||
These findings align with previous studies demonstrating that Gram-negative bacteria are generally more susceptible to silver nanoparticles.59–61 The exact mechanism of action of AgNPs remains an area of active research, but the most well-studied mechanism involves the ability of AgNPs to cause damage to bacterial cell membranes.62,63 This is mainly due to their high surface area relative to volume; smaller particle sizes result in greater silver penetration into the cells, causing bacterial cell death. In our experiment, the higher concentration of both test compounds needed to achieve 90% growth inhibition against Gram-positive S. aureus compared to Gram-negative bacteria may be linked to differences in the structural composition of their cell walls. Cell walls of Gram-positive bacteria have a thicker peptidoglycan layer (∼20–80 nm thick), whereas Gram-negative bacteria possess a much thinner peptidoglycan layer (∼2–7 nm thick). The dense, multilayered peptidoglycan layer of Gram-positive bacteria acts as a protective barrier, reducing the diffusion of AgNPs into the cells.64–66
Another possible mechanism explaining the lower MIC of AgNP-containing compounds against Gram-negative bacteria is their ability to penetrate bacterial cells through porin channels in the outer membrane of Gram-negative bacteria. Porins are transmembrane proteins primarily involved in the passive transport of molecules of various sizes and charges across the membrane.67 This provides an additional pathway for AgNP entry into Gram-negative bacterial cells, while Gram-positive bacteria rely solely on diffusion through their thick peptidoglycan layer—a much slower and less efficient process.
Additionally, the results showed that AgNPs were more potent across all bacterial strains compared to AgNP/ToCNF. This can be explained by the presence of ToCNFs: the in situ loading of AgNPs on ToCNFs limits the accessibility of the AgNPs to directly contact bacterial cells, which is crucial for their antibacterial activity while free AgNPs are more potent, their immobilization on ToCNF reduces their potency but serves an essential purpose—mitigating cytotoxicity while providing a slow release of AgNPs for a sustained antibacterial activity.68 This trade-off is further discussed in the LDH cytotoxicity subsection.
log reduction in CFU mL−1 within 24 hours, while bacteriostatic agents resulted in less than a 3log reduction. Ciprofloxacin (20 μg mL−1) served as the positive control, demonstrating reliable antimicrobial efficacy, whereas untreated bacteria acted as the negative control.For S. aureus, AgNPs exhibited bactericidal effects at concentrations of 62.5 μg mL−1 (MIC), 125 μg mL−1 (2× MIC), and 250 μg mL−1 (4× MIC), while AgNPs loaded on ToCNF required higher concentrations—250 μg mL−1 (MIC), 500 μg mL−1 (2× MIC), and 1000 μg mL−1 (4× MIC)—to achieve similar effects. Conversely, for the Gram-negative bacteria E. coli and P. aeruginosa, both free AgNPs and AgNP/ToCNF demonstrated effective bactericidal activity at lower concentrations, reflecting the generally higher susceptibility of these strains.
The data depicted in Fig. 13–15 illustrate that both forms of AgNPs exerted potent bactericidal activity against all tested strains. Free AgNPs at 2× and 4× MIC rapidly reduced bacterial counts by 6logs within just 3 hours, while the MIC concentration achieved the same reduction by 6 hours without any regrowth observed over the subsequent 24 hours. AgNP/ToCNF also demonstrated strong bactericidal properties, though with slightly delayed kinetics, reaching a 6log reduction at 4× MIC within 3 hours and at lower concentrations by 6 hours. This concentration-dependent killing pattern underscores the sustained antimicrobial potential of both treatments. Importantly, the absence of bacterial regrowth during the 24-hour observation period, confirmed by an additional 24-hour incubation of culture plates, supports the conclusion that bacterial death rather than temporary inhibition was responsible for the observed effects.
 | ||
| Fig. 13 Killing kinetics profile of AgNP and AgNP/ToCNF against S. aureus (n = 5). Representative culture plates of S. aureus are shown at (a) 3 h, (b) 6 h, (c) 12 h, and (d) 24 h following treatment. | ||
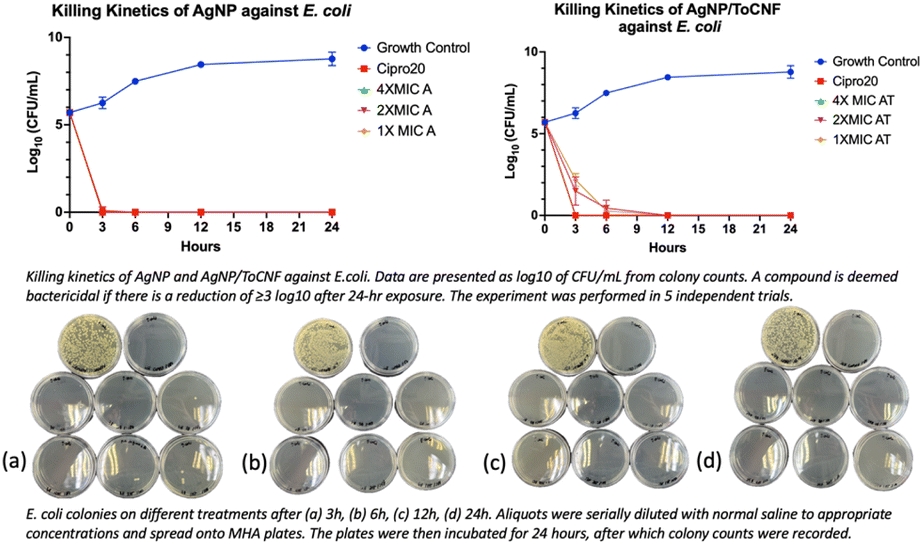 | ||
| Fig. 14 Killing kinetics profile of AgNP and AgNP/ToCNF against E. coli (n = 5). Representative culture plates of E. coli are shown at (a) 3 h, (b) 6 h, (c) 12 h, and (d) 24 h following treatment. | ||
 | ||
| Fig. 15 Killing kinetics profile of AgNP and AgNP/ToCNF against P. aeruginosa (n = 5). Representative culture plates of P. aeruginosa are shown at (a) 3 h, (b) 6 h, (c) 12 h, and (d) 24 h following treatment. | ||
Both test compounds were effective at eliminating 99.9999% bacterial populations. However, the more rapid and potent bactericidal action of free AgNPs compared to AgNP/ToCNF can be attributed to the greater immediate bioavailability and direct contact of silver nanoparticles with bacterial cells. Immobilization of AgNPs onto ToCNFs likely moderates their release, resulting in a slower but sustained antimicrobial effect.34 This controlled release may offer advantages in reducing cytotoxicity and prolonging antibacterial efficacy, a trade-off that warrants consideration in therapeutic applications.
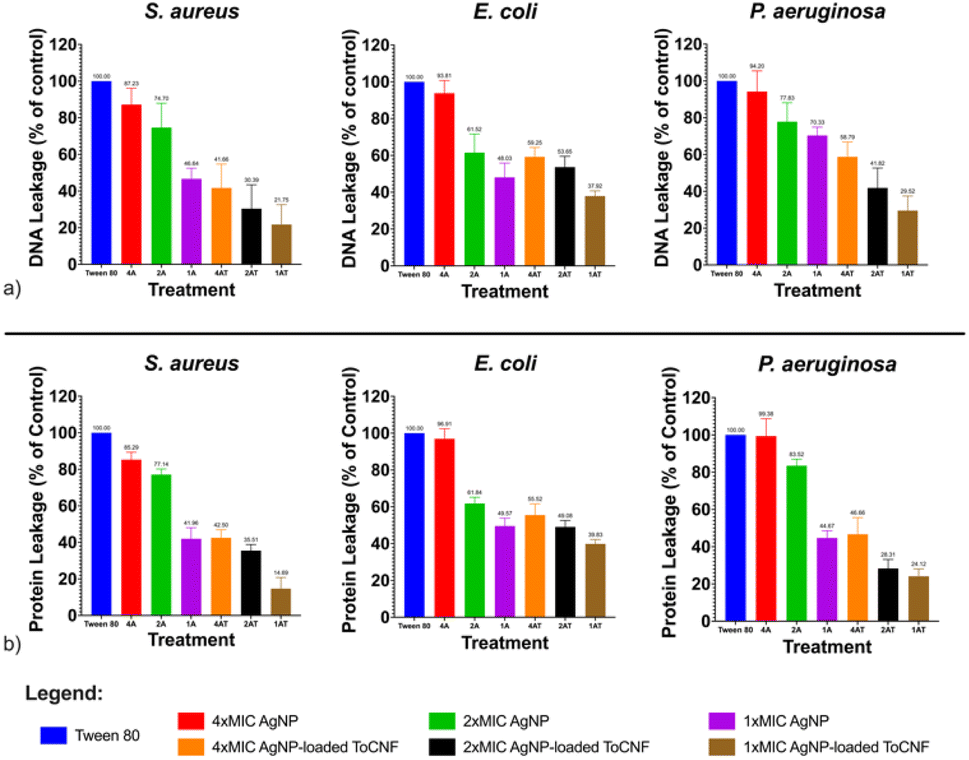 | ||
| Fig. 16 Relative leakage of bacterial intracellular materials (a) DNA, at 260 nm; (b) proteins, at 280 nm for AgNPs and AgNP/ToCNF, n = 9. | ||
Moreover, the slightly lower levels of DNA and protein leakage observed in S. aureus compared to E. coli and P. aeruginosa further support the role of bacterial cell wall structure in modulating susceptibility to AgNP-based treatments. The thick, multilayered peptidoglycan wall of Gram-positive S. aureus likely acts as a barrier, limiting the penetration of AgNPs and reducing their subsequent interaction with the cytoplasmic membrane. In contrast, the thinner peptidoglycan layer and outer membrane of Gram-negative bacteria may facilitate easier access and internalization of AgNPs, resulting in greater membrane disruption and leakage.65
The cell membrane disruption induced by silver-containing compounds, particularly at the nanoscale, is closely associated with the particle size of AgNPs and their affinity for sulfur-containing proteins in the bacterial cell wall. Numerous studies have demonstrated that smaller AgNPs exhibit greater antibacterial activity, primarily due to their higher surface area-to-volume ratio, which enhances their interaction with bacterial membranes and intracellular targets.69–72 This increased surface reactivity enables more effective attachment to the bacterial cell surface and facilitates penetration through the cell envelope.
Additionally, AgNPs are known to interact strongly with thiol (–SH) groups in sulfur-containing proteins present in the bacterial cell wall and membrane. This interaction leads to the denaturation of these proteins and irreversible structural alterations in the cell wall, ultimately compromising membrane integrity and function.62,73 These combined properties—enhanced surface interaction due to small size and specific affinity for thiol groups—allow AgNPs to overcome bacterial defenses, disrupt the membrane, and induce cell lysis. Such mechanisms underpin the potent bactericidal activity observed in silver nanoparticles and their nanocomposites.
In vitro proliferative effects and cytotoxicity
The Alamar Blue assay was employed to assess the proliferative response of fibroblasts to AgNP and AgNP/ToCNF exposure. This assay measures cellular metabolic activity based on the reduction of resazurin, a non-fluorescent dye, to resorufin, a fluorescent and colorimetric compound. A decrease in resorufin signal indicates reduced metabolic activity, which corresponds to compromised cell viability or proliferation.74,75 Fig. 17 illustrates the effects of the different treatments on fibroblast proliferation. | ||
| Fig. 17 Cell proliferation percentages at different time points relative to untreated cells for AgNP and AgNP/ToCNF (n = 6). | ||
ToCNFs were tested for their proliferative effects on HDFs, showing little to no difference in cell proliferation compared to untreated controls. This biocompatibility aligns with previous findings.76,77 Both untreated and ToCNF-treated cells exhibited high Alamar Blue (AB) reduction, indicating healthy metabolic activity and viability. In contrast, treatment with free-state AgNPs resulted in a dose- and time-dependent decrease in AB reduction, suggesting reduced metabolic activity at higher concentrations and longer exposure durations. A statistically significant reduction in metabolic activity was observed at concentrations of 125 μg mL−1 and above, likely due to cytotoxic effects. Time-course analysis further confirmed that prolonged exposure to AgNPs progressively decreased AB reduction. This decline in cell proliferation may be attributed to AgNP-induced disruption of cellular metabolism, possibly via oxidative stress, mitochondrial damage, and depletion of intracellular antioxidants such as reduced glutathione.78 Interestingly, the combination treatment (AgNP/ToCNF) did not significantly affect HDF metabolic activity or proliferation under the tested conditions, suggesting that the ToCNF matrix may mitigate the cytotoxic effects of AgNPs by moderating their release and limiting direct interaction with cells.
However, it is important to note that the Alamar Blue assay evaluates cell viability based on metabolic activity, which is an indirect indicator of cytotoxicity. To complement this, a lactate dehydrogenase (LDH) assay was performed to assess membrane integrity and quantify actual cell damage.
LDH is a stable cytoplasmic enzyme present in all cells and is released into the extracellular medium upon loss of membrane integrity. Its release serves as a hallmark of cellular damage due to apoptosis, necrosis, or other cytotoxic events. The LDH assay quantifies cell death by detecting NADH production during the conversion of lactate to pyruvate, which then reduces a tetrazolium salt (INT) into a red, water-soluble formazan product. The absorbance of formazan is directly proportional to the number of damaged or lysed cells.79,80
A concentration- and time-dependent increase in LDH release was observed in AgNP- and AgNP/ToCNF-treated cells, whereas ToCNFs induced only a slight, concentration-dependent increase in LDH release, with minimal variation across 72 hours (Fig. 18). Notably, AgNPs exhibited significant cytotoxicity at concentrations above 250 μg mL−1 after 24 hours of exposure, and marked LDH release was observed at concentrations of 125 μg mL−1 and higher from 48 to 72 hours. This trend suggests a cumulative cytotoxic effect, where extended exposure leads to progressive membrane damage.
 | ||
| Fig. 18 Cytotoxicity of AgNP, AgNP/ToCNF, and ToCNFs relative to the lysis control (n = 6). | ||
In contrast, AgNP/ToCNF-treated cells showed no significant LDH release at concentrations up to 500 μg mL−1 across all time points, indicating reduced cytotoxicity. Furthermore, ToCNFs alone did not induce significant toxicity at any concentration tested, further supporting their biocompatibility.
As shown in Fig. 19, cells treated with free-state AgNPs exhibited noticeable deposition of nanoparticles on the cell surface, whereas this phenomenon was not observed in cells treated with AgNP/ToCNF. These findings suggest that the cytotoxic effects of free AgNPs are primarily mediated by disruption of cell membrane integrity, likely due to the accumulation of free silver particles on the cytoplasmic membrane. This accumulation may trigger a cascade of cellular responses, including oxidative stress, inflammatory signaling, DNA damage, lipid peroxidation, and ultimately, cell death via apoptosis and necrosis.81–84 The significantly lower LDH release observed for AgNP/ToCNF across all concentrations and time points suggests that immobilizing AgNPs on the surface of ToCNFs reduces their bioavailability and limits direct interactions with the cell membrane. This indicates that the ToCNF carrier plays a modulating role by regulating the release of AgNPs, resulting in a slower, more controlled interaction with the membrane and thereby reducing cytotoxicity.85 The demonstrated non-toxicity of ToCNFs reinforces their potential as a safe and biocompatible carrier for biomedical applications. This intrinsic biocompatibility likely plays a key role in mitigating the cytotoxic effects of the AgNP/ToCNF composite, contributing to its improved cellular compatibility compared to free-state AgNPs.
 | ||
| Fig. 19 Microscopic images of HDF captured under inverted microscope at 400× magnification at each time point of the LDH cytotoxicity assay. | ||
The precise mechanism governing the release of Ag+ ions from ToCNFs remains incompletely understood and warrants further investigation. Current hypotheses suggest that the release may occur through multiple pathways, including the desorption of loosely bound AgNPs from the nanofiber surface, the diffusion of Ag+ ions from the nanofibers via oxidation of metallic silver (Ag0) in the presence of oxygen and protons, and partial degradation and swelling of the ToCNF matrix.60,86,87 These processes may collectively contribute to the gradual exposure and controlled release of silver from the composite material, but more detailed kinetic and mechanistic studies are needed, particularly under physiological conditions.
Fig. 20 illustrates the relationship between the antibacterial activity and cytotoxicity of the test compounds. While free-state AgNPs exhibit superior antibacterial efficacy compared to AgNP/ToCNF, their higher cytotoxicity restricts their suitability for biomedical applications. Conversely, AgNP/ToCNF shows slightly reduced antibacterial potency but significantly lower cytotoxicity relative to the free nanoparticles. The low cytotoxicity observed in AgNP/ToCNF, combined with its strong antibacterial activity—particularly at concentrations between 125 and 500 μg mL−1—is likely due to the controlled release of AgNPs from the ToCNF matrix.
 | ||
| Fig. 20 Graphical representation of the relationship of antibacterial activity vs. cytotoxicity of ToCNF, AgNP, and AgNP/ToCNF. | ||
This controlled release mechanism enables effective penetration and disruption of bacterial membranes while minimizing harmful effects on mammalian cells. The differential susceptibility between bacterial and mammalian cells may be attributed to structural and functional differences; for instance, mammalian cells like HDF possess more robust repair and defense mechanisms that help mitigate damage from AgNP exposure.88 However, further in vivo studies are necessary to validate these findings. Future research should focus on elucidating the precise release kinetics of AgNPs from the ToCNF matrix and assessing antibacterial efficacy and cytotoxicity within complex biological environments. Additionally, optimizing AgNP loading on the ToCNF scaffold could improve antibacterial performance while maintaining biocompatibility, thereby expanding its potential for diverse biomedical applications.
Conclusion
A green and sustainable method for synthesizing silver nanoparticles (AgNPs) in situ on TEMPO-oxidized cellulose nanofibers (ToCNFs) derived from spent mushroom (SMS), an underutilized agro-industrial residue, was successfully develop. The resulting AgNP/ToCNF nanocomposite exhibited potent, dose-dependent antibacterial activity (125–500 μg mL−1) against both Gram-positive and Gram-negative bacterial strains, while demonstrating minimal cytotoxicity towards human dermal fibroblasts (HDFs).Compared to free-state AgNPs, which showed rapid bactericidal effects but higher cytotoxicity, AgNP/ToCNF achieved comparable antibacterial efficacy with significantly reduced toxicity. This is attributed to the controlled release of silver ions from the ToCNF matrix, which limits direct nanoparticle–cell interaction, thus preserving mammalian cell viability. The observed membrane-disruptive mode of action and selective cytotoxicity underline the potential of the composite to selectively target microbial cells while sparing human cells. These findings highlight the potential of the nanocomposite synthesized from agricultural waste for biomedical applications, particularly in antimicrobial coatings and wound healing materials.
Future research should focus on evaluating the long-term stability and controlled release behavior of AgNP/ToCNF composites under physiological conditions. In vivo studies are essential to assess pharmacokinetics, biodistribution, efficacy, and biocompatibility. Scalability, reproducibility, cost-effectiveness, and environmental sustainability must also be addressed for clinical translation.
Additionally, integrating AgNP/ToCNF into formats such as wound dressings, implant coatings, medical textiles, and biodegradable packaging could enable eco-friendly antimicrobial solutions. Optimizing AgNP loading and release kinetics will be key to enhancing antibacterial efficacy while minimizing cytotoxicity across biomedical and environmental applications.
Data availability
The data presented in the study is available in the article.Author contributions
Conceptualization, H. K.; methodology, C. M. K.; software, C. M. K.; validation, C. M. K.; formal analysis, C. M. K.; investigation, C. M. K.; resources, C. M. K.; data curation, C. M. K.; writing—original draft preparation, C. M. K.; writing—review and editing, H. K.; visualization, C. M. K.; supervision, H. K.; project administration, H. K.; funding acquisition, H. K.Conflicts of interest
There are no conflicts to declare. The authors alone are responsible for the content and writing of this article.Acknowledgements
The authors gratefully acknowledge the Faculty of Pharmacy, Universiti Kebangsaan Malaysia for their support in providing the materials necessary to conduct this research and the invaluable contributions made by the Department of Science and Technology-Science Education Institute (DOST-SEI) and University of San Agustin. At the time of writing, C. M. K. was a recipient of the DOST-SEI scholarship.Notes and references
- B. Koul, M. Yakoob and M. P. Shah, Environ. Res., 2022, 206, 112285 CrossRef CAS PubMed.
- R. Phiri, S. Mavinkere Rangappa and S. Siengchin, J. Cleaner Prod., 2024, 434, 139989 CrossRef.
- M. Duque-Acevedo, I. Lancellotti, F. Andreola, L. Barbieri, L. J. Belmonte-Ureña and F. Camacho-Ferre, Environ. Sci. Eur., 2022, 34, 70 CrossRef.
- C. Martín, G. I. Zervakis, S. Xiong, G. Koutrotsios and K. O. Strætkvern, Bioengineered, 2023, 14, 2252138 CrossRef.
- A. Ravlikovsky, M. N. C. Pinheiro, L. Dinca, V. Crisan and L. Symochko, Recycling, 2024, 9, 44 CrossRef.
- K. N. Finney, C. Ryu, V. N. Sharifi and J. Swithenbank, Bioresour. Technol., 2009, 100, 310–315 CrossRef CAS PubMed.
- J. M. Marín-Benito, M. J. Sánchez-Martín and M. S. Rodríguez-Cruz, Toxics, 2016, 4, 17 Search PubMed.
- J. Blair and S. Mataraarachchi, Environments, 2021, 8, 73 Search PubMed.
- M. Dedousi, E.-M. Melanouri, D. Karayannis, E.-I. Kaminarides and P. Diamantopoulou, Carbon Resour. Convers., 2024, 7, 100196 CrossRef.
- A. Siddiqua, J. N. Hahladakis and W. Al-Attiya, Environ. Sci. Pollut. Res. Int., 2022, 29, 58514–58536 Search PubMed.
- I. R. Abubakar, K. M. Maniruzzaman, U. L. Dano, F. S. AlShihri, M. S. AlShammari, S. M. S. Ahmed, W. A. G. Al-Gehlani and T. I. Alrawaf, Int. J. Environ. Res. Public Health, 2022, 19, 12717 CrossRef CAS PubMed.
- R. Grover, A. Goel, L. Wati and K. Raj, Pollut. Res., 2015, 34, 121–124 Search PubMed.
- H. Li, S. Yoshida, N. Mitani, M. Egusa, M. Takagi, H. Izawa, T. Matsumoto, H. Kaminaka and S. Ifuku, Carbohydr. Polym., 2022, 284, 119233 Search PubMed.
- Z. Lou, Y. Sun, X. Zhou, S. A. Baig, B. Hu and X. Xu, Geoderma, 2017, 307, 30–37 Search PubMed.
- S. J. Klausen, A. B. Falck-Ytter, K. O. Strætkvern and C. Martin, Molecules, 2023, 28, 5140 CrossRef CAS PubMed.
- G. Koutrotsios, K. C. Mountzouris, I. Chatzipavlidis and G. I. Zervakis, Food Chem., 2014, 161, 127–135 CrossRef CAS PubMed.
- A. Shakir, B. Azahari, A. Salehabadi, Y. Yusup, M. Yhaya and M. Ahmad, Int. J. Fluid Mech. Therm. Sci., 2020, 75, 113–124 Search PubMed.
- G. Vasilakis, E. M. Rigos, N. Giannakis, P. Diamantopoulou and S. Papanikolaou, Microorganisms, 2023, 11(2), 532 CrossRef CAS PubMed.
- S. Acharya, S. Liyanage, P. Parajuli, S. S. Rumi, J. L. Shamshina and N. Abidi, Polymers, 2021, 13, 4344 CrossRef CAS PubMed.
- M. Madhushree, P. Vairavel, G. T. Mahesha and K. S. Bhat, J. Nat. Fibers, 2024, 21, 2418357 Search PubMed.
- A. Suresh Khurd and B. Kandasubramanian, Carbohydr. Polym. Technol. Appl., 2022, 4, 100234 Search PubMed.
- J. Grondahl, K. Karisalmi and J. Vapaavuori, Soft Matter, 2021, 17, 9842–9858 RSC.
- P. Kaur, N. Sharma, M. Munagala, R. Rajkhowa, B. Aallardyce, Y. Shastri and R. Agrawal, Front. Nanotechnol., 2021, 3, 747329 CrossRef.
- B. Thomas, M. C. Raj, A. K. B, R. M. H, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625 Search PubMed.
- S. Ventura-Cruz and A. Tecante, Food Hydrocolloids, 2021, 118, 106771 CrossRef CAS.
- A. Zeng, B. Wang, M. N. Yiasmin, R. Yang, Y. Tong and W. Zhao, Int. J. Biol. Macromol., 2024, 282, 136897 Search PubMed.
- S. Rashki, N. Shakour, Z. Yousefi, M. Rezaei, M. Homayoonfal, E. Khabazian, F. Atyabi, F. Aslanbeigi, R. Safaei Lapavandani, S. Mazaheri, M. R. Hamblin and H. Mirzaei, Front. Bioeng. Biotechnol., 2021, 9, 732461 CrossRef PubMed.
- A. Babaei-Ghazvini, R. Patel, B. Vafakish, A. F. A. Yazdi and B. Acharya, Int. J. Biol. Macromol., 2024, 278, 135200 Search PubMed.
- K. Patel, J. Shaikh and T. Khan, in Handbook of Nanocelluloses: Classification, Properties, Fabrication, and Emerging Applications, ed. A. Barhoum, Springer International Publishing, Cham, 2020, pp. 1–33, DOI:10.1007/978-3-030-62976-2_42-1.
- S. Suleman Ismail Abdalla, H. Katas, J. Y. Chan, P. Ganasan, F. Azmi and M. Fauzi Mh Busra, RSC Adv., 2020, 10, 4969–4983 Search PubMed.
- M. Scampicchio, J. Wang, A. J. Blasco, A. Sanchez Arribas, S. Mannino and A. Escarpa, Anal. Chem., 2006, 78, 2060–2063 CrossRef CAS PubMed.
- H. Zamani and A. Moradshahi, Mol. Biol. Res. Commun., 2013, 47–55 CAS.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- J. U. Shin, J. Gwon, S. Y. Lee and H. S. Yoo, ACS Omega, 2018, 3, 16150–16157 CrossRef CAS PubMed.
- F. C. Tenover, L. M. Weigel, P. C. Appelbaum, L. K. McDougal, J. Chaitram, S. McAllister, N. Clark, G. Killgore, C. M. O'Hara, L. Jevitt, J. B. Patel and B. Bozdogan, Antimicrob. Agents Chemother., 2004, 48, 275–280 Search PubMed.
- O. Romruen, T. Karbowiak, W. Tongdeesoontorn, K. A. Shiekh and S. Rawdkuen, Polymers, 2022, 14(9), 1830 Search PubMed.
- R. S. Abolore, S. Jaiswal and A. K. Jaiswal, Carbohydr. Polym. Technol. Appl., 2024, 7, 100396 CAS.
- F. Hamzah, A. Idris and T. K. Shuan, Biomass Bioenergy, 2011, 35, 1055–1059 CrossRef CAS.
- M. Adi, T. N. Tengku Izhar, N. Mohamad Ibrahim, N. Abd Aziz, H. Hadiyanto and M. Matei, IOP Conf. Ser. Earth Environ. Sci., 2023, 1216, 012015 Search PubMed.
- N. Huynh, J. J. Valle-Delgado, W. Fang, S. Arola and M. Österberg, Carbohydr. Polym., 2023, 317, 121095 CrossRef CAS PubMed.
- J. Levanič, V. P. Šenk, P. Nadrah, I. Poljanšek, P. Oven and A. Haapala, ACS Sustain. Chem. Eng., 2020, 8, 17752–17762 CrossRef.
- O. Nechyporchuk, M. N. Belgacem and J. Bras, Ind. Crops Prod., 2016, 93, 2–25 Search PubMed.
- G. Lowry, R. Hill, S. Harper, A. Rawle, C. Hendren, F. Klaessig, U. Nobbmann, P. Sayre and J. Rumble, Environ. Sci.: Nano, 2016, 3, 953–965 RSC.
- G. Chinga-Carrasco, E. Pasquier, A. Solberg, I. Leirset, J. S. Stevanic, J. Rosendahl and J. Håkansson, Carbohydr. Polym., 2023, 314, 120923 CrossRef CAS PubMed.
- A. Isogai and Y. Zhou, Curr. Opin. Solid State Mater. Sci., 2019, 23, 101–106 CrossRef CAS.
- I. Besbes, S. Alila and S. Boufi, Carbohydr. Polym., 2011, 84, 975–983 CrossRef CAS.
- X. Xu, F. Liu, L. Jiang, J. Y. Zhu, D. Haagenson and D. P. Wiesenborn, ACS Appl. Mater. Interfaces, 2013, 5, 2999–3009 CrossRef CAS.
- E. Afra, H. Yousefi, M. M. Hadilam and T. Nishino, Carbohydr. Polym., 2013, 97, 725–730 Search PubMed.
- Y. Wang, T. Pääkkönen, L. Solhi, N. Yousefi and E. Kontturi, Carbohydr. Polym., 2025, 354, 123315 CrossRef CAS.
- T. M. Oyinloye and W. B. Yoon, Processes, 2020, 8, 354 CrossRef CAS.
- M. Sastry, K. Mayya and K. Bandyopadhyay, Colloids Surf., A, 1997, 127, 221–228 CrossRef CAS.
- K. Nabeela, R. T. Thomas, J. B. Nair, K. K. Maiti, K. G. K. Warrier and S. Pillai, ACS Appl. Mater. Interfaces, 2016, 8, 29242–29251 Search PubMed.
- E. Smiechowicz, B. Niekraszewicz and P. Kulpinski, Materials, 2021, 14, 4126 CrossRef CAS PubMed.
- A. W. Jatoi, I. S. Kim and Q. Q. Ni, Mater. Sci. Eng., C, 2019, 98, 1179–1195 CrossRef CAS PubMed.
- L. Wei, J. Lu, H. Xu, A. Patel, Z. S. Chen and G. Chen, Drug Discovery Today, 2015, 20, 595–601 Search PubMed.
- Z. Mat Lazim, S. Salmiati, M. Marpongahtun, N. Z. Arman, M. R. Mohd Haniffah, S. Azman, E. L. Yong and M. R. Salim, Water, 2023, 15, 1349 CrossRef CAS.
- H. Ito, M. Sakata, C. Hongo, T. Matsumoto and T. Nishino, Nanocomposites, 2018, 4, 167–177 Search PubMed.
- N. Jiang, Y. Hu and Y. Cheng, Polymers, 2024, 16, 1016 Search PubMed.
- E. D. Cavassin, L. F. P. de Figueiredo, J. P. Otoch, M. M. Seckler, R. A. de Oliveira, F. F. Franco, V. S. Marangoni, V. Zucolotto, A. S. S. Levin and S. F. Costa, J. Nanobiotechnol., 2015, 13, 64 CrossRef PubMed.
- D. Dechojarassri, K. Komatsu, A. Sawara, H. Tamura and T. Furuike, Fibers, 2023, 11, 69 CrossRef CAS.
- A. S. Dove, D. I. Dzurny, W. R. Dees, N. Qin, C. C. Nunez Rodriguez, L. A. Alt, G. L. Ellward, J. A. Best, N. G. Rudawski, K. Fujii and D. M. Czyż, Front. Microbiol., 2023, 13, 1064095 CrossRef PubMed.
- T. C. Dakal, A. Kumar, R. S. Majumdar and V. Yadav, Front. Microbiol., 2016, 7, 1831 Search PubMed.
- E. O. Mikhailova, Antibiotics, 2025, 14, 5 CrossRef CAS PubMed.
- S. Anees Ahmad, S. Sachi Das, A. Khatoon, M. Tahir Ansari, M. Afzal, M. Saquib Hasnain and A. Kumar Nayak, Mater. Sci. Energy Technol., 2020, 3, 756–769 Search PubMed.
- P. R. More, S. Pandit, A. D. Filippis, G. Franci, I. Mijakovic and M. Galdiero, Microorganisms, 2023, 11, 369 Search PubMed.
- I. X. Yin, J. Zhang, I. S. Zhao, M. L. Mei, Q. Li and C. H. Chu, Int. J. Nanomed., 2020, 15, 2555–2562 CrossRef CAS PubMed.
- E. O. Mikhailova, J. Funct. Biomater., 2020, 11, 84 Search PubMed.
- S. Elayaraja, K. Zagorsek, F. Li and J. Xiang, Carbohydr. Polym., 2017, 166, 329–337 CrossRef CAS PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- L. F. Espinosa-Cristóbal, G. A. Martínez-Castañón, R. E. Martínez-Martínez, J. P. Loyola-Rodríguez, N. Patiño-Marín, J. F. Reyes-Macías and F. Ruiz, Mater. Lett., 2009, 63, 2603–2606 CrossRef.
- S.-H. Kim, H.-S. Lee, D.-S. Ryu and S.-J. Choi, Microbiol. Biotechnol. Lett., 2011, 39, 77–85 CAS.
- J. Li, K. Rong, H. Zhao, F. Li, Z. Lu and R. Chen, J. Nanosci. Nanotechnol., 2013, 13, 6806–6813 CrossRef PubMed.
- T. Bruna, F. Maldonado-Bravo, P. Jara and N. Caro, Int. J. Mol. Sci., 2021, 22, 7202 CrossRef CAS PubMed.
- M. N. Dinh, M. Hitomi, Z. A. Al-Turaihi and J. G. Scott, MethodsX, 2024, 13, 103024 Search PubMed.
- E. M. Longhin, N. El Yamani, E. Rundén-Pran and M. Dusinska, Front. Toxicol., 2022, 4, 981701 CrossRef PubMed.
- H. He, X. Shi, W. Chen, R. Chen, C. Zhao and S. Wang, J. Agric. Food Chem., 2020, 68, 7425–7433 CrossRef CAS PubMed.
- R. Kummala, D. Soto Véliz, Z. Fang, W. Xu, T. Abitbol, C. Xu and M. Toivakka, Biomacromolecules, 2020, 21, 1560–1567 CrossRef CAS PubMed.
- M. J. Piao, K. A. Kang, I. K. Lee, H. S. Kim, S. Kim, J. Y. Choi, J. Choi and J. W. Hyun, Toxicol. Lett., 2011, 201, 92–100 CrossRef CAS PubMed.
- P. Kumar, A. Nagarajan and P. D. Uchil, Cold Spring Harb. Protoc., 2018, 2018 Search PubMed.
- T. Riss, A. Niles, R. Moravec, N. Karassina and J. Vidugiriene, Cytotoxicity Assays: in Vitro Methods to Measure Dead Cells, Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda (MD), 2004 Search PubMed.
- N. S. Alharbi and A. I. Felimban, J. King Saud Univ., Sci., 2023, 35, 102972 CrossRef.
- S. Arora, J. Jain, J. M. Rajwade and K. M. Paknikar, Toxicol. Lett., 2008, 179, 93–100 CrossRef CAS PubMed.
- M. M. Rohde, C. M. Snyder, J. Sloop, S. R. Solst, G. L. Donati, D. R. Spitz, C. M. Furdui and R. Singh, Part. Fibre Toxicol., 2021, 18, 37 Search PubMed.
- T. Zhang, L. Wang, Q. Chen and C. Chen, Yonsei Med. J., 2014, 55, 283–291 CrossRef CAS PubMed.
- J. Wu, Y. Zheng, W. Song, J. Luan, X. Wen, Z. Wu, X. Chen, Q. Wang and S. Guo, Carbohydr. Polym., 2014, 102, 762–771 CrossRef CAS PubMed.
- G. Sabarees, V. Velmurugan, G. P. Tamilarasi, V. Alagarsamy and V. Raja Solomon, Polymers, 2022, 14, 3994 CrossRef CAS PubMed.
- Z. Adamczyk, M. Oćwieja, H. Mrowiec, S. Walas and D. Lupa, J. Colloid Interface Sci., 2016, 469, 355–364 CrossRef CAS PubMed.
- Y. Lu, G. Pan, Z. Wei, Y. Li and X. Pan, Exp. Gerontol., 2024, 196, 112559 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |