
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting apoptotic pathways in cancer: design, synthesis, and molecular docking studies of 1,3,5-trisubstituted-1H-pyrazole derivatives with Bcl-2 inhibition and DNA damage potential†
Ahmed Temirak *a,
Ahmed A. F. Solimanb,
Mohamed B. Shalabyc,
Mariam G. Eshakd,
Wagdy K. B. Khalild,
Zahid Shafiqe and
Nader M. Boshta*f
*a,
Ahmed A. F. Solimanb,
Mohamed B. Shalabyc,
Mariam G. Eshakd,
Wagdy K. B. Khalild,
Zahid Shafiqe and
Nader M. Boshta*f
aChemistry of Natural and Microbial Products Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, Giza, 12622, Egypt. E-mail: amtemirak@hotmail.com
bDrug Bioassay-Cell Culture Laboratory, Pharmacognosy Department, National Research Center, Giza 12622, Egypt
cToxicology Research Department, Research Institute of Medical Entomology (RIME), General Organisation of Teaching Hospitals and Institutes (GOTHI), Ministry of Health and Population (MoHP), Dokki, P. O. Box 12311, Cairo, Egypt
dDepartment of Cell Biology, National Research Centre, 12262 El-Bohouth St., Cairo, Egypt
eInstitute of Chemical Sciences, Bahauddin Zakariya University, Multan, Pakistan
fChemistry Department, Faculty of Science, Menoufia University, Shebin El-Koam 32511, Egypt. E-mail: nboshta2@gmail.com
First published on 6th June 2025
Abstract
The search for new anticancer agents targeting apoptotic and autophagic pathways is crucial due to their roles in cellular homeostasis and cancer cell elimination. In this study, we synthesized and evaluated a series of 1,3,5-trisubstituted-1H-pyrazole derivatives as potential inhibitors of Bcl-2, a key regulator of apoptosis and autophagy. Several compounds activated pro-apoptotic proteins Bax, p53, and Caspase-3. Structure–activity relationship (SAR) studies assessed the cytotoxic effects of the compounds on MCF-7, A549, and PC-3 cancer cell lines. Compounds 4, 5, 6b, 6c, 7, 8, 10b, 10c, and 12b showed significant cytotoxicity against MCF-7 cells (IC50: 3.9–35.5 μM), with similar activity observed against A549 and PC-3 cell lines. Compounds 6c, 8, 10b, and 10c also induced DNA damage, as evidenced by increased comet tail length, suggesting they cause genotoxic stress through DNA strand breaks. SAR analysis highlighted the importance of chlorophenyl, thiazole, and sulfonamide groups in enhancing cytotoxicity. Molecular docking confirmed high binding affinity of compounds 10b and 10c to Bcl-2 through key hydrogen bonding interactions. These findings suggest that the 1,3,5-trisubstituted-1H-pyrazole derivatives effectively target Bcl-2, activate apoptotic pathways, and induce DNA damage, making them promising candidates for further anticancer investigation.
Introduction
Cancer is a significant global health burden and ranks as the second-leading cause of death, following cardiovascular diseases.1,2 It remains one of the most challenging diseases to treat, characterized by uncontrolled cell proliferation and the ability to avoid programmed cell death mechanisms. The toxicity of existing anticancer drugs and the limited effectiveness of current chemotherapies are major obstacles in the fight against cancer. Consequently, the primary goal in the field of organic medical chemistry, particularly in targeted therapeutic strategies, is to design and discover effective and selective antitumor agents.3 Such strategies involve inducing apoptosis and autophagy.Apoptosis, a form of programmed cell death, is essential for maintaining cellular homeostasis and eliminating damaged cells. Tumor cells are known to have dysregulated apoptotic machinery, enabling tumor cells to survive longer than they should.4 Autophagy, a cellular degradation process, plays a dual role in cancer. While autophagy can provide nutrients to support cancer cell survival under stress, it can also promote cell death when overly activated.5 The balance between these processes is crucial in determining cell fate. One of the key regulators of both apoptosis and autophagy is the B-cell lymphoma 2 (Bcl-2) protein family.
Bcl-2 as a therapeutic target, Bcl-2 is an anti-apoptotic protein that is overexpressed in various cancers, contributing to tumorigenesis and resistance to chemotherapy.6 Therefore, targeting Bcl-2 to promote apoptosis and autophagy represents a promising strategy for cancer therapy.7 Bcl-2 proteins inhibit apoptosis by binding to pro-apoptotic proteins such as BCL-2-associated X protein (BAX) and also modulate autophagy by interacting with Beclin 1, a critical autophagy protein.8 Bcl-2 inhibitors can disrupt the interaction between Bcl-2 and pro-apoptotic proteins, thereby inducing cell death.9 Moreover, inhibiting the interaction between Bcl-2 and Beclin 1 can promote autophagy, which may enhance the anti-tumor effects of Bcl-2 inhibition (Fig. 1).10,11 By leveraging these insights, our current study aims to further explore the therapeutic potential of Bcl-2 inhibitors.
 | ||
| Fig. 1 Examples of reported 1,3,5-trisubstituted-1H-pyrazole derivatives with anticancer activities.11–14 | ||
Recent studies have highlighted the potential of 1,3,5-trisubstituted-1H-pyrazole derivatives as promising anticancer agents. These compounds have shown significant antiproliferative activity against various cancer cell lines, making them a focus of medicinal chemistry research. For instance, compound I exhibited remarkable antiproliferative effects against MCF-7, B16-F10, and HCT-116 cancer cell lines. Furthermore, this compound demonstrated potent inhibition of key oncogenic proteins, including epidermal growth factor receptor (EGFR) and HER-2, which are critical targets in cancer therapy.12 Additionally, compound II, identified from a separate study, was found to have a selective anticancer influence on colon cancer cell lines, particularly HT-29, where it displayed a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) GI50 value of −6.37, indicating its efficiency against this cancer type.13
GI50 value of −6.37, indicating its efficiency against this cancer type.13
Another potent derivative, compound III, has shown broad-spectrum anticancer activity with a mean GI50 of 0.071 μM and a TGI of 0.76 μM. It exhibited its highest antiproliferative effects on the non-small cell lung cancer line HOP-92, where the GI50 was less than 0.01 μM. This compound was also active against other lines, including HCT-116 (GI50 = 0.018 μM), CNS cancer line SNB-75 (GI50 = 0.0159 μM), ovarian cancer NCI/ADR-RES (GI50 = 0.0169 μM), and renal cancer RXF 393 (GI50 = 0.0197 μM).14 In the realm of Bcl-2 inhibition, compound IV (SW076956) emerged as a potent inhibitor, targeting the Bcl-2 protein involved in apoptosis.11 Structure–activity relationship studies on this compound led to the development of more selective Bcl-2 inhibitors, such as compound V, which specifically disrupts the Bcl-2-Beclin 1 interaction responsible for autophagy regulation while sparing the Bcl-2-Bax interaction critical for apoptosis. This specificity opens new avenues for cancer therapy by promoting autophagic cell death without triggering apoptosis.11
In our previous research, we synthesized 1,3,5-trisubstituted-1H-pyrazole derivatives and evaluated their anticancer activities, particularly focusing on ERK and RIPK3 kinase inhibition. These compounds were active against ERK and RIPK3 and demonstrated significant cytotoxicity.15 Building on this success, we synthesized new analogues and investigated their activity, particularly targeting ERK and RIPK3. However, these new compounds were found to be less active in those assays. Despite this reduced activity, they exhibited promising cytotoxicity on several cancer cell lines, that encouraged us to explore their activity on other targets related to apoptosis and autophagy.
Our subsequent screening efforts, including in vitro assays for different targets and compound similarity searches in the literature, identified Bcl-2 as a novel target for our pyrazole derivatives. The promising cytotoxicity data of these compounds against cancer cells, combined with their structural similarity to known Bcl-2 inhibitors, specifically compound SW076956 encouraged us to investigate their ability to inhibit Bcl-2 to develop effective anticancer agents (Fig. 2).11 The aim of this study is to develop small molecule inhibitors of Bcl-2 with dual functionality in promoting both autophagy and apoptosis. We synthesized different derivatives of our initial compounds to study the structure–activity relationship (SAR) and enhance the potency of Bcl-2 inhibition. Our findings suggest that these novel Bcl-2 inhibitors could serve as potential therapeutic agents for cancer treatment, offering a new strategy to induce cancer cell death through both apoptotic and autophagic pathways (Fig. 2).
 | ||
| Fig. 2 Design of the new 1,3,5-trisubstituted-1H-pyrazole derivatives with proposed Bcl-2 inhibition activities. | ||
Results and discussion
Chemistry
Chalcones are chemical compounds consisting of two aromatic/heterocyclic rings linked by three carbon atoms, forming an α, β-unsaturated carbonyl system. These compounds are abundant in edible plants and are frequently utilized in traditional folk remedies, such as Angelica, Glycyrrhiza, Humulus, and Scutellaria. Moreover, chalcones play a crucial role as precursors in the biosynthesis of flavonoids and isoflavonoids. The key characteristic of chalcones lies in their conjugated double bond system and completely delocalized π-electron system present in both aromatic rings. As a result, they exhibit relatively low redox potentials and a high tendency to participate in electron transfer reactions.16The starting material 3 can be prepared using a specific type of aldol-condensation reaction called the base-catalyzed Claisen–Schmidt condensation.14 This reaction involves the reaction of the commercially available 1-methyl-1H-pyrrole-2-carbaldehyde 1 with the commercially available ketone 1-(3,4-dichlorophenyl)ethan-1-one 2. The reaction takes place in the presence of 10% NaOH as a base, using ethanol as the solvent, and stirring at room temperature for 4 hours. As a result, the final compounds are obtained in the form of yellow powder (Scheme 1). The key intermediate 3-(3,4-dichlorophenyl)-5-(1-methyl-1H-pyrrol-2-yl)-4,5-dihydro-1H-pyrazole-1-carbothioamide 5 was obtained by heating to reflux with Thiosemicarbazide in ethanol for 8 h in the presence of catalytic amounts of sodium hydroxide.
 | ||
| Scheme 1 Synthesis of target compounds 4, and 6a–e. Reagents and conditions: (a) NaOH (10% aq.), EtOH, 0 °C–rt, 4 h; (b) diethyl azodicarboxylate, Ph3P, toluene, rt, 7h, 60%; (c) thiosemicarbazide, NaOH, EtOH, 100 °C, 8 h, 87%; (d) phenacyl bromide derivative, EtOH, 100 °C, 3h, 49–88%. | ||
In order to gain a deeper understanding of the SARs of the thiazole derivatives, we incorporated various aromatic and heteroaromatic residues at position 4 of the thiazole ring. The synthesis of the target compounds 6a–e were by reaction of the last the key intermediate 5 with the appropriate phenacyl bromide derivative in ethanol. On the other hand, the starting material 3 on reacting with diethyl azodicarboxylate in the presence of triphenyl phosphine provides pyrazole-1,2(3H)-dicarboxylate derivative 4 as depicted in Scheme 1. The structures of all the final products were verified using 1H and 13C NMR spectroscopy.
With more focus on studying the effect of different residues at position 4 of the thiazole ring, the suitable intermediate 7 was synthesized from the starting material 5 via the reaction with α-bromopyruvate. Subsequently, we used hydrazine hydrate under reflux conditions to obtain the hydrazide derivative 8. Further, the last compound was reacted with cyanogen bromide to afford the aminooxadiazole derivative 9. Moreover, 8 reacted with aryl sulfonyl chloride derivatives to afford the corresponding benzene sulfonohydrazide derivatives 10a–c, as outlined in Scheme 2.
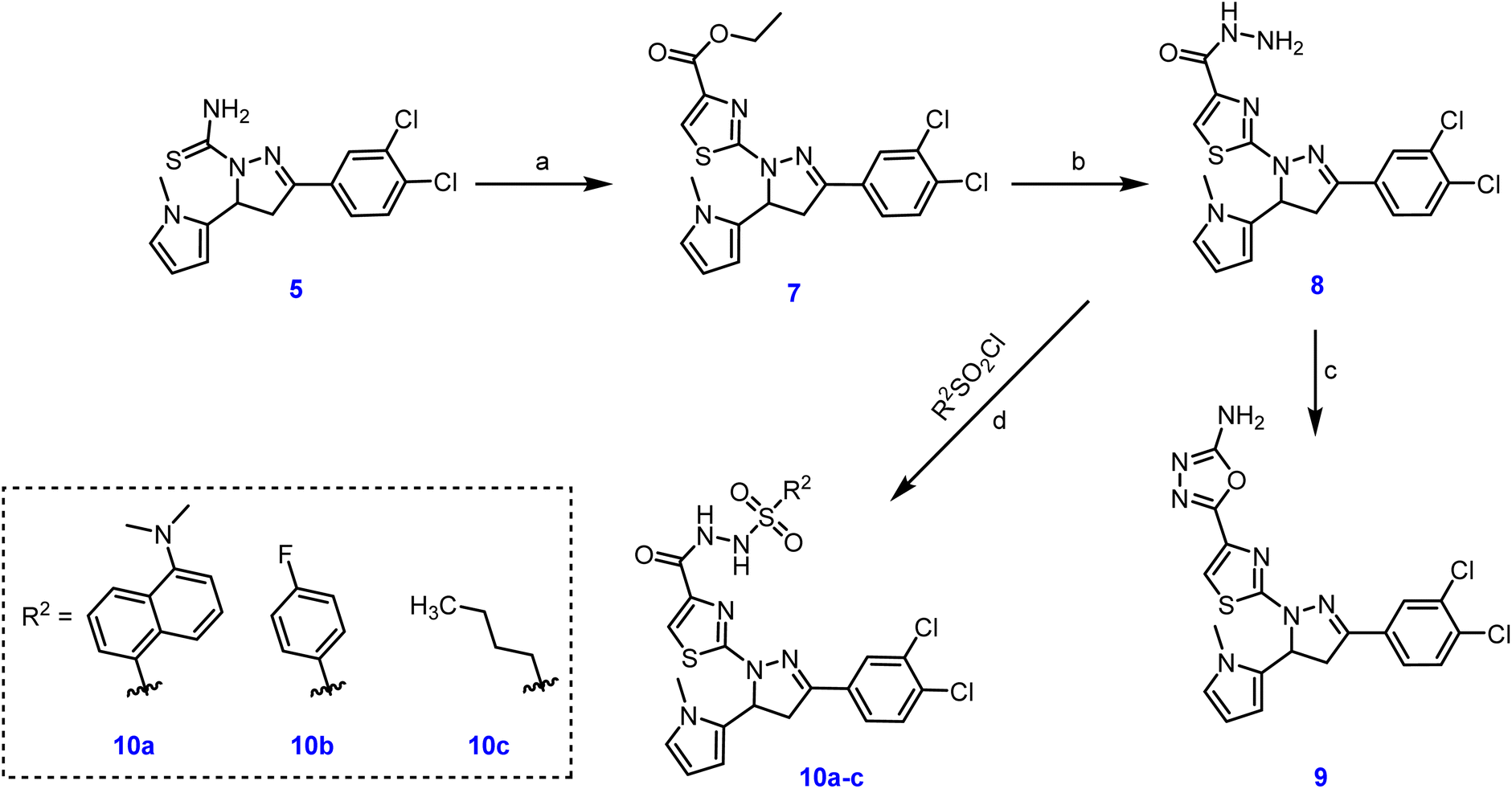 | ||
| Scheme 2 Synthesis of target compounds 9, and 10a–c. Reagents and conditions: (a) α-bromopyruvate, EtOH, 60 °C, 4 h, 88%; (b) hydrazine hydrate, EtOH, 100 °C, 5h, 89%; (c) (1) cyanogen bromide, MeOH, 1,4-dioxane, rt, 1 h (2) NaHCO3, rt, 18 h, 73%; (d) aryl sulfonyl chloride, DMF, 0 °C–rt, 4–10 h, 65–84%. | ||
Starting from the key intermediate 5, we also decided to study incorporating the thiazolone ring and its derivatives at position 1 of the 1H-pyrazole ring. To begin, we synthesized the thiazolone derivative 11 through a reaction between 5 and ethyl bromoacetate in ethanol. Subsequently, to explore the derivatization at the thiazolone ring, we employed various aldehydes and condensed them with 11 in the presence of piperidine in ethanol to afford the target compounds 2-amino-5-benzylidenethiazol-4-one derivatives 12a–c, as outlined in Scheme 3.
 | ||
| Scheme 3 Synthesis of target compounds 12a–c. Reagents and conditions: (a) ethyl bromoacetate, EtOH, 100 °C, 3 h, 83%; (b) benzaldehyde derivative, piperidine, EtOH, 100 °C, 4 h, 71–81%. | ||
Biological screening
| Compound | IC50 (μM) | ||
|---|---|---|---|
| MCF-7 | PC3 | A549 | |
| 4 | 67.8 ± 0.12 | 90.8 ± 0.166 | 28.4 ± 0.08 |
| 5 | 94.3 ± 0.122 | 81.0 ± 0.139 | 92.1 ± 0.112 |
| 6b | 96.1 ± 0.135 | 91.2 ± 0.144 | 84.0 ± 0.14 |
| 6c | 58.5 ± 0.10 | 95.3 ± 0.175 | 92.3 ± 0.184 |
| 7 | 92.3 ± 0.184 | 40.2 ± 0.110 | 44.4 ± 0.09 |
| 8 | 54.8 ± 0.09 | 92.1 ± 0.132 | 90.3 ± 0.155 |
| 10b | 41.4 ± 0.08 | 62.2 ± 0.132 | 30.0 ± 0.10 |
| 10c | 34.5 ± 0.157 | 54.1 ± 0.128 | 38.9 ± 0.10 |
| 12b | 86.1 ± 0.145 | 95.4 ± 0.155 | 66.3 ± 0.10 |
| Doxorubicin | 45.02 ± 1.60 | 32.4 ± 0.70 | 35.4 ± 0.90 |
The newly developed pyrazole derivatives demonstrated notable cytotoxic effects on the PC3, MCF-7 and A549 cancer cell lines highlighting their potential as anticancer agents. Specifically, compounds 4, 5, 6b, 6c, 7, 8, 10b, 10c, and 12b demonstrated significant cytotoxic effects on the MCF-7 breast cancer cell line. Against the PC-3 cell line, compounds 5, 7, 10b, and 10c demonstrated significant cytotoxic effects. For the A549 lung cancer cell line, compounds 4, 5, 6b, 7, 10b, 10c, and 12b showed significant cytotoxicity. The consistent presence of a chlorophenyl group in all compounds significantly contributes to their baseline cytotoxicity due to its strong electron-withdrawing properties (Fig. 3).
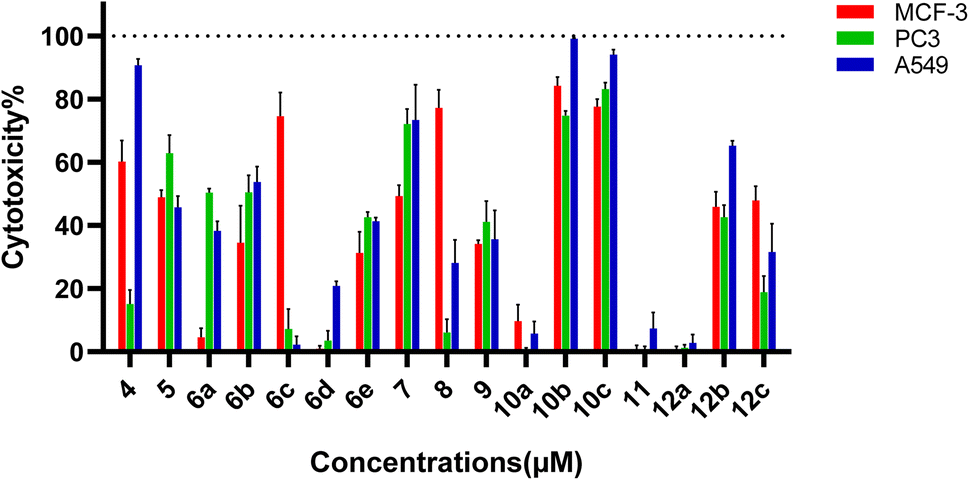 | ||
| Fig. 3 Cytotoxicity% of our compounds against human prostate cancer (PC-3), human breast cancer (MCF-7) and human lung cancer (A549) at 100 μM concentration. Results are represented as the mean of three independent experiments (n = 3). | ||
Compounds featuring a thiazole ring, such as 6c, 8, 10b, and 10c, exhibit enhanced cytotoxic activity. Additional ring systems like the pyridine ring in 6c and the acylhydrazone group in 8 further boost the bonding interactions, respectively. Sulfonamide groups with substituents, particularly the fluorophenyl group in 10b and the aliphatic chain in 10c, significantly enhance cytotoxic effects, with 10c showing the highest activity against MCF-7 cells.
The variation in IC50 values across different compounds indicates that specific structural modifications can significantly influence their anticancer activity. This variability underscores the importance of understanding the SAR to optimize these compounds for better efficacy. Against the PC-3 cell line, 7 showed the highest cytotoxicity with an IC50 of 40.2 ± 0.110 μM, followed by 10c with 54.1 ± 0.128 μM and 10b with 62.2 ± 0.132 μM. Compound 5 displayed the lowest activity with an IC50 of 81.0 ± 0.139 μM, reinforcing the need for potent substituents to enhance efficacy. Against the MCF-7 breast cancer cell line, the IC50 values of the compounds ranged widely, reflecting the impact of different structural features. Compound 10c showed the highest cytotoxicity with an IC50 value of 34.5 ± 0.157 μM which is better than the reference compound doxorubicin with an IC50 vale of 45.02 ± 1.60 μM, indicating that the presence of a sulfonamide group with an aliphatic chain substantially enhances activity. 10b also displayed significant activity with an IC50 of 41.4 ± 0.08 μM, attributed to its sulfonamide group with a fluorophenyl substituent.
Moderate activity was observed for 6c and 8, with IC50 values of 58.5 ± 0.10 μM and 54.8 ± 0.09 μM, respectively, highlighting the beneficial effects of the acylhydrazone and pyridine ring systems. Compound 5 exhibited the lowest activity against MCF-7, with an IC50 of 96.1 ± 0.135 μM, suggesting that additional potent substituents are necessary to enhance efficacy. For the A549 lung cancer cell line, the compounds also displayed varied cytotoxic effects. Compound 4 demonstrated the highest activity with an IC50 value of 28.4 ± 0.08 μM, indicating that the ester groups significantly improve the activity. 10b showed strong activity with an IC50 of 30.0 ± 0.10 μM, likely due to the presence of the sulfonamide group with a fluorophenyl substituent. 10c had an IC50 value of 38.9 ± 0.10 μM, suggesting that the aliphatic chain on the sulfonamide group enhances cytotoxicity. Compound 5 exhibited the lowest activity against A549, with an IC50 of 92.1 ± 0.112 μM.
 | ||
| Fig. 4 The alterations of Bcl-2, BAX, P53, and Caspase-3 gene expression in three different cancer cell lines (MCF-7, A549 and PC3) treated with compounds 6c, 8, 10b, 10c and doxorubicin (Doxo). Data are presented as mean ± SEM. a,b,c,d Mean values within tissue with unlike superscript letters were significantly different (P < 0.05). | ||
Bcl-2 is known to prevent apoptosis by inhibiting the release of cytochrome c from mitochondria, thereby blocking the apoptotic cascade. The observed downregulation of Bcl-2 implies that our compounds can disrupt this protective mechanism, making cancer cells more susceptible to programmed cell death. This is particularly significant as overexpression of Bcl-2 is often associated with resistance to chemotherapy and poor prognosis in various cancers. In contrast, the expression levels of the pro-apoptotic genes BAX, p53, and Caspase-3 increased significantly in the treated cancer cell lines. Compounds 6c, 10b, and 10c demonstrated a promising increase in the expression of these pro-apoptotic genes when compared to Doxorubicin. Specifically, these compounds enhanced the expression of BAX and Caspase-3 in MCF-7 and A549 cancer cell lines, with a slightly lesser effect observed in the PC3 cells. This suggests that these compounds can trigger apoptosis more effectively in breast and lung cancer cells.
BAX promotes apoptosis by promoting the release of cytochrome c from the mitochondria, leading to the activation of caspases. Caspase-3 is a critical executioner of apoptosis, responsible for the cleavage of various cellular substrates leading to cell death. The increased expression of these genes suggests that our compounds can effectively initiate and propagate the apoptotic signal within cancer cells. Regarding p53, the highest increase in gene expression was observed in PC3 cells. p53, often referred to as the “guardian of the genome,” plays a crucial role in regulating the cell cycle and inducing apoptosis in response to DNA damage. The upregulation of p53 indicates that our compounds may enhance the DNA damage response, leading to increased cell cycle arrest and apoptosis in prostate cancer cells.
The differential effects of our compounds on various cell lines underscore the complexity of their action mechanisms. While all compounds were effective in downregulating BCL-2 and upregulating BAX, p53, and Caspase-3, the extent of these effects varied among the cell lines. This variability suggests that the efficacy of our compounds may depend on the specific genetic and molecular context of each cancer type. These findings highlight the potential of compounds 6c, 10b, and 10c as powerful anticancer agents capable of modulating key apoptotic pathways. Their ability to decrease BCL-2 and increase BAX, p53, and Caspase-3 expression provides a strong rationale for further investigation and development of these compounds as targeted therapies for cancer treatment. Future studies should focus on elucidating the precise molecular interactions and pathways involved, as well as evaluating the in vivo efficacy and safety of these compounds in animal models.
Compounds 6c, 8, 10b, and 10c induced substantial DNA damage, as evidenced by the increased comet tail length and percentage of DNA in the tail. The presence of DNA strand breaks, as visualized by the comet assay, suggests that these compounds effectively induce genotoxic stress in cancer cells. This genotoxicity is a critical aspect of their anticancer activity, leading to apoptosis and cell death. In detail, the DNA damage induced by compound 6c was the most pronounced, with values surpassing those observed with Doxorubicin. This suggests that compound 6c is particularly effective in causing DNA strand breaks, thereby triggering cell death pathways. Similarly, compounds 10b and 10c also showed significant DNA damage, indicating their potential as strong anticancer agents. Compound 8, while slightly less effective than 6c, still demonstrated considerable DNA damage, supporting its role in cancer cell cytotoxicity. The comet assay results highlight the mechanism by which our compounds exert their anticancer effects. By causing significant DNA damage, they likely activate the DNA damage response pathways, leading to cell cycle arrest and apoptosis. This mechanism is supported by the observed increase in pro-apoptotic gene expression (BAX, p53, and Caspase-3) and the decrease in the anti-apoptotic gene Bcl-2, as discussed previously.
Molecular docking studies
Compounds 6c, 8, 10b, and 10c, among the newly synthesized 1,3,5-trisubstituted-1H-pyrazole derivatives, showed the highest cytotoxic activities towards MCF-7, A549, and PC3 cancer cell lines. These compounds significantly decreased Bcl-2 gene expression while increasing the expression of pro-apoptotic genes Bax, P53, and Caspase-3. This implies their anticancer mechanism may involve disrupting Bcl-2's interactions with pro-apoptotic and autophagy-related proteins. Based on these observations, we used molecular docking to study the binding affinities and interactions of these compounds with the Bcl-2 protein.The X-ray crystallographic structure of the Bcl-2 protein, along with its native ligand phenylacyl sulfonamide derivative (N,N-dibutyl-4-chloranyl-1-[2-(3,4-dihydro-1H-isoquinolin-2-ylcarbonyl) -4-[(7-iodanylnaphthalen-2-yl)sulfonylcarbamoyl]phenyl]-5-methyl-pyrazole-3-carboxamide), was obtained from the Protein Data Bank (PDB) under the accession code 4AQ3. The receptor preparation for docking was carried out using Biovia Discovery Studio. Initially, all water molecules and non-essential chains were removed from the protein structure to avoid any interference during the docking process. Polar hydrogen atoms were then added to the protein to accurately simulate hydrogen bonding interactions.
Subsequently, partial charges were adjusted to ensure proper electrostatic interactions within the protein structure. Energy minimization was performed to optimize the protein conformation, ensuring a stable and realistic receptor model. This meticulous preparation of the Bcl-2 protein was crucial for achieving accurate and reliable docking simulations, which were conducted using the Autodock 4.2 software package. The validation phase of the docking software began with the redocking of the native ligand into the Bcl-2 protein's binding pocket. This process yielded an RMSD (Root Mean Square Deviation) value of 0.75 Å, reflecting a high degree of accuracy in reproducing the native ligand's position. Furthermore, the redocked ligand displayed a binding free energy (ΔG) of −14.55 kcal mol−1 (Fig. 5). According to the interactions documented by Perez H. L. et al., the sulfonamide moiety of the native ligand engages in critical interactions with the ARG66 and TYR67 residues. Remarkably, our compounds 10b and 10c, which also feature a sulfonamide core, exhibited similar interactions (Fig. 6 and 7). This observation suggests that the sulfonamide group contributes to the stabilization of these compounds within the Bcl-2 protein pocket, enhancing their binding affinity.
 | ||
| Fig. 5 (A) Superimposition of the naturally co-crystallized phenylacyl sulfonamide native ligand with the docked one within the active site of Bcl-2 protein (PDB ID: 4AQ3). The carbon atoms of the original ligand are depicted in violet, while those of the docked ligand are represented in orange; (B) 2D interactions between the docked ligand and Bcl-2 protein. | ||
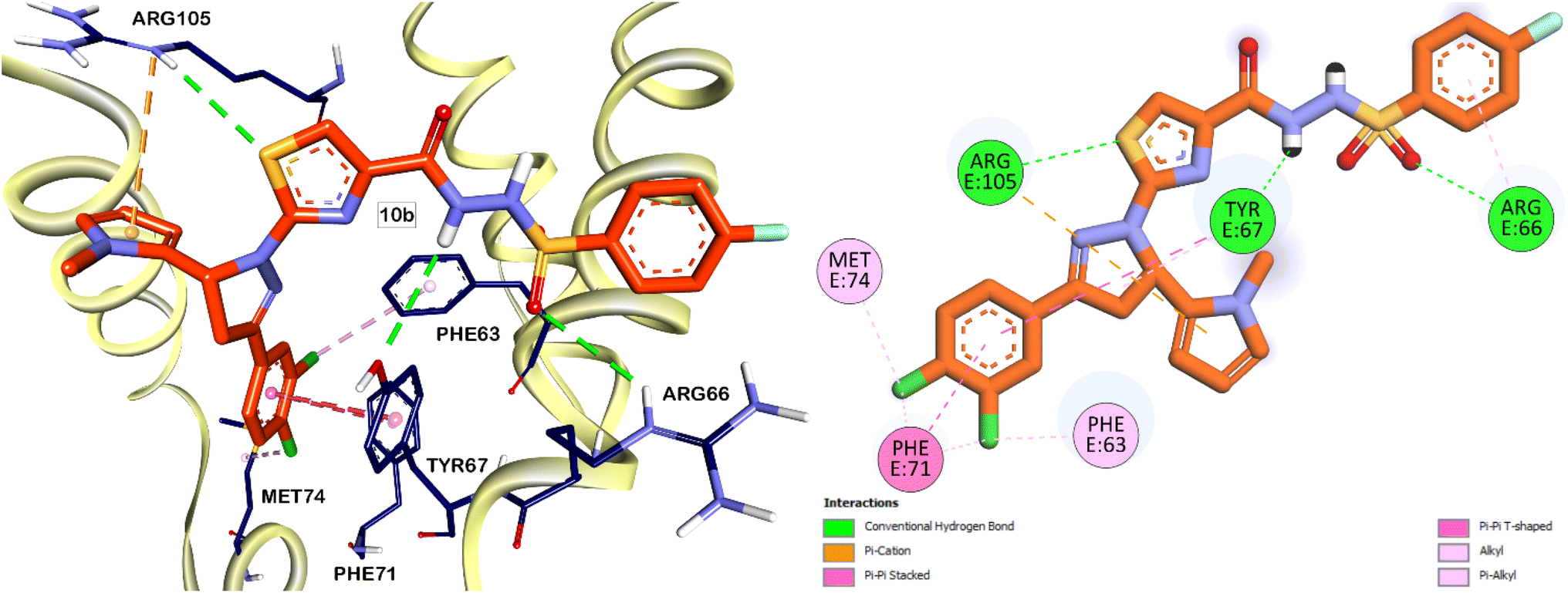 | ||
| Fig. 6 Predicted binding interactions (3D and 2D) of compound 10b within the active site of Bcl-2 protein. The carbon atoms of compound 10b are colored in orange. | ||
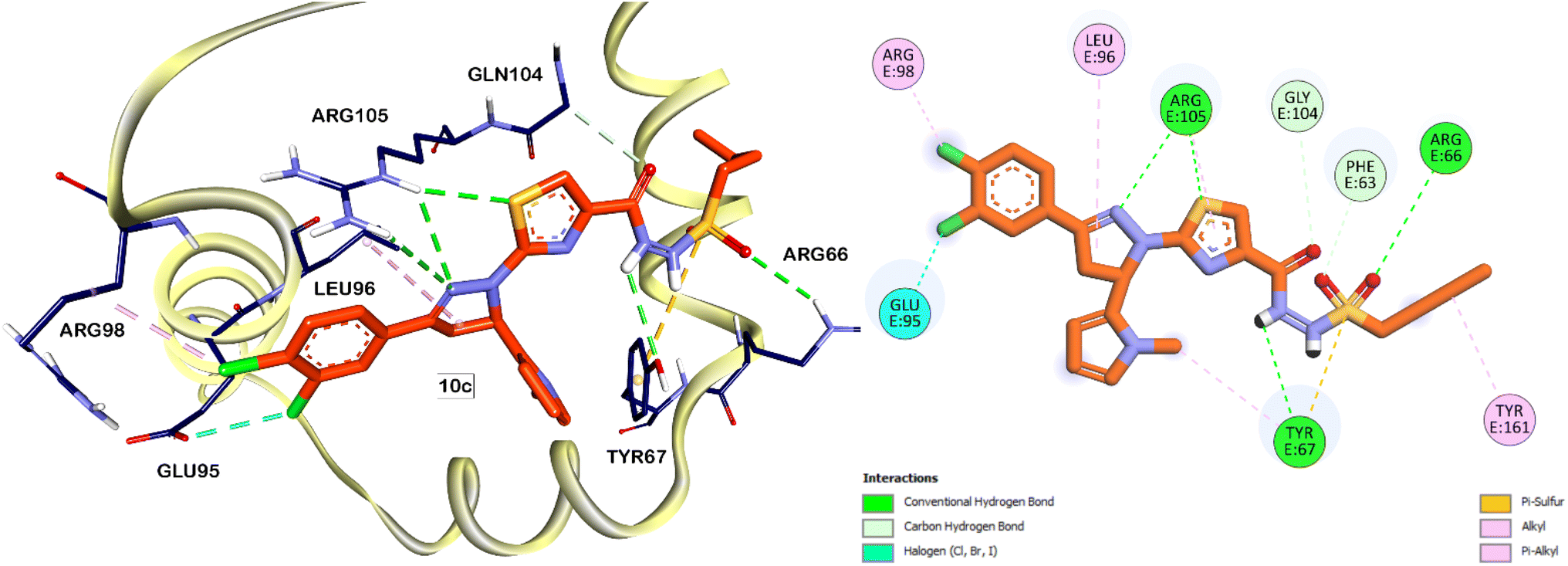 | ||
| Fig. 7 Predicted binding interactions (3D and 2D) of compound 10c within the active site of Bcl-2 protein. The carbon atoms of compound 10c are colored in orange. | ||
In analogy to the observed halogen-bond interaction of the chloro group in the pyrazole moiety of the native ligand with GLU95 residue (Fig. 5), the chloro group on the terminal phenyl ring of compound 10c (Fig. 7) displayed a similar interaction. This specific interaction type is crucial within protein binding sites, as it can contribute significantly to the stability and specificity of ligand–protein interactions. The similarity in binding interactions observed with our compounds and the native ligand, particularly through halogen bonding with GLU95, underscores the structural mimicry and potential efficacy of our compounds in targeting the Bcl-2 protein.
The phenyl ring of the native ligand is engaged in a pi–pi stacked hydrophobic interaction with the PHE63 residue, highlighting its aromatic interactions within the binding pocket. Additionally, the isoquinoline moiety formed pi–pi T-shaped interactions with the PHE71 residue and Pi-sulfur interactions with the MET74 residue (Fig. 5). Similarly, the terminal phenyl ring of compound 10b also participates in pi–pi stacked and pi–pi T-shaped hydrophobic interactions with the PHE71 residue. These interactions play a crucial role in stabilizing the ligand within the binding site of the Bcl-2 protein, thereby enhancing its overall binding affinity and specificity. In addition to the previously mentioned interactions, the sulphur atom of the thiazole ring in compounds 10b and 10c is involved with H-bonding with ARG105 residue. This interaction was not observed with the native ligand which have a phenyl ring instead of the thiazole ring in its core, therefore, having heterocycle in this position would contribute to improved affinities of the Bcl-2 inhibitors.
Conclusion
In conclusion, the pursuit of novel anticancer agents targeting apoptotic and autophagic pathways is of paramount importance due to their critical roles in maintaining cellular homeostasis and eradicating cancer cells. In this study, a comprehensive exploration was conducted on a series of 1,3,5-trisubstituted-1H-pyrazole derivatives designed as potential inhibitors of the Bcl-2 protein, a pivotal regulator of apoptosis and autophagy. Our investigation revealed that several compounds exhibited significant cytotoxicity against MCF-7, A549, and PC-3 cancer cell lines, with notable potency observed in compounds 10b and 10c. These compounds demonstrated robust binding affinity to Bcl-2 and effectively activated pro-apoptotic proteins including Bax, p53, and Caspase-3. The structural–activity relationship (SAR) analysis highlighted the importance of chlorophenyl groups, thiazole rings, and sulfonamide substituents in enhancing cytotoxic effects. Molecular docking studies provided further insights, confirming strong hydrogen bonding interactions for compounds 10b and 10c with the Bcl-2 protein. Overall, these findings underscore the promising potential of our synthesized derivatives as effective anticancer agents, warranting continued investigation for future therapeutic applications in oncology.Experimental
Chemistry
All chemicals and solvents were sourced from Acros, BLD, TCI, Aldrich, Fluka, Merck, and Sigma, and were used as received without further purification. Analytical and spectral analyses of the synthesized compounds were performed at the Microanalytical Labs of the National Research Centre and the Microanalytical Laboratory Center at the Faculty of Science, Cairo University, Egypt. 1H and 13C Nuclear Magnetic Resonance (NMR) spectra were obtained on Joel ECA 500 MHz spectrometers, using DMSO-d6 as the solvent. Chemical shifts are reported in parts per million (ppm) relative to the tetramethylsilane (TMS) standard in the solvent. Coupling constants are expressed in Hertz (Hz). The abbreviations for the splitting patterns are: singlet (s), doublet (d), triplet (t), and multiplet (m). Melting points were measured using a Stuart SMP30 Melting Point Apparatus and are uncorrected. Reaction progress was monitored via thin layer chromatography (TLC) using Merck Silica Gel 60 F254 plates.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 8.07 (dd, J = 8.4, 2.1 Hz, 1H, CHCH), 7.86 (s, 2H, Ar-H), 7.83 (d, J = 8.4 Hz, 1H, Ar-H), 7.74–7.64 (m, 2H, Ar-H), 3.80 (s, 3H, NCH3). 13C NMR (151 MHz, DMSO-d6) δ 186.32, 142.38, 137.81, 135.76, 134.03, 131.89, 131.07, 130.68, 130.10, 129.18, 128.32, 117.94, 31.75. HPLC-UV (254 nm) for C14H11Cl2NO ESI-MS, purity: 96.5%. LC-MS (m/z): 280.10 [M + H]+.
CH), 8.07 (dd, J = 8.4, 2.1 Hz, 1H, CHCH), 7.86 (s, 2H, Ar-H), 7.83 (d, J = 8.4 Hz, 1H, Ar-H), 7.74–7.64 (m, 2H, Ar-H), 3.80 (s, 3H, NCH3). 13C NMR (151 MHz, DMSO-d6) δ 186.32, 142.38, 137.81, 135.76, 134.03, 131.89, 131.07, 130.68, 130.10, 129.18, 128.32, 117.94, 31.75. HPLC-UV (254 nm) for C14H11Cl2NO ESI-MS, purity: 96.5%. LC-MS (m/z): 280.10 [M + H]+.General procedure for the synthesis of compounds 6a–e
To a solution of pyrazol-1-ylthiocarboxamide 5 (0.05 g, 0.141 mmol) in 2 mL of ethanol, phenacyl bromide derivative (0.143 mmol) was added to the solution and then refluxed at 100 °C for 3 h. After cooling, the product was obtained by filtration, washed with cold ethanol, and dried. The resulting compound was recrystallized using hot ethanol to obtain the desired compounds 6a–e.General procedure for the synthesis of compounds 10a–c
To a solution of acid hydrazide 8 (100 mg, 0.23 mmol) in DMF (10 mL) and cooled to 0 °C, sulfonyl chloride derivative (0.23 mmol) was added portion wise and then the reaction mixture was stirred at room temperature for 4 h. When the reaction is finished, distilled water (25 mL) was added and the formed precipitate that formed was filtered, washed with water and dried under high vacuum. The formed precipitate was recrystallized from ethanol yielding compounds 10a–c.General procedure for the synthesis of compounds 12a–c
To a flask containing compound 11 (0.02 g, 0.051 mmol) dissolved in 1 mL ethanol, a suitable aldehyde (0.007 g, 0.051 mmol), and piperidine (0.05 mL) were added and the reaction mixture was heated under reflux at 100 °C for 4 h. After cooling, the resulting powder was filtered, washed with methanol, and recrystallized with ethanol yielding compounds 12a–c.Gene expression analysis
For cDNA synthesis, 5 μg of total RNA from MCF-7, A549, and PC3 cell lines was reverse transcribed using the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Germany) in a 20 μL reaction volume. The master mix included 50 mM MgCl2, 10× RT buffer (50 mM KCl; 10 mM Tris–HCl; pH 8.3), 10 mM of each dNTP, 50 μM oligo-dT primer, 20 IU ribonuclease inhibitor, and 50 IU MuLV reverse transcriptase. Each sample mixture was centrifuged for 30 seconds at 1000 g before transferring to a thermocycler. The RT reaction proceeded at 25 °C for 10 minutes, 42 °C for 1 hour, and concluded with a denaturation step at 99 °C for 5 minutes. The reaction tubes were then flash-cooled on ice and stored until use for cDNA amplification via quantitative Real Time PCR (qRT-PCR).
For the docking studies, we utilized the Autodock 4.2 software package. The process began with preparing coordinate files for both the ligands and the target protein, followed by the calculation of an affinity grid for the target. The grid box was set at dimensions of 50 × 50 × 50 with a grid point spacing of 0.375 Å, centered on the position of the co-crystallized ligand. The Lamarckian Genetic Algorithm (LGA) was employed to explore possible conformations, running ten iterations with up to 27000 generations per iteration. The mutation rate was set at 0.02, and the crossover rate was 0.8. Multiple docking runs were performed using AutoDock 4.2 (ref. 28) to generate a variety of docked conformations. The best docking results were determined by evaluating the predicted binding energies and the consistency of the docking poses.
Ethical statement
This experimental study was carried out according to recommendations and under the regulations of the Medical Research Ethics Committee (MREC) at the National Research Centre in Egypt.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no financial or commercial conflicts of interest.Acknowledgements
The authors would like to thank National Research Centre, Cairo, Egypt and Menoufia University, Menoufia, Egypt for the financial support of this study.References
- M. S. Litwin and H.-J. Tan, The diagnosis and treatment of prostate cancer: A review, JAMA, J. Am. Med. Assoc., 2017, 317, 2532–2542 CrossRef PubMed.
- R. J. Rebello, C. Oing, K. E. Knudsen, S. Loeb, D. C. Johnson, R. E. Reiter, S. Gillessen, T. Van der Kwast and R. G. Bristow, Prostate cancer, Nat. Rev. Dis. Primers, 2021, 7, 1–27 CrossRef PubMed.
- R. Elancheran, K. Saravanan, S. Divakar, S. Kumari, V. L. Maruthanila, S. Kabilan, M. Ramanathan, R. Devi and J. Kotoky, Design, synthesis and biological evaluation of novel 1,3-thiazolidine-2,4-diones as anti-prostate cancer agents. Anti-Cancer Agents, Med. Chem., 2017, 17, 1756–1768 CAS.
- S. Elmore, Apoptosis: A review of programmed cell death, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- B. Levine and G. Kroemer, Autophagy in the pathogenesis of disease, Cell, 2008, 132, 27–42 CrossRef CAS PubMed.
- J. C. Reed, Bcl-2-family proteins and hematologic malignancies: History and future prospects, Blood, 2008, 111, 3322–3330 CrossRef CAS PubMed.
- K. J. Campbell and S. W. G. Tait, Targeting BCL-2 regulated apoptosis in cancer, Open Biol., 2018, 8, 180002 CrossRef PubMed.
- S. Pattingre, A. Tassa, X. Qu, R. Garuti, X. H. Liang, N. Mizushima, M. Packer, M. D. Schneider and B. Levine, Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy, Cell, 2005, 122, 927–939 CrossRef CAS PubMed.
- M. Vogler, D. Dinsdale, M. J. Dyer and G. M. Cohen, Bcl-2 inhibitors: Small molecules with a big impact on cancer therapy, Cell Death Differ., 2009, 16, 360–367 CrossRef CAS PubMed.
- R. T. Marquez and L. Xu, Bcl-2:Beclin 1 complex: Multiple mechanisms regulating autophagy/apoptosis toggle switch, Am. J. Cancer Res., 2012, 2, 214–221 CAS.
- X. Dong, Q. Liang, Y. Z. Pan, X. Wang, Y. C. Kuo, W. C. Chiang, X. Zhang, N. S. Williams, J. Rizo, B. Levine and J. K. De Brabander, Novel Bcl-2 inhibitors selectively disrupt the autophagy-specific Bcl-2-Beclin 1 protein-protein interaction, ACS Med. Chem. Lett., 2022, 13, 1510–1516 CrossRef CAS PubMed.
- K.-M. Qiu, H.-H. Wang, L.-M. Wang, Y. Luo, X.-H. Yang, X.-M. Wang and H.-L. Zhu, Design, Synthesis and Biological Evaluation of Pyrazolyl-Thiazolinone Derivatives as Potential EGFR and Her-2 Kinase Inhibitors, Bioorg. Med. Chem., 2012, 20(6), 2010–2018 CrossRef CAS PubMed.
- D. Havrylyuk, B. Zimenkovsky, O. Vasylenko, L. Zaprutko, A. Gzella and R. Lesyk, Synthesis of Novel Thiazolone-Based Compounds Containing Pyrazoline Moiety and Evaluation of Their Anticancer Activity, Eur. J. Med. Chem., 2009, 44(4), 1396–1404 CrossRef CAS PubMed.
- D. Havrylyuk, B. Zimenkovsky, O. Vasylenko, A. Gzella and R. Lesyk, Synthesis of New 4-Thiazolidinone-, Pyrazoline-, and Isatin-Based Conjugates with Promising Antitumor Activity, J. Med. Chem., 2012, 55(20), 8630–8641 CrossRef CAS PubMed.
- N. M. Boshta, A. Temirak, Z. A. El-Shahid, Z. Shafiq and A. A. F. Soliman, Design, synthesis, molecular docking and biological evaluation of 1,3,5-trisubstituted-1H-pyrazole derivatives as anticancer agents with cell cycle arrest, ERK and RIPK3-kinase activities, Bioorg. Chem., 2024, 143, 107058 CrossRef CAS PubMed.
- G. Rajendran, D. Bhanu, B. Aruchamy, P. Ramani, N. Pandurangan, K. N. Bobba, E. J. Oh, H. Y. Chung, P. Gangadaran and B.-C. Ahn, Chalcone: A Promising Bioactive Scaffold in Medicinal Chemistry, Pharmaceuticals, 2022, 15, 1250 CrossRef CAS PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part A: Structure and Mechanisms, Springer Science, New York, 5th edn, 2007 Search PubMed.
- S. A. A. Linjawi, E. A. Sharafeldin, W. K. B. Khalil and H. F. Booles, Potential role of Jasonia montana and Jasonia candicans against Alzheimer's disease: assessment of oxidative stress and gene expression changes in AD-induced rats, Egypt. J. Genet. Cytol., 2011, 40, 61–80 CrossRef.
- S. Y. Elateek, L. M. Salem, E. S. Ahmed and W. K. B. Khalil, Staphylococcus aureus isolates from hospital clinics induce ROS-mediated DNA damage, apoptosis, and gene expression alterations in male mice, Gene Rep., 2021, 23, 101028 CrossRef CAS.
- A. F. Brito, A. M. Abrantes, C. Pinto-Costa, A. R. Gomes, A. C. Mamede, J. Casalta-Lopes, A. C. Gonçalves, A. B. Sarmento-Ribeiro, J. G. Tralhão and M. F. Botelho, Hepatocellular carcinoma and chemotherapy: The role of p53, Chemotherapy, 2012, 58, 381–386 CrossRef CAS PubMed.
- M. A. Ramadan, A. E. Shawkey, M. A. Rabeh and A. O. Abdellatif, Expression of P53, BAX, and BCL-2 in human malignant melanoma and squamous cell carcinoma cells after tea tree oil treatment in vitro, Cytotechnology, 2019, 71, 461–473 CrossRef CAS PubMed.
- M. A. Elhinnawi, R. M. Mohareb, H. M. Rady, W. K. B. Khalil, M. M. Abd Elhalim and G. A. Elmegeed, Novel pregnenolone derivatives modulate apoptosis via Bcl-2 family genes in hepatocellular carcinoma in vitro, J. Steroid Biochem. Mol. Biol., 2018, 183, 125–136 CrossRef CAS PubMed.
- W. K. B. Khalil, W. Zarouk, G. Nour Eldeen, A. Ramadan, A. Fayez, N. Esmaiel, B. Foda, K. Hamed, S. M. Kassem and H. El-Bassyouni, Apoptosis, reactive oxygen species and DNA damage in familial Mediterranean fever patients, Gene Rep., 2019, 14, 76–80 CrossRef.
- P. L. Olive, J. P. Banáth and R. E. Durand, Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay, Radiat. Res., 2012, 178, AV35–AV42 CrossRef CAS PubMed.
- A. Collins, M. Dusinska, M. Franklin, M. Somorovska, H. Petrovska, S. Duthie, L. Fillion, M. Panayiotidis, K. Raslova and N. Vaughan, Comet assay in human biomonitoring studies: Reliability, validation, and applications, Environ. Mol. Mutagen., 1997, 30, 139–146 CrossRef CAS PubMed.
- H. L. Perez, P. Banfi, J. Bertrand, Z. W. Cai, J. W. Grebinski, K. Kim, J. Lippy, M. Modugno, J. Naglich, R. J. Schmidt, A. Tebben, P. Vianello, D. D. Wei, L. Zhang, A. Galvani, L. J. Lombardo and R. M. Borzilleri, Identification of a phenylacylsulfonamide series of dual Bcl-2/Bcl-xL antagonists, Bioorg. Med. Chem. Lett., 2012, 22, 3946–3950 CrossRef CAS PubMed.
- BIOVIA and Dassault Systèmes, BIOVIA Discovery Studio Visualizer, 2021, Dassault Systèmes, San Diego, 2023, https://www.3ds.com/ Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02046h |
| This journal is © The Royal Society of Chemistry 2025 |