
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
S-decorated Mo2C as efficient catalyst for Li–O2 battery system
Yanhong Ding ,
Zhichao Gao,
Rongpeng Lin,
Yong Cao,
Haoyang Liu,
Yulin Zhou,
Haifeng Xu,
Jiayi Liu,
Fangqi Ren and
Yirong Zhu*
,
Zhichao Gao,
Rongpeng Lin,
Yong Cao,
Haoyang Liu,
Yulin Zhou,
Haifeng Xu,
Jiayi Liu,
Fangqi Ren and
Yirong Zhu*
College of Materials and Advanced Manufacturing, Hunan University of Technology, Zhuzhou 412007, People's Republic of China. E-mail: zhuyirong2004@163.com
First published on 8th July 2025
Abstract
Lithium–oxygen (Li–O2) batteries are considered an important candidate for the next generation of energy storage systems due to their ultra-high theoretical energy density (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 586 mA h g−1), but their slow kinetic reactions, high overpotential and cyclic instability seriously limit their practical applications. In this study, sulfur modified Mo2C (S@Mo2C) cathode materials were prepared by hydrothermal synthesis by sulfur (S) doping to optimize the electronic structure and catalytic activity of Mo2C (Mo2C). Experiments show that S@Mo2C exhibits significantly improved electrochemical performance compared to commercial Mo2C: its specific capacity is up to 3955 mA h g−1 (commercial material only 508 mA h g−1), the charge and discharge overpotential is reduced to 0.26 V (53.6%), and the capacity retention rate remains 77.8% after 250 cycles. X-ray diffraction (XRD), transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS) analysis showed that the introduction of sulfur induced the formation of a heterostructure of MoS2/MoS3 in the Mo2C lattice, which enhanced the conductivity and oxygen reduction/precipitation (ORR/OER) activity of the material. In addition, sulfur doping promotes the formation of highly conductive amorphous Li2O2 and effectively inhibits the accumulation of insulating ring Li2O2, thus significantly improving the cycle stability and energy efficiency of the battery. This study provides a new structural regulation strategy for the design of high efficiency lithium oxygen battery catalysts.
586 mA h g−1), but their slow kinetic reactions, high overpotential and cyclic instability seriously limit their practical applications. In this study, sulfur modified Mo2C (S@Mo2C) cathode materials were prepared by hydrothermal synthesis by sulfur (S) doping to optimize the electronic structure and catalytic activity of Mo2C (Mo2C). Experiments show that S@Mo2C exhibits significantly improved electrochemical performance compared to commercial Mo2C: its specific capacity is up to 3955 mA h g−1 (commercial material only 508 mA h g−1), the charge and discharge overpotential is reduced to 0.26 V (53.6%), and the capacity retention rate remains 77.8% after 250 cycles. X-ray diffraction (XRD), transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS) analysis showed that the introduction of sulfur induced the formation of a heterostructure of MoS2/MoS3 in the Mo2C lattice, which enhanced the conductivity and oxygen reduction/precipitation (ORR/OER) activity of the material. In addition, sulfur doping promotes the formation of highly conductive amorphous Li2O2 and effectively inhibits the accumulation of insulating ring Li2O2, thus significantly improving the cycle stability and energy efficiency of the battery. This study provides a new structural regulation strategy for the design of high efficiency lithium oxygen battery catalysts.
1 Introduction
The energy sector has undergone transformative changes aligned with modern developmental paradigms, driven by accelerated societal progress and escalating global environmental degradation. Among emerging energy storage technologies, lithium–oxygen (Li–O2) batteries have garnered substantial scientific interest due to their exceptional theoretical energy density of 11586 mA h g−1 – approximately 3–5 times greater than conventional lithium-ion systems.1,2 This characteristic positions Li–O2 batteries as a highly promising next-generation energy storage solution.
However, the practical implementation of this technology faces critical challenges, primarily stemming from elevated overpotentials that impede reaction kinetics and compromise cycling efficiency.3,4 Current research identifies the insulating nature of lithium peroxide (Li2O2), the predominant discharge product, as a principal contributor to these high overpotentials. Although the more conductive lithium superoxide (LiO2) is also generated during the reaction process, its inherent instability prevents it from persisting as the primary discharge product.5,6 This limitation underscores the necessity for advanced catalytic materials capable of effectively decomposing Li2O2. Consequently, the development of high-performance catalysts has become a pivotal research focus, as such materials could substantially mitigate overpotential challenges and enhance battery performance through optimized reaction pathways.
Molybdenum carbide (Mo2C) is widely used as high effective catalysts. With abundant active sites, however, typical β-type Mo2C with a stable structure still does not show satisfactory performance in practice and needs more adjustment and optimization, and structural optimization is a very effective method.7–10 Metal sulfides are also widely used as electrode materials.11,12 The use of Mo2S as a catalyst in HER reactions has been widely investigated due to its excellent catalytic performance. Crystalline Mo2S and amorphous Mo2Sx are commonly used, however, the stacking layer structure causes it to exhibit a high impedance, resulting in a decrease of its conductivity, which implies a decrease in catalytic efficiency. The catalytic efficiency of Mo2Sx can be effectively improved by reconstructing the structure, which makes Mo2Sx feasible as a catalyst for Li–O2 batteries.13,14
While previous studies have established the catalytic potential of Mo2C- and MoS2-based cathodes in Li–O2 systems,15–17 critical limitations persist in addressing key electrochemical challenges. Conventional Mo2C cathodes often suffer from insufficient electrical conductivity and poor Li2O2 decomposition kinetics, typically manifesting in high charge overpotentials (>0.4 V) and rapid capacity fading (<50 cycles) due to parasitic side reactions.8,18 Although sulfur incorporation strategies (e.g., MoS2 heterostructures) have been explored to enhance surface reactivity,19 most reported systems exhibit suboptimal phase stability under deep cycling conditions, with inevitable MoS2/Mo2C phase segregation leading to active site passivation.20 Furthermore, existing sulfur-doped architectures predominantly focus on single-phase modifications, neglecting the synergistic effects of multivalent Mo species (e.g., Mo3+/Mo4+ redox couples) in regulating Li2O2 nucleation pathways.21
In this work, the S@Mo2C structure with efficient, conductive structure and active sites were optimized successfully by using commercial Mo2C with thioacetamide, and the S element was successfully doped during calcination, and used as the cathode to explore the synergistic effect of Li–O2 battery. As a result, the S-modified Mo2C structure can effectively improve the performance of the Li–O2 battery system with more excellent specific capacity (3955 mA h g−1), lower overpotential (0.26 V), and improved cycling performance (over 250 cycles) compared with the conventional commercial Mo2C.
In view of the reasons for the abnormally high specific capacity of our work: our analysis is composed of the following: Potential Factors Contributing to Enhanced Capacity in Lithium–Oxygen Battery Systems.
1.1 Material-specific reaction mechanisms
1.2 Nanostructural optimization effects
2 Experimental section
2.1 Materials and chemicals
Commercial Mo2C (Sigma, 99.0%), deionized water, thioacetamide (Aladdin), lithium bis(trifluoromethanesulfony) imide (LiTFSI, Aladdin), tetraethylene glycol dimethyl ether (TEGDME, Aladdin), polyvinylidene fluoride (PVDF, Aladdin) and N-methylpyrrolidone (NMP, Aladdin) are all analytical pure.2.2 Preparation of S-decorated Mo2C
Commercial Mo2C (120 mg) was added to 50 ml deionized water, and thioacetamide (636.76 mg) was further added. After electromagnetic stirring for 10 min, the mixture was placed in a 100 ml teflon-lined stainless steel autoclave, and heated to 473 K and the temperature maintained for 10 h. After cooling down, the mixture precursor was obtained. After centrifugation (7800 rpm) and dry (60 °C for 12 h), the black target specimen was obtained.2.3 Preparation of Li–O2 cells
Commercial Mo2C (or S@Mo2C) was mixed with polyvinylidene fluoride (PVDF) and N-methylpyrrolidone (NMP) binder in a weight ratio of 8:1:1 to form a slurry, which was placed on carbon paper discs (14 mm diameter) and then dried in a vacuum oven at 120 °C for 12 h. The amount of mass loading is about 2 mg. The button cell used for electrochemical testing consisted of a cathode, 0.1 ml electrolyte (1 M LiTFSI in TEGDME) permeated to a glass fiber septum (Whatman GF/D microfiber filter paper, pore size 2.7 μm) and a lithium metal anode.
3 Results and discussion
Fig. 1a shows the process of obtaining S-decorated Mo2C by doping with sulfur for commercial Mo2C by hydrothermal method. The commercial Mo2C (Fig. 1b) was added to deionized water and thioacetamide (TAA) was added to the mixture to obtain the precursor solution, which was further heated over 200 °C to obtain S-decorated Mo2C (Fig. 1c). | ||
| Fig. 1 (a) Schematic synthesis process of S-decorated Mo2C; (b) the SEM image of commercial Mo2C; (c) the SEM image of S@Mo2C. | ||
Fig. 2a presents the X-ray diffraction (XRD) patterns of commercial Mo2C and S@Mo2C. Significant strong peaks are observed at 34.5, 38.0, 39.6, 52.3, 61.9, 69.8, and 75.0°, corresponding to the (100), (002), (101), (102), (110), (103), and (112) planes of the typical β-Mo2C crystal structure, respectively. Further analysis indicates that the original crystal structure is preserved after sulfur modification. Compared with the Mo2C sample before sulfidation, the diffraction peak intensities on the (002), (101), (102), (110), (103), and (112) planes have increased to varying degrees, and the peak widths have narrowed, suggesting an increase in grain size. This phenomenon can be attributed to the sulfur atom radius (0.104 nm) being larger than that of carbon (0.077 nm) during the sulfidation process, leading to lattice expansion and reconstruction of the crystal structure, thereby causing changes in the crystal diffraction characteristics. On the other hand, the decrease in the intensity of the (100) peak of Mo2C may be due to sulfur replacing carbon atoms on the (100) plane, forming MoS2 with a 2H structure, which reduces the number of Mo2C structures on the (100) plane (see Fig. 2b). The enhanced intensity of the (002) plane is attributed to sulfur doping optimizing the interlayer stacking of Mo2C, promoting the preferential growth of the S@Mo2C (002) plane.
 | ||
| Fig. 2 (a) XRD patterns of commercial Mo2C and S-decorated Mo2C; (b) crystal structure of S-decorated Mo2C. | ||
To deeply explore the microstructural characteristics of S-modified Mo2C, this study employed transmission electron microscopy (TEM) techniques for detailed observation and analysis. The high-resolution TEM image of S-modified Mo2C (Fig. 3a) reveals the uniform distribution of the S-modified Mo2C structure. The TEM image clearly shows a thin nanometer-thick film layer with a thickness of about 5 nanometers on the outer layer of Mo2C. By magnified scanning transmission electron microscopy (STEM) images, significant changes in the Mo2C lattice spacing can be observed, where the complete Mo2C lattice spacing distorts from 0.26 nanometers (corresponding to Mo2C (100), PDF#35-0787) to 0.21 nanometers (corresponding to MoS2 (006), PDF#73-1508). In the X-ray photoelectron spectroscopy (XPS) S 2p spectrum, the double peaks at 162.02 eV and 163.32 eV correspond to the 2p3/2 and 2p1/2 orbitals of s2−, confirming the presence of sulfur in the form of s2− (Fig. 4c). Additionally, in the Mo 3d spectrum of XPS, the peaks at 228.85 eV and 231.8 eV shift, indicating the formation of Mo–S bonds. This result further verifies that the change in layer spacing is caused by the formation of MoS2 structures due to sulfur doping in the Mo2C lattice, consistent with the analysis results of the X-ray diffraction (XRD) spectrum. Fig. 3b shows the scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDX) surface scanning image of S-doped Mo2C, and Fig. 3c–f display the content and distribution of Mo, S, C, and O elements, respectively. Mo, S, C, and O elements are uniformly distributed throughout the sample, further confirming the formation of S@Mo2C. Notably, throughout the sample, especially in Fig. 3f, the distribution of O elements can be clearly observed, which may be due to the oxidation reaction of the sample with oxygen in the air under exposure conditions, leading to the formation of some MoOx. However, the MoOx signal in the XPS analysis is weak, so the impact of MoOx contamination on the experimental results can be ignored. Moreover, a trace amount of MoOx may act as a protective layer to inhibit the decomposition of the electrolyte.
 | ||
| Fig. 3 (a) The TEM and magnified STEM images of S-decorated Mo2C; (b–f) elemental mapping analysis of S-decorated Mo2C. | ||
 | ||
| Fig. 4 (a and b) XPS images of S@Mo2C sample; (c) XPS spectra of S element of S@Mo2C; (d and e) XPS images of different ion valences of Mo element in commercial Mo2C and S@Mo2C. | ||
To further analyze the composition of the chemical states on the surface of S@Mo2C, X-ray photoelectron spectroscopy (XPS) was used. The XPS spectrum (Fig. 4) shows that S@Mo2C is comprised of Mo, C, O and S elements. From Fig. 4a, Mo 3d peaks and S 2s, S 2p peaks can be clearly observed, indicating that S element is present in large quantities of the S@Mo2C sample, with a mass fraction of 14.86% corresponding to Table 1, which is highly close to previous research.22 Strong peaks were observed at 227.75 eV (3d5/2) and 230.9 eV (3d3/2), and the binding energy characteristics were consistent with β-Mo2C. Compared with the typical Mo 3d peak (∼229 eV) of 2H–MoS2, the Mo 3d peak in S@Mo2C shifts 0.15 eV towards the high binding energy, which corresponds to the lattice expansion caused by the incorporation of S in the XRD pattern (Δd = 1.36%). It is indicated that Mo and S may form a complex coordination structure of crystalline MoS2 and amorphous MoS3, MoSx.
| C 1s | Mo 3d | S 2p | |
|---|---|---|---|
| At% | 79.89% | 9.77% | 10.34% |
| Wt% | 43.05% | 42.09% | 14.86% |
Fig. 4b shows the high-resolution spectrum of the binding energy in the range of 220–237.5 eV, where the presence of Mo2C and Mo2Sx with different valence Mo elements can be clearly observed. The peak of S 2 s is located at around 225.7 eV. Mo 3d exhibits strong peaks at 227.75 and 230.9 eV, respectively, showing a doublet with β-Mo2C characteristics.23 The Mo 3d peaks at 228.85 and 231.8 eV has shifted ∼0.15 eV compared with the normal value of 2H-MoS2 (229 eV), which can be certified by the characteristic double peaks of the Mo element of MoSx in the Mo4+ state.24 As shown in the high-resolution S 2p spectrum (Fig. 4c), the peaks of S12− and S22− ligands at 162.02 and 163.32 eV corresponding to S12− 2p3/2 and S12− 2p1/2, respectively, can be observed. The ∼1.2 eV energy separation between S12− and S22− is the characteristic of S2− in MoS2. Moreover, the peaks at 163.52 and 164.62 eV corresponding to S22 2p3/2 and S22− 2p1/2 are also observed, respectively. The energy separation of doublet is close to 1.2 eV, which is characteristic of S2− in MoS3 species.25 Thus, the poorly crystalline “MoSx” in S@Mo2C is identified as a mixture of crystalline MoS2 and non-crystalline MoS3. The XPS spectra of Mo elements show the presence of different chemical valence states on the Mo surface, as shown in Fig. 4 (d, commercial Mo2C) and (e, S@Mo2C). The Mo–C bond in Mo2C contributes to the Mo2+ state and the partially low oxidation state of Mo3+, while other reports have shown that the Mo–O bond can also be assigned to Mo3+ and Mo5+ in MoO2, as well as Mo6+ in MoO3 due to air contamination.26 In Fig. 4d, Mo 3d5/2 peaked at 229.0 eV, and Mo 3d3 peaked at 232.1 eV, corresponding to Mo3+ (mainly MO–C bond). A weak peak at 235.5 eV was attributed to Mo6+ (MoO3) formed by surface oxidation. In Figure e, the Mo 3d5/2 peak migrates towards the low binding energy to 227.75 eV and the Mo 3 d5/2 peak is located at 230.9 eV, indicating a reduction in the oxidation state of Mo (Mo3+, Mo2+). The peak strength of Mo6+ decreased significantly (peak weakening at 235.5 eV), indicating that S doping inhibited surface oxidation and induced partial reduction of high-priced Mo (Mo6+, Mo5+) to low-priced Mo (Mo3+, Mo2+). The phenomena of lower valence states of eMo in Fig. 4c and d are both due to the incorporation of S, and the electronegativity of S is higher than that of C, resulting in S atoms attracting electrons from Mo and reducing the electron density of D orbital of Mo (increasing binding energy). Moreover, the introduction of S simultaneously provides electronic compensation by forming Mo–S bonds. It can be observed that the peaks of Mo6+ disappear, the intensity of Mo5+ peaks decreases, and the intensity of Mo3+ and Mo2+ peaks increases, indicating that Mo6+ and part of Mo5+ are reduced to lower Mo3+ and Mo2+ due to S doping. The appearance of a slight amount of S 2s peaks may be due to the oxidation of a slight amount of S2− to SO42− during the preparation process.27
The plateau fraction of carbon (79.89%) was mainly derived from the carbon paper substrate used in electrode preparation (Fig. 3c SEM-EDX surface scan shows uniform distribution of carbon). Sulfur doping (10.34 at%) leads to a decrease in molybdenum content (9.77 at%) due to the partial substitution of sulfur atoms for carbon atoms in the Mo2C lattice to form the Mo2C1−x Sx hybrid structure. The reduction of Mo–C bond strength in the XPS analysis (Fig. 4) further supports this substitution mechanism.
To verify the properties of kinetics reaction of S@Mo2C, a coin-type Li–O2 battery consisting of a lithium metal anode was used for the electrochemical test. The galvanostatic full discharge curves of Mo2C and S@Mo2C at a 100 mA g−1 current density with 0.1 mV s−1 scan rate from 2.0 to 4.5 V (vs. Li/Li+) were obtained in Fig. 5a. Obviously, the coexistence of S and Mo2C as a cathode for Li–O2 battery presents the largest capacity (3955 mA h g−1), which shows a much higher specific capacity compared to commercial Mo2C cathode (508 mA h g−1). Moreover, the discharge overpotential is significantly decreases from 560 mV to 260 mV for the S@Mo2C cathode, indicating that the more completely ORR (oxidation–reduction reaction) have occurred at the positive electrode and the discharge products have grown more fully on the S@Mo2C cathode than commercial Mo2C. S@Mo2C with better electrical conductivity and ORR performance can be attributed to lower overpotential and more fully grown discharge products.28
 | ||
| Fig. 5 (a) Voltage profile of the full discharge for commercial Mo2C and S@Mo2C in Li–O2 batteries; (b) charge and discharge curves of commercial Mo2C at a current density of 100 mA g−1; (c) charge and discharge curves of S@Mo2C at a current density of 100 mA g−1. | ||
The discharge and charge curves of Mo2C and S@Mo2C cathodes with a cut off capacity of 500 mA h g−1 at a current density of 100 mA g−1 were investigated in Fig. 5b and c. It can be observed clearly that commercial Mo2C cathode presents a good OER (oxygen evolution reaction) kinetics in Fig. 5b. As the charge/discharge proceeds, the overpotential continues to increase after 5 and 10 cycles rapidly (∼1.5 V overpotential, ∼630 mV increased after 10 cycles), while the round-trip efficiency degrades from 75.1% down to 63.2%. Compared with commercial Mo2C, S@Mo2C cathode presents a better, stable and reversible charge/discharge characteristic in Fig. 5c. The charge/discharge curves from the 1st to even 250th cycle of S@Mo2C presents a great overpotential difference (∼0.8 V overpotential, ∼250 mV increased after 250 cycles) and a smooth charge/discharge plateau with little overpotential difference (3.7% degradation of round-trip efficiency, 77.8% remained after 250 cycles). Lower overpotential allows the discharge products Li2O2 decomposing effectively, OER occurring more fully at the cathode and avoiding the generation of by-products effectively.29,30 Besides, charging plateaus below 3.5 V is very beneficial to the cycling performance for the Li–O2 battery, as carbon materials decompose above 3.8 V.31 In order to better reveal the mechanism, the charge/discharge characteristics of S@Mo2C cathode will be analyzed next. A direct comparison with the previous material properties is tabulated as shown in Table 2 below.
| Catalyst | Specific capacity (mA h g−1) | Overpotential (V) | Cycle stability |
|---|---|---|---|
| S@Mo2C | 3955 | 0.26 | 250 cycles |
| Mo2C | 508 | 0.56 | <50 cycles |
| Co3O4HPNT | 4146 | 0.099 | 40 |
| Mo2C/MoO2@RGO | 2365 | 0.56 | |
| Mo2C/C | 7500 | 1.2 | 100 cycles |
| MoN | 7400 | 0.19 |
In lithium–oxygen batteries, S@Mo2C is used as the cathode material, and its charge–discharge process is monitored using a scanning electron microscope (SEM), corresponding to the different states shown in Fig. 6a (Fig. 6b–d). The charge–discharge reactions of the battery follow the following chemical eqn (1) and (2):32
| Discharge: 2Li+ + O2 + 2e− → Li2O2 | (1) |
| Charge: Li2O2 → 2Li+ + O2 + 2e− | (2) |
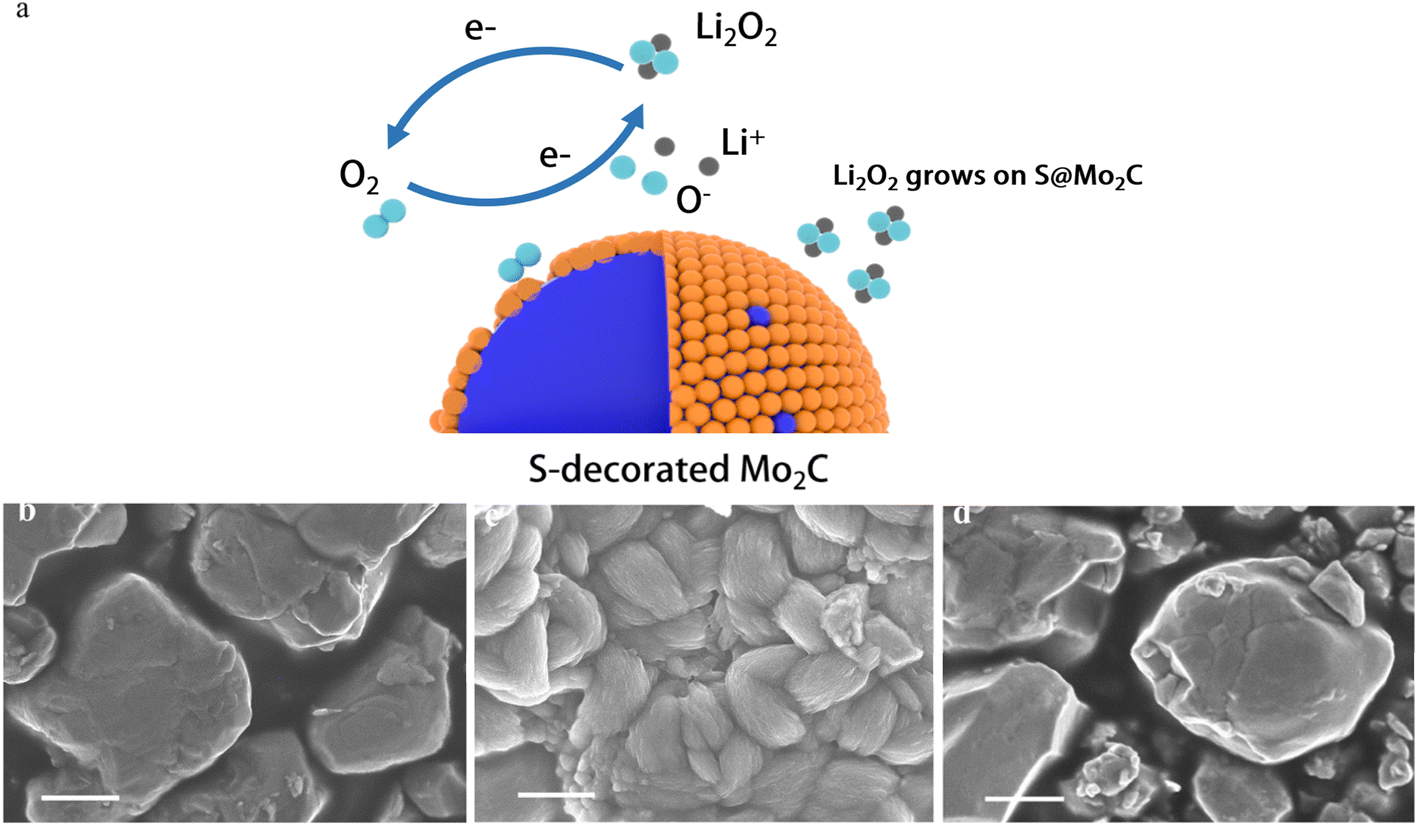 | ||
| Fig. 6 (a) The schematic diagram of S@Mo2C electrode Li–O2 battery; (b) pristine S@Mo2C cathode with uniform nanoparticles. (c) post-discharge: toroidal Li2O2 and amorphous Li2O2 deposited on the surface. (d) Post-recharge: complete decomposition of Li2O2, restoring the cathode morphology. The (c) and (d) two figures are TEM images after 250 cycles. | ||
The initial state of S@Mo2C presents a flat and undamaged surface, as shown in Fig. 6b. As the discharge process progresses, a small-sized, high-crystallinity annular film forms on the cathode surface, a phenomenon clearly visible in Fig. 6c. This annular film is considered the typical discharge product Li2O2. Under normal circumstances, the annular Li2O2 is the main factor causing the high overpotential of lithium–oxygen batteries.33 However, during the recharging process, the decomposition of Li2O2 is observed, and S@Mo2C returns to its original flat and undamaged state, which is very similar to Fig. 6b, indicating that S@Mo2C can return to its initial form after charging. After 250 cycles of electrochemical stability testing, the capacity retention rate reaches 77.8%, and the overpotential only increases by 250 mV. The low overpotential (0.8 V) indicates a high reversibility of Li2O2 decomposition, reducing structural stress, thus indicating that the nanosheets did not undergo structural collapse or peeling during the charge–discharge cycles, showing good crystal structure stability. This suggests that not only is annular Li2O2 formed, but also amorphous Li2O2 with higher conductivity. Under high current density conditions, the formation of small annular Li2O2 is more easily observed.34 Typically, amorphous Li2O2 promotes rapid electron tunneling during the charging process, resulting in rapid and efficient decomposition of Li2O2.35,36 Interestingly, although S doping may adversely affect the number of active sites on the Mo2C cathode, leading to an increase in cathode impedance, S doping also improves the quality of the effective active sites, significantly enhancing the conductivity of S@Mo2C.37 High-quality active sites may also more effectively break the O–O bond, thereby significantly reducing the activation energy required for the reaction. While experimental evidence including XPS analysis of Mo4+/Mo6+ ratios strongly supports the proposed mechanism of Mo valence tuning and O–O bond activation, future density-functional theory (DFT) calculations will contribute to a comprehensive understanding of the mechanism. Such computational studies can quantitatively verify synergistic effects between Co/Mo sites, accurately map reaction pathways (e.g., O–O cleavage energies), and reveal electronic structure modifications at the atomic level. Nevertheless, the consistency between our structural characterization (XRD, XPS, TEM) and the excellent electrochemical properties (low overpotential, high stability) provides a reliable empirical validation of the material's design strategy.38–40
4 Conclusions
In this study, we successfully modified commercial Mo2C (S@Mo2C) cathode material by sulfur doping strategy, and prepared sulfur modified Mo2C (S@Mo2C) cathode material. The system characterization indicated that the introduction of sulfur not only optimized the crystal structure of Mo2C and formed a heterogeneous interface of MoS2/MoS3, but also significantly increased the conductivity and catalytic activity of the material. Electrochemical tests show that the S@Mo2C-based lithium–oxygen battery achieves a high specific capacity of 3955 mA h g−1 at a current density of 100 mA g−1, reduces the charge and discharge overpotential to 0.26 V, and exhibits excellent cycle stability (capacity retention rate of 77.8% after 250 cycles). The mechanism study shows that sulfur doping can improve battery performance in the following ways: (1) enhance electron transport capacity and promote ORR/OER reaction kinetics; (2) induce the formation of highly conductive amorphous Li2O2 and inhibit the irreversible accumulation of insulating ring Li2O2; (3) optimize the distribution of active sites and reduce the reaction energy barrier. In addition, the S@Mo2C stable charge and discharge platform (<3.5 V) effectively avoids the decomposition of carbon-based materials, further extending the battery life. This study provides an important theoretical and experimental basis for the development of efficient and long-lived lithium–oxygen battery catalysts. Subsequent studies can be extended to explore the generalizability of the sulfur doping strategy in other transition metal carbide systems and large-scale preparation techniques, while incorporating density-functional theory (DFT) modeling in order to establish a clear conformational relationship and guide the further optimization of the bimetallic catalysts.Author contributions
Yanhong Ding was responsible for the experimental design and writing the first draft of the paper, Zhichao Gao was responsible for writing or revising the chapters, Rongpeng Lin was responsible for the experimental part, Yong Cao assisted in completing the experiments, Haoyang Liu assisted in completing the experiments or data analysis, Yulin Zhou assisted in completing the data analysis, Haifeng Xu was responsible for finding the related papers on Li–O2, Jiayi Liu was involved in the XRD part of the revisions, Fangqi Ren was involved in the XPS part of the revisions, Yirong Zhu was responsible for the paper submission, response to review comments and communication with journals.Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.Acknowledgements
This work is financially supported by Hunan Provincial Natural Science Foundation of China (2022JJ50059).References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- Z. Lyu, Y. Zhou, W. Dai, X. Cui, M. Lai, L. Wang, F. Huo, W. Huang, Z. Hu and W. Chen, Chem. Soc. Rev., 2017, 46, 6046–6072 RSC.
- Y. Meng, J.-K. Zhang, H.-Y. Lu, X.-H. Chen and J.-T. Xu, Rare Met., 2022, 41, 4027–4033 CrossRef CAS.
- J. Wang, L. Liu, S. Chou, H. Liu and J. Wang, J. Mater. Chem. A, 2017, 5, 1462–1471 RSC.
- L. Liu, H. Guo, Y. Hou, J. Wang, L. Fu, J. Chen, H. Liu, J. Wang and Y. Wu, J. Mater. Chem. A, 2017, 5, 14673–14681 RSC.
- X. Bi, K. Amine and J. Lu, J. Mater. Chem. A, 2020, 8, 3563–3573 RSC.
- J. Wu, M. Jing, T. Wu, M. Yi, Y. Bai, W. Deng, Y. Zhu, Y. Yang and X. Wang, J. Colloid Interface Sci., 2023, 641, 831–841 CrossRef CAS PubMed.
- W. Jiao, Q. Su, J. Ge, S. Dong, D. Wang, M. Zhang, S. Ding, G. Du and B. Xu, Mater. Res. Bull., 2021, 133, 111020 CrossRef CAS.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407–6410 CrossRef CAS PubMed.
- H. Zhou, Z. Chen, E. Kountoupi, A. Tsoukalou, P. M. Abdala, P. Florian, A. Fedorov and C. R. Müller, Nat. Commun., 2021, 12, 5510 CrossRef CAS PubMed.
- Y. Ding, R. Lin, S. Xiong, Y. Zhu, M. Yu and X. Duan, Molecules, 2023, 28, 2487 CrossRef CAS PubMed.
- D. Sarkar, D. Das, S. Das, A. Kumar, S. Patil, K. K. Nanda, D. D. Sarma and A. Shukla, ACS Energy Lett., 2019, 4, 1602–1609 CrossRef CAS.
- D. Escalera-López, Y. Niu, S. J. Park, M. Isaacs, K. Wilson, R. E. Palmer and N. V. Rees, Appl. Catal., B, 2018, 235, 84–91 CrossRef.
- J. Li, B. Zhang, Q. Song, X. Xu and W. Hou, Ceram. Int., 2020, 46, 14178–14187 CrossRef CAS.
- X. Mu, C. Xia, B. Gao, S. Guo, X. Zhang, J. He, Y. Wang, H. Dong, P. He and H. Zhou, Energy Storage Mater., 2021, 41, 650–655 CrossRef.
- W.-J. Kwak, K. C. Lau, C.-D. Shin, K. Amine, L. A. Curtiss and Y.-K. Sun, ACS Nano, 2015, 9, 4129–4137 CrossRef CAS PubMed.
- Z. Zhu, A. Mosallanezhad, D. Sun, X. Lei, X. Liu, Z. Pei, G. Wang and Y. Qian, Energy Fuels, 2021, 35, 5613–5626 CrossRef CAS.
- B. Ge, J. Wang, Y. Sun, J. Guo, C. Fernandez and Q. Peng, ACS Appl. Energy Mater., 2020, 3, 3789–3797 CrossRef CAS.
- M. Wu, D. Y. Kim, H. Park, K. M. Cho, J. Y. Kim, S. J. Kim, S. Choi, Y. Kang, J. Kim and H.-T. Jung, RSC Adv., 2019, 9, 41120–41125 RSC.
- Y.-M. Zhang, Z.-R. Zhou, L.-L. Zhao, Y.-B. Li, R.-F. Li, Y. Zhou, S. Shoukat, A.-A.-A. Adam and J. Wang, Rare Met., 2024, 43, 3383–3390 CrossRef CAS.
- D. Sun, Y. Shen, W. Zhang, L. Yu, Z. Yi, W. Yin, D. Wang, Y. Huang, J. Wang, D. Wang and J. B. Goodenough, J. Am. Chem. Soc., 2014, 136, 8941–8946 CrossRef CAS PubMed.
- C. Tang, W. Wang, A. Sun, C. Qi, D. Zhang, Z. Wu and D. Wang, ACS Catal., 2015, 5, 6956–6963 CrossRef CAS.
- Z. Fang, L.-C. Wang, Y. Wang, E. Sikorski, S. Tan, K. D. Li-Oakey, L. Li, G. Yablonsky, D. A. Dixon and R. Fushimi, ACS Catal., 2020, 10, 1894–1911 CrossRef CAS.
- H. Wang, S. Liu and K. J. Smith, J. Catal., 2019, 369, 427–439 CrossRef CAS.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671–2681 CrossRef CAS PubMed.
- C. Wu, Y. Hou, J. Jiang, H. Guo, H.-K. Liu, J. Chen and J. Wang, J. Power Sources, 2020, 470, 228317 CrossRef CAS.
- S. Yuan, Y. Liu, J. Zheng, M. Cui, K. Wang and N. Li, J. Alloys Compd., 2023, 933, 167664 CrossRef CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef PubMed.
- M. Balaish, J.-W. Jung, I.-D. Kim and Y. Ein-Eli, Adv. Funct. Mater., 2020, 30, 1808303 CrossRef CAS.
- Z. Chang, J. Xu and X. Zhang, Adv. Energy Mater., 2017, 7, 1700875 CrossRef.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- Z.-Z. Shen, C. Zhou, R. Wen and L.-J. Wan, Chin. J. Chem., 2021, 39, 2668–2672 CrossRef CAS.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772–1778 RSC.
- H.-D. Lim, B. Lee, Y. Bae, H. Park, Y. Ko, H. Kim, J. Kim and K. Kang, Chem. Soc. Rev., 2017, 46, 2873–2888 RSC.
- Q.-C. Liu, J.-J. Xu, D. Xu and X.-B. Zhang, Nat. Commun., 2015, 6, 7892 CrossRef CAS PubMed.
- M. Zhou, H. A. Doan, L. A. Curtiss and R. S. Assary, J. Phys. Chem. C, 2020, 124, 5636–5646 CrossRef CAS.
- D. Cao, X. Shen, A. Wang, F. Yu, Y. Wu, S. Shi, S. A. Freunberger and Y. Chen, Nat. Catal., 2022, 5, 193–201 CrossRef CAS.
- X. Wu, X. Wang, Z. Li, L. Chen, S. Zhou, H. Zhang, Y. Qiao, H. Yue, L. Huang and S.-G. Sun, Nano Lett., 2022, 22, 4985–4992 CrossRef CAS PubMed.
- C. Y. Zhang, C. Zhang, J. L. Pan, G. W. Sun, Z. Shi, C. Li, X. Chang, G. Z. Sun, J. Y. Zhou and A. Cabot, eScience, 2022, 2, 405–415 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |