
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiochar-mediated carbon nitride and covalent organic framework photocatalyst for enhanced tetracycline degradation†
Qi Weia,
Baojun Yi *ab,
Zewen Huaa,
Zhengshuai Suna and
Feng Guoa
*ab,
Zewen Huaa,
Zhengshuai Suna and
Feng Guoa
aCollege of Engineering, Huazhong Agricultural University, No. 1, Shizishan Street, Hongshan District, Wuhan, 430070, PR China. E-mail: bjyi@mail.hzau.edu.cn
bKey Laboratory of Agricultural Equipment in the Mid-lower Yangtze River, Ministry of Agriculture, Wuhan, 430070, PR China
First published on 27th May 2025
Abstract
The accumulation of tetracycline (TC) poses a significant challenge to human health and ecological systems. Photocatalytic degradation of TC has been a focus of research, heterojunctions receiving particular attention due to their superior charge separation and transfer properties. This study explores the structural characteristics of heterojunctions and their efficacy in degrading TC in aqueous solutions. We synthesized various combinations of biochar (BC), carbon nitride (CN), and covalent organic frameworks (COF) to form heterojunctions and characterized their morphological, structural, and optical properties using SEM, XRD, XPS, and UV-Vis DRS. These analyses helped elucidate the mechanisms underlying TC degradation. The CN10/COF2-12, synthesized in situ, showed significantly improved degradation efficiency, outperforming CN-10 and COF-2 by factors of 2.02 and 1.96, respectively. Furthermore, the all-solid-state Z-scheme heterojunction photocatalyst BC10–CN10/COF2-22, in which BC serves as the electron mediator, achieved a 3.13-fold increase in TC degradation compared to BC10–CN10-20. The BC-mediated all-solid-state Z-scheme heterojunction effectively facilitated the separation and transfer of photogenerated electron–hole pairs. This study combines the advantages of CN, COF, and BC, thereby providing a novel approach to the development of high-performance photocatalysts.
1 Introduction
Tetracycline (TC) is a broad-spectrum antibiotic extensively used due to its favorable antimicrobial efficacy and low cost. However, the unregulated overuse of TC has led to significant residues entering water bodies through excretory routes, resulting in water contamination and the spread of antibiotic resistance genes.1 Therefore, it is crucial to promptly develop effective degradation technologies for TC. Conventional water treatment methods, such as physical adsorption2 and biological treatment,3 often fail to completely eliminate TC. In contrast, advanced oxidation processes, particularly photocatalytic technologies,4,5 show promise in water treatment. These technologies efficiently harness solar energy and convert it into chemical energy under ambient conditions, generating reactive free radicals that facilitate TC degradation.In the field of photocatalysis, heterostructures have gained significant interest due to their ability to facilitate the separation of photogenerated charge carriers.6 Particularly, van der Waals (vdW) heterostructures provide a means of bypass the stringent lattice matching demands of epitaxial growth, while enabling precise interfacial control through non-covalent interactions.7,8 2D/2D vdW heterojunctions have demonstrated enhanced photocatalytic performance due to their large interfacial area, strong interactions, extended photo-response, and accelerated interfacial charge separation.9 Compared to 1D–2D and 0D–2D heterostructures, the face-to-face structure offers distinct advantages in increasing interfacial contact area and accelerating charge transfer rates.10 For example, Qiao et al.11 synthesized a 2D/2D vdW heterostructure of metal-free phosphorene and g-C3N4, which demonstrated enhanced efficiency in photocatalytic hydrogen production under visible light. Li et al.8 combined atomically thin g-C3N4 with Bi9O7.5S6 nanoplates to form a 2D/2D vdW heterostructure for CO2 photoreduction, exhibiting enhanced photocatalytic activity. CN, with its 2D layered structure, suitable band gap, mild synthesis method, and good stability, is ideal for constructing vdW heterostructures.12–14 COF, a novel class of two-dimensional materials, have potential applications in catalysis, gas storage and optoelectronics due to their structural tunability.15 They possess excellent specific surface area and tunable structural properties, providing more active sites for photocatalytic reactions. In particular, imine-conjugated COF not only have efficient electron mobility, but also significantly enhance the separation efficiency of photogenerated carriers through the off-domain of π-electrons, thus improving the photocatalytic performance.16,17 The combination of COF and g-C3N4 in a 2D/2D vdW heterojunction is important for the photocatalytic degradation of pollutants. Luo et al.18 modified g-C3N4 with COF for imine bonding, preparing β-ketoamine-bonded COF:TP–TTA@g-C3N4 with hydrogen production up to 10.1 mmol g−1 h−1, significantly enhancing the visible photocatalytic hydrogen evolution performance of g-C3N4. Wang et al.19 prepared a novel vdW heterojunction composite combining g-C3N4 with COF in nitrogen vacancies, which exhibits an effective interfacial contact area and excellent photocatalytic CO2 reduction performance. Furthermore, the efficacy of composites in applications is significantly influenced by the preparation methods. For instance, Liu et al.20 demonstrated the in situ synthesis of chalcogenides on imide-based COF, which enhanced the stability of chalcogenides and the crystallinity of COF. Wang et al.21 prepared CsPbBr3/CTF-1 composites by electrostatic self-assembly with a simple mechanical stirring method. This invented a covalent triazine framework, which enhanced the efficiency of visible light harvesting and CO2 photoreduction activity.
However, due to interface resistance, the interface contact of these binary composites is poor, which reduces the transfer efficiency of photogenerated electrons.22 Therefore, conductive solid electronic dielectrics, such as metal nanoparticles, graphene, and nanocarbon, are incorporated to facilitate carrier transfer.23 For example, Liu et al.24 synthesized the photocatalyst g-C3N4/Ag/TiO2, where Ag nanoparticles were strategically positioned at the g-C3N4 and TiO2 interface to serve as an electron transport bridge, thereby accelerating charge transfer. Jiang et al.25 constructed an all-solid-state Z-scheme heterojunction consisting of CN, CNT, and Bi2WO6 nanosheets using carbon nanotubes as an electronic mediator. This configuration effectively promoted the separation and transfer of photogenerated charge carriers, thereby enhancing the photocatalytic activity for TC degradation. However, the widespread application of electronic mediators, including heavy metals and carbon nanotubes, is constrained by their high cost. Consequently, BC, which is cost-effective and exhibits excellent adsorption properties, an abundant pore structure, and favorable electrical conductivity and electron storage capacity, was employed as an electronic mediator.26,27 Ma et al.28 constructed a novel ternary CdS–WPB–g-C3N4 heterojunction photocatalyst, using BC as a green electron transport medium to significantly enhance the carrier separation efficiency. They achieved efficient degradation of methyl orange under visible light (99% degradation in 80 minutes) and ultraviolet light (complete degradation in 60 minutes). Furthermore, there are few reports on the use of BC in g-C3N4/COF composites or even ternary heterojunctions.
To address the challenges in current photocatalytic systems, this study aims to develop photocatalysts with excellent photocatalytic performance and cost-effectiveness by synergistically integrating the structural and electronic advantages of CN, COF and BC. Specifically, BC is adopted as a sustainable electron transfer medium, which overcomes the limitations of traditional high-cost metal media. A 2D/2D vdW heterostructure is constructed between COF and g-C3N4. Through atomic-level π-orbital overlap, the interface charge separation is maximized. These physicochemical and chemical characterization of photocatalysts to reveal the relationship between their structure and photocatalytic performance. The findings provide novel insights for the development of efficient photocatalysts and propose innovative solutions for the effective degradation of TC.
2 Experimental methods
2.1 Materials
TC, dicyandiamide, anhydrous ethanol, sodium chloride, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)triphenylamine (TTA), 1,3,5-tricarbonylresorcinol (TP), dimethylsulfoxide (DMSO), and methanol were purchased from China National Pharmaceutical Chemical Reagent Co. Wood chips, obtained from Anhui Zhengjie Feed Co., Ltd, China, were passed through a 60-mesh sieve and subsequently dried at 105 °C for 48 h to ensure complete removal of moisture.The preparation of CN involved the dissolution of 4.5 g of dicyandiamide in 500 mL of ethanol. Subsequently, a saturated aqueous solution of sodium chloride was added dropwise to the mixture under constant stirring. The ethanol and water were removed by rotary evaporation, thereby obtaining a solid residue. The residue was then heated for 2 h at 550 °C with a heating rate of 5 °C min−1. After cooling, the product was washed extensively with deionized water and dried at 120 °C to obtain the final CN material.
The preparation of COF was initiated by dissolving 0.07 mmol of TTA and 0.13 mmol of TP in 5 mL of DMSO, followed by sonicated to ensure complete dissolution. The resulting solution was then poured into an autoclave and heated at 120 °C for 12 h. The product was then washed with methanol and subsequently dried at 105 °C, thereby obtaining the COF.19
10 mg of BC and 10 mg of CN were thoroughly stirred under an inert atmosphere, and the product was labeled as BC10–CN10-20. The ‘–’ symbol stands for the physical compounding method.
A solution of 82.98 mg of BC10–CN10-20 in 5 mL of DMSO was prepared and sonicated for 2 minutes to disperse the material. Subsequently, 0.07 mmol of TTA and 0.13 mmol of TP were added, and the solution was sonicated and mixed again to provide the reaction conditions for the generation of COF. The mixture was then heat-treated at 120 °C in an autoclave for 12 h. After this period, the product was washed with methanol and recorded as BC10–CN10/COF2-22. The synthesis of BC10/COF2–CN10-22 involved the complete stirring of the reactants under an inert atmosphere. Similarly, the synthesis of CN10/COF2–BC10-22 entailed the complete stirring of CN10/COF2-12 and BC under an inert atmosphere. The naming correspondence table of photocatalysts with fixed component masses is shown in Table S1.†
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, while BC3/COF2-5 was prepared in a mass ratio of BC to COF of 3:2, following the preparation method detailed in Section 2.1.2.
:2, while BC3/COF2-5 was prepared in a mass ratio of BC to COF of 3:2, following the preparation method detailed in Section 2.1.2.The samples, designated as BC3–CN3/COF4-5 and BC3/COF3–CN4-5, were prepared by combining BC, CN, and COF in a mass ratio of 3:3:4, in accordance with the methodology described in Section 2.1.2. The naming correspondence table of photocatalysts with fixed total mass is shown in Table S2.†
2.2 Adsorption and degradation experiments
A 50 mL of aqueous solution of TC at a concentration of 20 mg L−1 was prepared and transferred into a 50 mL reactor. The catalyst was then added, followed by thorough stirring to achieve uniform dispersion throughout the solution. The mixture was continuously stirred for an additional 30 minutes in the dark to establish a dynamic equilibrium between the catalyst and TC molecules. Subsequently, the light source was initiated, with the light intensity adjusted to approximately 7000 Lux to control the photocatalytic reaction rate, thereby facilitating observation and detailed analysis of the reaction process. At regular intervals, 1 mL of the solution was removed and filtered through a 0.45 μm aqueous membrane to eliminate catalyst particles. The concentration of TC in the filtered samples was subsequently quantified using high-performance liquid chromatography (HPLC). Each experiment was independently repeated three times, and the experimental data is presented as the mean, with the coefficient of variation provided in Table S3.†2.3 Characterization methods
The crystal structures of the samples were analyzed using a Bruker D8 Advance X-ray diffraction (XRD) from Germany. The elemental composition and the chemical bonding states of the photocatalysts were investigated using a Thermo Fisher Scientific Escalab 250Xi photoelectron spectrometer (XPS) from the USA. Microscopic morphology was visualized with a high-resolution, cold field-emission scanning electron microscope (SEM), model Regulus-8100, from Hitachi, Japan. Pore properties were investigated using a Quadrasorb evoTM fully automated specific surface area and porosity analyzer, manufactured by Kantar Instruments, USA. The visible light absorption properties of the photocatalysts were determined using a UV-3600 UV-Vis spectrophotometer from Shimadzu, Japan. Photoluminescence (PL) properties were characterized using a FLS-1000 instrument from Edinburgh Instruments Ltd, UK, with an excitation wavelength of 350 nm. The photoelectric properties were evaluated using a CHI-660E electrochemical workstation from Shanghai Chenhua Instrument Co.3 Results and discussion
3.1 Photocatalytic degradation performance
 | ||
| Fig. 1 Single and dual-component photocatalysts: (a) photocatalytic degradation of TC. (b) Kinetic fitting curve. | ||
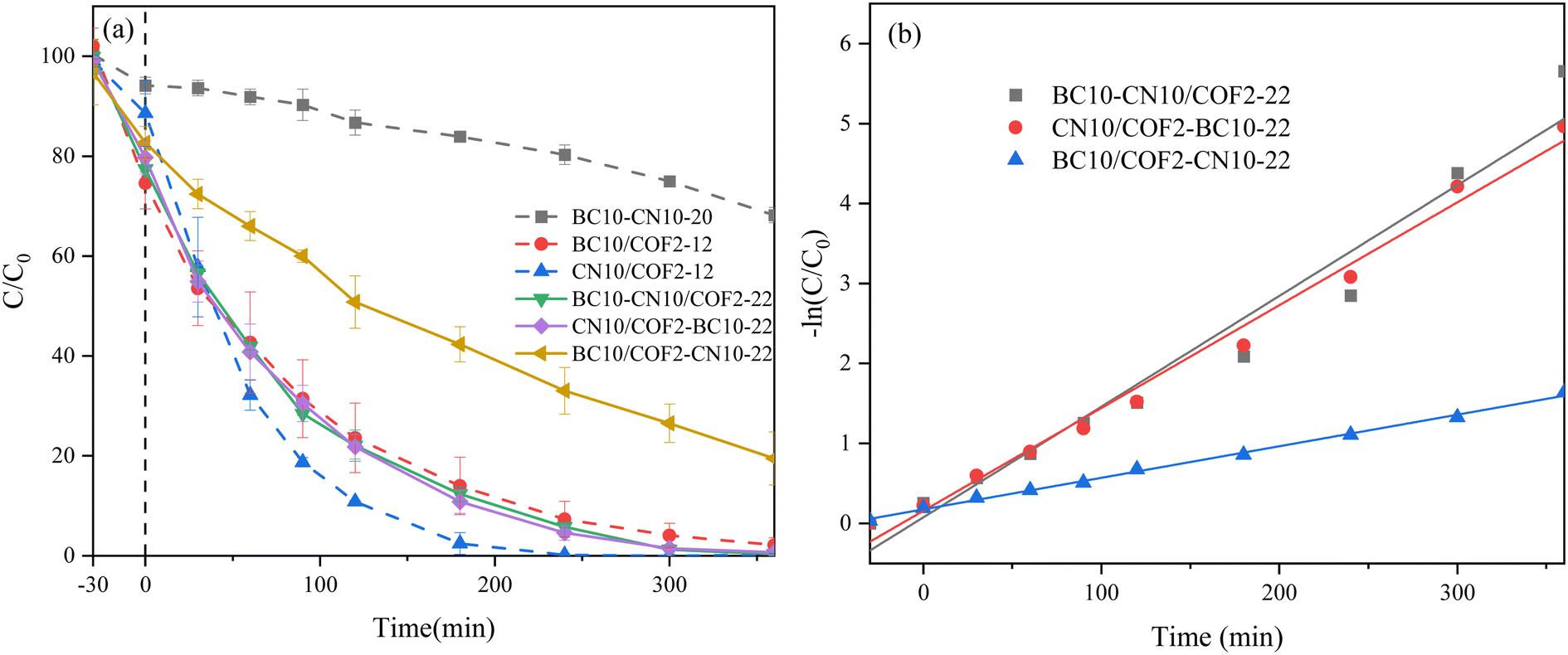 | ||
| Fig. 2 Three-component photocatalysts: (a) photocatalytic degradation of TC. (b) Kinetic fitting curve. | ||
 | ||
| Fig. 3 Quantitative photocatalysts: (a) photocatalytic degradation of TC. (b) Kinetic fitting curve. | ||
3.2 Material characterization
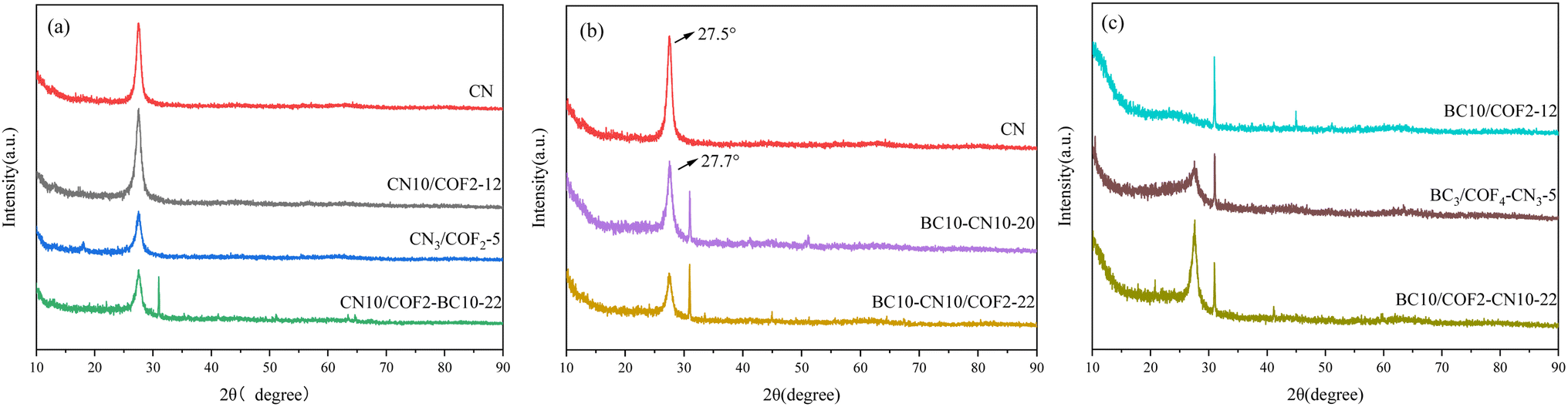 | ||
| Fig. 4 XRD patterns of photocatalyst: (a) CN, CN10/COF2-12, CN3/COF2-5, and CN10/COF2–BC10-22, (b) CN, BC10–CN10-20, BC10–CN10/COF2-22, and (c) BC10/COF2-12, BC3/COF4–CN3-5, and BC10/COF2–CN10-22. | ||
Fig. 5 depicts the surface elemental composition and chemical forms of the photocatalysts. The N 1s high-resolution spectrum of COF, after fitting, revealed two distinct peaks at 398.2 eV and 399.8 eV, corresponding to the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and –N–C groups, respectively.36 In CN, three N 1s peaks were observed at 398 eV, 399.5 eV, and 400.6 eV, respectively. The peak at 398 eV is attributed to an aromatic nitrogen carbon atom (CN–C) hybridized by sp2 in the triazine ring. The peak at 399.5 eV is assigned to a nitrogen atom in (N–(C)3), and the peak at 400.6 eV is indicative of the presence of a C–N–H group.37 For CN10/COF2-12, these peaks shifted to higher binding energies, suggesting changes in the chemical state or coordination conditions of the N atoms. This shift is proposed to result from electron transfer from CN to COF upon coupling, indicating the formation of an intimate heterojunction between CN and COF.19 As illustrated in Fig. 5b, the N 1s high-resolution spectrum of BC10–CN10-20 exhibits three fitted peaks at 397.9 eV, 399.4 eV, and 400.5 eV. The peaks at 397.9 eV and 400.5 eV are indicative of the C–N and N–H bonds in the CN triazine ring, respectively. However, the peaks in BC10–CN10-20 are shifted toward lower binding energies, indicating a decrease in electron cloud density and weakened electron binding ability.38 The characteristic peak of the BC3–CN3/COF4-5, observed at 399.5 eV, is attributed to the –N–C group in COF. This observation suggests that the complexation of BC10–CN10-20 with the COF material results in the formation of the –N–C group. Furthermore, in comparison to the BC10–CN10-20, the characteristic peaks of BC3–CN3/COF4-5 at 398.2 eV and 400.6 eV, exhibiting a shift towards higher binding energies. This phenomenon may be attributed to interfacial charge transfer, which results in the nitrogen atoms experiencing a loss of electron density.39 It is hypothesized that BC3–CN3/COF4-5 forms a Z-scheme heterostructure.
C and –N–C groups, respectively.36 In CN, three N 1s peaks were observed at 398 eV, 399.5 eV, and 400.6 eV, respectively. The peak at 398 eV is attributed to an aromatic nitrogen carbon atom (CN–C) hybridized by sp2 in the triazine ring. The peak at 399.5 eV is assigned to a nitrogen atom in (N–(C)3), and the peak at 400.6 eV is indicative of the presence of a C–N–H group.37 For CN10/COF2-12, these peaks shifted to higher binding energies, suggesting changes in the chemical state or coordination conditions of the N atoms. This shift is proposed to result from electron transfer from CN to COF upon coupling, indicating the formation of an intimate heterojunction between CN and COF.19 As illustrated in Fig. 5b, the N 1s high-resolution spectrum of BC10–CN10-20 exhibits three fitted peaks at 397.9 eV, 399.4 eV, and 400.5 eV. The peaks at 397.9 eV and 400.5 eV are indicative of the C–N and N–H bonds in the CN triazine ring, respectively. However, the peaks in BC10–CN10-20 are shifted toward lower binding energies, indicating a decrease in electron cloud density and weakened electron binding ability.38 The characteristic peak of the BC3–CN3/COF4-5, observed at 399.5 eV, is attributed to the –N–C group in COF. This observation suggests that the complexation of BC10–CN10-20 with the COF material results in the formation of the –N–C group. Furthermore, in comparison to the BC10–CN10-20, the characteristic peaks of BC3–CN3/COF4-5 at 398.2 eV and 400.6 eV, exhibiting a shift towards higher binding energies. This phenomenon may be attributed to interfacial charge transfer, which results in the nitrogen atoms experiencing a loss of electron density.39 It is hypothesized that BC3–CN3/COF4-5 forms a Z-scheme heterostructure.
 | ||
| Fig. 5 High-resolution XPS spectra of N 1s in photocatalyst: (a) COF, CN, and CN10/COF2-12 and (b) CN, BC10–CN10-20, and BC3–CN3/COF4-5. | ||
As illustrated in Fig. S3a,† BC displays a conventional type IV isothermal curve with H4 hysteresis loops, indicative of a uniform pore size distribution. This is attributed to the gradual release of volatiles during the carbonization of the BC at lower pyrolysis temperatures, which contributes to the formation of micropores and a larger specific surface area.41 With the exception of BC, all other samples display type IV isothermal curves with evident H3 hysteresis loops, indicating pronounced mesoporous structures. The COF material, characterized by a well-ordered crystalline and pore structure, results in a high specific surface area. Additionally, the presence of hollow space within the material contributes to a large total pore volume, as illustrated in Fig. S2b.† The notable enlargement of the pore size in the CN10/COF2-12 composite enhances the adsorption capacity, although its specific surface area is markedly lower in comparison to the other samples. The lower specific surface area of CN10/COF2–BC10-22 in comparison to CN10/COF2-12 can be attributed to the physical agitation approach, causing the formation of particle agglomerates of BC and CN10/COF2-12, which limits the effective formation of a CN10/COF2-12 and BC composite. Fig. S3b† illustrates that BC has a considerable abundance of mesopores and a paucity of macropores. In contrast, CN10/COF2-12 composites display a relatively high proportion of macropores. Consequently, BC3–CN3/COF4-5 composites possess a substantial proportion of both mesopores and macropores, which is conducive to enhanced adsorption capacity. It is particularly emphasized that the presence of macropores plays a beneficial role in the photocatalytic process. In addition to providing more adsorption sites,42 they also facilitate the mass transfer efficiency of the photocatalytic reaction. In comparison to BC10–CN10/COF2-22, the proportion of mesopores and macropores in CN10/COF2–BC10-22 and BC10/COF2–CN10-22 was significantly diminished, indicating that different preparation techniques significantly affect the pore structure of the samples. The photocatalytic degradation efficiency of CN10/COF2–BC10-22 (99.3%) and BC10/COF2–CN10-22 (80.53%) was lower than that of BC10–CN10/COF2-22 (99.65%), indicating that the presence of mesopores and macropores is favorable for the photocatalytic (Table 1).
| Sample | SBET (m2 g−1) | VTotal (cm3 g−1) | VMicroa (cm3 g−1) | VExtb (cm3 g−1) | DAverage (nm) |
|---|---|---|---|---|---|
| a Pore volume of micropore, calculated by t-plot.b Calculated by difference (VExt = VBET − VMicro). | |||||
| BC | 250.50 | 0.12 | 0.02 | 0.10 | 1.93 |
| COF | 239.56 | 0.23 | 0.16 | 0.07 | 3.88 |
| CN10/COF2-12 | 43.37 | 0.10 | 0.09 | 0.01 | 9.09 |
| BC3–CN3/COF4-5 | 131.23 | 0.18 | 0.10 | 0.08 | 5.37 |
| CN10/COF2–BC10-22 | 32.68 | 0.06 | 0.05 | 0.01 | 7.23 |
| BC10/COF2–CN10-22 | 33.22 | 0.06 | 0.05 | 0.01 | 7.47 |
 | ||
| Fig. 6 Analysis of optical properties on photocatalysts: (a) UV-Vis spectra of CN, CN10/COF2-12, and BC10–CN10/COF2-22. (b) Plot of (ahν)2 vs. hν for the band gap energy of CN, CN10/COF2-12, and BC10–CN10/COF2-22. (c) Valence band XPS spectra of CN and COF. (d) Mechanism of CN10/COF2-12 composite photocatalyst. | ||
The photoluminescence (PL) spectra of heterojunction materials serve as an indicator of the separation efficiency of photogenerated electron–hole pairs. In general, a higher PL peak intensity correlates with a faster recombination rate of photogenerated electrons and holes in photocatalysts.46 Fig. 7a illustrates the PL spectra of CN, CN10/COF2-12, and BC10–CN10/COF2-22. Notably, CN exhibits a higher intensity of typical emission peaks at an excitation wavelength of 461 nm, suggesting rapid recombination of photogenerated e−–h+, which is the primary factor limiting its photocatalytic activity. This phenomenon may be attributed to the n–π* electron leaps.47 The PL peak intensity of CN10/COF2-12 is significantly lower compared to CN, indicating that the heterojunction between CN and COF effectively suppresses the recombination of photogenerated e−–h+, facilitating an enhancement in photocatalytic activity. The PL peak intensity of BC10–CN10/COF2-22 is observed to decrease further, indicating that the all-solid-state Z-scheme heterojunction more effectively inhibits the recombination of photogenerated e−–h+. The separation effect of photogenerated charges at the material interface is illustrated in Fig. 7b, with CN and COF exhibiting considerable impedance radii. In comparison to CN and COF, CN10/COF2-12 exhibits a reduced impedance radius, and BC10–CN10/COF2-22 shows an even lower impedance radius, suggesting lower resistance and faster interfacial charge transfer.48 These results demonstrate that heterojunctions, particularly all-solid-state Z-scheme heterojunctions, can enhance the separation efficiency of photogenerated carriers and accelerate the interfacial charge transfer, thereby facilitating the photocatalytic degradation of TC.
 | ||
| Fig. 7 Analysis of photogenerated charge carrier on photocatalysts: (a) photoluminescence of CN, CN10/COF2-12, and BC10–CN10/COF2-22. (b) Electrochemical impedance spectroscopy of CN, COF, CN10/COF2-12, and BC10–CN10/COF2-22. | ||
3.3 Mechanism of photocatalytic degradation
This study conducted capture experiments to identify the primary active species in the photocatalytic process. P-Benzoquinone (BQ), triethanolamine (TEOA), and isopropanol (IPA) at a concentration of 0.01 mol L−1 were utilized as trapping agents to eliminate ·O2−, h+, and ·OH radicals, respectively, with the results depicted in Fig. 8. The degradation efficiency of TC in the system with the addition of BQ was found to be significantly reduced, reaching approximately 57% compared to the control without a trapping agent. In contrast, the addition of TEOA and IPA had a less pronounced effect, with TC degradation rates of 94.18% and 94.89%, respectively. The results demonstrate that ·O2− radical played a pivotal role in the photocatalytic degradation of TC, while the contributions of h+ and ·OH were comparatively less significant. | ||
| Fig. 8 Effect of different scavengers on the BC10–CN10/COF2-22 photocatalytic degradation of TC. | ||
Xu et al.49 proposed a classification of Z-scheme heterojunctions into three distinct categories: conventional Z-scheme heterojunctions, all-solid-state Z-scheme heterojunctions, and direct Z-scheme heterojunctions. In a conventional Z-scheme heterostructure, the photo-induced holes in the VB of the photocatalyst 1 are captured by the electron donor, while the photo-induced electrons in the CB of the photocatalyst 2 are absorbed by the electron acceptor.50 In the all-solid-state Z-scheme heterostructure, upon light activation of the component photocatalysts, the light-induced electrons in the CB of photocatalyst 2 are transferred to the conducting material, subsequently reaching the VB of photocatalyst 1. Unlike other heterostructures, direct Z-scheme heterostructures do not involve a charge-transfer medium. Instead, the formation of charge complex is achieved through direct electron and hole transfer between the semiconductors.51
This study elucidates the photo-induced charge transfer mechanism of the catalyst under visible light irradiation, highlighting the crucial role of the Z-scheme heterojunction in photocatalytic. The absorption edges of CN10/COF2-12 and BC10–CN10/COF2-22 were obtained by UV-Vis spectroscopy, from which direct band gaps of 2.31 eV and 2.01 eV were calculated using the Tauc equation. The mechanism diagram is presented in Fig. 9. The CN10/COF2-12 heterostructure improves the photocatalytic efficiency. The all-solid-state Z-scheme heterostructure in BC10–CN10/COF2-22 broadens the absorption spectrum of visible light, optimizes the separation efficiency of the photogenerated e−–h+ and inhibits their recombination effectively, thus improving the photocatalytic efficiency. Specifically, under visible light excitation, electrons transition from the conduction band of the COF to the valence band of the CN, while holes accumulating in the valence band of the COF and electrons in the conduction band of the CN. The migration of electrons and holes is facilitated by the electron mediator BC, which enhances the catalytic effect due to its high conductivity. Electrons accumulated on the CN conduction band are trapped by O2, resulting in the generation of ·O2−. This is due to the fact that the CB edge potential of CN is lower than the standard Eθ(O2/·O2−) (−0.33 V vs. NHE). Simultaneously, the collective defects on the valence band of COF can oxidize OH− to produce ·OH. This is due to the fact that the VB edge potential of COF is higher than the standard Eθ(·OH/OH−) (1.99 V vs. NHE). In conclusion, the BC-mediated all-solid-state Z-scheme heterojunction mechanism facilitates efficient carrier separation, the generation of reactive oxygen species, and an improvement in material stability, collectively enhancing the photocatalytic activity of BC10–CN10/COF2-22.
 | ||
| Fig. 9 Photocatalytic degradation mechanism of photocatalyst. | ||
4 Conclusion
This study successfully constructed a multifunctional heterojunction photocatalytic system for the efficient photocatalytic degradation of TC. Comparative experiments indicated that the optimal binary photocatalysts, CN10/COF2-12 achieved a TC degradation rate of 99.79% after 360 minutes of photocatalytic reaction, exhibiting 2.02-fold and 1.96-fold enhancement compared to individual components CN-10 and COF-2, respectively. The ternary all-solid-state Z-scheme heterojunction BC10–CN10/COF2-22, constructed by further introducing BC, achieved a TC degradation rate of 99.65%, representing a 3.13-fold improvement over BC10–CN10-20. Radical trapping experiments revealed that ·O2− was the primary active species with a contribution rate of 83.18% in the photocatalytic degradation of TC, while h+ and ·OH played a less significant role. The BC-mediated all-solid-state Z-scheme heterojunction promotes efficient charge carrier separation, was the principal reason for the enhanced photocatalytic activity of BC10–CN10/COF2-22. This study presents a novel approach for the photocatalytic degradation of TC, which is crucial for effective environmental remediation.Data availability
The data used to support the findings of this study are available from the corresponding author upon request.Author contributions
Qi Wei: experiments, data processing, writing-original draft. Baojun Yi (corresponding author): general framework, data processing, writing-review and editing, supervision, financial support. Zewen Hua: experiments, data processing. Zhengshuai Sun: data processing, writing-review and editing. Feng Guo: drawing, writing-reviewing and editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by Key Research and Development Program of Hubei Province (2023BBB001), National Key Research and Development Program of China (2021YFD1600503), and Fundamental Research Funds for the Central Universities (2662024GXPY004).References
- Z. Sun, Q. Wei, W. Hua, M. Chen, Q. Yuan, B. Yi and Y. S. Ok, J. Environ. Chem. Eng., 2024, 12, 114290 CrossRef CAS.
- K. L. A. Cao, A. M. Rahmatika, Y. Kitamoto, M. T. T. Nguyen and T. Ogi, J. Colloid Interface Sci., 2021, 589, 252–263 CrossRef CAS PubMed.
- J.-Y. Zhang, C. Xia, H.-F. Wang and C. Tang, J. Energy Chem., 2022, 67, 432–450 CrossRef CAS.
- Y. Li, M. Fu, R. Wang, S. Wu and X. Tan, Chem. Eng. J., 2022, 444, 136567 CrossRef CAS.
- Ö. Hanay, B. Yıldız, S. Aslan and H. Hasar, Environ. Sci. Pollut. Res., 2014, 21, 3774–3782 CrossRef PubMed.
- X. He, T. Kai and P. Ding, Environ. Chem. Lett., 2021, 19, 4563–4601 CrossRef CAS PubMed.
- M.-Q. Zhu and X.-F. Wang, J. Phys. D: Appl. Phys., 2024, 57, 335104 CrossRef CAS.
- J. Li, B. Huang, Q. Guo, S. Guo, Z. Peng, J. Liu, Q. Tian, Y. Yang, Q. Xu, Z. Liu and B. Liu, Appl. Catal., B, 2021, 284, 119733 CrossRef CAS.
- J. E. Ellis, D. C. Sorescu, S. I. Hwang, S. C. Burkert, D. L. White, H. Kim and A. Star, ACS Appl. Mater. Interfaces, 2019, 11, 41588–41594 CrossRef CAS PubMed.
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S.-Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- C. Yang, Z. Xue, J. Qin, M. Sawangphruk, S. Rajendran, X. Zhang and R. Liu, J. Phys. Chem. C, 2019, 123, 4795–4804 CrossRef CAS.
- I. Papailias, N. Todorova, T. Giannakopoulou, N. Ioannidis, N. Boukos, C. P. Athanasekou, D. Dimotikali and C. Trapalis, Appl. Catal., B, 2018, 239, 16–26 CrossRef CAS.
- C. Feng, J. Rong, Y. Zhang, X. Zheng, X. Li, S. Xu and Z. Li, Appl. Catal., B, 2023, 337, 123005 CrossRef CAS.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1970011 CrossRef.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- H. Ran, Q. Xu, Y. Yang, H. Li, J. Fan, G. Liu, L. Zhang, J. Zou, H. Jin and S. Wang, ACS Catal., 2024, 14, 11675–11704 CrossRef CAS.
- M. Luo, Q. Yang, K. Liu, H. Cao and H. Yan, Chem. Commun., 2019, 55, 5829–5832 RSC.
- J. Wang, Y. Yu, J. Cui, X. Li, Y. Zhang, C. Wang, X. Yu and J. Ye, Appl. Catal., B, 2022, 301, 120814 CrossRef CAS.
- Y. Liu, Y. Zhu, S. B. Alahakoon and E. Egap, ACS Mater. Lett., 2020, 2, 1561–1566 CrossRef CAS.
- Q. Wang, J. Wang, J.-C. Wang, X. Hu, Y. Bai, X. Zhong and Z. Li, ChemSusChem, 2021, 14, 1131–1139 CrossRef CAS PubMed.
- H. Li, W. Tu, Y. Zhou and Z. Zou, Adv. Sci., 2016, 3, 1500389 CrossRef PubMed.
- X. Wu, J. Zhao, L. Wang, M. Han, M. Zhang, H. Wang, H. Huang, Y. Liu and Z. Kang, Appl. Catal., B, 2017, 206, 501–509 CrossRef CAS.
- Y. Liu, Y. Yuan, S. Ni, J. Liu, S. Xie and Y. Liu, Water Sci. Technol., 2022, 85, 2639–2651 CrossRef CAS PubMed.
- D. Jiang, W. Ma, P. Xiao, L. Shao, D. Li and M. Chen, J. Colloid Interface Sci., 2018, 512, 693–700 CrossRef CAS PubMed.
- J. Zou, J. Liu, Y. Xie, G. Peng, L. Duan, D. Hu, S. Chen, F. Qu and L. Lu, Chem. Eng. J., 2023, 468, 143849 CrossRef CAS.
- W. Xiang, X. Zhang, J. Chen, W. Zou, F. He, X. Hu, D. C. W. Tsang, Y. S. Ok and B. Gao, Chemosphere, 2020, 252, 126539 CrossRef CAS PubMed.
- C. Ma, Y. Zhang, B. Yin, J. F. Chen, M. H. Guo and X. Gao, J. Environ. Chem. Eng., 2023, 11, 109135 CrossRef CAS.
- Q. Tang, Q. Cheng and Z. Pan, Sustainable Mater. Technol., 2024, 41, e01115 CrossRef CAS.
- L. Xiao, S. Zhang, B. Chen, P. Wu, N. Feng, F. Deng and Z. Wang, J. Environ. Chem. Eng., 2023, 11, 109693 CrossRef CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- M. Zebardast, A. F. Shojaei and K. Tabatabaeian, Iran. J. Catal., 2018, 8, 297–309 CAS.
- X. Du, G. Zou, Z. Wang and X. Wang, Nanoscale, 2015, 7, 8701–8706 RSC.
- Q. Zhao, B. Zhao, X. Long, R. Feng, M. Shakouri, A. Paterson, Q. Xiao, Y. Zhang, X.-Z. Fu and J.-L. Luo, Nano-Micro Lett., 2024, 16, 80 CrossRef CAS PubMed.
- M.-Y. Zhang, J.-K. Li, R. Wang, S.-N. Zhao, S.-Q. Zang and T. C. W. Mak, Adv. Sci., 2021, 8, 2101884 CrossRef CAS PubMed.
- J. Du, Z. Xu, H. Li, H. Yang, S. Xu, J. Wang, Y. Jia, S. Ma and S. Zhan, Appl. Surf. Sci., 2021, 541, 148487 CrossRef CAS.
- K. Qi, M. Song, X. Xie, Y. Wen, Z. Wang, B. Wei and Z. Wang, Chemosphere, 2022, 287, 132192 CrossRef CAS PubMed.
- Y.-N. Zang, S.-S. Yang, J. Ding, S.-Y. Zhao, C.-X. Chen, L. He and N.-Q. Ren, RSC Adv., 2021, 11, 15106–15117 RSC.
- H. Dai, H. Li and Q. Yang, Microporous Mesoporous Mater., 2022, 342, 112121 CrossRef CAS.
- Z. Tan, J. Zou, L. Zhang and Q. Huang, J. Mater. Cycles Waste Manage., 2018, 20, 1036–1049 CrossRef CAS.
- Q. Xue, B. Song, Q. Feng, Z. Yu, K. Hu, Y. Yang and X. Shen, Appl. Surf. Sci., 2024, 645, 158869 CrossRef CAS.
- D. Jiang, T. Wang, Q. Xu, D. Li, S. Meng and M. Chen, Appl. Catal., B, 2017, 201, 617–628 CrossRef CAS.
- H. Zhang, L.-c. Nengzi, X. Li, Z. Wang, B. Li, L. Liu and X. Cheng, Chem. Eng. J., 2020, 386, 124011 CrossRef CAS.
- Y. Yao, Y. Hu, H. Hu, L. Chen, M. Yu, M. Gao and S. Wang, J. Colloid Interface Sci., 2019, 554, 376–387 CrossRef CAS PubMed.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- S. Zhang, X. Rong, T. Sun, P. Gao, J. Liu, X. Qiu, X. Zhou and Z. Wu, Diamond Relat. Mater., 2023, 138, 110167 CrossRef CAS.
- F.-J. Zhang, F.-Z. Xie, S.-F. Zhu, J. Liu, J. Zhang, S.-F. Mei and W. Zhao, Chem. Eng. J., 2013, 228, 435–441 CrossRef CAS.
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- I. J. Ani, U. G. Akpan, M. A. Olutoye and B. H. Hameed, J. Cleaner Prod., 2018, 205, 930–954 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01986a |
| This journal is © The Royal Society of Chemistry 2025 |