
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization, and in vitro and in silico α-glucosidase inhibitory evolution of novel N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide derivatives†
Ram Reddy Mudireddyab,
Rambabu Gundla*a,
Baji Baba Shaikc,
Anoop Bodapatid,
Panasa Mahesha,
Shiva Sravan Naidue,
Damodar Tirumalasettif and
Naresh Kumar Katari*c
aDepartment of Chemistry, GITAM School of Science, GITAM (Deemed to be University) Hyderabad, Telangana 502 329, India. E-mail: rgundla@gitam.edu
bB.V.Raju Institute of Technology, Vishnupur, Narsapur, Medak Dist, Telangana 502313, India
cSchool of Chemistry & Physics, College of Agriculture, Engineering & Science, University of KwaZulu-Natal, Westville Campus, P Bag X 54001, Durban 4000, South Africa. E-mail: KatariN@ukzn.ac.za
dAnalytical Research & Development, Hikma Pharmaceuticals USA Inc, Columbus, Ohio 43228, USA
eR&D, Vertex Pharmaceuticals, 316 Northern Avenue, Boston, MA 02210, USA
fDepartment of Chemistry, University of Pennsylvania, New Town, PA 18940, USA
First published on 21st May 2025
Abstract
To discover potential α-glucosidase inhibitory agents, a new series of N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide derivatives were designed and synthesized as α-glucosidase inhibitors. The newly synthesized compounds were characterized using 1H, 13C NMR, and mass spectroscopy analysis and evaluated for their in vitro α-glucosidase inhibitory effects. All the tested compounds displayed significant α-glucosidase inhibitory activity compared to the standard drug acarbose. Among all, compounds 7b, 7d and 6g exhibited the strongest inhibition with IC50 values of 14.48 nmol, 18.88 nmol and 28.51 nmol, respectively. Molecular docking analysis was conducted to identify the important binding interactions responsible for inhibition activity of a-glucosidase. The compounds 7b and 7d exhibit the highest docking energies, with same value of −10.1 kcal mol−1 with crucial hydrogen bonding interactions with HIS:280 and ASN:415, respectively. Furthermore, computational drug likeness/ADME/toxicity analysis was conducted on the compounds, which indicated that these compounds exhibit drug-like properties and possess favourable ADME and toxicity profiles.
1 Introduction
A well-known metabolic disease, diabetes mellitus, has emerged as a major public health concern in the modern era owing to the devastating consequences it can have on an individual's wellness over time.1,2 Specifically, type 2 diabetes (T2DM) accounts for the most encountered form of diabetes.3,4 Major risk factors for developing diabetes include abnormalities in glucose metabolism.5 The hallmark of type 2 diabetes is an irregular rise in blood glucose levels.6,7 Acute hyperglycaemia damages blood vessels by acting directly on the vascular endothelium; this damages the patient's quality of life and eventually leads to heart attacks, strokes, retinopathy, and coronary heart disease.8,9 Currently, the US FDA has approved five main classes of oral antidiabetic drugs: biguanides, thiazolidinediones, sulfonylureas, meglitinides and α-glucosidase inhibitors.10–12 Unfortunately, some patients experience intolerable adverse effects from these drugs, while others see their effectiveness wane with time.13,14 Consequently, there has been and will be immense interest in the development for novel anti-diabetic drugs. Among these drugs, oral anti-diabetic drugs such as α-glucosidase inhibitors reduce postprandial hyperglycemia by inhibiting the enzyme a-glucosidase, which is responsible for the carbohydrate's degradation.15–17 The Third Asia-Pacific Region Diabetes Treatment Guidelines have recommended α-glucosidase inhibitors as the first line for decreasing postprandial hyperglycemia due to their several advantages.18α-Glucosidase is an essential enzyme in the amylase family, significantly contributing to carbohydrate metabolism in organisms.19–21 So, by limiting α-glucosidase activity, intestinal glycolysis can be diminished, leading to a reduction in postprandial hyperglycemia.22,23 At now, α-glucosidase inhibitors, namely acarbose, voglibose, and miglitol (Fig. 1A), have been developed in the clinical treatment of T2DM.24,25 However, prolonged use of these drugs may result in gastrointestinal adverse effects.26–29 Therefore, it is essential to develop novel α-glucosidase inhibitors to tackle diabetes. To date, many research groups are developed novel α-glucosidase inhibitors and some of these compounds are under preclinical evaluation.30–34
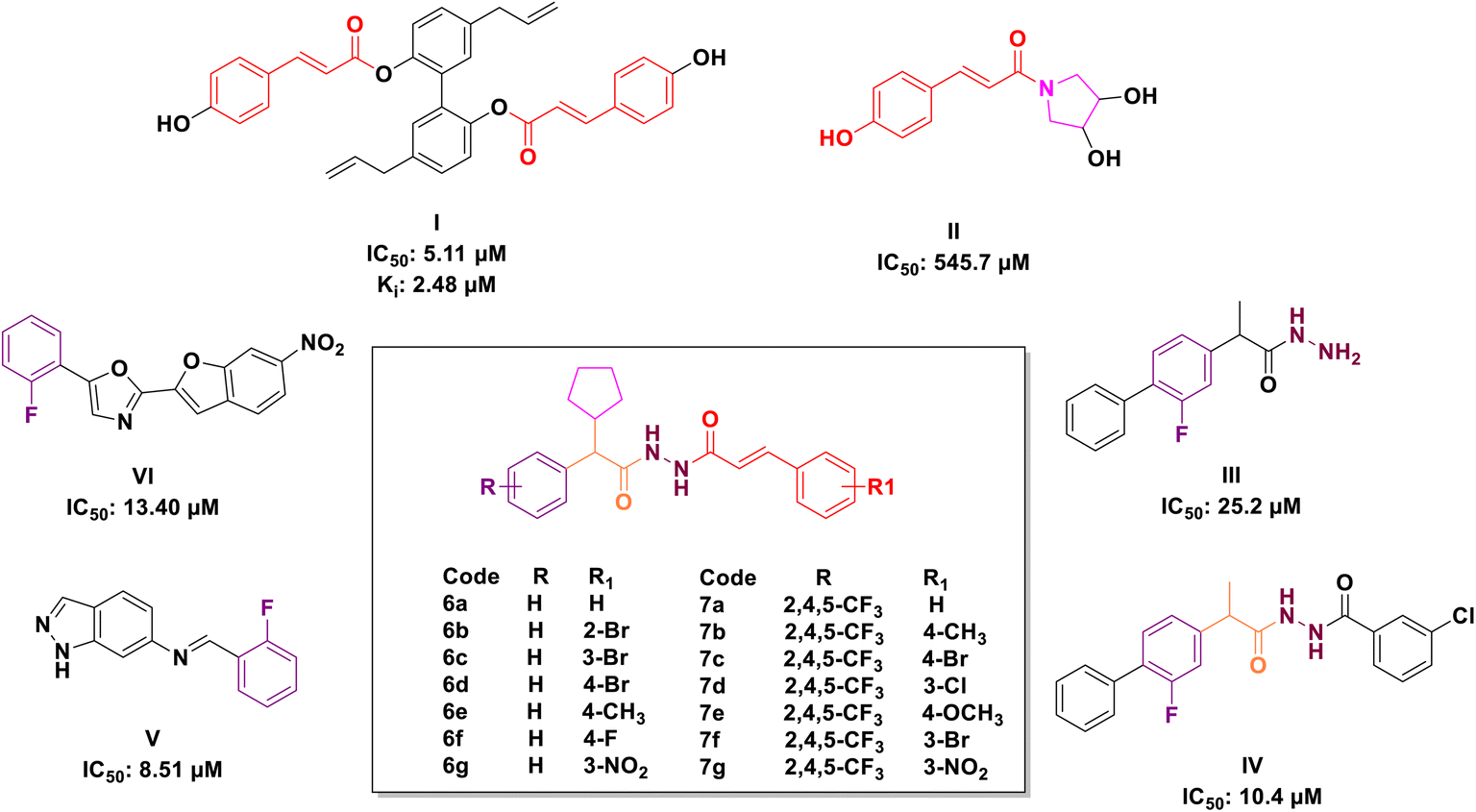 | ||
| Fig. 1 Rationale design of targeted compounds 6a–g and 7a–g. | ||
Cinnamic acid, a significant component of Cinnamomum cassia Presl, exhibits several biological activities,35 including anticancer,36–42 antioxidative,43 cardioprotective,44,45 antibacterial46,47 and antidiabetic48,49 properties. The structural alteration of cinnamic acid is garnering increasing attention to generate more potent molecules.36 Cinnamic acid derivatives (Fig. 1) and cinnamic acid ester derivatives (Fig. 1) found to have significant inhibitory activity against α-glucosidase. Chun et al. reported twenty novel cinnamic acid magnolol derivatives and screened for their anti-hyperglycaemic potency (I, IC50: 5.11 μM).50 Siva Prasad et al. reported novel deacetylsarmentamide derivatives (II) and evaluated for their in vitro α-glucosidase inhibitory potency with an excellent result.51
On the other hand, hydrazine's (III & IV, Fig. 1) bonds are prevalent in bioactive and drug candidates and emerged as a favourable ligand in drug discovery because of their unique properties, which include polarity, protein binding, and the proton exchange rate.52 Similarly, compounds with phenyl rings were evaluated for their α-glucosidase inhibitory properties and identified as potential lead agents for type II diabetes mellitus (V & VI, Fig. 1).50,53 Taha et al. described several compounds containing fluorine atoms linked to the benzene ring, demonstrating significant α-glucosidase inhibitory potential, and demonstrated that the fluorine group influences the potency of the compounds.54
The molecular hybridisation strategy involves the combination of two or more pharmacophores into a single molecule to enhance pharmacological activity.55,56 Following the analysis of marketed drugs and prior research in the synthesis of α-glucosidase inhibitors derived from cinnamic acid derivatives, hydrazone and fluorophenyls, we fused these compounds into a single molecule to develop a series of novel α-glucosidase inhibitors (Fig. 1).
2 Results and discussion
2.1. Chemistry
The final derivatives 6a–g and 7a–g were prepared by using conventional synthesis route as depicted in Scheme 1. The esterification of commercially available substituted phenyl acetic acid in methanol and catalytic amount of sulfuric acid at 60–70 °C to yielded corresponding methyl ester (2a & 2b). Compound 2a & 2b was alkylated with cyclopentyl bromide in presence of potassium tert-butoxide in DMF solvent offered substituted methyl 2-cyclopentyl-2-phenylacetate (3a & 3b). Methyl ester was converted to hydrazide with 98% hydrazine hydrate and ethanol at reflux temperature to give substituted 2-cyclopentyl-2-phenylacetohydrazide (4a & 4b) with appreciable yield. The key intermediates (4a & 4b) was treated with different substituted cinnamic acids to yielded the N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide derivatives. The structures of these derivatives were characterized by using IR, 1H, 13C NMR and mass spectroscopy analysis.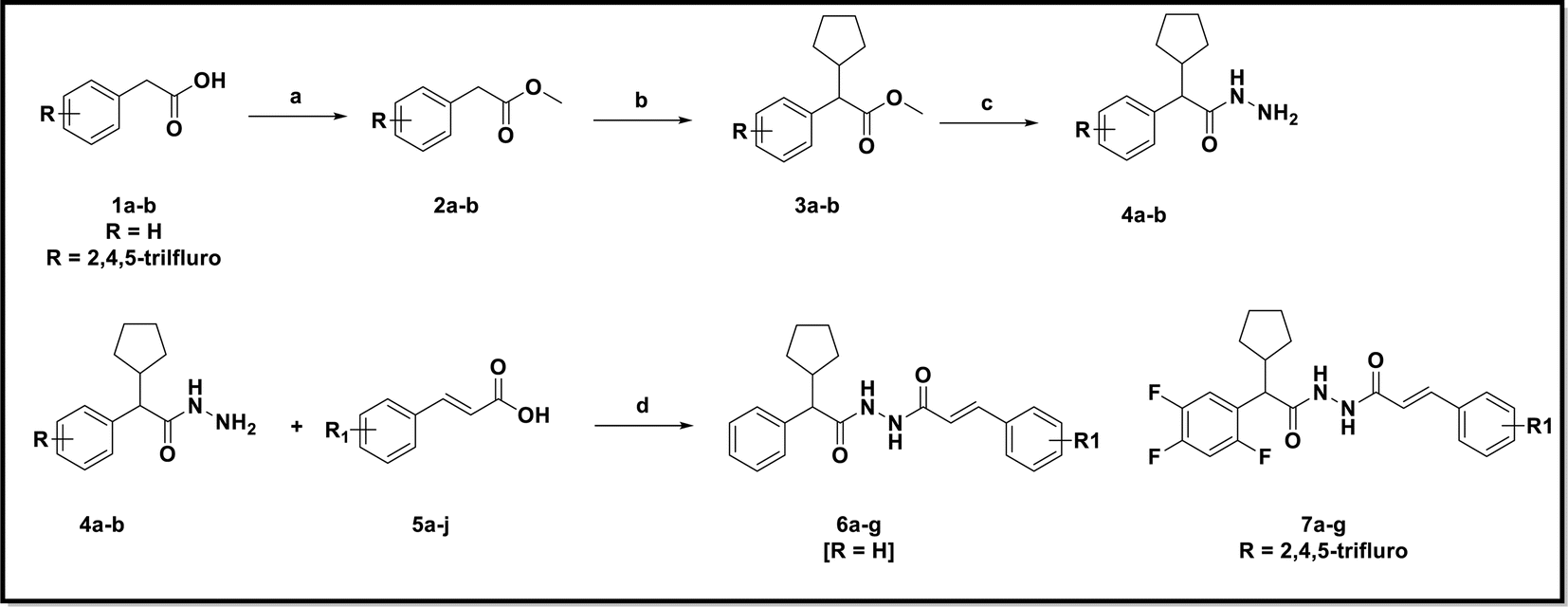 | ||
| Scheme 1 Reagents and conditions: (a) sulfuric acid, methanol, 60–70 °C, yield 95%; (b) potassium tert-butoxide, bromo cyclopentane, DMF, 20–30 °C, yield 86%; (c) 98% hydrazine hydride, ethanol, reflux 70 °C, yield 90%; (d) HATU, DIPERA, DMF, rt, 16 h. | ||
2.2. α-Glucosidase inhibitory assay
All the final compound 6a–g and 7a–g were evaluated for their in vitro α-glucosidase inhibitory activity. The results were displayed in Table 1. The results displayed that fluoro substitution of phenyl ring A (7a–g) exhibited potential activity compared to unsubstituted phenyl ring A (6a–g). The structure–activity relationship (SAR) was investigated based on the nature and locations of substitutions on the phenyl rings A and B of the N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide system. The α-glucosidase activities of all synthesized N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide-based candidates, specifically compounds 6a–g and 7a–g, were superior (IC50 = 14.48 to 232.72 nmol) in comparison to the standard acarbose (IC50 = 35.91 nmol). The unsubstituted phenyl group (6a and 7a) shows the moderate inhibition with an IC50 of 79.75 and 85.16 nmol, suggesting that the unsubstituted phenyl ring does not have much effective in key interactions within the enzyme's active site. Among all (7a–g), The most potent compound 7b (IC50 = 14.48 nmol) containing electron-withdrawing groups (2,4,5-trifluro) on ring A and electron donating (para-methyl) on ring B found to be increased inhibition and most promising α-glucosidase inhibitor among all the synthetic derivatives due to better interaction with the active pockets of enzymes. Furthermore, compound 7d (IC50 = 18.88 nmol) with m-Cl substitution on ring B and a 2,4,5-trifluro substituent on aromatic ring A showed better inhibitory activity as compared to the standard acarbose (IC50 = 35.91 nmol). Halogenated substituent 4-bromophenyl (7c) and 3-bromophenyl (7f) displayed moderate activity (IC50: 49.34 and 33.22 nmol), indicating that bromo substitutions do not enhance binding affinity for α-glucosidase inhibition. A considerable a-glucosidase inhibition was observed by 7g, with an IC50 of 54.78 nmol. However, simple aromatic ring substrate (7a) exhibited the moderate inhibition. Compound 7e, introducing a methoxy substituent at para position have no effect on inhibition.| S. no | R | R1 | Compound code | IC50 (nmol) |
|---|---|---|---|---|
| 1 | H | H | 6a | 79.75 |
| 2 | H | 2-Br | 6b | 104.44 |
| 3 | H | 3-Br | 6c | 46.80 |
| 4 | H | 4-Br | 6d | 64.80 |
| 5 | H | 4-CH3 | 6e | 232.72 |
| 6 | H | 4-F | 6f | No inhibition |
| 7 | H | 3-NO2 | 6g | 28.51 |
| 8 | 2,4,5-Trifluro | H | 7a | 85.16 |
| 9 | 2,4,5-Trifluro | 4-CH3 | 7b | 14.48 |
| 10 | 2,4,5-Trifluro | 4-Br | 7c | 49.34 |
| 11 | 2,4,5-Trifluro | 3-Cl | 7d | 18.88 |
| 12 | 2,4,5-Trifluro | 4-OCH3 | 7e | No inhibition |
| 13 | 2,4,5-Trifluro | 3-Br | 7f | 33.22 |
| 14 | 2,4,5-Trifluro | 3-NO2 | 7g | 54.78 |
| 15 | Acarbose | 35.91 |
In the series of 6a–g, Compound 6c and 6d had significant inhibition against α-Glucosidase with an IC50 of 46.80 and 64.80 nmol, respectively. This indicates that bromo substitution had considerable effect in both cases (6 and 7). Compound 6a, which is simple aromatic group exhibited an IC50 value of 79.75 nmol. Further, substituting the R1 position to a nitro group at meta position in 6g enhanced the inhibition (IC50 = 28.51 nmol). Compound 6b and 6e showed moderate inhibition. However, 6f and 7e had no inhibition effect. From the results, it could be concluded that the introduction of methyl and halogen in the para or ortho position of phenyl ring leads to an improved increase of the inhibitory activity. The SAR analysis of final derivatives was shown in Fig. 2.
 | ||
| Fig. 2 SAR of the synthesized target compounds 6a–g and 7a–g. | ||
2.3. Docking studies
Molecular docking is an effective technique for elucidating the interaction mechanism between a ligand and its receptor, as well as forecasting the potential binding site of the ligand.57,58 To further improve and confirm the inhibitory profile synthesized compounds, a thorough screening procedure was carried out using AutoDock Vina (RSC PDD:3WY1) (Fig. 3). Among the compounds that were docked, 7b, and7d had the highest docking energy, whereas 6c had the lowest docking energy. Hybrid 7b and 7d sticks an efficient conformation in the α-glucosidase binding pocket. Compound 7b had a docking energy of −10.1 kcal mol−1. Whereas, compounds 7d and 7a exhibited docking energies of −10.1 kcal mol−1 and −9.9 kcal mol−1, respectively. The 7b formed a conventional hydrogen bonding with HIS:280 and ASN:415. The 7d compound forms conventional hydrogen bonds with HIS:280 and ASN:415. The compound 6c exhibited the lowest docking energy, with −8.9 kcal mol−1. In addition, the presence of a conventional hydrogen bond with PRO:312 and ARG:315 were found. These findings were supported to the in vitro assay. | ||
| Fig. 3 3D & 2D Ligand interaction diagrams for docking compounds A: 7b; B: 7d (PDB ID: 3WY1). | ||
2.4. Evaluation of physico-chemical properties
Literature review indicated that drugs with lower molecular weights and more lipophilicity can be absorbed and excreted via para cellular and trans cellular routes with greater ease. Possible side effects include mild toxicity and increase renal excretion. A molecule with drug-like qualities (DLM) can be precisely characterized using the rule of five (RO5).59 The pharmacokinetic characteristics of N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide derivatives 6a–g and 7a–g, such as permeability of the blood–brain barrier (BBB) and gastrointestinal absorption (GIA) have also calculated (Table 2). As illustrated in Fig. 4 and 5, the intestinal or brain estimated permeation technique (BOILED Egg) was computed using the lipophilicity, indicated by the WLOGP, and the topological polar surface area (TPSA). All molecules are expected to be inside the white ellipse and grey region. This implies that certain substances may exhibit poorer BBB properties but superior GIA. All drugs whose ability to transport out of the Central Nervous System (CNS) is dependent on P-glycoprotein (PGP).60,61 Compounds 7b and 7d have the highest docking energy with −10.1 kcal mol−1 and −10.1 kcal mol−1.| Compound | Physico-chemical properties | Pharmacokinetic properties | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW (g mol−1) | HA | AHA | RBs | HBA | HBD | MR | TPSA | iLOGP | Violation | GI absorption | BBB permeant | PgP substrate | |
| 6a | 348.44 | 26 | 12 | 8 | 2 | 2 | 103.37 | 58.20 | 3.13 | 0 | High | Yes | No |
| 6b | 427.33 | 27 | 12 | 8 | 2 | 2 | 111.07 | 58.20 | 3.15 | 1 | High | Yes | No |
| 6c | 427.33 | 27 | 12 | 8 | 2 | 2 | 111.07 | 58.20 | 2.88 | 1 | High | Yes | No |
| 6d | 427.33 | 27 | 12 | 8 | 2 | 2 | 11.07 | 58.20 | 3.15 | 1 | High | Yes | No |
| 6e | 362.46 | 27 | 12 | 8 | 2 | 2 | 108.33 | 58.20 | 2.97 | 0 | High | Yes | No |
| 6f | 366.43 | 27 | 12 | 8 | 3 | 2 | 103.32 | 58.20 | 3.18 | 0 | High | Yes | No |
| 6g | 393.44 | 29 | 12 | 9 | 4 | 2 | 112.19 | 104.02 | 2.77 | 0 | High | No | No |
| 7a | 402.41 | 29 | 12 | 8 | 5 | 2 | 103.24 | 58.20 | 3.24 | 1 | High | Yes | No |
| 7b | 416.44 | 30 | 12 | 8 | 5 | 2 | 108.21 | 58.20 | 3.81 | 1 | High | No | No |
| 7c | 481.31 | 30 | 12 | 8 | 5 | 2 | 110.94 | 58.20 | 3.37 | 1 | High | No | No |
| 7d | 427.33 | 27 | 12 | 8 | 2 | 2 | 111.07 | 58.20 | 3.15 | 1 | High | Yes | No |
| 7e | 432.44 | 31 | 12 | 9 | 6 | 2 | 109.43 | 67.43 | 3.65 | 1 | High | No | No |
| 7f | 481.31 | 30 | 12 | 8 | 5 | 2 | 110.94 | 58.20 | 3.76 | 1 | High | No | No |
| 7g | 447.41 | 32 | 12 | 9 | 7 | 2 | 112.06 | 104.02 | 3.03 | 1 | High | No | No |
 | ||
| Fig. 4 Boiled egg of N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide derivatives. | ||
 | ||
| Fig. 5 3D representation and web representation of physico-chemical properties of molecule 7b & 7d. | ||
3 Conclusion
In conclusion, we have synthesized a new series of N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide derivatives in moderate to excellent yields and characterized by using FT-IT, 1H NMR, 13C NMR and HRMS spectral analysis. Thereafter, the target compounds 6a–g and 7a–g were assessed for in vitro activity in inhibiting α-glucosidase. Most of the synthesized compounds exhibited α-glucosidase inhibitory potency with IC50 values ranging from IC50 of 14.48 to 232.72 nmol as compared to standard acarbose (IC50 = 35.91 nmol). Compounds 7b and 7d showed the highest potency with IC50 values of 14.48 and 18.88 μM, respectively, as compared to standard acarbose, followed by 6g and 7f with IC50 values of 28.51 and 33.22 nmol, respectively. SAR of synthesized target hybrids were established. SAR indicated that compounds containing halogen and methyl substituents on the phenyl ring of cinnamohydrazide exhibited promising α-glucosidase as compared to those with electron-withdrawing groups. Molecular docking results agreed with in vitro biological assay data by forming key conventional hydrogen bonding interactions with HIS:280 and ASN:415 within the α-glucosidase binding pocket. Further, all the novel compounds are under druglike and conform the rules of ADME and toxicity profile. In summary, this study has identified a new family of N′-(2-cyclopentyl-2-phenylacetyl)cinnamohydrazide derivatives that could serve as lead compounds for the development of novel α-glucosidase inhibitors.4 Experimental section
4.1. General information
4.2. Chemistry
4.2.1.1 Methyl 2-(2,4,5-trifluorophenyl)acetate (2b). Liquid mass: yield 25 g, 94%, 1H NMR (300 MHz, CDCl3): δ (ppm): 7.16–7.08 (m, 1H), 6.97–6.89 (m, 1H), 3.72 (s, 3H), 3.61 (s, 2H). HRMS (ESI) m/z calcd. For [C9H7F3O2]+: 204.15 [M + H]+, found 203.
4.2.2.1 Methyl 2-cyclopentyl-2-phenylacetate (3a). Liquid mass: yield 25 g, 86%. 1H NMR (300 MHz, DMSO-d6): δ (ppm): 7.32–7.23 (m, 5H), 3.57 (s, 3H), 3.38–3.35 (d, J = 11.1 Hz 1H), 2.50–2.42 (m, 1H), 1.83–1.74 (m, 1H), 1.65–1.13 (m, 6H), 1.03–0.94 (m, 1H). HRMS (ESI) m/z calcd. For [C14H18O2]+: 218.3 [M + H]+, found 219.7.
4.2.2.2 Methyl 2-cyclopentyl-2-(2,4,5-trifluorophenyl)acetate (3b). Viscous liquid: yield 30.5 g, 84%. 1H NMR (300 MHz, CDCl3): δ (ppm): 7.39–7.31 (s, 1H), 6.97–6.58 (s, 1H), 3.72 (s, 1H), 3.68 (s, 3H), 2.45–2.42 (s, 1H), 1.90–1.86 (m, 1H), 1.66–1.45 (m, 6H), 1.30–1.23 (m, 1H). HRMS (ESI) m/z calcd. For [C14H15F3O2]+: 272.27 [M + H]+, found 272.27.
4.2.3.1 2-Cyclopentyl-2-phenylacetohydrazide (4a). Viscous liquid: yield 22.3 g, 89%, 1H NMR (300 MHz, DMSO-d6): δ (ppm): 9.18 (s, 1H), 7.34–7.16 (m, 5H), 4.22 (b, 2H), 3.06–3.02 (d, J = 11.1 Hz, 1H), 2.57–2.54 (m, 1H), 1.72–1.27 (m, 7H), 0.93–0.86 (m, 1H). HRMS (ESI) m/z calcd. For [C13H18N2O]+: 218.3 [M + H]+, found 219.8.
4.2.3.2 2-Cyclopentyl-2-(2,4,5-trifluorophenyl)acetohydrazide (4b). Viscous liquid: yield 22 g, 88%, 1H NMR (300 MHz, CDCl3): δ (ppm): 5.78 (s, 1H), 5.29 (s, 1H), 3.69 (s, 3H), 2.21 (t, J = 2.4 Hz, 6H). HRMS (ESI) m/z calcd. For [C13H15F3N2O]+: 272.27 [M + H]+, found 273.1.
4.2.4.1 N′-(2-Cyclopentyl-2-phenylacetyl)cinnamohydrazide (6a). Off-white solid, 113 mg, 71% yield, IR (KBr, cm−1): 3200, 3058, 3025, 2945, 2862, 1640, 1590, 1494, 1462, 1329, 1184, 1157, 1072, 1030, 967, 855, 756, 735, 695, 656, 562, 531. 1H NMR (300 MHz, DMSO-d6): δ (ppm): 10.3–10.14 (b, 1H), 7.58–7.12 (m, 10H), 6.66–6.61 (d, J = 15.9 Hz, 1H), 2.58–2.2 (m, 1H), 1.98–1.28 (m, 7H), 0.99–0.92 (m, 1H). 13C NMR spectrum, δc, ppm, DMSO-d6: 171.26, 163.45, 140.06, 140.01, 134.57, 129.76, 128.98, 128.11, 127.64, 126.68, 119.40, 55.05, 42.41, 30.76, 30.23, 24.65, 24.46. HRMS (ESI) m/z calcd. For [C22H24N2O2]+: 348.45 [M + H]+, found 349.5.
4.2.4.2 3-(2-Bromophenyl)-N′-(2-cyclopentyl-2-phenylacetyl)acrylohydrazide (6b). Off-white solid, 137 mg, 70% yield, IR (KBr, cm−1): 3201, 2947, 2859, 1639, 1589, 1472, 1324, 1268, 1200, 1183, 1023, 964, 853, 796, 755, 743, 723, 700, 658, 582, 543. 1H NMR (300 MHz, DMSO-d6): δ (ppm): 10.29–10.25 (d, J = 9.9 Hz, 2H), 7.77–7.67 (m, 3H), 7.47–7.19 (m, 8H), 6.66–6.61 (d, J = 15.6 Hz, 1H), 2.56–2.55 (m, 1H), 1.81–1.31 (m, 8H), 0.99–0.92 (m, 1H). 13C NMR spectrum, δc, ppm, DMSO-d6: 171.27, 162.89, 140, 137.89, 134.09, 133.23, 131.42, 128.4, 128.12, 127.73, 126.7, 124.26, 122.57, 55.06, 42.38, 30.77, 30.23, 24.64, 24.45. HRMS (ESI) m/z calcd. For [C22H23BrN2O2]+: 427.34 [M + H]+, found 429.6.
4.2.4.3 3-(3-Bromophenyl)-N′-(2-cyclopentyl-2-phenylacetyl)acrylohydrazide (6c). Off-white solid, 156 mg, 80% yield, IR (KBr, cm−1): 3180, 3030, 2948, 2858, 1661, 1640, 1588, 1464, 1368, 1270, 1200, 1180, 1071, 1032, 966, 854, 784, 719, 698, 663, 552. 1H NMR (300 MHz, DMSO-d6): δ (ppm): 10.29 (s, 1H), 10.16 (s, 1H), 7.78 (s, 1H), 7.6–7.55 (dd, J = 8.1 Hz, 2H), 7.49–7.09 (m, 7H), 6.7–6.65 (d, J = 15.9 Hz, 1H), 2.56–2.5 (m, 1H), 1.76–1.3 (m, 8H), 0.96–0.94 (m, 1H). 13C NMR spectrum, δc, ppm, DMSO-d6: 171.19, 168.93, 163.02, 156.08, 140.55, 140.03, 138.38, 137.14, 132.26, 131.06, 130.25, 128.4, 128.11, 128.03, 127.96, 126.68, 126.54, 126.42, 122.26, 121.08, 55.15, 55.02, 51.94, 42.50, 42.41, 30.75, 30.28, 30.23, 30.15, 24.87, 24.64, 24.45. HRMS (ESI) m/z calcd. For [C22H23BrN2O2]+: 427.34 [M + H]+, found 425.1.
4.2.4.4 3-(4-Bromophenyl)-N′-(2-cyclopentyl-2-phenylacetyl)acrylohydrazide (6d). Off-white solid, 147 mg, 75% yield, IR (KBr, cm−1): 3192, 3025, 2946, 2864, 1637, 1591, 1488, 1461, 1322, 1202, 1181, 1073, 1011, 972, 859, 815, 743, 697, 645, 558, 527. 1H NMR (300 MHz, DMSO-d6): δ (ppm): 10.26–10.16 (d, J = 29.7 Hz, 2H), 7.63–7.05 (m, 10H), 6.67–6.62 (d, J = 15.6 Hz, 1H), 2.58–2.49 (m, 1H), 1.81–1.30 (m, 8H), 1.01–0.92 (m, 1H). 13C NMR spectrum, δc, ppm, DMSO-d6: 171.24, 163.23, 140.04, 138.75, 133.86, 131.94, 129.6, 128.11, 126.68, 122.95, 120.25, 55.03, 42.4, 30.75, 30.23, 24.65, 24.45. HRMS (ESI) m/z calcd. For [C22H23BrN2O2]+: 427.34 [M + H]+, found 429.6.
4.2.4.5 N′-(2-Cyclopentyl-2-phenylacetyl)-3-(p-tolyl)acrylohydrazide (6e). Off-white solid, 118 mg, 71% yield, IR (KBr, cm−1): 3193, 3024, 2945, 2864, 2819, 1693, 1635, 1590, 1512, 1456, 1386, 1203, 1180, 1144, 1110, 1034, 973, 861, 810, 771, 725, 697, 653, 561, 530.1H NMR (300 MHz, DMSO-d6): δ (ppm): 10.24 (b, 1H), 10.09 (b, 1H), 9.01 (m, 1H), 7.57–7.21 (m, 9H), 6.6–6.54 (d, J = 15.9 Hz, 1H), 3.32–3.2 (m, 1H), 2.33 (s, 3H), 1.82–1.31 (m, 8H), 0.98–0.96 (m, 1H). 13C NMR spectrum, δc, ppm, DMSO-d6: 171.28, 163.64, 140.07, 139.97, 139.58, 131.83, 129.57, 128.1, 127.62, 126.66, 118.32, 55.05, 45.14, 44.78, 42.41, 30.76, 30.23, 24.64, 24.45, 20.95. HRMS (ESI) m/z calcd. For [C23H26N2O2]+: 362.47 [M + H]+, found 321.
4.2.4.6 3-(4-Fluoro)-N′-(2-cyclopentyl-2-phenylacetyl)acrylohydrazide (6f). Off-white solid, 139 mg, 83% yield, IR (KBr, cm−1): 3747, 3196, 3026, 2951, 2864, 1639, 1589, 1507, 1457, 1327, 1229, 1199, 1179, 1157, 1096, 1032, 967, 871, 827, 779, 726, 696, 638, 530.1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.26 (b, 1H), 10.13 (b, 1H), 7.66–7.65 (m, 2H), 7.39–7.41 (dd, 2H), 7.53–7.50 (d, J = 15 Hz, 1H), 7.16–7.32 (m, 5H), 6.61–6.55 (d, J = 15.9 Hz, 1H), 3.31–3.33 (d, J = 10 Hz, 1H), 2.5–2.51 (m, 1H), 1.33–1.82 (m, 7H), 0.95–0.99 (m, 1H). 13C NMR spectrum, δc, ppm, DMSO-d6: 171.73, 162.31–164.3, 163.89, 140.54, 139.29, 131.71, 130.36, 128.6, 128.57, 127.14, 116.51, 119.78, 55.53, 42.89, 31.24, 30.71, 25.12, 24.93. HRMS (ESI) m/z calcd. For [C23H23FN2O2]+: 366.44 [M + H]+, found 367.2.
4.2.4.7 3-(3-Nitrophenyl)-N′-(2-cyclopentyl-2-phenylacetyl)acrylohydrazide (6g). Pale yellow solid, 140 mg, 78% yield, IR (KBr, cm−1): 3747, 3167, 3028, 2952, 2866, 1642, 1590, 1530, 1468, 1346, 1267, 1206, 1184, 1074, 1031, 973, 902, 861, 834, 805, 751, 732, 712, 654, 562, 545, 517. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.33 (S, 1H), 10.24 (s, 1H), 8.4 (s, 1H), 8.23–8.21 (d, J = 8 Hz, 1H), 8.04–8.02 (d, J = 8.5 Hz, 1H), 7.73–7.72 (t, J = 8 Hz, 1H), 7.65–7.62 (d, J = 16 Hz, 1H), 7.38–7.21 (m, 5H), 6.84–6.81 (d, J = 15.5 Hz, 1H), 2.57–2.53 (m, 1H), 1.80–1.3 (m, 8H), 0.99–0.94 (m, 1H). 13C NMR spectrum, δc, ppm, DMSO-d6: 171.16, 162.77, 148.3, 140.02, 137.73, 136.45, 133.82, 130.52, 128.12, 126.7, 124.01, 122.32, 121.91, 55.02, 42.43, 30.76, 30.23, 24.64, 24.45. HRMS (ESI) m/z calcd. For [C22H23N3O4]+: 393.44 [M + H]+, found 394.4.
4.2.4.8 N′-(2-cyclopentyl-2-(2,4,5-trifluorophenyl)acetyl)cinnamohydrazide (7a). Off-white solid, 96 mg, 65% yield, IR (KBr, cm−1): 3747, 3184, 2953, 2869, 1682, 1642, 1610, 1513, 1423, 1335, 1257, 1207, 1185, 1148, 976, 878, 857, 761, 708, 615, 518. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.36 (s, 1H), 10.13 (s, 1H), 7.67–7.62 (m, 1H), 7.59–7.49 (m, 4H), 7.44–7.40 (m, 3H), 6.65–6.62 (d, J = 15.5 Hz, 1H), 3.68–3.66 (d, J = 11 Hz, 1H), 2.44–2.42 (d, J = 10.5 Hz, 1H), 1.83–1.79 (m, 1H), 1.67–1.65 (m 1H), 1.58–1.49 (m, 6H), 0.99–0.91 (m, 1H). 19F NMR (500 MHz, DMSO-d6): δ (ppm): −118.79, −136.45, −142.94. 13C NMR spectrum, δc, ppm, DMSO-d6: 170.04, 163.68, 140.26, 134.51, 129.82, 128.99, 127.68, 119.25, 117.15, 105.7, 45.47, 43, 30.52, 29.99, 24.48, 24.27. HRMS (ESI) m/z calcd. For [C22H21F3N2O2]+: 402.2 [M + H]+, found 403.1.
4.2.4.9 N′-(2-Cyclopentyl-2-(2,4,5-trifluorophenyl)acetyl)-3-(p-tolyl)acrylohydrazide (7b). Off-white solid, 104 mg, 68% yield, IR (KBr, cm−1): 3186, 3029, 2948, 2867, 1666, 1632, 1587, 1513, 1461, 1424, 1330, 1255, 1197, 1184, 1156, 973, 884, 862, 807, 758, 709, 646, 524, 514. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.34 (s, 1H), 10.07 (s, 1H), 7.67–7.61 (m, 1H), 7.55–7.45 (m, 4H), 7.24–7.22 (d, J = 7.5 Hz, 1H), 6.58–6.55 (dd, J = 15.5 Hz, 1H), 3.67–3.65 (d, J = 2 Hz, 11H), 2.44–2.42 (m, 1H), 2.32 (s, 3H), 1.81–1.78 (m, 1H), 1.67–1.65 (m, 1H), 1.58–1.49 (m, 6H), 0.99–0.91 (m, 1H). 19F NMR (500 MHz, DMSO-d6): δ (ppm): 118.79, 136.46, 142.95.13C NMR spectrum, δc, ppm, DMSO-d6: 170.04, 163.5, 156.04, 140.2, 139.56, 131.78, 129.56, 127.65, 123.72, 118.16, 114.75, 105.67, 45.45, 43.02, 30.52, 24.47, 22.2. HRMS (ESI) m/z calcd. For [C23H23 F3N2O2]+: 416.44 [M + H]+, found 417.2.
4.2.4.10 3-(4-Bromophenyl)-N′-(2-cyclopentyl-2-(2,4,5-trifluorophenyl)acetyl)acrylohydrazide (7c). Off-white solid, 131 mg, 74% yield, IR (KBr, cm−1): 3747, 3176, 3030, 2945, 2866, 1633, 1587, 1514, 1488, 1463, 1424, 1330, 1256, 1200, 1157, 1074, 1012, 971, 884, 863, 814, 771, 709, 695, 646, 518. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.38 (s, 1H), 10.16 (s, 1H), 7.63–7.61 (m, 3H), 7.54–7.53 (m, 4H), 6.66–6.63 (d, J = 16 Hz, 1H), 3.68–3.65 (d, J = 10.5 Hz, 1H), 2.49–2.1 (m, 1H), 1.9–1.78 (m, 1H), 1.68–1.64 (m, 1H), 1.58–1.45 (m, 6H), 0.99–0.91 (m, 1H). 19F NMR (500 MHz, DMSO-d6): δ (ppm): 118.78, 136.42, 142.93. 13C NMR spectrum, δc, ppm, DMSO-d6: 170.02, 163.46, 139.01, 133.81, 131.94, 129.64, 123.02, 120.09, 117.11, 105.46, 45.47, 43, 30.52, 29.98, 24.48, 24.27. HRMS (ESI) m/z calcd. For [C22H20Br F3N2O2]+: 481.31 [M + H]+, found 478.9.
4.2.4.11 3-(3-Chlorophenyl)-N′-(2-cyclopentyl-2-(2,4,5-trifluorophenyl)acetyl)acrylohydrazide (7d). Off-white solid, 128 mg, 80% yield, IR (KBr, cm−1): 3747, 3186, 2951, 2869, 1685, 1590, 1514, 1475, 1423, 1331, 1256, 1203, 1150, 1078, 971, 879, 851, 784, 703, 683, 640, 523. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.39 (s, 1H), 10.15 (s, 1H), 7.67–7.61 (m, 2H), 7.56–7.45 (m, 5H), 6.7–6.67 (d, J = 16 Hz, 1H), 3.68–3.66 (d, J = 11 Hz, 1H), 2.49–2.41 (m, 1H), 1.82–1.78 (m, 1H), 1.68–1.64 (m, 1H), 1.58–1.49 (s, 6H), 0.99–0.96 (m, 1H). 19F NMR (500 MHz, DMSO-d6): δ (ppm): 118.79, 136.42, 142.92. 13C NMR spectrum, δc, ppm, DMSO-d6: 169.99, 163.31, 138.72, 136.83, 133.7, 130.81, 129.43, 127.44, 126.11, 120.95, 105.3, 45.46, 42.99, 30.52, 29.99, 24.48, 24.27. HRMS (ESI) m/z calcd. For [C22H20Cl F3N3O2]+: 436.86 [M + H]+, found 437.1.
4.2.4.12 N′-(2-Cyclopentyl-2-(2,4,5-trifluorophenyl)acetyl)-3-(4-methoxyphenyl)acrylohydrazide (7e). Off-white solid, 141 mg, 89% yield, IR (KBr, cm−1): 3747, 3201, 3025, 2954, 2868, 2838, 1667, 1630, 1583, 1512, 1458, 1423, 1331, 1306, 1287, 1259, 1196, 1174, 1157, 1110, 1030, 973, 884, 864, 830, 809, 783, 757, 698, 645, 550, 525. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.31 (s, 1H), 10.01 (s, 1H), 7.67–7.61 (m, 1H), 7.54–7.43 (m, 4H), 6.99–6.97 (d, J = 8.5 Hz, 1H), 6.49–6.46 (d, J = 16 Hz, 1H), 3.8 (s, 3H), 3.67–3.63 (d, J = 11 Hz, 1H), 2.49–2.44 (m, 1H), 1.83–1.77 (m, 1H), 1.69–1.64 (m, 1H), 1.59–1.4 (m, 6H), 0.99–0.95 (m, 1H). 19F NMR (500 MHz, DMSO-d6): δ (ppm): 118.8, 136.48, 142.96. 13C NMR spectrum, δc, ppm, DMSO-d6: 170.06, 164.05, 160.6, 139.97, 129.32, 127.09, 116.66, 114.43, 55.28, 45.46, 43.01, 30.52, 29.98, 24.48, 24.27. HRMS (ESI) m/z calcd. For [C23H23F3N2O3]+: 432.44 [M + H]−, found 433.1.
4.2.4.13 3-(3-Bromophenyl)-N′-(2-cyclopentyl-2-(2,4,5-trifluorophenyl)acetyl)acrylohydrazide (7f). Off-white solid, 152 mg, 86% yield, IR (KBr, cm−1): 3747, 3178, 3034, 2962, 2869, 1646, 1598, 1513, 1472, 1424, 1335, 1258, 1204, 1157, 1072, 970, 890, 860, 844, 809, 781, 741, 719, 697, 663, 642, 603, 522. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.39 (s, 1H), 10.14 (s, 1H), 7.79–7.67 (t, J = 1.5 Hz, 1H), 7.65–7.57 (m, 3H), 7.53–7.4 (m, 2H), 7.38–7.32 (t, J = 8 Hz, 1H), 6.69–6.66 (m, J = 16 Hz, 1H), 3.68–3.66 (d, J = 11 Hz, 1H), 2.49–2.44 (m, 1H), 1.81–1.78 (m, 1H), 1.67–1.66 (m, 1H), 1.58–1.44 (m, 6H), 0.99–0.96 (m, 1H). 19F NMR (500 MHz, DMSO-d6): δ (ppm): 118.79, 136.42, 142.92. 13C NMR spectrum, δc, ppm, DMSO-d6: 169.98, 163.28, 138.64, 137.09, 132.33, 131.07, 130.3, 126.47, 122.27, 120.93, 105.54, 45.46, 42.99, 30.52, 29.99, 24.48, 24.27. HRMS (ESI) m/z calcd. For [C22H20BrF3N2O2]+: 481.31 [M + H]+, found 481.2.
4.2.4.14 3-(3-Nitrophenyl)-N′-(2-cyclopentyl-2-(2,4,5-trifluorophenyl)acetyl)acrylohydrazide (7g). Brown colored solid, 145 mg, 89% yield, IR (KBr, cm−1): 3198, 3073, 2948, 2865, 1641, 1586, 1530, 1462, 1426, 1354, 1253, 1205, 1157, 1099, 961, 886, 856, 808, 775, 751, 718, 700, 660, 634, 603, 523. 1H NMR (500 MHz, DMSO-d6): δ (ppm): 10.43 (s, 1H), 10.22 (s, 1H), 8.41 (d, J = 2 Hz, 1H), 8.23–8.22 (dd, J = 1.5, 1 Hz, 1H), 8.21 (d, J = 0.5 Hz, 1H), 7.73–7.7 (t, J = 8 Hz, 1H), 7.66–7.63 (m, 2H), 7.53–7.5 (m, 1H), 6.84–6.81 (d, J = 16 Hz, 1H), 3.69–3.67 (d, J = 11 Hz, 1H), 2.49–2.43 (m, 1H), 1.86–1.8 (m, 1H), 1.67–1.65 (m, 1H), 1.58–1.4 (m, 6H), 0.99–0.96 (m, 1H). 19F NMR (500 MHz, DMSO-d6): δ (ppm): 118.79, 136.41, 142.92. 13C NMR spectrum, δc, ppm, DMSO-d6: 169.93, 163.01, 148.29, 146.93, 137.96, 136.4, 133.83, 130.5, 123.79, 122.18, 121.95, 117.25, 105.69, 45.73, 43.11, 30.51, 24.88. HRMS (ESI) m/z calcd. For [C22H20F3N3O4]+: 447.41 [M + H]+, found 445.9.
4.3. α-Glucosidase inhibitory assay
The test compounds were evaluated for their α-glucosidase inhibitory activity following the methodology outlined by Pistia et al.62 and Bhatia et al.63 The test compounds and Acarbose were both dissolved in phosphate buffer at a pH of 6.8. The compounds at varied concentrations ranging from 50 to 200 μg mL−1 were combined with 50 μL of α-glucosidase (maltase) enzyme solution (1 U mL−1) from Yeast (Sisco Research Laboratories Pvt. Ltd). The mixer was placed at a temperature of 37 °C for duration of 15 minutes, after the addition of 0.1 M phosphate buffer (pH 6.8). Afterward, 25 μl of substrate buffer was introduced into the system to initiate the reaction, and the incubation was prolonged at a temperature of 37 °C for a length of 15 minutes. Finally, the reaction was stopped by adding 50 μl of a 0.2 M sodium carbonate solution, thereby terminating the processes. The measurement was determined at a wavelength of 450 nm. Acarbose was used as a standard chemical and given at various doses ranging from 50 to 200 μg mL−1. The measurement of enzyme activity was carried out as follows.The formula for calculating the percentage is (ODblank − ODsample) divided by ODblank, multiplied by 100.
A unit of enzyme may be accurately defined as the amount of α-glucosidase enzyme required to generate one micromole of p-nitrophenol (product) from p-nitrophenyl-α-D-glucopyranoside (substrate) for a time of one minute. The IC50, representing the concentration required to inhibit 50% of the enzyme activity, was determined by fitting a regression equation to a graph that plotted concentration (varying from 50–200 μg mL−1) on the X-axis and percentage of inhibition on the Y-axis for various fractions and extracts.
4.4. Molecular docking
To ascertain the most effective way of binding a ligand to a target protein, molecular docking is utilized. When the ligand forms a stable combination with other potential ligands, this is very beneficial. A highly effective technique for examining the molecular interactions between ligands and proteins is docking.4.5. Swiss ADME
Data availability
All data generated or analyzed during this study are included in this published article and its ESI files.†Ethics approval
This article does not contain any studies with animals performed by any of the authors.Consent for publication
We authorize to publish the article without any conflict.Author contributions
Ram Reddy Mudireddy: investigation, formal analysis, methodology, and writing – original draft. Rambabu Gundla: project administration and supervision. Baji Baba Shaik: investigation, and software. Anoop Bodapati: validation, and resources. Panasa Mahesh: conceptualisation, and writing – original draft. Shiva Sravan Naidu: data curation, and funding acquisition. Damodar Tirumalasetti: formal analysis, and writing – review & editing. Naresh Kumar Katari: visualization and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest regarding the publication of this manuscript.Acknowledgements
Dr Rambabu Gundla acknowledges DBT-BIRAC (BT/AIR01566/PACE-27/22) for financial assistance and GITAM for the facility.References
- E. Dale Abel, A. L. Gloyn, C. Evans-Molina, J. J. Joseph, S. Misra, U. B. Pajvani, J. Simcox, K. Susztak and D. J. Drucker, Diabetes mellitus—Progress and opportunities in the evolving epidemic, Cell, 2024, 187, 3789–3820, DOI:10.1016/j.cell.2024.06.029.
- N. D. Wong and N. Sattar, Cardiovascular risk in diabetes mellitus: epidemiology, assessment and prevention, Nat. Rev. Cardiol., 2023, 20, 685–695, DOI:10.1038/s41569-023-00877-z.
- S. Babu, An update on diagnosis and therapeutics for type-2 diabetes mellitus, Bioinformation, 2023, 19, 295–298, DOI:10.6026/97320630019295.
- C. Moran, R. Beare, T. G. Phan, D. G. Bruce, M. L. Callisaya and V. Srikanth, Type 2 diabetes mellitus and biomarkers of neurodegeneration, Neurology, 2015, 85, 1123–1130, DOI:10.1212/wnl.0000000000001982.
- H. Mills, R. Acquah, N. Tang, L. Cheung, S. Klenk, R. Glassen, M. Pirson, A. Albert, D. T. Hoang and T. N. Van, Type 2 Diabetes Mellitus (T2DM) and Carbohydrate Metabolism in Relation to T2DM from Endocrinology, Neurophysiology, Molecular Biology, and Biochemistry Perspectives, J. Evidence-Based Complementary Altern. Med., 2022, 2022, e1708769, DOI:10.1155/2022/1708769.
- S. N. Bogatyrev, Physical activity and type 2 diabetes mellitus risk: population studies review, Diabetes Mellitus, 2017, 19, 486, DOI:10.14341/dm8030.
- K. M. Alonge, D. A. D'Alessio and M. W. Schwartz, Brain control of blood glucose levels: implications for the pathogenesis of type 2 diabetes, Diabetologia, 2020, 64, 5–14, DOI:10.1007/s00125-020-05293-3.
- C. D. Maida, M. Daidone, G. Pacinella, R. L. Norrito, A. Pinto and A. Tuttolomondo, Diabetes and Ischemic Stroke: An Old and New Relationship an Overview of the Close Interaction between These Diseases, Int. J. Mol. Sci., 2022, 23, 2397, DOI:10.3390/ijms23042397.
- B. Menon, R. Syed, P. Kumar Yadav and M. Menon, Diabetes and Stroke—A Focused Review, J. Diabetol., 2024, 15, 247–257, DOI:10.4103/jod.jod_46_24.
- C. R. Sagandira, A. Z. Khasipo, M. B. Sagandira and P. Watts, An overview of the synthetic routes to essential oral anti-diabetes drugs, Tetrahedron, 2021, 96, 132378, DOI:10.1016/j.tet.2021.132378.
- F. Haddad, G. Dokmak, M. Bader and R. Karaman, A Comprehensive Review on Weight Loss Associated with Anti-Diabetic Medications, Life, 2023, 13, 1012, DOI:10.3390/life13041012.
- A. D. Dahlén, G. Dashi, I. Maslov, M. M. Attwood, J. Jonsson, V. Trukhan and H. B. Schiöth, Trends in Antidiabetic Drug Discovery: FDA Approved Drugs, New Drugs in Clinical Trials and Global Sales, Front. Pharmacol, 2022, 12, 807548, DOI:10.3389/fphar.2021.807548.
- E. Lazzaroni, M. Ben Nasr, C. Loretelli, I. Pastore, L. Plebani, M. E. Lunati, L. Vallone, A. M. Bolla, A. Rossi, L. Montefusco, E. Ippolito, C. Berra, F. D'Addio, G. V. Zuccotti and P. Fiorina, Anti-diabetic drugs and weight loss in patients with type 2 diabetes, Pharmacol. Res., 2021, 171, 105782, DOI:10.1016/j.phrs.2021.105782.
- Y. Zhang, Y. Zheng, Y. Yuan, S.-C. Chen and B. Xie, Effects of Anti-Diabetic Drugs on Fracture Risk: A Systematic Review and Network Meta-Analysis, Front. Endocrinol., 2021, 12, 735824, DOI:10.3389/fendo.2021.735824.
- A. Mushtaq, U. Azam, S. Mehreen and M. M. Naseer, Synthetic α-glucosidase inhibitors as promising anti-diabetic agents: Recent developments and future challenges, Eur. J. Med. Chem., 2023, 249, 115119, DOI:10.1016/j.ejmech.2023.115119.
- A. M. Dirir, M. Daou, A. F. Yousef and L. F. Yousef, A review of alpha-glucosidase inhibitors from plants as potential candidates for the treatment of type-2 diabetes, Phytochem. Rev., 2021, 21, 1049–1079, DOI:10.1007/s11101-021-09773-1.
- A. Singh, K. Singh, A. Sharma, K. Kaur, K. Kaur, R. Chadha and M. Singh, Recent developments in synthetic α-glucosidase inhibitors: A comprehensive review with structural and molecular insight, J. Mol. Struct., 2023, 1281, 135115, DOI:10.1016/j.molstruc.2023.135115.
- A. P. S. Kong, S. Lim, S. H. Yoo, L. Ji, L. Chen, Y. Bao, E. Yeoh, S. P. Chan, C. Wang, V. Mohan, N. Cohen, M. McGill and S. M. Twigg, Asia-Pacific consensus recommendations for application of continuous glucose monitoring in diabetes management, Diabetes Res. Clin. Pract., 2023, 201, 110718, DOI:10.1016/j.diabres.2023.110718.
- C. A. Garcia and J. G. Gardner, Bacterial α-diglucoside metabolism: perspectives and potential for biotechnology and biomedicine, Appl. Microbiol. Biotechnol., 2021, 105, 4033–4052, DOI:10.1007/s00253-021-11322-x.
- L. Han, T. Xie, Q. Wu, Z. Hu, Y. Luo and F. Luo, Alpha-Glucosidase Inhibitory Peptides: Sources, Preparations, Identifications, and Action Mechanisms, Nutrients, 2023, 15, 4267, DOI:10.3390/nu15194267.
- I. Ćorković, D. Gašo-Sokač, A. Pichler, J. Šimunović and M. Kopjar, Dietary Polyphenols as Natural Inhibitors of α-Amylase and α-Glucosidase, Life, 2022, 12, 1692, DOI:10.3390/life12111692.
- H. E. Lebovitz, Oral Antidiabetic Agents, Drugs, 1992, 44, 21–28, DOI:10.2165/00003495-199200443-00004.
- E. A. H. Mohamed, M. J. A. Siddiqui, L. F. Ang, A. Sadikun, S. H. Chan, S. C. Tan, M. Z. Asmawi and M. F. Yam, Potent α-glucosidase and α-amylase inhibitory activities of standardized 50% ethanolic extracts and sinensetin from Orthosiphon stamineus Benth as anti-diabetic mechanism, BMC Compl. Alternative Med., 2012, 12, 176, DOI:10.1186/1472-6882-12-176.
- A. S. Dabhi, N. R. Bhatt and M. J. Shah, Voglibose: An Alpha Glucosidase Inhibitor, J. Clin. Diagn. Res., 2013, 12, 3023–3027, DOI:10.7860/jcdr/2013/6373.3838.
- U. Ghani, Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack, Eur. J. Med. Chem., 2015, 103, 133–162, DOI:10.1016/j.ejmech.2015.08.043.
- U. Ghani, Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack, Eur. J. Med. Chem., 2015, 103, 133–162, DOI:10.1016/j.ejmech.2015.08.043.
- K. Aoki, T. Muraoka, Y. Ito, Y. Togashi and Y. Terauchi, Comparison of Adverse Gastrointestinal Effects of Acarbose and Miglitol in Healthy Men: A Crossover Study, Intern. Med., 2010, 49, 1085–1087, DOI:10.2169/internalmedicine.49.3218.
- S. Kurebayashi, H. Watada, Y. Tanaka, M. Kawasumi, R. Kawamori and T. Hirose, Efficacy and Adverse Effects of Nateglinide in Early Type 2 Diabetes. Comparison with Voglibose in a Cross-over Study, Endocr. J., 2006, 53, 213–217, DOI:10.1507/endocrj.53.213.
- J.-P. J. Sels, M. S. Huijberts and B. H. Wolffenbuttel, Miglitol, a new α-glucosidase inhibitor, Expert Opin. Pharmacother., 1999, 1, 149–156, DOI:10.1517/14656566.1.1.149.
- M. Li, H. Li, X. Min, J. Sun, B. Liang, L. Xu, J. Li, S.-H. Wang and X. Xu, Identification of 1,3,4-Thiadiazolyl-Containing Thiazolidine-2,4-dione Derivatives as Novel PTP1B Inhibitors with Antidiabetic Activity, J. Med. Chem., 2024, 67, 8406–8419, DOI:10.1021/acs.jmedchem.4c00676.
- B. Liang, D. Xiao, S.-H. Wang and X. Xu, Novel thiosemicarbazide-based β-carboline derivatives as α-glucosidase inhibitors: Synthesis and biological evaluation, Eur. J. Med. Chem., 2024, 275, 116595, DOI:10.1016/j.ejmech.2024.116595.
- C. Hu, B. Liang, J. Sun, J. Li, Z. Xiong, S.-H. Wang and X. Xuetao, Synthesis and biological evaluation of indole derivatives containing thiazolidine-2,4-dione as α-glucosidase inhibitors with antidiabetic activity, Eur. J. Med. Chem., 2024, 264, 115957, DOI:10.1016/j.ejmech.2023.115957.
- X. Min, S. Guo, Y. Lu and X. Xu, Investigation on the inhibition mechanism and binding behavior of cryptolepine to α-glucosidase and its hypoglycemic activity by multi-spectroscopic method, J. Lumin., 2024, 269, 120437, DOI:10.1016/j.jlumin.2024.120437.
- X.-Z. Wu, W.-J. Zhu, L. Lu, C.-M. Hu, Y.-Y. Zheng, X. Zhang, J. Lin, J.-Y. Wu, Z. Xiong, K. Zhang and X.-T. Xu, Synthesis and anti-α-glucosidase activity evaluation of betulinic acid derivatives, Arabian J. Chem., 2023, 16, 104659, DOI:10.1016/j.arabjc.2023.104659.
- I. Fotopoulos, Dimitra Hadjipavlou-Litina, Approaches for the discovery of cinnamic acid derivatives with anticancer potential, Expert Opin. Drug Discovery, 2024, 19, 1281–1291, DOI:10.1080/17460441.2024.2387122.
- H. Deng, Q. Xu, H.-Y. Guo, X. Huang, F. Chen, L. Jin, Z.-S. Quan and Q.-K. Shen, Application of cinnamic acid in the structural modification of natural products: A review, Phytochemistry, 2023, 206, 113532, DOI:10.1016/j.phytochem.2022.113532.
- L. Feng, J. Cheng, W. Su, H. Li, T. Xiao, D. Chen and Z. Zhang, Cinnamic acid hybrids as anticancer agents: A mini-review, Arch. Pharm. Chem. Life Sci., 2022, 355, e2200052, DOI:10.1002/ardp.202200052.
- G. Mallikarjuna Reddy, J. Raul Garcia, V. Hanuman Reddy, A. Meier de Andrade, A. Camilo Jr, R. A. P. Ribeiro and S. R. de Lazaro, Synthesis, antimicrobial activity and advances in structure-activity relationships (SARs) of novel tri-substituted thiazole derivatives, Eur. J. Med. Chem., 2016, 123, 508–513 CrossRef PubMed.
- R. Kanagaddi, S. S. Nannapaneni, J. K. Raghupathi, N. K. Katari, S. Salakolusu, J. N. S. R. C. Murty, M. Ranga and M. Kaliyaperumal, Isolation, Identification, and Characterization of Forced Degradation Products of Bosentan by Using Advanced Analytical Techniques, Sep. Sci. plus, 2024, 7, e202400194, DOI:10.1002/sscp.202400194.
- S. Salakolusu, N. K. Katari, G. V. R. Sharma, D. P. Rendedula, M. Ranga, M. Kaliyaperumal and J. K. Raghupathi, Utilizing cutting-edge analytical techniques for the identification, isolation, and structural characterization of donepezil HCl forced degradation products, Microchem. J., 2024, 207, 111665, DOI:10.1016/j.microc.2024.111665.
- K. K. Vunnam, N. K. Katari, N. Jeedimalla, R. Gundla and J. K. Raghupathi, Design, Synthesis, and Biological Evaluation of Novel Heterocyclic Derivatives of 2,4-Thiazolidine Dione as Anti-Cancer Agents, Appl. Res., 2024, e202400176, DOI:10.1002/appl.202400176.
- L. G. Raju bandaru, P. Raja Kanuparthy, N. Jeedimalla, B. V. Nayak, J. K. Raghupathi, N. Kumar Katari and R. Gundla, Stability indicating RP-HPLC method development and validation for quantification of impurities in gonadotropin-releasing hormone (Elagolix): Robustness study by quality by design, Biomed. Chromatogr., 2024, 38(12), e6036, DOI:10.1002/bmc.6036.
- R. S. Porto and V. A. Porto, Uncovering the Biological Applications of Cinnamic Acid Derivatives: A Patent Review, Lett. Drug Des. Discovery, 2023, 21, 2828–2837, DOI:10.2174/0115701808273623231009074241.
- P. Koczurkiewicz-Adamczyk, K. Klaś, A. Gunia-Krzyżak, K. Piska, K. Andrysiak, J. Stępniewski, S. Lasota, K. Wójcik-Pszczoła, J. Dulak, Z. Madeja and E. Pękala, Cinnamic Acid Derivatives as Cardioprotective Agents against Oxidative and Structural Damage Induced by Doxorubicin, Int. J. Mol. Sci., 2021, 22, 6217, DOI:10.3390/ijms22126217.
- M. Yazdi, A. Nafari, M. Azadpour, M. Alaee, F. H. Moradi, R. Choghakhori, M. Hormozi and H. Ahmadvand, Protective Effects of Cinnamic Acid Against Hyperglycemia Induced Oxidative Stress and Inflammation in HepG2 Cells, Rep. Biochem. Mol. Biol., 2023, 12, 1–12, DOI:10.52547/rbmb.12.1.1.
- S. Wang, J. Chen, J. Shi, Z. Wang, D. Hu and B. Song, Novel Cinnamic Acid Derivatives Containing the 1,3,4-Oxadiazole Moiety: Design, Synthesis, Antibacterial Activities, and Mechanisms, J. Agric. Food Chem., 2021, 69, 11804–11815, DOI:10.1021/acs.jafc.1c03087.
- M. Mingoia, C. Conte, A. Di Rienzo, M. Pia Dimmito, L. Marinucci, G. Magi, H. Turkez, M. C. Cufaro, P. Del Boccio, A. Di Stefano and I. Cacciatore, Synthesis and Biological Evaluation of Novel Cinnamic Acid-Based Antimicrobials, Pharmaceuticals, 2022, 15, 228, DOI:10.3390/ph15020228.
- C. I. Chukwuma, S. S. Mashele and S. S. Swain, Antidiabetic and antioxidative properties of novel Zn(II)-cinnamic acid complex, Med. Chem., 2020, 17, 913–925, DOI:10.2174/1573406416666200929143257.
- A. Nair, M. R. Preetha Rani, P. Salin Raj, S. Ranjit, K. Rajankutty and K. G. Raghu, Cinnamic acid is beneficial to diabetic cardiomyopathy via its cardioprotective, anti-inflammatory, anti-dyslipidemia, and antidiabetic properties, J. Biochem. Mol. Toxicol., 2022, 36, e23215, DOI:10.1002/jbt.23215.
- C. Hu, W. Wang, Y.-N. Ye, Y. Kang, J. Lin, P. Wu, D. Li, L. Bai, X. Xu, B.-Q. Li and K. Zhang, Novel cinnamic acid magnolol derivatives as potent α-glucosidase and α-amylase inhibitors: Synthesis, in vitro and in silico studies, Bioorg. Chem., 2021, 116, 105291, DOI:10.1016/j.bioorg.2021.105291.
- S. Kasturi, S. Surarapu, S. Uppalanchi, J. S. Anireddy, S. Dwivedi, H. S. Anantaraju, Y. Perumal, D. K. Sigalapalli, B. N. Babu and K. S. Ethiraj, Synthesis and α-glucosidase inhibition activity of dihydroxy pyrrolidines, Bioorg. Med. Chem. Lett., 2017, 27, 2818–2823, DOI:10.1016/j.bmcl.2017.04.078.
- A. Alam, M. Ali, N. U. Rehman, S. Ullah, S. A. Halim, A. Latif, Zainab, A. Khan, O. Ullah, S. Ahmad, A. Al-Harrasi and M. Ahmad, Bio-Oriented Synthesis of Novel (S)-Flurbiprofen Clubbed Hydrazone Schiff's Bases for Diabetic Management: In Vitro and In Silico Studies, Pharmaceuticals, 2022, 15, 672, DOI:10.3390/ph15060672.
- M. Taha, N. H. Ismail, S. Imran, A. Wadood, F. Rahim, S. M. Saad, K. M. Khan and A. Nasir, Synthesis, molecular docking and α-glucosidase inhibition of 5-aryl-2-(6′-nitrobenzofuran-2′-yl)-1,3,4-oxadiazoles, Bioorg. Chem., 2016, 66, 117–123, DOI:10.1016/j.bioorg.2016.04.006.
- M. Taha, N. Hadiani Ismail, S. Imran, A. Wadood, M. Ali, F. Rahim, A. A. Khan and M. Riaz, Novel thiosemicarbazide–oxadiazole hybrids as unprecedented inhibitors of yeast α-glucosidase and in silico binding analysis, RSC Adv., 2016, 6, 33733–33742, 10.1039/c5ra28012e.
- C. Viegas-Junior, E. J. Barreiro and C. A. M. Fraga, Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes, Curr. Med. Chem., 2007, 14, 1829–1852, DOI:10.2174/092986707781058805.
- G. Bérubé, An overview of molecular hybrids in drug discovery, Expert Opin. Drug Discovery, 2016, 11, 281–305, DOI:10.1517/17460441.2016.1135125.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256.
- L. Pinzi and G. Rastelli, Molecular Docking: Shifting Paradigms in Drug Discovery, Int. J. Mol. Sci., 2019, 20, 4331, DOI:10.3390/ijms20184331.
- A. Daina, O. Michielin and V. Zoete, iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach, J. Chem. Inf. Model., 2014, 54, 3284–3301, DOI:10.1021/ci500467k.
- T. P. Davis, L. Sanchez-Covarubias and M. E. Tome, P-glycoprotein Trafficking as a Therapeutic Target to Optimize CNS Drug Delivery, Pharmacology of the Blood Brain Barrier: Targeting CNS Disorders, 2014, pp. 25–44, DOI:10.1016/bs.apha.2014.06.009.
- M. F. Fromm, P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs, Int. J. Clin. Pharmacol. Ther., 2000, 38, 69–74, DOI:10.5414/cpp38069.
- G. Pistia-Brueggeman and R. I. Hollingsworth, A preparation and screening strategy for glycosidase inhibitors, Tetrahedron, 2001, 57, 8773–8778, DOI:10.1016/s0040-4020(01)00877-8.
- A. Bhatia, B. Singh, R. Arora and S. Arora, In vitro evaluation of the α-glucosidase inhibitory potential of methanolic extracts of traditionally used antidiabetic plants, BMC Compl. Alternative Med., 2019, 19, 74, DOI:10.1186/s12906-019-2482-z.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a Free web Tool to Evaluate pharmacokinetics, drug-likeness and Medicinal Chemistry Friendliness of Small Molecules, Sci. Rep., 2017, 7, 1–13, DOI:10.1038/srep42717.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01971k |
| This journal is © The Royal Society of Chemistry 2025 |