
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment and in vitro evaluation of 1,4,7-triazacyclononane-coupled β-lactams against metallo-β-lactamase producing bacteria†
Mbongeni Shungubea,
Nakita Reddy ab,
Terisha Ghazic,
Kimberleigh B. Govendera,
Ravesh Singhcd,
Afsana Kajeecd,
Anil Chuturgoonc,
Hendrik G. Krugera,
Per I. Arvidssonae,
Dileep Tiwariaf,
Thavendran Govender*g and
Tricia Naicker*a
ab,
Terisha Ghazic,
Kimberleigh B. Govendera,
Ravesh Singhcd,
Afsana Kajeecd,
Anil Chuturgoonc,
Hendrik G. Krugera,
Per I. Arvidssonae,
Dileep Tiwariaf,
Thavendran Govender*g and
Tricia Naicker*a
aCatalysis and Peptide Research Unit, University of KwaZulu-Natal, Durban, 4001, South Africa. E-mail: govenderthav@icloud.com; naickert1@ukzn.ac.za; Tel: +27 312601845
bOffice of AIDS and TB, South African Medical Research Council, 1 Soutpansberg Road, Pretoria, South Africa
cSchool of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban, 4041, South Africa
dDepartment of Medical Microbiology, KwaZulu-Natal Academic Complex, National Health Laboratory Service, Durban, South Africa
eScience for Life Laboratory, Drug Discovery & Development Platform & Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden
fHericure HealthCare Pvt Ltd, 412, Shree Ganesh Ace Arcade, Business Tower, NR Kokane Chowk Pimple Saudagar, Pune-17, India
gDepartment of Chemistry, University of Zululand, Private Bag X1001, KwaDlangezwa 3886, South Africa
First published on 7th July 2025
Abstract
Antimicrobial resistance (AMR) is a critical global issue, particularly against β-lactam antibiotics, which comprise over 60% of prescriptions. Metallo-β-lactamases (MBLs) are especially concerning as they inactivate nearly all β-lactams, except monobactams. Unlike serine-β-lactamases (SBLs), for which inhibitors exist, there are no clinically approved MBL inhibitors; only taniborbactam is in pre-registration. This study introduces eight new MBL inhibitors (13a–f, 14a-b), designed using a 1,4,7-triazacyclononane (NO3PY) chelator linked to a β-lactam. These inhibitors restored the efficacy of meropenem, reducing its minimum inhibitory concentration (MIC) against MBL-expressing pathogens to <2 mg L−1. Time-kill assays confirmed bactericidal activity, with this series being non-toxic and highly specific, these compounds hold promising potential as MBL inhibitors.
Introduction
The World Health Organization (WHO) reported that drug-resistant infections caused at least 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths worldwide in 2019,1 while another report in that same year estimated over 1.2 million deaths.2 This alarming number is estimated to increase to about 10 million by 2050, with continued usage of antimicrobials such as β-lactams and the emergence of multidrug-resistant pathogens in the clinical setting.3 The threat of β-lactams becoming obsolete is a global concern, for example, in the USA, β-lactam antibiotics (penicillins, cephalosporins, carbapenems, and monobactams)4 are the most commonly used, accounting for about 60% of all antibiotic prescriptions, and are even used to treat serious infections in the clinic.5
000 deaths worldwide in 2019,1 while another report in that same year estimated over 1.2 million deaths.2 This alarming number is estimated to increase to about 10 million by 2050, with continued usage of antimicrobials such as β-lactams and the emergence of multidrug-resistant pathogens in the clinical setting.3 The threat of β-lactams becoming obsolete is a global concern, for example, in the USA, β-lactam antibiotics (penicillins, cephalosporins, carbapenems, and monobactams)4 are the most commonly used, accounting for about 60% of all antibiotic prescriptions, and are even used to treat serious infections in the clinic.5
β-Lactamases, are enzymes produced by bacteria that specifically target and hydrolyze the β-lactam ring, rendering them ineffective against the bacterial infections.6 Four classes of β-lactamases exist; enzymes belonging to classes A, C, and D are referred to as serine β-lactamases (SBL). They are characterized by the presence of a serine residue at the enzymes' active site.7,8 Fortunately, several SBL inhibitors, such as sulbactam, clavulanic acid, and vaborbactam, have been developed as combination therapies that restore the activity of β-lactam antibiotics.9 In contrast, class B are the metallo-β-lactamases (MBLs) with the presence of one or two zinc ions at their catalytic active site.10 Clinically significant MBLs include imipenemases (IMP), Verona integron-encoded metallo-β-lactamases (VIM), and New Delhi metallo-β-lactamases (NDM). Notably, there are no FDA-approved MBL inhibitors available in the clinic, with only taniborbactam in the pre-registration phase of development.11
One promising strategy for developing MBL inhibitors involves the use of metal stripping agents (chelators), such as EDTA,12,13 NOTA,14 DOTA,14 Aspergillomarasmine A,15,16 o-phenanthroline,17,18 dipicolic acids,19 and L/D-captopril.20,21 In vitro studies of these chelators demonstrated high activity to potentiate meropenem (β-lactam antibiotic susceptible to MBL resistance)22 against metallo-β-lactamase enzymes.22 However, a significant drawback of using such metal chelators as potential MBL inhibitors is their off-target activity with metalloenzymes in the host, resulting in poor selectivity, high toxicity, and poor bioavailability.22–25 Also, the chelators as such are generally not bioavailable.14 To resolve these challenges, recent advancements have involved a few metal chelators that have been covalently linked to a modulator construct.26–28 The modulators are selected to reduce the off-target activity of the chelator and provide better selectivity toward bacterial cell enzymes and enhance the chelator's bioavailability.
Our group14,24,25 has previously reported on the potential of cyclic chelators such as NOTA, DOTA, and NO3PY as MBL inhibitors. In recent work, we successfully developed NOTA-derived chelators linked to β-lactam antibiotics, which demonstrated better physio-chemical properties, reduced toxicity, and high selectivity toward bacterial MBLs (Fig. 1).26–28
 | ||
| Fig. 1 Structural similarities of our current to previous work. | ||
To further advance this concept, we have developed a new construct 1,4,7-triazacyclononanes referred to as NO3PY (2) based chelators and covalently linked it to β-lactam antibiotics. We hypothesize that this novel chelator will exhibit superior binding affinity to the zinc ions due to the nitrogen donor atoms that are much softer than the hard oxygen donors of the NOTA analog,29 potentially leading to more effective strategies against β-lactam-resistant infections and improving clinical outcomes.
Materials and methods
General information
Antibiotics were purchased from Merck (Germany), DLD Scientific (South Africa), and Hangzhou Dayangchem Co., Ltd (China). Reagents and solvents were purchased from Merck. All solvents were dried using standard procedures. All the synthetic steps were monitored using LC-MS (Shimadzu 2020 UFLC-MS, Japan). The LC-MS method used a gradient of 5% ACN: H2O (0.1% formic acid) to 95% ACN: H2O (0.1% formic acid) over 9 minutes or 5% ACN: H2O (0.1% formic acid) to 70% ACN: H2O (0.1% formic acid) over 20 minutes on an XBridgeTM C18 5 μm 4.6 × 150 mm column, where the flow rate is 1 mL min−1. The intermediates were purified by either gravity column chromatography (mesh particle size, 40–63 μm) or preparatory supercritical fluid chromatography performed on a Sepiatec Prep SFC basic/basic 30 (Germany). High-resolution mass spectrometric (HRMS) data were obtained with a Bruker micrOTOF-Q II instrument that operated at ambient temperatures and a 1.0 μg mL−1 sample concentration. NMR data were recorded at room temperature using a Bruker AVANCE III 400 MHz. Chemical shifts are expressed in ppm. Optical rotations were recorded on a Bellingham & Stanley ADP440+ Polarimeter.Preparation of 1,4,7-tris(pyridin-2-ylmethyl)-1,4,7-triazonane (2)
1,4,7-Tris(pyridin-2-ylmethyl)-1,4,7-triazonane (2) also known as NO3PY was prepared following a literature procedure.30 1,4,7-triazacyclononane (200 mg, 1.55 mmol) was dissolved in dry acetonitrile (1.8 mL mmol−1) with excess Na2CO3 (3.5 equiv.) and 2-(chloromethyl)pyridine hydrochloride (766 mg, 4.7 mmol) which was added drop-wise at room temperature, the mixture was allowed to stir for 5 days. The reaction mixture was filtered, and the solvent was removed in vacuo. The crude residue was purified with neutral aluminium (DCM:MeOH) (100-98:1) to afford pure compound 2 in 46% yield (285 mg) as a dark brown oil. Confirmed by LC-MS: m/z [M + H]+ = 403 (Fig. S1, ESI†). 1H NMR (CDCl3): 3.08 (m, 12H), 4.10 (m, 6H), 7.18 (m, 3H), 7.56 (m, 3H), 7.62 (m, 3H), 8.47 (m, 3H) ppm (Fig. S2, ESI†). HRMS (ESI): m/z [M + H]+ calc. for C24H30N6: 403.2605; found: 403.2042 (Fig. S3, ESI†).
Synthesis of Zn(II) 1,4,7-tris(pyridin-2-ylmethyl)-1,4,7-triazonane complex (3), (Zn(NO3PY))
Compound 3 was prepared using a modified literature procedure for the Zn-NOTA complex formation.31 NO3PY (2) (13 mg, 0.033 mmol) was dissolved in 0.5 mL methanol. Thereafter, zinc perchlorate hexahydrate (1.0 equiv., 12.3 mg, 0.033 mmol) was also dissolved in 0.5 mL methanol and slowly added to the solution of NO3PY. A precipitate immediately formed, filtered, and washed with cold (3 × 1.0 mL) methanol to give the pure Zn–NO3PY complex (3) as an off-white powder in quantitative yield. LC-MS: m/z [M]2+ = 233 (Fig. S4, ESI†). HRMS (ESI): m/z [M]2+ calc. for C24H30N6Zn: 233.0906; found: 233.0917 (Fig. S5, ESI†).Synthesis of hexahydro-1H-2a,4a,6a-triazacyclopenta[cd]pentalene (4)
Compound 4 was prepared following a literature procedure,32–34 1,4,7-triazacyclononane (1000 mg, 7.74 mmol) was dissolved in 3 mL chloroform (2.6 mL mmol−1) and 9 mL toluene (0.86 mL mmol−1). N,N-dimethylformamide dimethyl acetal (1028 μL, 1.0 mol equiv.) was added, and the reaction mixture was refluxed for 2 hours. The solvent was removed in vacuo and further dried to afford the pure compound 4 in 92% (1075 mg) yield as a yellow oil. Compound 4 was confirmed by LC-MS: m/z [M + H]+ = 140, 1H NMR (CDCl3): 2.78 (m, 6H), 3.07 (m, 6H), 5.03 (s, 1H) ppm (Fig. S6, ESI†).Synthesis of 4a-(pyridin-2-ylmethyl)octahydro-2a,4a,6a-triazacyclopenta[cd]pentalen-4a-ium chloride (5)
2-(Chloromethyl)pyridine was initially extracted from the 2-(chloromethyl)pyridine hydrochloride salt, which was dissolved in 5–10 mL water and the pH adjusted to about 12 with NaOH (5 M) thereafter, extracted with 10 mL DCM or CHCl3 (3 times) and then dried to give an orange oil. Compound 5 was prepared following the literature procedure.35 Compound 4 (1000 mg, 7.19 mmol) was dissolved in dry THF (1.0 mL mmol−1). A solution of (1.0 equiv.) 2-(chloromethyl)pyridine in dry THF (1.0 mL mol−1) was added drop-wise over 15–30 minutes, thereafter, the reaction was allowed to proceed overnight at room temperature. The reaction mixture was then centrifuged (5000 rpm for 5 minutes), and the supernatant was decanted, the residuals were washed three times with cold THF (dry) to afford pure compound 5 in a 65% (1250 mg) yield as a maroon solid. Confirmed by LC-MS: m/z [M + H]+ = 231(Fig. S7, ESI†).Synthesis of 4-(pyridin-2-ylmethyl)-1,4,7-triazonane-1-carbaldehyde (6)
Compound 6 was prepared following the literature procedure.35 Compound 5 (1250 mg, 4.70 mmol) was dissolved in MilliQ water (4.7 mL, 1.0 mL mmol−1) and refluxed for 4 hours. Thereafter, the reaction was cooled to room temperature, and the pH was adjusted to about 12 with NaOH (5 M) thereafter, extracted with 10 mL DCM or CHCl3 (3 times), dried with anhydrous Na2SO4 and the solvent evaporated in vacuo to give compound 6 in 82% (950 mg) yield as orange oil. Confirmed by LC-MS: m/z [M + H]+ = 249 (Fig. S8, ESI†).Synthesis of 4,7-bis(pyridin-2-ylmethyl)-1,4,7-triazonane-1-carbaldehyde (7)
Compound 7 was prepared following the literature procedure.35 Compound 6 (900 mg, 3.63 mmol) was dissolved in dry acetonitrile (30 mL, 8.33 mL mmol−1) with excess K2CO3 (4.0 equiv.) and KI (10.0 equiv.) 2-(chloromethyl)pyridine hydrochloride (592 mg, 3.63 mmol) was dissolved in dry acetonitrile (30 mL, 8.33 mL mmol−1) and added drop-wise at room temperature, the reaction mixture was allowed to stir for 1 hour. Thereafter, the reaction mixture was refluxed overnight. The reaction mixture was cooled, filtered, and then the solvent was removed in vacuo to afford pure compound 7 in 86% (1060 mg) yield as a dark brown oil. Confirmed by LC-MS: m/z [M + H]+ = 340 (Fig. S9, ESI†).Synthesis of 1,4-bis(pyridin-2-ylmethyl)-1,4,7-triazonane (8)
Compound 8 was prepared following the literature procedure.35 Compound 7 (1000 mg, 2.95 mmol) was dissolved in 4 M HCl (3.0 mL, 1 mL mmol−1) and refluxed for 4 hours. Thereafter, the reaction mixture was cooled to room temperature. The pH was adjusted to about 12 with NaOH (5 M) thereafter, extracted with 10 mL DCM or CHCl3 (3 times), dried with anhydrous Na2SO4 and solvent evaporated in vacuo to give pure compound 8 in 75% (690 mg) yield as a dark brown oil. Confirmed by LC-MS m/z [M + H]+ = 312 (Fig. S10, ESI†).Synthesis of ethyl 6-(4,7-bis(pyridin-2-ylmethyl)-1,4,7-triazonan-1-yl)hexanoate (9)
Compound 9 was prepared following the literature procedure.35 Compound 8 (650 mg, 2.09 mmol) was dissolved in dry acetonitrile (17.4 mL, 8.33 mL mmol−1) with K2CO3 (0.4 equiv.) and KI (0.2 equiv.). Ethyl 6-bromohexanoate (372 μL, 2.09 mmol) was diluted in dry acetonitrile (17.4 mL, 8.33 mL mol−1) and was added dropwise at room temperature, the mixture was allowed to stir for 1 hour. Thereafter, the reaction mixture was refluxed overnight. The reaction mixture was cooled, filtered and solvent removed in vacuo to afford pure compound 9 in 86% (810 mg) yield as a dark brown oil. Confirmed by LC-MS: m/z [M + H]+ = 454 (Fig. S11, ESI†).Synthesis of 6-(4,7-bis(pyridin-2-ylmethyl)-1,4,7-triazonan-1-yl)hexanoic acid (10)
Compound 9 (800 mg, 1.77 mmol) was dissolved in 2 M HCl (2.65 mL, 3 equiv.) and microwaved at 100 °C for 1 hour. The reaction mixture was freeze-dried to afford the pure HCl salt of compound 10 in a quantitative yield (850 mg). Confirmed by LC-MS: m/z [M + H]+ = 426 (Fig. S12, ESI†).Synthesis of ethyl 2-(4,7-bis(pyridin-2-ylmethyl)-1,4,7-triazonan-1-yl)acetate (11)
Compound 11 was prepared following the literature procedure.35 Compound 8 (650 mg, 2.09 mmol) was dissolved in dry acetonitrile (17.4 mL, 8.33 mL mmol−1) with K2CO3 (0.4 equiv.) and KI (0.2 equiv.). Ethyl bromoacetate (231 μL, 2.09 mmol) was diluted in dry acetonitrile (17.4 mL, 8.33 mL mol−1) and was added dropwise at room temperature, the mixture was allowed to stir for 1 hour. Thereafter, the reaction mixture was refluxed overnight. The reaction mixture was cooled, filtered and solvent removed in vacuo to afford pure compound 11 (Scheme 2, ESI†) in 90% (750 mg) yield as a dark brown oil. Confirmed by LC-MS: m/z [M + H]+ = 398 (Fig. S13, ESI†).2-(4,7-Bis(pyridin-2-ylmethyl)-1,4,7-triazonan-1-yl)acetic acid (12)
Compound 11 (750 mg, 1.77 mmol) was used to prepare compound 12 similar to compound 10. A pure HCl salt of compound 12 was obtained in a quantitative yield (780 mg). Confirmed by LC-MS: m/z [M + H]+ = 370 (Fig. S14, ESI†).SFC purification method
Preparative SFC purification was done using a Sepiatec Prep SFC basic/30 system. All compounds were purified with the following parameters: sample concentration = 10–20 mg mL−1 in acetonitrile or methanol, injection volume = 100–200 μL, column = pentafluorophenyl (PFP) (250 × 10 mm, 5 Å) at 40 °C, mobile phase (gradient elution) = 10–50% MeOH: ACN (2:1) spike with 0.1% trifluoroacetate (TFA) or 0.3% DIEA (diisopropyl ethylamine) as the modifier (Pump A) with technical grade-wet CO2 with a flow at 10 mL min−1 (Pump B), flow = 10 mL min−1, BPR setting = 150 bar, monitoring and collection at 210 nm. All samples were injected in a multi-loop circle between 2–50 injections and the product fractions were collected and then concentrated in vacuo. The gradient elution was performed using the following modifier (Pump A): 10% at 0.00 min, held at 10% until 1.00 min, increased linearly to 30% at 3.00 min, followed by a sharp increase to 50% at 3.01 min. The composition was maintained at 50% until 6.00 min, then rapidly decreased back to 10% at 6.01 min, and held at 10% until 9.00 min.
Compound 10 (100 mg, 0.24 mmol) or a given amount was dissolved dry DMF (2 mL mmol−1) the base DIEA (6.0 equiv.). The coupling agent (100 mg, 2.6 mmol, 1.1 equiv.) and HATU were added and allowed to activate carboxylic acid over 2 minutes. Thereafter, 1.0 mol equivalent of the respective β-lactam, 7-aminocephaosporanic acid (7-aca), cefaclor, ampicillin, cephalexin, cefadroxil and 1-azetidinesulfonic acid were added respectively. The reactions were monitored by LC-MS and were completed between 30 minutes to an hour. Thereafter, 10 mL acetonitrile was added, which resulted in the precipitation of some by-products. The precipitate was removed by centrifugation (5000 rpm for 2 minutes) and the supernatant was purified using the SFC purification method above. Compound 13a was purified with a basic modifier (spiked with 0.3% DIEA) while 13b–f with an acid modifier (spiked with 0.1% TFA). Thereafter, the solvent was evaporated in vacuo to afford pure compounds 13a–f.
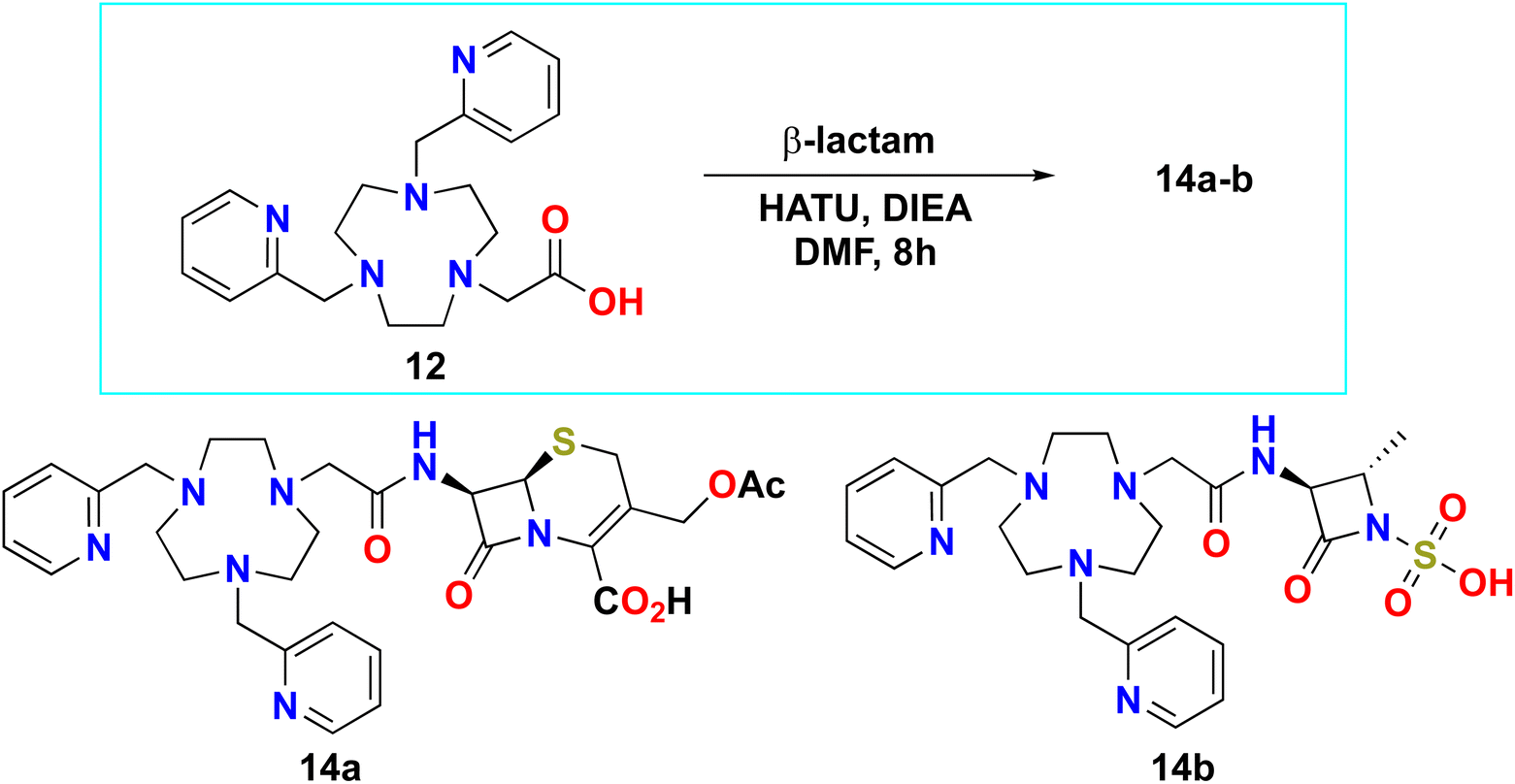
Compounds 14a and 14b (Scheme 4, ESI†) were prepared similarly to 13a–f. Compound 12 (100 mg, 0.27 mmol) was dissolved in dry DMF (0.54 mL, 2 mL mmol−1) with base DIEA (6.0 equiv.). The coupling agent (112 mg, 3.0 mmol, 1.1 equiv.) and HATU were added to activate carboxylic acid over 2 minutes. Thereafter, 1.0 mol equivalent of the respective β-lactam, 7-aca, or 1-azetidinesulfonic acid was added to afford compounds 14a or 14b, respectively. The reactions were monitored by LC-MS and were completed after 8 hours. Thereafter, the reactions were worked up similarly to 13b–f.
Biological evaluation
Cytotoxicity assay
000 cells per well) were seeded into a 96-well microtiter plate and allowed to adhere overnight (37 °C, 5% CO2). Thereafter, the cells were incubated (37 °C, 5% CO2) with a range of chelator concentrations (0, 1, 8, 10, 50, 100, and 200 μg mL−1) in triplicate for either 6 h (compounds 2, 10 and 12) or 24 h (compounds 13a, 13f, 14a, and 14b). After incubation, the cells were washed with 0.1 M phosphate-buffered saline (PBS) and incubated with 20 μL MTT salt solution (5 mg mL−1 in 0.1 M PBS) and 100 μL CCM for 4 h (37 °C, 5% CO2). The MTT salt solution was removed, and DMSO (100 μL per well) was added and incubated for 1 h. The optical density was measured using a spectrophotometer (SPECTROstar Nano) at 570/690 nm. Results are expressed as % cell viability versus MBL inhibitor concentration (μg mL−1).644793001, Sigma Aldrich) was added to each well. The plate was incubated for 30 minutes at room temperature in the dark. Optical density was read using a spectrophotometer (SPECTROstar Nano) at 500 nm. Results are represented as mean optical density compared to the untreated control.Results and discussions
We initially set out to explore the microbial activity of the chelator alone, NO3PY (2), and its zinc complex (3) against MBL-expressing bacteria (Scheme 1). NO3PY (2) was synthesised in 45% yield following a literature procedure42 and confirmed by LCMS and 1H-NMR spectroscopy. Zn(II)–NO3Py complex (3) was further prepared by dropwise addition of zinc perchlorate hexahydrate, dissolved in methanol, to compound 2 (also dissolved in methanol) in the same concentration, resulting in the pure complex 3 being obtained in quantitative yields. | ||
| Scheme 1 Synthesis of NO3PY and its complex Zn(II)–NO3PY; Py-Cl. HCl = 2-(chloromethyl) pyridine hydrochloride. | ||
We previously tested several cyclic chelators such as NODAGA, DOTA, and NOTA against MBL-harbouring bacteria; they showed excellent activities to restore the activity of meropenem to concentrations < 0.5 mg L−1 when co-administered with cyclic chelators in the range of 4–64 mg L−1.26–28 When NO3PY (2) was evaluated against K. pneumoniae NDM, it too demonstrated high activity to potentiate meropenem to 0.125 mg L−1 when co-administered with 4 mg L−1 of compound 2 (entry 3, Table 1). These biological activities were similar to NOTA (entry 2). When the corresponding zinc complex (3) of NO3PY (2) was tested, there was no inhibitory activity observed (entry 4), affirming the necessity of zinc chelation by the free ligand for inhibition (Table 1). Similar MIC values were observed when NOTA was precomplexed to zinc and evaluated as a potential MBL inhibitor.28,43 In those studies,28,43 molecular docking of NOTA coupled to a β-lactam construct showed favourable docking scores against the NDM-1 and VIM-2 enzymes, and showed the chelator region interacting with Zn2+ of the lactamase enzymes.
| Entry | Inhibitor | Minimum inhibitory concentration (mg L−1) | |
|---|---|---|---|
| K. pneumoniae (NDM) | |||
| Meropenem | Inhibitor | ||
| a All assays were conducted in triplicate. | |||
| 1 | None | >32 | 0 |
| 2 | NOTA | 0.06 | 4 |
| 3 | 2 | 0.125 | 4 |
| 4 | 3 | >32 | >32 |
Based on these results, we set out to expand the scope with NO3PY (2) derivatives linked to β-lactam antibiotics. To achieve this, we prepared two chelators with different linker lengths 6-(4,7-bis(pyridin-2-ylmethyl)-1,4,7-triazonan-1-yl)hexanoic acid (Hno1ha2py) (10) and 2-(4,7-bis(pyridin-2-ylmethyl)-1,4,7-triazonan-1-yl)acetic acid (Hno1a2py) (12) (Scheme 2). Compound 12 has been previously synthesized by Gasser et al.;43 for the synthesis of 8 we first prepared compound 4 following literature procedures.33,44 Compound 4 was obtained in qualitative yields and confirmed by NMR. Compound 4 was used to prepare intermediate 8 following a literature procedure.26 First, 2-(chloromethyl) pyridine (Py-Cl) oil (extracted with DCM from a basic aqueous solution of 2-(chloromethyl) pyridine hydrochloride Py-Cl. HCl adjusted with NaOH (5 M) to pH 12) was dissolved in dry THF. It was added slowly to a solution of compound 4 that was dissolved in THF at room temperature to afford compound 5. Compound 5 was hydrolyzed with Milli-Q water under reflux and then extracted with DCM at pH 12 to afford pure compound 6 in a 65% yield. Compound 6 was alkylated using Py-Cl. HCl in dry ACN under reflux to afford compound 7 in quantitative yields. Thereafter, it was deprotected using HCl (6 M) under reflux conditions to afford compound 8 in 75% yield. Compound 11 was prepared following the literature procedure.26 However, the final base hydrolysis to afford 12 used in the literature protocol was inefficient with multiple side-products. Therefore, we performed acid hydrolysis at 100 °C in a microwave reactor, which afforded compound 12. A pure HCl salt of compound 12 was obtained after lyophilisation and used without further purification in quantitative yields (Scheme 2). Compounds 9 and 10 were prepared similarly to 11 and 12, respectively.
 | ||
| Scheme 2 Synthesis of chelators 10 and 12; Py-Cl = 2-(chloromethyl) pyridine, EBA = ethyl bromoacetate, EBH = ethyl 6-bromohexananoate. | ||
With the chelators in hand, next, we attempted to couple compound 10 to various β-lactam antibiotics that were successful in our previous studies; details on coupling involving compound 12 are discussed in a subsequent section. We explored various coupling conditions to optimize the reaction conditions for alkyl acid with aryl or alkyl amines. Some of the conditions were adopted from MacMillan's report.45 Based on the solubility of the chelator and β-lactams, we tested several dry solvent conditions for the amide coupling such as acetonitrile (ACN), DMF, DMSO, and DCM. Furthermore, we also explored several coupling agents such as COMU, HATU, DIC/HOBt, EDC/HOBt, and Oxyma Pure. Through systematic optimization of the coupling conditions, we determined that all the β-lactams with the alkyl amine could be efficiently (100% conversion) coupled to compound 10 using six moles equivalents of a base (DIEA) in 2 mL mmol−1 of DMF in 30 minutes to an hour at room temperature. We initially coupled compound 10 to 7-aminocephaosporanic acid (7-aca) to afford compound 13a in 43% yield after supercritical fluid chromatography (SFC) purification (Scheme 3).
 | ||
| Scheme 3 Synthesis of β-lactam MBL inhibitor 13a. | ||
Initially, compound 13a, in combination with meropenem, was screened for microbial activity against several bacteria-producing MBLs (Table 2) using the checkerboard assay following a previously described protocol46 and as per CLSI antimicrobial susceptibility guidelines.47 Encouragingly, compound 13a was able to reactivate meropenem to between 4 mg L−1 and 0.06 mg L−1 against all MBL-harboring pathogens (Table 2). Compound 13a was active towards MBLs – NDM, IMP, VIM; however, it proved to be inactive when tested against SBLs (as expected).
| No. | Bacterial reference | Bacterial strain | MBL produced | Meropenem MIC mg L−1 | 13a + meropenem MIC mg L−1 |
|---|---|---|---|---|---|
| a All assays were conducted in triplicate. | |||||
| 1 | AUS-271 | Escherichia coli | NDM-1 | >32 | 16 + 0.25 |
| 2 | FEK | E. coli | NDM-4 | >32 | 16 + 0.25 |
| 3 | JAP | E. coli | IMP-1 | 32 | 16 + 0.25 |
| 4 | TWA | E. coli | IMP-8 | 8 | 32 + 0.25 |
| 5 | IR386 | Enterobacter cloacae | NDM-1 | 16 | 8 + 0.25 |
| 6 | KAR | E. cloacae | VIM-1 | 4 | 16 + 0.5 |
| 7 | USA-449 | Klebsiella pneumoniae | NDM | >32 | 16 + 0.5 |
| 8 | 6852 | K. pneumoniae | IMP-1 | >32 | 64 + 4 |
| 9 | BM-5 | E. cloacae | IMP-1 | >32 | 64 + 4 |
| 10 | BM-20 | Serratia marcescens | VIM-2 | >32 | 64 + 0.5 |
| 11 | IR-38 | Providencia rettgeri | NDM-1 | 16 | 32 + 0.5 |
| 12 | ENNES | K. pneumoniae | VIM-1 | >32 | 32 + 0.5 |
| 13 | BM-14 | E. coli | VIM-1 | 16 | 64 + 0.5 |
| 14 | TC CARF | E. coli | VIM-2 | 1 | 32 + 0.06 |
| 15 | FRANCE | S. marcescens | IMP-11 | >32 | 64 + 0.5 |
| 16 | PSTU | Providencia stuartii | NDM-1 | 16 | 64 + 0.5 |
| 17 | TWA | K. pneumoniae | IMP-8 | 16 | 64 + 0.5 |
| 18 | AFR-7 | K. pneumoniae | NDM-1 | >32 | 64 + 0.5 |
| 19 | FRANCE | K. pneumoniae | VIM-19 | >32 | 32 + 1 |
Based on the successful MIC results of compound 13a, we expanded the scope of the compounds with other β-lactams used in our previous studies.26–28 We employed the protocol used to prepare compound 13a to couple compound 10 with ampicillin, cefaclor, cephalexin, and cefadroxil, which afforded 13b–e in 11% to 36% yields (Scheme 4). The coupling of β-lactams with aryl amines (such as thiazole amines), namely, ceftiofur, ceftibuten, and cefotaxime, showed poor conversion of less than 5% and was not pursued further.
 | ||
| Scheme 4 Synthesis of β-lactam MBL inhibitors from chelator (10). | ||
Notably, for the first time, we included an aztreonam derivative (1-azetidinesulfonic acid) in the series of β-lactams. Aztreonam, in combination with avibactam, showed activity against metallo-β-lactamase enzymes and is currently in Phase 3 clinical trials.9 Following the same coupling protocol, compound 10 was successfully linked to the monobactam scaffold, 1-azetidinesulfonic acid to afford 13f in a 33% yield (Scheme 4). Currently, among the beta-lactam family of drugs, monobactams (e.g. aztreonam) remain unique in their resistance to hydrolysis by MBLs48 putting this series at a distinct advantage of over other β-lactam derivatives.
We evaluated the MICs of all the synthesized compounds against four selected MBL-harbouring bacteria: Escherichia coli (NDM-1), K. pneumoniae (NDM), E. cloacae (VIM-1), and E. coli (IMP-1) (Table 2). This was done to ensure the compounds were screened for efficacy against the most clinically relevant MBLs: NDM, IMP, and VIM.
Compound 10 (entry 1) which is a precursor to 13a–f, displayed excellent MIC outcomes for meropenem, generating MICs of <1 mg L−1 at a low administration concentration of 16 mg L−1. Interestingly, 12 which is also a precursor to our desired compounds (entry 2, Table 3), exhibited the best efficacy, utilizing concentrations as low as 4 mg L−1 with <1 mg L−1 meropenem. Compound 12 is therefore a potent chelator and was observed to produce inhibitory activity in the absence of meropenem, utilizing a low concentration of 64 mg L−1, with the inhibitory activity stable for at least 24 hours. The findings for precursor chelators, 10 and 12 are noteworthy and could benefit from additional exploration as a starting construct.
| Entry | Inhibitor | Minimum inhibitory concentration (mg L−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| E. coli (NDM-1) | K. pneumoniae (NDM) | E. cloacae (VIM-1) | E. coli (IMP-1) | ||||||
| Meropenem | Inhibitor | Meropenem | Inhibitor | Meropenem | Inhibitor | Meropenem | Inhibitor | ||
| a Bacteria ref: Escherichia coli (NDM-1) = Aus-271, Klebsiella pneumoniae (NDM) = USA 449, Enterobacter cloacae (VIM-1) = KAR, Escherichia coli (IMP-1) = JAP. All assays were conducted in triplicate. | |||||||||
| 1 | 10 | 0.25 | 16 | 0.5 | 16 | 0.5 | 16 | 0.25 | 16 |
| 2 | 12 | 0.06 | 4 | 0.125 | 4 | 1 | 4 | 0.03 | 4 |
| 3 | 13a | 0.5 | 16 | 0.125 | 16 | 0.25 | 16 | 0.03 | 16 |
| 4 | 13b | 0.25 | 16 | 0.125 | 32 | 2 | 64 | 0.125 | 32 |
| 5 | 13c | 0.5 | 32 | 0.25 | 64 | 0.5 | 16 | 0.5 | 16 |
| 6 | 13d | 0.25 | 32 | 0.5 | 32 | 1 | 64 | 0.25 | 32 |
| 7 | 13e | 0.5 | 64 | 0.25 | 32 | 1 | 64 | 0.25 | 32 |
| 8 | 13f | 0.25 | 16 | 0.5 | 8 | 0.5 | 8 | 0.25 | 8 |
| 9 | 14a | 0.25 | 16 | 0.125 | 16 | 0.5 | 16 | 0.03 | 16 |
| 10 | 14b | 0.5 | 8 | 0.5 | 8 | 0.25 | 16 | 0.03 | 16 |
Entries 3 to 8, employing inhibitory compounds 13a–f depict the MIC results of meropenem against four MBL-producing pathogens. Encouragingly, meropenem activity was restored when co-administered with the MBL inhibitor constructs. MBL inhibitors 13a and 13f entry 3 and 8 demonstrated slightly superior efficacy than the chelator with linker compound 10 (entry 1), achieving activity <0.5 mg L−1 and inhibitor concentration of <16 mg L−1 for all evaluated MBL-producing pathogens. These concentrations are therapeutically acceptable as they aim to produce low meropenem concentrations, generally <1 mg L−1, that correspond to the lowest possible inhibitor concentration. This combination ensures sustained and potent inhibition against the MBL pathogen.
Building on the optimized coupling conditions established for 13a–f, we applied this protocol to synthesize analogs based on intermediate 12, allowing us to evaluate the effect of the length of the linker between the chelator and β-lactams on the activity of the construct. Compound 12 was coupled to 7-aminocephaosporanic acid (7-aca) and 1-azetidinesulfonic acid to afford analogues 14a and 14b, respectively (Scheme 5).
 | ||
| Scheme 5 Synthesis of β-lactam MBL inhibitors from chelator (12). | ||
These reactions, however, proceeded more slowly than those with compound 10, taking approximately 8 hours to reach completion and yielding compounds 14a and 14b in 9% and 5% respectively, after prep-SFC purification. Attempts to couple compound 12 with other β-lactams listed in Scheme 3 resulted in poor conversion rates (below 5%) and were not pursued further.
Entries 1–10 in Table 3 showed significantly reduced meropenem MICs by at least 12-fold. Notably, compounds 13f and 14b (entries 8 and 10, Table 3) exhibited exceptional inhibitory activity, reducing the meropenem MIC to <0.5 mg L−1 when used at concentrations of 8 or 16 mg L−1 across all four evaluated MBL-expressing pathogens. Compounds 13f and 14b, derived from the monobactam 1-azetidinesulfonic acid with varying linker lengths, demonstrated outstanding efficacy. Similarly, compounds 13a and 14a showed potent inhibitory activity against all tested MBL-harbouring pathogens.
Both 13a and 14a were synthesized using the same cephalosporin β-lactam, 7-ACA. The variation in linker lengths between compounds 13a and 14a, as well as 13f and 14b, did not result in notable differences in their MICs (Table 3). Compounds 13a and 14a produced very similar MICs (Table 3), indicating minimal impact from linker length variation. However, a two-fold advantage was observed for 13f over 14b against E. cloacae VIM-1, while 14b demonstrated a two-fold advantage over 13f against E. coli NDM-1. Despite these differences, the MIC values for all compounds remained within the recommended EUCAST breakpoint of <2 mg L−1 for meropenem, suggesting that the observed variations do not significantly affect the overall efficacy of the compounds (Table 3). Entries 1–10 demonstrated inhibitory activity comparable to our previously successful compounds (albeit with a change in the type of chelator and linker), with differences of only one to two folds. The findings reported herein suggest that these compounds hold promise as potential MBL inhibitors, pending further biological evaluation.
In comparison to the MIC activities of the NO3PY derivatives and our NOTA analogues26–28 with the same β-lactam. We observed that compounds 13a and 14a showed superior activity compared to compound 15 (NOTA derivative) with at least a two-fold advantage against the two pathogens tested (E. coli (NDM-1) and K. pneumoniae NDM) (Fig. 2).28 This may be attributed to the better binding affinity of the softer nitrogen donor to the softer zinc atom as opposed to the hard oxygen donor of the NOTA derivative.29
 | ||
| Fig. 2 MIC comparison of NO3PY and NOTA analogues linked to lactam 7-ACA;28 a = E. coli (NDM-1), b = K. pneumoniae NDM, MIC = Meropenem + MBL inhibitor. | ||
Subsequently, the time required to achieve complete bactericidal activity was assessed for 13f, since this compound produced the best activity according to the MICs (Table 3). Compound 13a was also studied to evaluate the bactericidal effect exhibited, since this inhibitor was derived from 7-ACA, in comparison to 13f. The overall aim of this experiment was to restore meropenem's efficacy using the lowest possible carbapenem concentration, therefore, meropenem was fixed at 1 mg L−1. In the absence of the antibiotic and inhibitor combination, observations of exponential bacterial growth can be noted in Fig. 3. When using only the inhibitor, 13f, a slight decrease in the cfu mL−1 count of 2log10 units was observed, within the first 2 hours, post inoculation (Fig. 3). Thereafter, 13f was inactive against the pathogen. This initial activity could be attributed to the beta-lactam component of 13f. A > 3log10 decrease in the cfu mL−1 count was observed when meropenem monotherapy was used, however, this activity lasted for only 4 hours post inoculation (Fig. 3). Thereafter, K. pneumoniae NDM (USA-449) was unhindered by the effects of meropenem. The combination of meropenem with either 13a or 13f was highly effective in reducing the cfu mL−1 count of K. pneumoniae NDM (USA-449). Compound 13a exhibited excellent bactericidal activity from 2 hours post inoculation, reaching a cfu mL−1 count below the limit of detection, and maintaining this activity up until 24 hours (Fig. 3). Compound 13f exhibited superior activity as compared to 13a, since it reduced the cfu mL−1 count further. From 6 hours post inoculation up until 24 hours post inoculation, sterilizing activity was achieved, without any signs of bacterial re-growth, indicating that 13f successfully restored the potency of meropenem (Fig. 3). Therefore, 13a and 13f are both efficacious MBL inhibitors.
 | ||
| Fig. 3 Time kill curves of 13a and 13f + meropenem over 24 h. The bacterial control (green curve) utilized K. pneumoniae NDM (USA-449) as the carbapenem-resistant strain, without inclusion of any antibiotics. Compound 13f (orange curve) and meropenem (blue curve) were administered to K. pneumoniae NDM (USA-449) alone, at 16 mg L−1 and 1 mg L−1, respectively. The combination of 13f and meropenem at 16 + 1 mg L−1 is depicted by a purple curve and the combination of 13a and meropenem at 16 + 1 mg L−1 is shown by a grey curve. The limit of detection (LoD) used was 100 cfu mL−1 (1 × 102). | ||
The cytotoxicity of the MBL inhibitory compounds (10, 12, 13a/13f, 14a/14b; Fig. S31, ESI†), were studied using the cell viability assay (HepG2 cells) and the lactate dehydrogenase assay. The MBL inhibitory compounds were non-toxic up to a concentration of 200 μg mL−1. This indicated that the compounds are safe to use and can advance to in vivo studies.
Metal chelating agents such as 13a, 13f, 14a and 14b, have great potential for use as MBL inhibitors. However, they are known to have poor specificity and may chelate other human metallo-proteins that contain zinc,49 in addition to the MBL of interest. Recombinant human glyoxylase II is a zinc-containing protein that is actively involved in the detoxification of reactive dicarbonyls such as methylglyoxal via the metabolic pathway.50 Thus, an enzymatic assay was conducted to determine if inhibitors 13a, 13f, 14a and 14b possessed specific inhibition towards the MBL (Fig. 4). EDTA and TPEN, commercially available chelators, were included as controls to compare the specificity of 13a/13f and 14a/14b. The presence of EDTA and TPEN from concentrations of 150 μM and 300 μM, respectively, reduced the activity of glyoxylase II significantly to about 30% (Fig. 4). In contrast, 13a/13f and 14a/14b did not significantly interfere with the glyoxylase activity, as evidenced by a >90% activity rate (Fig. 4). This confirms that compounds 13a/13f and 14a/14b do not bind to the zinc ions at the active site of glyoxylase II, as compared to commercial chelators, EDTA and TPEN. This further indicates that 13a, 13f, 14a and 14b are specific inhibitors of MBL enzymes, with the potential to replace EDTA and TPEN.
 | ||
| Fig. 4 Glyoxylase II activity in the presence of varying inhibitor concentrations. Normal glyoxylase II activity was observed in the absence of any inhibitor. Compounds 13a, 13f, 14a and 14b did not reduce the activity of glyoxylase II significantly as compared to EDTA and TPEN. Statistical significance was denoted by p < 0.005 (**). The experiment was conducted in triplicate. | ||
Conclusion
We have successfully synthesized eight novel MBL inhibitors based on 1,4,7-triazacyclononanes linked to various β-lactams (compounds 13a–f and 14a-b). These compounds demonstrate the ability to restore the MIC of meropenem to an impressive range of 0.03–2 mg L−1 against MBL-harboring pathogens at concentrations of 8–64 mg L−1. They also exhibited superior MIC activity compared to their NOTA analogues against E. coli NDM-1 and K. pneumoniae NDM, indicating a potential advantage attributed to the better binding affinity of the pyridyl arms in contrast to the oxygen donors of the NOTA derivatives. Interestingly, MIC values remained consistent despite variations in linker length between the chelator and β-lactam (from a six-carbon chain in 13a and 13f to a two-carbon chain in 14a and 14b), indicating a robust and stable inhibitory performance. Time-kill assays indicated that 13a and 13f, each in combination with meropenem, achieved excellent bactericidal activity over 24 hours, without bacterial regrowth, suggesting effective synergistic potential. Cytotoxicity assessments in HepG2 cells confirmed these compounds are non-toxic and safe to use in biological studies. Additionally, specific MBL-inhibitory activity was directed by 13a/13f and 14a/14b, demonstrating non-interference with the activity of zinc-containing enzyme, glyoxylase II. Thus, suggesting that 13a/13f and 14a/14b do not possess off-target specificity. These promising findings position compounds 13a/13f and 14a/14b as promising MBL inhibitory candidates for further pre-clinical evaluation. Ongoing studies aim to extend these evaluations by assessing acute toxicity, pharmacokinetics, and bioavailability to further advance these inhibitors toward therapeutic application.Data availability
Experimental procedures and data supporting the results or analyses presented in the paper can be found as ESI.†Author contributions
MS, TN and TG (conception and design), MS, NR, TGh, KG, RS, AK, AC, HK, PA, DT, TG and TN (data collection, analysis and interpretation of the data) MS and NR (drafting and writing of the paper), MS, NR, TGh, KG, RS, AK, AC, HK, PIA, DT, TG and TN (revising it critically for intellectual content), HK, PIA, TG and TN (funding, supervision and resources), TG and TN (final approval of the version to be published). All authors agree to be accountable for all aspects of the work.Conflicts of interest
The authors report there are no competing interests to declare.Acknowledgements
The South African National Research Foundation grant no. 120419, 137979, 145774, 105236, 105216, 105303, SAMRC BRICS JAF 2021/033, and the Technology Innovation Agency of South Africa (UKZN_17-18_1). The authors wish to thank Patrice Nordmann and David P. Nicolau for the CRE and NDM strain, respectively.References
- World Health Organization(WHO), New report calls for urgent action to avert antimicrobial resistance crisis, https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis, accessed 05 June 2023.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, S. Achalapong, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F.-X. Babin, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, J. A. Berkley, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, R. Clotaire Donatien, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Duong Bich, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, C. Garcia, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, Y. Hsia, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, A. W. J. Jenney, M. Khorana, S. Khusuwan, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, K. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, A. Manoharan, F. Marks, J. May, M. Mayxay, N. Mturi, T. Munera-Huertas, P. Musicha, L. A. Musila, M. M. Mussi-Pinhata, R. N. Naidu, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, T. J. Ochoa, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, P. Ounchanum, G. D. Pak, J. L. Paredes, A. Y. Peleg, C. Perrone, T. Phe, K. Phommasone, N. Plakkal, A. Ponce-De-Leon, M. Raad, T. Ramdin, S. Rattanavong, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, V. D. Rosenthal, K. E. Rudd, N. Russell, H. S. Sader, W. Saengchan, J. Schnall, J. A. G. Scott, S. Seekaew, M. Sharland, M. Shivamallappa, J. Sifuentes-Osornio, A. J. Simpson, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Tigoi, C. Turner, P. Turner, H. R. Van Doorn, S. Velaphi, A. Vongpradith, M. Vongsouvath, H. Vu, T. Walsh, J. L. Walson, S. Waner, T. Wangrangsimakul, P. Wannapinij, T. Wozniak, T. E. M. W. Young Sharma, K. C. Yu, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655, DOI:10.1016/s0140-6736(21)02724-0.
- M. E. A. De Kraker, A. J. Stewardson and S. Harbarth, PLoS Med., 2016, 13, e1002184, DOI:10.1371/journal.pmed.1002184.
- B. Mandal, P. K. Ghosh and B. Basu, ChemInform, 2010, 42, 261–311 Search PubMed.
- S. Alfei and A. M. Schito, Pharmaceuticals, 2022, 15, 476, DOI:10.3390/ph15040476.
- D. Kim, S. Kim, Y. Kwon, Y. Kim, H. Park, K. Kwak, H. Lee, J. H. Lee, K.-M. Jang and D. Kim, Biomol. Ther., 2023, 31, 141, DOI:10.4062/biomolther.2023.008.
- K. Bush, Expert Rev. Anti-infect. Ther., 2023, 21, 513–522, DOI:10.1080/14787210.2023.2194633.
- A. Akhtar, N. Fatima and H. M. Khan, Beta-Lactam Resistance in Gram-Negative Bacteria: Threats and Challenges, 2022, pp. 25–33, DOI:10.1007/978-981-16-9097-6.
- M. F. Mojica, M.-A. Rossi, A. J. Vila and R. A. Bonomo, Lancet Infect. Dis., 2022, 22, e28–e34, DOI:10.1016/S1473-3099(20)30868-9.
- G. Bahr, L. J. González and A. J. Vila, Chem. Rev., 2021, 121, 7957–8094, DOI:10.1021/acs.chemrev.1c00138.
- World Health Organization(WHO), Antibacterial products in clinical development for priority pathogens, https://www.who.int/observatories/global-observatory-on-health-research-and-development/monitoring/antibacterial-products-in-clinical-development-for-priority-pathogens, accessed 11 October, 2024.
- N. Aoki, Y. Ishii, K. Tateda, T. Saga, S. Kimura, Y. Kikuchi, T. Kobayashi, Y. Tanabe, H. Tsukada, F. Gejyo and K. Yamaguchi, Antimicrob. Agents Chemother., 2010, 54, 4582–4588, DOI:10.1128/aac.00511-10.
- D. L. Wannigama, A. M. S. Shein, C. Hurst, P. N. Monk, P. Hongsing, P. Phattharapornjaroen, W. G. F. Ditcham, P. Ounjai, T. Saethang and N. Chantaravisoot, iScience, 2023, 26(7), 1–24, DOI:10.1016/j.isci.2023.107215.
- A. M. Somboro, D. Tiwari, L. A. Bester, R. Parboosing, L. Chonco, H. G. Kruger, P. I. Arvidsson, T. Govender, T. Naicker and S. Y. Essack, J. Antimicrob. Chemother., 2015, 70, 1594–1596, DOI:10.1093/jac/dku538.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503–506, DOI:10.1038/nature13445.
- X. Li, J. Zhao, B. Zhang, X. Duan, J. Jiao, W. Wu, Y. Zhou and H. Wang, Front. Microbiol., 2022, 13, 959107, DOI:10.3389/fmicb.2022.959107.
- Y. Yang and K. Bush, FEMS Microbiol. Lett., 1996, 137, 193–200, DOI:10.1111/j.1574-6968.1996.tb08105.x.
- K. Nagshetty, B. Shilpa, S. A. Patil, C. Shivannavar and N. Manjula, Adv. Microbiol., 2021, 11, 37, DOI:10.4236/aim.2021.111004.
- A. Y. Chen, P. W. Thomas, A. C. Stewart, A. Bergstrom, Z. Cheng, C. Miller, C. R. Bethel, S. H. Marshall, C. V. Credille, C. L. Riley, R. C. Page, R. A. Bonomo, M. W. Crowder, D. L. Tierney, W. Fast and S. M. Cohen, J. Med. Chem., 2017, 60, 7267–7283, DOI:10.1021/acs.jmedchem.7b00407.
- F. M. Klingler, T. A. Wichelhaus, D. Frank, J. Cuesta-Bernal, J. El-Delik, H. F. Müller, H. Sjuts, S. Göttig, A. Koenigs, K. M. Pos, D. Pogoryelov and E. Proschak, J. Med. Chem., 2015, 58, 3626–3630, DOI:10.1021/jm501844d.
- Y. Yusof, D. T. C. Tan, O. K. Arjomandi, G. Schenk and R. P. McGeary, Bioorg. Med. Chem. Lett., 2016, 26, 1589–1593, DOI:10.1016/j.bmcl.2016.02.007.
- R. A. Bonomo, Cold Spring Harb. Perspect. Med., 2017, 7, a025239, DOI:10.1101/cshperspect.a025239.
- A. Prandina, S. Radix, M. Le Borgne, L. P. Jordheim, Z. Bousfiha, C. Fröhlich, H.-K. S. Leiros, Ø. Samuelsen, E. Frøvold, P. Rongved and O. A. H. Åstrand, Tetrahedron, 2019, 75, 1525–1540, DOI:10.1016/j.tet.2019.02.004.
- K. F. Omolabi, N. Reddy, S. Mdanda, S. Ntshangase, S. D. Singh, H. G. Kruger, T. Naicker, T. Govender and S. Bajinath, FEMS Microbiol. Lett., 2023, 370, 1–7, DOI:10.1093/femsle/fnac122.
- R. Azumah, J. Dutta, A. M. Somboro, M. Ramtahal, L. Chonco, R. Parboosing, L. A. Bester, H. G. Kruger, T. Naicker, S. Y. Essack and T. Govender, J. Appl. Microbiol., 2016, 120, 860–867, DOI:10.1111/jam.13085.
- B. K. Peters, N. Reddy, M. Shungube, L. Girdhari, S. Baijnath, S. Mdanda, L. Chetty, T. Ntombela, T. Arumugam, L. A. Bester, S. D. Singh, A. Chuturgoon, P. I. Arvidsson, G. E. M. Maguire, H. G. Kruger, T. Naicker and T. Govender, ACS Infect. Dis., 2023, 9, 486–496, DOI:10.1021/acsinfecdis.2c00485.
- N. Reddy, L. Girdhari, M. Shungube, A. C. Gouws, B. K. Peters, K. K. Rajbongshi, S. Baijnath, S. Mdanda, T. Ntombela, T. Arumugam, L. A. Bester, S. D. Singh, A. Chuturgoon, P. I. Arvidsson, G. E. M. Maguire, H. G. Kruger, T. Govender and T. Naicker, Antibiotics, 2023, 12, 633, DOI:10.3390/antibiotics12040633.
- M. Shungube, A. K. Hlophe, L. Girdhari, V. T. Sabe, B. B. Peters, N. Reddy, K. F. Omolabi, L. Chetty, T. Arumugam, A. Chuturgoon, H. G. Kruger, P. I. Arvidsson, H.-L. Qin, T. Naicker and T. Govender, RSC Adv., 2023, 13, 18991–19001, 10.1039/D3RA02490C.
- R. Cammack and M. N. Hughes, Considerations for the specification of enzyme assays involving metal ions, in Proceedings of the 3rd Beilstein ESCEC Symposium Experimental Standard Conditions Of Enzyme Characterization, Rüdesheim, Germany, 2008 Search PubMed.
- A. Guillou, L. M. P. Lima, M. Roger, D. Esteban-Gómez, R. Delgado, C. Platas-Iglesias, V. Patinec and R. Tripier, Eur. J. Inorg. Chem., 2017, 2017, 2435–2443, DOI:10.1002/ejic.201700176.
- I. Pereira-García, A. Macías, R. Bastida and L. Valencia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 65, m84–m85, DOI:10.1107/s1600536808041895.
- M. Roger, L. M. P. Lima, M. Frindel, C. Platas-Iglesias, J.-F. Gestin, R. Delgado, V. Patinec and R. Tripier, Inorg. Chem., 2013, 52, 5246–5259, DOI:10.1021/ic400174r.
- T. J. Atkins, J. Am. Chem. Soc., 1980, 102, 6364–6365, DOI:10.1021/ja00540a044.
- J. M. Erhardt, E. R. Grover and J. D. Wuest, J. Am. Chem. Soc., 1980, 102, 6365–6369, DOI:10.1021/ja00540a045.
- G. Gasser, L. Tjioe, B. Graham, M. J. Belousoff, S. Juran, M. Walther, J.-U. Kuenstler, R. Bergmann, H. Stephan and L. Spiccia, Bioconjugate Chem., 2008, 19, 719–730, DOI:10.1021/bc700396e.
- P. Nordmann, L. Poirel and L. Dortet, Emerg. Infect. Dis., 2012, 18, 1503, DOI:10.3201/Feid1809.120355.
- S. H. MacVane, J. L. Crandon, W. W. Nichols and D. P. Nicolau, Antimicrob. Agents Chemother., 2014, 58, 7007–7009 CrossRef PubMed.
- M. H. Hsieh, M. Y. Chen, L. Y. Victor and J. W. Chow, Diagn. Microbiol. Infect. Dis., 1993, 16, 343–349, DOI:10.1016/0732-8893(93)90087-N.
- Clinical and Laboratory Standards Institute, Performance Standards for Antimicrobial Susceptibility Testing: Approved Twenty: Document M100-S28, 2018, https://clsi.org/media/1930/m100ed28_sample.pdf Search PubMed.
- S. C. Sosibo, A. M. Somboro, D. G. Amoako, J. Osei Sekyere, L. A. Bester, J. C. Ngila, D. D. Sun and H. M. Kumalo, Microb. Drug Resist., 2019, 25, 439–449, DOI:10.1089/mdr.2018.0272.
- Ø. Samuelsen, O. A. H. Åstrand, C. Fröhlich, A. Heikal, S. Skagseth, T. J. O. Carlsen, H.-K. S. Leiros, A. Bayer, C. Schnaars and G. Kildahl-Andersen, Antimicrob. Agents Chemother., 2020, 64(6), e02415, DOI:10.1128/aac.02415-02419.
- A. Guillou, L. Lima, M. Roger, D. Esteban-Gómez, R. Delgado, C. Platas-Iglesias, P. Veronique and R. Tripier, Eur. J. Inorg. Chem., 2017, 2017, 2435, DOI:10.1002/ejic.201700176.
- G. Gasser, L. Tjioe, B. Graham, M. J. Belousoff, S. Juran, M. Walther, J. U. Künstler, R. Bergmann, H. Stephan and L. Spiccia, Bioconjug. Chem., 2008, 19, 719–730, DOI:10.1021/bc700396e.
- J. M. Erhardt, E. R. Grover and J. D. Wuest, J. Am. Chem. Soc., 1980, 102, 6365–6369, DOI:10.1021/ja00540a045.
- D. S. Macmillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596, 10.1039/c2gc36900a.
- M. H. Hsieh, C. M. Yu, V. L. Yu and J. W. Chow, Diagn. Microbiol. Infect. Dis., 1993, 16, 343–349, DOI:10.1016/0732-8893(93)90087-n.
- P. U. Wayne, Performance Standards for Antimicrobial Susceptibility Testing: Approved Twenty [Document M100-S28], https://clsi.org/media/1930/m100ed28_sample.pdf.
- K. H. M. E. Tehrani and N. I. Martin, MedChemComm, 2018, 9, 1439–1456, 10.1039/C8MD00342D.
- N. Wade, K. H. Tehrani, N. C. Brüchle, M. J. van Haren, V. Mashayekhi and N. I. Martin, ChemMedChem, 2021, 16, 1651–1659, DOI:10.1002/cmdc.202100042.
- A. Scirè, L. Cianfruglia, C. Minnelli, B. Romaldi, E. Laudadio, R. Galeazzi, C. Antognelli and T. Armeni, Antioxidants, 2022, 11, 2131, DOI:10.3390/antiox11112131.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01842k |
| This journal is © The Royal Society of Chemistry 2025 |