
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient formation of C3 and C4 hydrocarbons from cellulose over Pt/Mg-doped ZrO2 catalysts without hydrogen addition†
Yukino Ofuchia,
Mihiro Hosokawaa,
Naruki Horieb,
Sae Doia,
Shuhei Ogo b,
Ayumu Ondab,
Tatsuya Hamaguchic,
Takafumi Saikic and
Yasushi Sekine*a
b,
Ayumu Ondab,
Tatsuya Hamaguchic,
Takafumi Saikic and
Yasushi Sekine*a
aWaseda University, Tokyo, Japan. E-mail: ysekine@waseda.jp
bKochi University, Kochi, Japan
cAstomos Energy Corporation, Tokyo, Japan
First published on 20th May 2025
Abstract
At low temperatures, C3 and C4 hydrocarbons (equivalent to propane gas) were produced using only water, a solid catalyst, and cellulose, employing no enzymes or expensive, valuable hydrogen. High C3 + C4 yields were achieved using a catalyst doped with Mg as a base site in ZrO2, which has Lewis acid and base properties, supported with Pt, which has high hydrocarbon production capacity. After screening and characterisation of this catalyst using various values of parameters such as the amount of Mg doping and the amount of Pt loading, findings indicated that 1 wt% Pt caused moderate decarbonylation and dehydration, and that the Zr0.5Mg0.5O2−δ support promoted cellulose degradation by its Lewis acid–base properties. The reaction mechanism was investigated, clarifying the C3 and C4 hydrocarbon formation mechanisms.
1. Introduction
For the transition away from the use of fossil resources to alleviate global environmental difficulties, high hopes persist that biomass, a renewable resource, will be rationally convertible into hydrocarbons that are useful as fuel.1 Among biomass resources, the use of cellulose is being considered. It is the main component of woody biomass and cannot be digested by humans. For those reasons, it does not compete with food production. Unfortunately, cellulose has strong beta-1,4 glycosidic bonds. Therefore, its hydrolysis is complex.2 Thus, homogeneous catalysts such as strong acids have been proposed for the chemical conversion of cellulose. However, these methods have disadvantages such as difficult separation, high cost, and corrosion of equipment.3 Moreover, direct conversion of cellulose to hydrocarbons necessitates the progression of various reactions such as dehydration, hydrogenation, and decarbonation, making it a complicated reaction.4,5The use of solid catalysts for cellulose hydrolysis has been extensively investigated.6–9 Takegaki et al. reported that layered metal oxides, which are both strongly acidic and water-resistant, exhibit high catalytic activity in the hydrolysis of sugars.8 Onda et al. showed that carbon sulfonate catalysts exhibit high yield and selectivity in the hydrolysis of cellulose to glucose.6 Furthermore, it has been reported that the hydrolysis of cellulose and its reduction to sugar alcohols is promoted by adding pressurized hydrogen and using metal-supported catalysts.10,11 Shrotri et al. reported that Ru- and Ni-supported carbon catalysts were highly active in the conversion of cellulose to sorbitol.10 Fukuoka et al. synthesized sugar alcohols from cellulose in 31% yield using Pt/γ-Al2O3.11 Sugar alcohols can be converted further to hydrocarbons by reducing them under hydrogen pressure.12 These approaches, however, necessitate a multi-step process to convert cellulose to hydrocarbons. Therefore, direct conversion of cellulose to hydrocarbon has also been investigated.13–17 Murata et al. performed a hydrothermal reaction of pretreated cellulose with Pt/H-ZSM-5 under hydrogen pressurized conditions and obtained C2–C9 alkanes in 89% yield.13 Although, these research examples require pressurised hydrogen, which entails high costs. Using a Pt-loaded zeolite catalyst, we have produced hydrocarbons from cellulose successfully in an environmentally friendly way without pressurised hydrogen.18–20 However, zeolite is prone to clogging, resulting in limited reusability, other supports must be considered.
Brønsted and Lewis acids are thought to be effective for converting bio-derived sugars.21,22 Although, it is known that the Lewis acid sites of metal oxides lose their functionality due to the coordination of water molecules in water.23 On the other hand, it has been reported that the dehydration reaction is promoted in water by a water-resistant Lewis acid.23–27 For example, Nakajima et al. performed glucose conversion in water using Nb2O5·nH2O, a water-resistant Lewis acid, and obtained HMF in 52.1% yield. Similarly, ZrO2 is also known as a water-resistant Lewis acid.28,29 In addition, it has been reported that the introduction of base sites by doping with different metals can promote dehydration and isomerization reactions.26 Mg2+ is a strong Lewis acid among group II elements, and doping is expected to generate strong base sites.30 Mg2+ has also been reported to promote solid hydrolysis of cellulose.31 Moreover, the Lewis acidity can be changed by doping cations with different ionic radii from the matrix, causing distortion in the bonds between the metal oxides.32
To conduct a direct cellulose conversion reaction to hydrocarbons without pressurising hydrogen, we used a Pt-loaded ZrO2 catalyst doped with a base site for this study. Among hydrocarbons, C3 and C4 hydrocarbons have high potential for use as liquefied petroleum gas and chemical raw materials. Therefore, for this study, we specifically examined C3 and C4 hydrocarbons. When a Pt-loaded Mg-doped ZrO2 catalyst was used, high C3 and C4 selectivity in the gas phase was achieved. This high selectivity was suggested to be attributable to enhancement of the Lewis acidity and basicity of ZrO2 and to suppression of the over-degradation of sugars by Mg doping. In addition, we clarified the possible hydrocarbon formation pathway that was followed when using this catalyst.
2. Experimental
2.1 Preparation of catalysts
For this study, we prepared catalyst supports with Lewis acids and bases such as ZrO2, Zr0.5M0.5O2−δ (M = Mg, Ca, or Sr), and Mg-doped ZrO2 with different doping ratios, using the citric acid complex polymerization method. As precursors, ZrO(NO3)2·2H2O and various metal nitrates were used. Citric acid and ethylene glycol were added to an aqueous solution of metal nitrates at a molar ratio of metal cation: citric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylene glycol of 1:3:3. After the solution was dried and calcined for 10 h at 673 K, Pt was loaded on the support using Pt(NH4)3(NO3)2 as a metal precursor with an impregnation method. Calcination was conducted at 773 K for 1 h. Catalysts were used with no pre-treatment.
:ethylene glycol of 1:3:3. After the solution was dried and calcined for 10 h at 673 K, Pt was loaded on the support using Pt(NH4)3(NO3)2 as a metal precursor with an impregnation method. Calcination was conducted at 773 K for 1 h. Catalysts were used with no pre-treatment.
2.2 Characterization
The catalytic structures and cellulose were examined using powder X-ray diffraction (XRD, Smart Lab III; Rigaku Corp.) with Cu Kα radiation under the conditions of 40 kV and 40 mA, with a 10–90° measurement range. To ascertain the specific surface area of the prepared catalysts, analyses were conducted using an automatic specific surface area measuring instrument (Gemini VII 2390; Micromeritics Inst. Corp.). The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) method from N2 adsorption results after 1 h of He purges at 473 K before the measurement. The metal amount of catalysts was ascertained using inductively coupled plasma (ICP; Agilent Technologies Inc.). The Pt particle diameter and dispersion were measured using CO pulse adsorption (BEL-CAT; Microtrac-Bel Corp.) after pretreatment in H2 at 773 K for 30 min. Acid and base amounts of catalysts were examined respectively using NH3-TPD and CO2-TPD (temperature-programmed desorption of NH3 and CO2, BEL-CAT; Microtrac-Bel Corp.). Metal distribution in the catalyst was confirmed using transmission electron microscopy (TEM, JEM-2100F; JEOL Ltd). The fine structures of Zr and supported Pt were observed using XAFS for Zr K-edge and Pt L3-edge at BL14B2 station of SPring-8 in Japan. Before XAFS measurement, the sample was pressed into a pellet (ϕ 10 mm). Measurements were taken at room temperature using transmission mode. Fourier transformation of k3-weighted EXAFS spectra was obtained in the Δk range of 3–14 Å−1. Subsequently, the spectra after Fourier transformation were fitted with standard samples: Zr foil for Zr–Zr bond and ZrO2 for Zr–O bond, Pt foil for Pt–Pt bond, and PtO2 for Pt–O bond. EXAFS analyses were done using software: xTunes.2.3 Catalytic activity test
Catalytic reactions were performed using a batch-wise autoclave (30 mL, SUS316; Fig. S1 in ESI†) with the following reaction conditions: water:cellulose:catalyst = 20 mL:0.25 g:0.25 g; 443–493 K temperature; 0–168 h time; 600 rpm stirring rate. For the starting material change test, 9.251 C mmol (0.25 g for cellulose) of raw material was used for the reaction. Cellulose reportedly improves solubility by decreasing crystallinity.33 Therefore, before the catalytic reaction, cellulose powder (microcrystalline; MP Biomedicals) was pre-treated using a planetary ball mill (Pulverisette 6 classic line; Fritsch GmbH) to break the crystal structure as shown in Fig. S2 in ESI.† The ball mill rotation speed was 600 rpm for 180 min (30 min × 6 times). The catalytic reactions were conducted under N2 atmosphere. The reaction starting time was recorded when the reaction temperature reached the preset temperature. After the reaction, the reactor was cooled. After it reached room temperature, the increase in the amount of gas was measured quantitatively. Specifically, after a large syringe was attached to the end of the valve (1) in Fig. S1,† valve (1) was opened. The amount of gas which entered the syringe was measured. The total volume of gas was calculated by combining the volume of space in the reaction tube and this increase in gas volume. After valve (2) in Fig. S1† was opened, gas was collected from the sampling port. Gaseous products were assessed using two gas chromatographs: a GC-FID (GC-8A, CP-SilicaPLOT, CP8567; Agilent Technologies Inc.) and a GC-TCD (GC-2014, Shincarbon-ST 50-80; Shimadzu Corp.). Then, the reaction mixture was separated using filtration. The filtrate solution was examined using HPLC (L-2490 RI detector; Hitachi Ltd/Diode Array Detector; Hitachi Ltd/L-2350 column; Hitachi Ltd). The total amount of liquiform products in the resultant solution after reactions was measured using a total organic carbon (TOC) analyser (TOC-V CPN; Shimadzu Corp.). In gaseous products, H2, CO, CO2, and C1–C4 hydrocarbons were detected. In liquiform products, glucose, cellobiose, furfural, 5-hydroxymetyl-furfural (5-HMF), C1–C4 alcohols, ketones, and organic acids were detected. Cellulose conversion and product yield were calculated on a carbon basis and were defined as presented below (eqn (1) and (2)).
 | (1) |
 | (2) |
The weight of unreacted solid cellulose was ascertained by subtracting the weight of charged catalyst from the weight of the remaining solid. Carbon balance was calculated based on eqn (3).
 | (3) |
The selectivity of the target product, C3 + C4 hydrocarbon, in gas was calculated from eqn (4).
 | (4) |
3. Results and discussion
3.1 Catalytic activities for various catalysts
Earlier reports have described that Lewis acids in metal oxides are effective for the conversion reactions of bio-derived sugars.21,22 Therefore, we conducted cellulose conversion tests using a catalyst in which Pt, which has been reported to have high hydrocarbon generation ability,18 was loaded onto metal oxides with Lewis acids such as ZrO2 and TiO2. The results are presented in Fig. 1. The amount of gas produced was in the order of TiO2 > ZrO2 > Ta2O5 > CeO2 for the supports used, but the amounts of target C3 and C4 hydrocarbons were similar for all supports except CeO2. However, the Ta2O5 support produced a large amount of C4 hydrocarbons, ZrO2 produced a good balance of hydrocarbons, and TiO2 produced a large amount of CO2 and C1 and C2 hydrocarbons. Therefore, the selectivity of C3+C4 in the gas was high for ZrO2 and Ta2O5 supports. | ||
| Fig. 1 (a) Gaseous product yield and (b) hydrocarbon yield obtained using 1 wt% Pt/support (TiO2, ZrO2, Ta2O5, CeO2), cellulose, 443 K, 12 h. | ||
The NH3-TPD and CO2-TPD results for these catalysts are shown in Fig. S3 in ESI.† The CO2-TPD profiles showed relatively large desorption peaks for TiO2 and CeO2 supports. These catalysts exhibited high CO2 selectivity, indicating that basic sites might promote the decarbonylation reaction. Furthermore, NH3-TPD measurements indicated that the amount of acid sites increased in the order: CeO2 < ZrO2 < Ta2O5 < TiO2 for the supports used. The 1 wt% Pt/CeO2 catalyst, characterized by the lowest acidity and highest basicity, yielded the minimum amount of hydrocarbons. This finding implies that acid sites are required for hydrocarbon production.
Based on the results of this screening, we decided on further studies of ZrO2 support, which had high selectivity for C3 + C4 in the gas and which produced a good balance of hydrocarbons.
Results showed that ZrO2, which has a water-resistant Lewis acid–base,28,29 can convert cellulose to hydrocarbons by loading Pt,18 which reportedly has high hydrocarbon production capacity. Furthermore, based on a report describing that introduction of base sites promotes the dehydration reaction,26 cellulose conversion was conducted at 443 K for 12 h using a catalyst in which basic sites were introduced by doping a Group II element into ZrO2: 1 wt% Pt/Zr0.5M0.5O2−δ (M = Zr, Mg, Ca, Sr). The results are presented in Fig. 2. The highest C3 + C4 yield and gas selectivity were obtained when Mg was doped.
 | ||
| Fig. 2 (a) Gaseous product yield and (b) hydrocarbon yield obtained using 1 wt% Pt/Zr0.5M0.5O2−δ (M = Zr, Mg, Ca, Sr), cellulose, 443 K, 12 h. Liquid products are shown in Fig. S4 in ESI.† | ||
When ZrO2 is doped with different cations, they interact with ZrO2 to produce acid–base sites of various strengths.34 Among the Group II elements doped in this experiment, Mg is the strongest Lewis acid, and the doping product exhibits relatively strong Lewis acid–base, which probably promoted the target reaction.
To confirm the Mg doping effects, 1 wt% Pt/Zr1−xMgxO2−δ (x = 0, 0.1, 0.3, 0.5, 1) with varying doping amounts were prepared. The results of XAFS measurements focusing on the Zr-K edge for these catalysts are shown in Fig. S5 in ESI.† The chemical bonding states of Zr was analyzed from the Fourier transforms of the obtained EXAFS oscillations. As a result, the peak of the second coordination (Zr–Zr or Zr–Mg) was observed to shift towards shorter bond distances, whereas the first coordination (Zr–O bond) did not change markedly with increasing Mg doping. This result, which is consistent with earlier reported results,35 also suggests that Mg was doped into the ZrO2 crystal.
The results using 1 wt% Pt/Zr1−xMgxO2−δ (x = 0, 0.1, 0.3, 0.5, 1) are depicted in Fig. 3. The amount of hydrocarbon produced increased as the degree of Mg doping increased. The C3 + C4 selectivity in the gas phase was maximum at x = 0.5. However, when 1 wt% Pt/MgO (x = 1) was used, the amount of light hydrocarbons such as methane and ethane produced increased. The C3 + C4 selectivity decreased.
 | ||
| Fig. 3 (a) Gaseous product yield and (b) hydrocarbon yield obtained using 1 wt% Pt/Zr1−xMgxO2−δ (x = 0, 0.1, 0.3, 0.5, 1), cellulose, 443 K, 12 h. Liquid products are shown in Fig. S7 in ESI.† | ||
Furthermore, the results of comparing Mg-doped catalysts and MgO mixed catalysts, in which the amount of Zr and Mg in the catalyst was adjusted using 1 wt% Pt/Zr0.5Mg0.5O2−δ and 1 wt% Pt/(ZrO2 + MgO), are portrayed in Fig. 4. The results showed that the Mg-doped catalysts produced a higher proportion of C3 hydrocarbons, whereas the mixed catalysts and MgO supported catalysts produced a higher proportion of C1 and C2 hydrocarbons.
 | ||
| Fig. 4 (a) Gaseous product yield and (b) hydrocarbon yield obtained using 1 wt% Pt/support (ZrO2, MgO, mixed, doped), cellulose, 443 K, 12 h. Liquid products are shown in Fig. S8 in ESI.† | ||
To investigate the acidity and basicity of catalysts, NH3-TPD and CO2-TPD were conducted. As Fig. S6 in ESI† shows, the amount of acid and base sites was 1 wt% Pt/ZrO2 < 1 wt% Pt/Zr0.5Mg0.5O2−δ < 1 wt% Pt/MgO. The MgO support had both weak and strong sites. The peaks were split into two components, whereas the Zr0.5Mg0.5O2−δ had a peak with a broad distribution, probably because the acid and base sites of ZrO2 are strengthened by Mg doping. Simultaneously, acid and base sites of various strengths are generated.
Base catalysis has been reported to promote the conversion of sugars through the reverse aldol reaction,36,37 and the reverse aldol reaction can cause excessive degradation of the carbon structure. Therefore, when MgO-supported catalyst with very strong base sites was used, the retro-aldol reaction was promoted, resulting in a higher proportion of light hydrocarbons. Moreover, when the Zr0.5Mg0.5O2−δ with acid–base sites of different strengths was used, it is possible that its various active sites promoted reaction pathways other than the retro-aldol reaction, such as dehydration reactions, which probably resulted in high C3 + C4 yields.
Furthermore, the results of changing the supported metal using the Zr0.5Mg0.5O2−δ support are presented in Fig. 5. Even when using this support, only when Pt was used was the production of a large amount of hydrocarbons confirmed, as described in earlier reports.18 When using Pt, CO2 and H2 were produced, but when using Pd, CO and a smaller amount of H2 were confirmed. These products suggest that on Pt, the decarbonylation reaction (–CO) occurs first; then the water gas shift reaction (CO + H2O → CO2 + H2) is more likely to occur than on Pd. More H2 is thought to be produced when Pt is used in this reaction. Moreover, this H2 is thought to act as a hydrogen source for the production of hydrocarbons, leading to a higher hydrocarbon production rate. In addition, several monoalcohols that were not produced when other metals were used were confirmed in the solution after the reaction when Pt was used, suggesting promotion of the hydrogenation dehydration reaction, which removes the OH group from the sugar.
 | ||
| Fig. 5 (a) Gaseous product yield and (b) hydrocarbon yield using 1 wt% M/Zr0.5Mg0.5O2−δ (M = Pt, Pd, Ru), cellulose, 443 K, 12 h. Liquid products are shown in Fig. S9 in ESI.† | ||
Results of conversion tests using X wt% Pt/Zr0.5Mg0.5O2−δ (X = 0, 1, 3, 5, 10) with varying Pt loadings are depicted in Fig. 6. The amount of gas produced increased as the Pt loading increased, but the C3 + C4 selectivity in the gas decreased, especially with increased CO2, methane, and ethane.
 | ||
| Fig. 6 (a) Gaseous product yields and (b) hydrocarbon yield using X wt% Pt/Zr0.5Mg0.5O2−δ (X = 0, 1, 3, 5, 10), cellulose, 443 K, 12 h. Liquid products are shown in Fig. S10 in ESI.† | ||
For X wt% Pt/Zr0.5Mg0.5O2−δ (X = 0, 1, 3, 5, 10), XAFS measurements were performed focusing on the Pt-L edge (Fig. S11 in ESI†). As the Pt loading increased, the proportion of Pt oxide decreased, whereas the proportion and coordination number of Pt–Pt increased. Similar to the results of particle size analysis by CO pulse measurement, it is clear that Pt particles are growing with increasing Pt loading (Table S1†).
The reason for the increase in CO2, C1 and C2 hydrocarbons with higher Pt loading is thought to be that the increase in Pt loading accelerates the decarbonylation reaction. The decarbonylation reaction was accelerated for three main reasons. The first is that the frequency of contact between Pt and the raw material increased. The second is that a larger amount of loaded Pt is associated with larger Pt particle size, and that the decarbonylation reaction occurred as consecutive reactions with the reactants adsorbed onto Pt. The third is that a larger particle size is associated with a greater exposed surface area of the Pt, which contributes to the decarbonylation reaction. The (311) and (331) planes are thought to be exposed on Pt particles of 5 nm or greater size.38 Moreover, it is possible that the decarbonylation reaction was promoted on such planes. Table S2† shows the results of specific surface area measurements for the catalysts used in the experiment. 1 wt% Pt/Zr0.5M0.5O2−δ (M = Zr, Mg, Ca, Sr) had a higher specific surface area when Mg was doped and with increasing doping amount. On the other hand, specific surface area did not change significantly when the metal loading was changed, and there was no clear correlation between the loading amount and specific surface area. The results of characterization for 1 wt% Pt/Zr0.5Mg0.5O2−δ are shown. The ICP analysis results are shown in Table S3 in ESI.† Regarding the percentages of the metals present, the theoretical values and experimental values obtained by ICP were in general agreement. TEM and EDS mapping images of the catalyst are shown in Fig. S12 in ESI.† Results showed that the Pt, Zr, Mg, and O elements were uniformly distributed.
3.2 Testing under different reaction conditions
Using a 1 wt% Pt/Zr0.5Mg0.5O2−δ catalyst with high C3 + C4 selectivity in the gas phase, we conducted tests under different reaction conditions to achieve the highest yield. Results of the tests, in which the temperature, time, pressure, water volume and amount of raw material were varied, are shown in the Table S4 in ESI.† When the tests were conducted at 443–493 K, the CO2 content increased rapidly at temperatures higher than 473 K. This rapid increase is probably attributable to the accelerated thermal decomposition of cellulose and its derivatives. Therefore, a reaction temperature of 443–463 K is suitable. When the reaction time was varied from 12 h to 168 h, the amounts of hydrocarbons produced were not proportional to the time, suggesting that hydrocarbon production is higher in early stages of the reaction. Therefore, repeating short reactions of around 12 h was inferred as effective for yield improvement. When the pressure at the start of heating was changed to 1–2.5 atm by pressurization with nitrogen, the results demonstrated that the hydrocarbon production amount decreased as the pressure increased. Because gas production was suppressed at higher pressures, it was thought that yield could be improved by increasing the space volume or reducing raw materials. Therefore, the amount of water was reduced first to increase the volume of space inside the reaction tube. The results showed that yield improved. This improvement is probably attributable not only to the increase in the volume of space, but also to the increased frequency of contact between the raw materials and intermediate substances and the catalyst. Furthermore, results showed that the yield could be improved even by further reduction of the amount of raw materials. This improvement is probably achieved because the pressure of the generated gas at the same yield decreased as the amount of raw materials decreased, and also because the amount of catalyst per unit of raw material increased.Based on the results of the reaction condition change tests, the raw material amount was reduced to 0.05 g. Repeated tests were conducted at 453 K for 12 h. Cumulative results of hydrocarbon production are presented in Fig. S13 in ESI.† As a result of this test, it was possible to increase the yield by 6.34 times from the first 12 h test while maintaining high C3 + C4 selectivity in the gas phase. Particularly, the amount of C4 production increased as the reaction was repeated. This increase is probably attributable to changes in the composition of the liquid product, which is an intermediate, and because the amounts of mono-alcohol and di-alcohol, which are more likely to become C4 hydrocarbons, increase during the latter half of the test. Although these tests are conducted in batches using an autoclave, based on these test results, the possibility exists of further improving the yield by carrying out the reaction in a continuous system.
Results of tests using fresh 1 wt% Pt/Zr0.5Mg0.5O2−δ catalysts and heat-treated catalysts after use in the reaction are shown in Fig. S14.† Almost no change was observed in the hydrocarbon product distribution, confirming the reusability of the catalyst. XRD patterns of the Mg-doped ZrO2 catalyst before the reaction are shown in Fig. S15–S16 in ESI.† The crystal structure of the catalyst was monoclinic ZrO2. A small peak derived from MgO was observed in the Zr0.5Mg0.5O2−δ support, suggesting precipitation of undoped Mg as MgO. On the other hand, the XRD pattern of the solid residue after the reaction is shown in Fig. S17 in ESI.† After the reaction, the MgO peak had disappeared and a carbon peak was confirmed which is thought to be derived from unreacted cellulose. When this solid residue was heat-treated at 673 K, the carbon-derived peak disappeared; the MgO-derived peak also reappeared. This result suggests that the MgO peak disappeared as a result of the transformation of MgO into hydrated magnesium carbonate in the presence of H2O and CO2 in the solid residue after the reaction,39,40 and the catalyst was regenerated by heat treatment. The results of ICP analysis of the catalyst and the liquid phase after the reaction are shown in Table S5.† The amount of Mg in the catalyst was slightly reduced by the reaction, and this reduction was generally consistent with the amount of Mg ions in the solution after the reaction.
3.3 Elucidation of the reaction mechanism
To investigate which properties of the catalyst are effective for which reactions, we compared the results obtained from reactions conducted without a catalyst, reactions using only a support, and reactions using a catalyst with a trace amount of Pt (0.1 wt% Pt) or a catalyst with a large amount of Pt (1 wt% Pt). The results are presented in Fig. 7. Only when Pt was present was hydrocarbon formation observed clearly. The liquid component was dominated by organic acids such as lactic acid and formic acid. However, when using the 1 wt% Pt catalyst, monohydric alcohols such as butanol and propanol were also produced. These results clarified that Pt promoted the hydrogenation dehydration reaction, which removes the OH group from the sugar, as occured during the metal change tests. In addition, the amount of products formed in the liquid increased in the order: without catalyst < ZrO2 support < Mg-containing support, suggesting that the Lewis acids and bases of the support promoted cellulose degradation.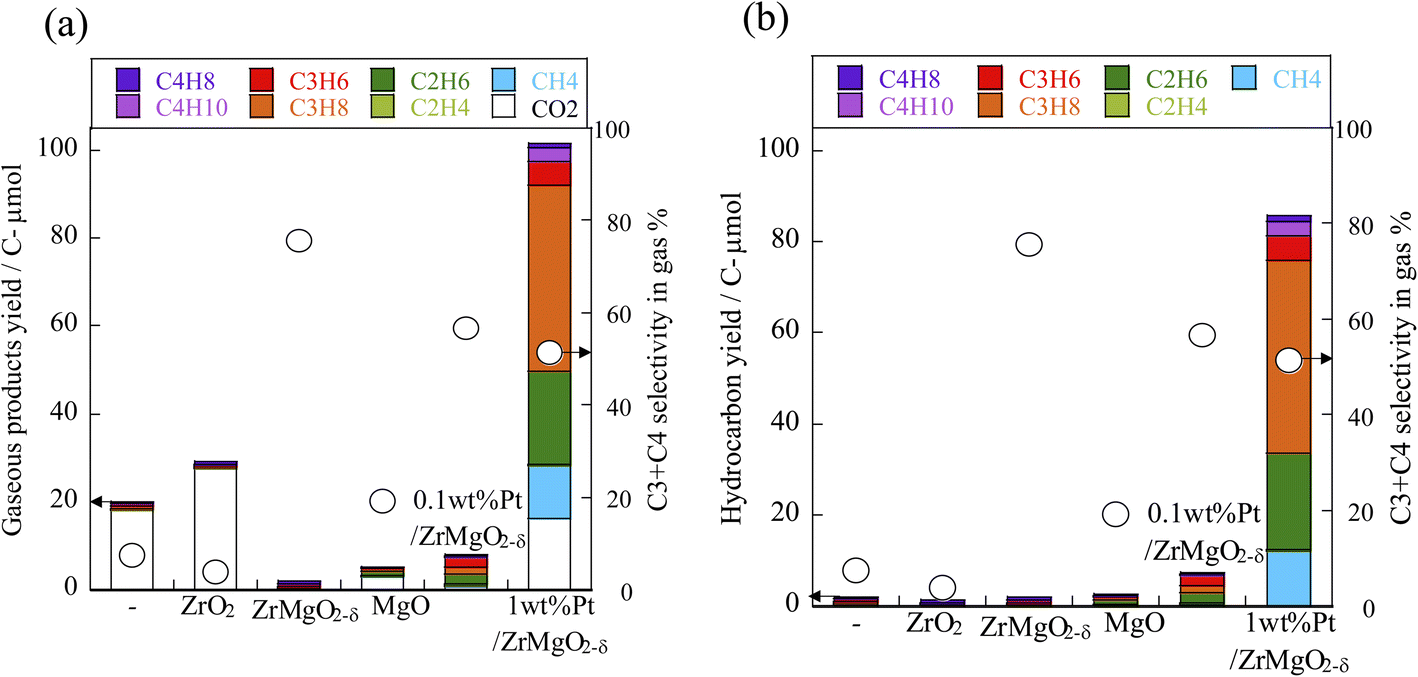 | ||
| Fig. 7 (a) Gaseous product yield and (b) hydrocarbon yield using X wt% Pt/Zr1−xMgxO2−δ (X = 0, 0.1, 1) (x = 0, 0.5, 1), cellulose, 443 K, 12 h. Liquid products are shown in Fig. S18 in ESI.† | ||
Reportedly, there are numerous by-products of the cellulose degradation reaction and the degradation reaction of glucose, a monosaccharide.41–43 Determining whether these substances are reaction intermediates, or not, and investigating their reactivity are difficult. For this study, we investigated the reactivity of mono-alcohols and di-alcohols having similar structure to that of the target hydrocarbons. Furthermore, we clarified the reactions which produce hydrocarbons. Results of conversion from hydrocarbon-like liquid substances are presented in Fig. 8. During conversion from 1-butanol, a primary alcohol, approximately five times as much hydrocarbon were produced during conversion from 2-butanol, a secondary alcohol. Therefore, results demonstrated that the hydroxy group reactivity is much higher in primary alcohols than in secondary alcohols. In addition, the main product from 1-butanol was propane, which illustrates clearly that the primary hydroxy group was removed together with the carbon. In other words, results demonstrated that the decarbonylation reaction (–CO) is more likely to occur preferentially than the production of hydrocarbons by the hydrogenation dehydration reaction of alcohols. Next, butenol, which has one double bond and one OH group in the molecule, produced far more hydrocarbons than butanol. Also, 1-butene-3-ol mainly produced C4 hydrocarbons, suggesting that the presence of a double bond made the desorption of the OH group by dehydration more likely to occur. In addition, 3-butene-1-ol produced very large quantities of C3 and C4 hydrocarbons in equal amounts. This is due to the formation of C4 hydrocarbons by dehydration in the presence of a double bond and C3 hydrocarbons by decarbonylation of primary alcohols, indicating that both processes are equally probable and are strongly promoted by the presence of a double bond in the molecule. C2 hydrocarbons were obtained from 1,2-butanediol in large quantities; also, C3 hydrocarbons were obtained from 1,3-butanediol in large quantities. Two decarbonylation reactions are thought to have occurred in the former case. In the latter case, because almost no hydrocarbons were produced from the conversion of 2-butanol, it was assumed that only a decarboxylation reaction occurred, leaving a secondary alcohol. However, because many C3 hydrocarbons were produced in which the OH group had been removed, the dehydration reaction is thought to have occurred simultaneously with the decarbonylation reaction. Furthermore, to confirm the reactivity of organic acids such as formic acid, acetic acid, and butyric acid, which were found in the liquid products of cellulose conversion, we also conducted conversion tests using butyric acid (a C4 organic acid) and levulinic acid (a metabolite of glucose). Results indicated that, although C3 hydrocarbons were obtained in small quantities from butyric acid, it was not very reactive. Moreover, almost no hydrocarbons were produced from levulinic acid. Even if a carboxylic acid were obtained from levulinic acid, as in the case of butyric acid, the ketone group would remain. Also, hydrocarbons are not formed, probably because of the low reactivity of ketones.
 | ||
| Fig. 8 Conversion of C3 and C4 hydrocarbon-like liquid compounds using 1 wt% Pt/Zr0.5Mg0.5O2−δ catalyst (1-B: 1-butanol, 2-B: 2-butanol, 1B3ol: 1-butene-3-ol, 3B1ol: 3-butene-1-ol, 1,2-BD: 1,2-butanediol, 1,3-BD: 1,3-butanediol, BA: butyric acid, LA: levulinic acid). | ||
From the test results presented above, we inferred that the hydrocarbon formation reaction occurs as a result of a decarbonylation reaction or a dehydration reaction in the presence of a double bond, and that the dehydration reaction progresses simultaneously with the decarbonylation reaction. The reaction mechanisms which can be inferred from these findings are shown in Fig. 9. Path 1 represents a reaction that starts with a decarbonylation reaction. Path 2 represents a reaction that starts with a retro-aldol reaction that has been reported as promoted by a water-resistant Lewis acid–base catalyst.22
 | ||
| Fig. 9 Estimated reaction mechanisms: Path 1, reactions initiated by decarbonyl reaction; Path 2, reactions initiated by retro-aldol reaction. | ||
Based on this assumption, the results of conversion from the candidate intermediate substances are presented in Fig. 10. The results were as expected, with C2, C3, and C4 hydrocarbons being obtained from the 3,4-dihydroxy-1-butene which was estimated as produced by Path 1. Results demonstrated that a large amount of methane was produced from erythrose, which appears in Path 2. A large amount of methane was also produced from glucose. More methane was produced from cellobiose than from cellulose. From these results, it was inferred that the retro-aldol reaction progressed from disaccharides and monosaccharides, and that light hydrocarbons were produced via erythrose. However, because little methane was produced from cellulose, the C3 production reaction was thought to be more likely to occur preferentially in cellulose conversion. Because reactions are likely to progress at the ends of cellulose,44 as presented in Fig. 11, it was thought that reactions progressed from the open-ringed terminal sugar because of the connected sugar chains. This presumed reaction mechanism is consistent with results obtained from various earlier tests, a possible reaction, and a highly probable C3 and C4 hydrocarbon formation mechanism.
 | ||
| Fig. 10 Conversion of candidate intermediates using 1 wt% Pt/Zr0.5Mg0.5O2−δ, 443 K, 12 h. | ||
 | ||
| Fig. 11 Estimated reaction mechanisms from cellulose over Pt/Zr0.5Mg0.5O2−δ catalyst. | ||
4. Conclusion
In conclusion, high C3 + C4 hydrocarbon yields were obtained when 1 wt% Pt/Zr0.5Mg0.5O2−δ was used, probably because Pt caused decarbonylation and hydrogenation dehydration reactions and because the Zr0.5Mg0.5O2−δ support promoted cellulose degradation with its broad strength of Lewis acids and bases. The reaction proceeds from the end of the cellulose starting from decarbonylation. The composition of the produced hydrocarbons is thought to be controllable by changing the amount of Pt loaded and the basicity of the catalyst. For this study, direct conversion of cellulose to hydrocarbons was achieved successfully using no sacrificing agent such as hydrogen, enzymes, or strong acids. Further yield improvement can be expected by conducting the reaction in a continuous process. The results of this study are expected to be useful for the effective utilization of biomass toward a sustainable society, especially for converting cellulose into LPG and supplying LPG to dispersed locations such as households.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yukino Ofuchi: methodology, investigation, visualization, writing-original draft preparation, Mihiro Hosokawa: investigation, visualization, writing (revising the manuscript), Naruki Horie: investigation (analysis of liquid products), Sae Doi: methodology, investigation, Shuhei Ogo: methodology, conceptualization, Ayumu Onda: methodology, conceptualization, Tatsuya Hamaguchi: methodology, Takafumi Saiki: methodology, Yasushi Sekine: writing-reviewing and editing, conceptualization, methodology, supervision.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors thank Ms. Chinatsu Matsuda for her dedicated contribution to this research. Also, we thank Mr Hiromu Akiyama (Waseda University) for his excellent assistance with XAFS analysis. The TEM measurement experiment was performed at the Joint Research Center for Environmentally Conscious Technologies in Materials Science at ZAIKEN, Waseda University (Grant No. JPMXS0440500024, MEXT, Japan). A part of this work was supported by Demonstration Project of Innovative Catalyst Technology for Decarbonization through Regional Resource Recycling, the Ministry of the Environment, Government of Japan.References
- S. Hansen, A. Mirkouei and L. Diaz, Renewable Sustainable Energy Rev., 2020, 118, 109548 CrossRef CAS.
- M. Zeng and X. Pan, Catal. Rev., 2022, 64, 445–490 CrossRef CAS.
- R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS.
- W. Liu and H. Yu, ACS Environ. Au, 2022, 2, 98–114 CrossRef CAS PubMed.
- P. Mortensen, J. Grunwaldt, P. Jensen, K. Knudsen and A. Jensen, Appl. Catal. A, 2011, 407, 1–19 CrossRef CAS.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS PubMed.
- A. Takagaki, C. Tagusagawa and K. Domen, Chem. Commun., 2008, 5363–5365 RSC.
- A. T. To, P. W. Chung and A. Katz, Angew. Chem., Int. Ed., 2015, 54(38), 11050–11053 CrossRef CAS PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51(3), 761–768 CrossRef CAS PubMed.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- K. Murata, Y. Liu, M. Inaba and I. Takahara, Catal. Lett., 2010, 140, 8–13 CrossRef CAS.
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustain. Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal. B, 2010, 100, 184–189 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- Y. Kato and Y. Sekine, Catal. Lett., 2013, 143(5), 418–423 CrossRef CAS.
- S. Ogo, T. Nishio, H. Sekine, A. Onda and Y. Sekine, Fuel Process. Technol., 2016, 141, 123–129 CrossRef CAS.
- S. Ogo, Y. Okuno, H. Sekine, S. Manabe, T. Yabe, A. Onda and Y. Sekine, ChemistrySelect, 2017, 2(22), 6201–6205 CrossRef CAS.
- S. Yamaguchi and S. Imamura, J. Jpn. Inst. Energy, 2017, 96(10), 408–416 CrossRef CAS.
- L. Lin, X. Han, B. Han and S. Yang, Chem. Soc. Rev., 2021, 50, 11270–11292 RSC.
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS PubMed.
- N. K. Gupta, A. Fukuoka and K. Nakajima, ACS Catal., 2017, 7(4), 2430–2436 CrossRef CAS.
- R. Noma, K. Nakajima, K. Kamata, M. Kitano, S. Hayashi and M. Hara, J. Phys. Chem. C, 2015, 119(30), 17117–17125 CrossRef CAS.
- M. Kim, S. Ronchetti, B. Onida, N. Ichikuni, A. Fukuoka, H. Kato and K. Nakajima, ChemCatChem, 2019, 12, 350–359 CrossRef.
- D. Padovan, K. Endo, T. Matsumoto, T. Yokoi, A. Fukuoka, H. Kato and K. Nakajima, Small Struct., 2022, 4, 2200224 CrossRef.
- T. Komanoya, K. Nakajima, M. Kitano and M. Hara, J. Phys. Chem. C, 2015, 119, 26540–26546 CrossRef CAS.
- P. Wattanapaphawong, P. Reubroycharoen and A. Yamaguchi, RSC Adv., 2017, 7, 18561–18568 RSC.
- S. Bailleul, I. Yarulina, A. E. J. Hoffman, A. Dokania, E. Abou-Hamad, A. D. Chowdhury, G. Pieters, J. Hajek, K. D. Wispelaere, M. Waroquier, J. Gascon and V. V. Speybroeck, J. Am. Chem. Soc., 2019, 141, 14823–14842 CrossRef CAS PubMed.
- N. Shimada, H. Kawamoto and S. Saka, Carbohydr. Res., 2007, 342, 1373–1377 CrossRef CAS PubMed.
- P. Sudarsanam, H. Li and T. V. Sagar, ACS Catal., 2020, 10(16), 9555–9584 CrossRef CAS.
- A. Onda, J. Jpn. Pet. Inst., 2012, 55, 73–86 CrossRef CAS.
- M. Miyamoto, A. Hamajima, Y. Oumi and S. Uemiya, Int. J. Hydrogen Energy, 2018, 43, 730–738 CrossRef CAS.
- V. V. Kriventsov, J. Surf. Invest., 2023, 17, 1483–1489 CrossRef CAS.
- D. Esposito and M. Antonietti, ChemSusChem, 2013, 6, 989–992 CrossRef CAS.
- X. Yan, F. Jin, K. Tohji, A. Kishita and H. Enomoto, AIChE J., 2010, 56, 2727–2733 CrossRef CAS.
- A. Nakano, S. Manabe, T. Higo, H. Seki, S. Nagatake, T. Yabe, S. Ogo, T. Nagatsuka, Y. Sugiura, H. Iki and Y. Sekine, Appl. Catal. A, 2017, 543, 75–81 CrossRef CAS.
- L. Hopkinson, P. Kristova, K. Rutt and G. Cressey, Geochim. Cosmochim. Acta, 2012, 76, 1–13 CrossRef CAS.
- C. Rincke, H. Schmidt, G. Buthb and W. Voigta, Acta Crystallogr., Sect. C:Struct. Chem., 2020, 76, 741–745 CrossRef CAS.
- Q. Wang, H. Song, S. Pan, N. Dong, X. Wang and S. Sun, Sci. Rep., 2020, 10, 3626 CrossRef CAS PubMed.
- A. Bayu, A. Abudula and G. Guan, Fuel Process. Technol., 2019, 196, 106162 CrossRef CAS.
- H. Kawamoto, J. Wood Sci., 2015, 61, 1–24 CrossRef CAS.
- T. Hosoya and S. Sakaki, ChemSusChem, 2013, 6, 2356–2368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01826a |
| This journal is © The Royal Society of Chemistry 2025 |