
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast charge transfer dynamics in excited-state donor–acceptor benzylideneaniline†
Prajoy Kumar Mitra ,
Preetika Verma and
Yapamanu Adithya Lakshmanna*
,
Preetika Verma and
Yapamanu Adithya Lakshmanna*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Vithura, Thiruvananthapuram 695551, India. E-mail: adithya@iisertvm.ac.in
First published on 17th April 2025
Abstract
Imine-based molecular systems have served as promising semi-conductive materials for applications in light-emitting diodes and photovoltaics. Benzylideneaniline, an imine-based molecular system, acts as a bioisostere of diarylethylenes such as stilbene and resveratrol, and plays a crucial role in pharmaceutical and biological applications. However, the excited-state dynamics of benzylideneanilines is less explored. In this context, we investigated 4′-(dimethylamino)benzylidene-4-nitroaniline (DMABNA), an imine-based donor–acceptor system, to unravel the excited-state dynamics. Here, we explored excited-state charge transfer dynamics and associated structural dynamics in various solvent environments through femtosecond transient absorption (fs-TA) and femtosecond fluorescence spectroscopic methods. The fs-TA measurements revealed distinct spectral and temporal features from cyclohexane to acetonitrile indicating that DMABNA essentially undergoes significant configurational changes in the excited state, leading to intramolecular charge transfer (ICT), particularly in polar environments. The fs-fluorescence measurements reveal the ultrashort lifetime (a few ps) of fluorescence across all the solvents indicating an effective non-radiative relaxation in the excited electronic state. These studies are corroborated by the computational analysis through DFT and TDDFT methods, wherein we predicted that DMABNA undergoes significant structural changes in the excited state (S1) due to varying solvent polarity and viscosity. Such excited state molecular activity can play a significant role in the context of photo-initiated applications.
Introduction
Semiconducting organic materials have garnered significant attention due to their inherent electrical, optoelectronic, and processing advantages, making them highly desirable for the development of a new class of electronic devices.1,2 These materials are utilized in advanced technologies, including organic light-emitting diodes, solar cells, field-effect transistors, electrophotographic systems, photoreceptors, and electrochromic devices.3–6 One of the fundamental phenomena driving the functionality of these materials is photoinduced charge transfer, a process observed both in natural systems like photosynthesis and in artificial photonic devices such as photovoltaics.7–9 During photoinduced charge transfer, light energy is converted into chemical energy stored within photoactive molecules by forming charge transfer states. A wide range of research efforts have been directed towards understanding the molecular mechanisms underlying these charge transfer processes at the molecular level.10–14In charge transfer systems such as donor–acceptor stilbenes, the intramolecular charge transfer (ICT) process is accompanied by twisting of either the electron donor or the acceptor group, facilitating charge separation and preventing back-ICT reactions.15–20 Twisting of the electron donor or acceptor groups in a molecule can indeed improve charge separation, help in minimizing the donor and acceptor orbital overlap, and facilitate the charge transfer, in a process known as the twisted intramolecular charge transfer (TICT).21–28 The formation of a planarized intramolecular charge transfer (PICT) state is crucial for maximizing the efficiency of charge separation processes within molecules.29–35 However, the excited dynamics of donor–acceptor benzylideneanilines remains relatively underexplored to date as compared to stilbene derivatives.
Herein, we utilized a model system 4′-(dimethylamino)benzylidene-4-nitroaniline (DMABNA), an imine-based donor–acceptor system capable of exhibiting an extended π-conjugation as shown in Chart 1. The N,N-dimethylamino group (–N(CH3)2) acts as the electron donor, while the nitro group (–NO2) serves as the electron acceptor, facilitating intramolecular charge transfer (ICT). Such benzylidene derivative exhibits tunable electronic properties, making it a promising candidate for optoelectronic applications.36–39 Compared to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond found in stilbene, a notable distinction is the presence of lone pair of electrons on the nitrogen atom in the –CN bond of benzylideneaniline, which significantly affects the conformation of the compound. The π-conjugation from the donor moiety (dimethylamino benzene) to the acceptor group (nitrobenzene) via the –CN link might be disrupted owing to the non-planar conformation of DMABNA. However, conformational changes in the geometry may increase the π-conjugation and can restore the conjugation across the molecule.40–42 The transition from a non-planar geometry is analyzed with respect to rotations across the four possible single bonds (1, 2, 3, 4 as numbered in Chart 1) in the excited state (S1) geometry of DMABNA for varying solvent polarity. In this article, we also elucidate systematically the roles of ICT, PICT, and TICT processes through their distinct spectral characteristics, providing a comprehensive understanding of their contributions to the excited-state structural dynamics. The study expands our fundamental understanding of this molecule, which has not been investigated to date from the ultrafast dynamics perspective, and lays the groundwork for future applications in electronic device technologies.
C bond found in stilbene, a notable distinction is the presence of lone pair of electrons on the nitrogen atom in the –CN bond of benzylideneaniline, which significantly affects the conformation of the compound. The π-conjugation from the donor moiety (dimethylamino benzene) to the acceptor group (nitrobenzene) via the –CN link might be disrupted owing to the non-planar conformation of DMABNA. However, conformational changes in the geometry may increase the π-conjugation and can restore the conjugation across the molecule.40–42 The transition from a non-planar geometry is analyzed with respect to rotations across the four possible single bonds (1, 2, 3, 4 as numbered in Chart 1) in the excited state (S1) geometry of DMABNA for varying solvent polarity. In this article, we also elucidate systematically the roles of ICT, PICT, and TICT processes through their distinct spectral characteristics, providing a comprehensive understanding of their contributions to the excited-state structural dynamics. The study expands our fundamental understanding of this molecule, which has not been investigated to date from the ultrafast dynamics perspective, and lays the groundwork for future applications in electronic device technologies.
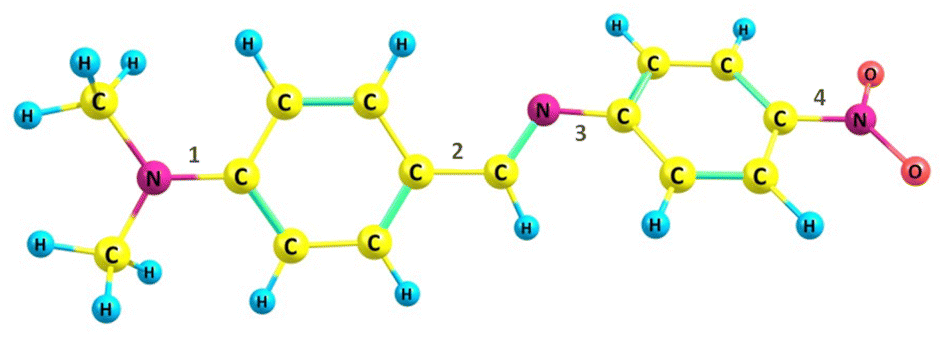 | ||
| Chart 1 Molecular structure of DMABNA. | ||
Experimental methods
Sample preparation
4′-(Dimethylamino)benzylidene-4-nitro aniline (DMABNA) was purchased from TCI and was used without any further purification. All the solvents, such as cyclohexane (CHX), toluene (TOL), 1,4-dioxane (DOX), ethyl acetate (EA), acetone (AC), and acetonitrile (MeCN), used in the experiments are from Spectrochem (HPLC grade) and polyethylene glycol 300 (PEG 300) obtained from Sigma-Aldrich and used without further purification.Steady-state spectroscopic measurements
Steady-state absorption spectra were measured using a Shimadzu UV-3600 ultraviolet-visible-near-infrared (UV-vis-NIR) spectrophotometer, and fluorescence emission spectra were measured using a Horiba Jobin Yvon-Fluorolog 3 spectrofluorometer. The steady-state measurements were performed using a cuvette of 1 cm path length.Femtosecond transient absorption (fs-TA) measurements
The setup consists of a Ti-sapphire-based amplifier system from Spectra-Physics, which produces fundamental laser light having a central wavelength at 800 nm, a pulse width of 100 fs, a pulse energy of ∼5 mJ, and a repetition rate of 1 kHz. The output beam is split into two parts: one beam is used to pump an optical parametric amplifier (OPA), and a small portion of the remaining energy is used to generate a white-light continuum from a sapphire plate that is further split into two parts, one of which acts as a probe while the other acts as a reference beam. OPA generates a pump beam in the ultraviolet region of 380 nm with beam parameters of 100 fs, ∼10 μJ, and 1 kHz to create excited-state molecules. The fs-TA measurements are performed by a spatiotemporal overlapping of the pump and the probe beams in a 1.0 mm thick cuvette with a continuous XY-axes motorized movement (beam propagation direction is chosen as z-axis) to avoid sample degradation. The probe beams are dispersed in the spectrometer and are detected by a dual-array detector. The excited-state absorption signal is obtained by modulating the pump beam with a mechanical chopper at 500 Hz for each time delay point. The TA measurements were carried out by using pump energy at ∼800 nJ with a focal spot of the beam at the sample is of 100 μm. The sample concentrations of 1.5 mM of DMABNA were employed for transient absorption measurements.Femtosecond fluorescence up-conversion measurements
The femtosecond fluorescence up-conversion measurements were carried out using the FOG-100 system from CDP systems. The excitation of the sample was achieved through the second harmonic generation of the fundamental output originating from Mai Tai HP (with a central wavelength of ∼800 nm, pulse width of 100 fs, and repetition rate of 80 MHz) at 400 nm. The resulting fluorescence and residual fundamental beams were spatially overlapped within a nonlinear β-barium borate (BBO) crystal to generate a sum frequency signal, generating an upconverted fluorescence signal. Subsequently, the upconverted signal traversed through a double monochromator before being detected by a highly sensitive photomultiplier tube coupled with a photon counter. The sample concentrations of 1.5 mM of DMABNA were employed for fluorescence up-conversion measurements.Computational studies
Theoretical calculations were carried out by using density functional theory (DFT) in the Gaussian 16 suite.43 The B3LYP functional with the 6-311G(d,p) split-valence basis set was employed to perform these calculations. Optimization of the structures has been performed in the solvent phases using the Integral Equation Formalism Polarizable Continuum Model (IEFPCM). The Franck–Condon vertical excitation energies and oscillator strengths were obtained using time-dependent density functional theory (TDDFT). Further, we analyzed the frontier orbitals (HOMO and LUMO) to understand the distribution of the electron density and performed dihedral scans to analyse the potential energy profiles.Results and discussion
Steady-state measurements
The steady-state absorption and emission measurements of DMABNA in solvents of varying polarity were recorded, and the spectra are shown in Fig. 1. The absorption spectrum consists of two bands, one at ∼300 nm and the other at ∼390 nm, that are attributed to π–π* transition and the n–π* transitions, respectively. The absorption maximum exhibits a slight red-shift from 381 nm in CHX to ∼397 nm in polar solvent, MeCN. Such a red-shift in absorption clearly indicates the polar nature of DMABNA. | ||
| Fig. 1 Steady-state (a) absorption, and (b) emission spectra of DMABNA in solvents of varying polarities with a sample concentration of 25 μM. | ||
The emission spectra in all the solvents were measured by exciting at a wavelength corresponding to their absorption maximum. The fluorescence spectrum of DMABNA in CHX exhibits a structured band possessing a maximum of ∼455 nm with a Stokes shift of 4300 cm−1. The emission spectra are noticed to be broad and featureless in the case of polar solvents, EA, AC, and MeCN, having maxima at ∼600 nm (Stokes shift of 8000 cm−1), ∼695 nm (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1), and ∼730 nm (11200 cm−1), respectively. A large Stokes shift in polar solvents is a clear indication that DMABNA undergoes significant intramolecular charge transfer (ICT) upon excitation and stabilization of such a state in polar environments. Furthermore, the emission spectrum of DMABNA in MeCN exhibits dual-band with maxima at ∼520 nm and ∼730 nm. In comparison with the emission spectrum in a non-polar solvent, the bands at ∼520 nm band and ∼730 nm in MeCN can be assigned to the emission from the locally excited-state and relaxed ICT states, respectively. The spectral features associated with the absorption and emission spectra of DMABNA in all the solvents are summarized in Table 1.
000 cm−1), and ∼730 nm (11200 cm−1), respectively. A large Stokes shift in polar solvents is a clear indication that DMABNA undergoes significant intramolecular charge transfer (ICT) upon excitation and stabilization of such a state in polar environments. Furthermore, the emission spectrum of DMABNA in MeCN exhibits dual-band with maxima at ∼520 nm and ∼730 nm. In comparison with the emission spectrum in a non-polar solvent, the bands at ∼520 nm band and ∼730 nm in MeCN can be assigned to the emission from the locally excited-state and relaxed ICT states, respectively. The spectral features associated with the absorption and emission spectra of DMABNA in all the solvents are summarized in Table 1.
| Solvents | ε | λabs (nm) | λfl (nm) | FWHM of the fluorescence band (nm) |
|---|---|---|---|---|
| CHX | 2.02 | 380 | 455 | 65.2 ± 0.9 |
| TOL | 2.38 | 395 | 515 | 77.0 ± 0.9 |
| DOX | 2.21 | 391 | 550 | 88.4 ± 1.0 |
| EA | 6.02 | 393 | 600 | 114.9 ± 2.1 |
| AC | 20.56 | 396 | 695 | 136.1 ± 0.8 |
| MeCN | 35.94 | 397 | 730 | 123.5 ± 1.7 |
Further, the Lippert–Mataga plot was constructed based on the parameters obtained in these solvents using eqn (1), and depicted in Fig. 2. The plot exhibits a linear relationship between solvent polarity and the Stokes shift. It clearly indicates that excited-state species possess higher dipole moments, resulting in an effective ICT process in a polar environment.
 | (1) |
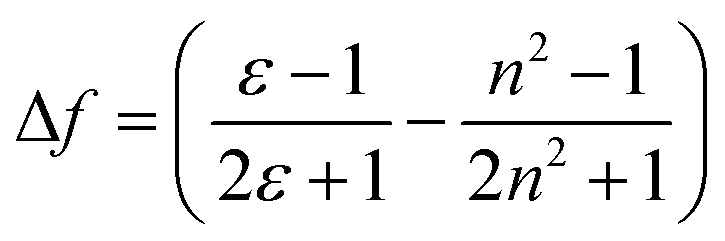 .
.
 | ||
| Fig. 2 Lippert–Mataga plot of DMABNA in solvents of different polarity. | ||
Here, Δν−1 is the Stokes shift in wavenumbers (cm−1), Δμ is the difference in the dipole moment between the ground (μg) and excited (μe) states. c, h and a are the speed of light, Planck's constant, and the radius of the Onsager cavity around the fluorophore, respectively. The solvent dielectric constant (ε) and refractive index (n) are included in the term Δf, which is known as the solvent polarity parameter.
Furthermore, DMABNA exhibits solvent polarity-dependent fluorescence quantum yields. The fluorescence quantum yield in CHX is notably minimal, of the order 10−3. A marginal increase in solvent polarity in EA results in a substantial increase in the fluorescence quantum yield by more than two orders of magnitude.15,17 However, with an increase in solvent polarity, i.e., in case of AC, the fluorescence yield experiences an increment. The molecule also exhibits a markedly weak fluorescence quantum yield in MeCN, as a result, the emission spectrum is burred when compared to the relatively smooth spectra in other solvents. Such solvent polarity-dependent emission yields suggest that the excited-state evolves through diverse relaxation pathways, particularly non-radiative relaxation pathways that play a significant role in the excited-state dynamics of DMABNA. To further analyze the excited-state dynamics of DMABNA, we carried out femtosecond transient absorption and femtosecond fluorescence up-conversion measurements in various solvents.
Femtosecond transient absorption (fs-TA) measurements
 | ||
| Fig. 3 The fs-TA spectra of DMABNA in CHX at different time-delays up to (a) 150 ps, (b) 1 ns, (c) species-associated spectra, SAS3 and SAS4 are plotted separately as depicted in the inset, and (d) kinetics of the decay-associated spectra. The time constants are provided in the figures. | ||
 | ||
| Fig. 4 (a) The fs-TA spectra of DMABNA in MeCN at different time delays, (b) species-associated spectra, and (c) kinetics of the decay-associated spectra. | ||
We carried out single wavelength amplitude kinetics associated with various regions of the TA spectra in CHX and MeCN, and the corresponding plots are depicted in Fig. 5a and b, respectively. In CHX, wavelengths were chosen at 500 nm, 580 nm, and 700 nm for the analysis. The kinetics at these wavelengths were best fitted with bi- and tri-exponential fittings, and we observed that for 500 nm and 580 nm, the amplitude decays (represented by ‘d’) with a time scale of ∼50 and ∼100 ps. Whereas at 700 nm, the evolution of the amplitude is described by the sequential rise and decay components with time constants of ∼2.4 ps, ∼89 ps, respectively. Further, it exhibits a rise component for longer time delays.
 | ||
| Fig. 5 Single wavelength kinetics of TA spectra of DMABNA along with the best-fit in (a) CHX at 500 nm, 580 nm, and 700 nm, and in (b) MeCN at 500 nm, 600 nm, and 700 nm. The lifetimes are shown in the figures, and ‘d’ and ‘r’ represent decay and rise components, respectively. | ||
In the case of MeCN, single-wavelength kinetic traces at 500 nm, 600 nm, and 700 nm were analyzed and depicted in Fig. 5b. The kinetics at these wavelengths were optimally fitted using single and bi-exponential models, with time constants of approximately ∼0.3 ps and ∼4.0 ps. The time constant values obtained are in good agreement with the global fitting parameters. The fs-TA spectra in other solvents, including TOL, DOX, EA, and AC, are presented in Fig. S1 of ESI.† In DOX, the transient absorption spectrum is predominantly centered at ∼500 nm for the initial delay times. It is also apparent from the spectra that, within a time-delay of ∼0.3 ps, a negative band with significant amplitude appears at ∼540 nm and a shoulder band appears at ∼590 nm. For subsequent time delays of ∼2 ps, the amplitude at ∼540 experiences a decrease, while the amplitude at ∼590 nm exhibits an increment. The transient absorption is characterized by a positive band centered at ∼500 nm and a broad negative band centered at ∼590 nm. The SE features match with the steady-state emission of DMABNA in DOX. The ESA band exhibits a blue shift in a time scale of ∼2 ps, while the SE band exhibits a red shift from ∼540 nm to ∼590 nm. The substantial shift in the SE maximum from ∼540 nm to ∼590 nm in DOX is a clear indication of the stabilization of the polar excited species by the polar DOX. The fs-TA data was globally analyzed, and it was best fitted with three time constants having ∼1.0 ps, 2.6 ps, and ∼75 ps. The fast time component of ∼1.0 ps is attributed to the relaxation of the Franck–Condon (FC) state, the time-component of ∼2.6 ps is attributed to the lifetime of the relaxed excited-state species/relaxed charge-transferred state, and ∼75 ps is the lifetime of the S1 state. The single wavelength kinetics at a few selective wavelengths of 500 nm, 540 nm, and 600 nm with the best-fits are shown in Fig. S2(b).† It is evident from the plots that amplitudes are best fit with bi-exponentials, and they decay with a time constant of ∼80 ps. In a moderate polarity solvent (EA), DMABNA exhibits the highest fluorescence quantum yield of ∼7.55% as compared to other solvents, including polar solvents, indicating that excited-state relaxation to the ground-state through the radiative channel is prominent. The fs-TA spectra of DMABNA in EA are shown in Fig. S1(c).† Upon photoexcitation, TA spectra are characterized by ESA centered at ∼500 nm, and broad SE band centered at ∼580 nm. The SE band exhibits a red-shift for the initial time delays and possesses broad bands centered at ∼600 nm at longer time delays. For subsequent time delays, the amplitudes of the ESA and SE bands exhibit a systematic decrease. The fs-TA spectra were globally analyzed, and the data was best fitted with three time constants of ∼0.9 ps, 2.4 ps and 548 ps. The time components of ∼0.9 ps, 2.4 ps and 548 ps are attributed to solvent relaxation, vibrational relaxation/structural relaxation of the charge-transferred (CT) state, and lifetime of the relaxed CT state, respectively.
A detailed summary of the time constants obtained through global analysis in solvents of varying polarity is presented in Table 3. For DMABNA, the excited-state lifetime in the non-polar solvent CHX is notably longer compared to solvents with different polarities. Interestingly, the non-radiative rate constant in CHX is also found to be quite high (Table 2), suggesting the possibility of a non-radiative relaxation pathway from the S1 excited state. In contrast, the decay time in TOL is significantly shorter (∼21 ps), likely due to a decrease in the non-radiative decay time. As the solvent polarity transitions from low (TOL) to moderate (EA), a gradual increase in lifetime is observed, rising substantially from ∼21 ps in TOL to ∼548 ps in EA. This extended lifetime in EA may be attributed to its higher quantum yield, which facilitates enhanced relaxation in the CT state.
| Solvents | Φfl (10−2) | τ1 (ps) | τ2 (ps) | kr (108 s−1) | knr (1011 s−1) |
|---|---|---|---|---|---|
| CHX | 0.16 | 0.3 | 1.5 | 10.7 | 6.66 |
| TOL | 0.25 | 0.9 | 7.5 | 3.33 | 1.33 |
| DOX | 0.48 | 3.3 | 33.7 | 1.42 | 0.29 |
| EA | 7.55 | 13.6 | 198.7 | 3.80 | 0.04 |
| AC | 0.43 | 0.9 | 15.4 | 2.79 | 0.64 |
| MeCN | 0.02 | 0.2 | 2.1 | 1.05 | 4.76 |
| Solvents | η (cP) | τ1 (ps) | τ2 (ps) | τ3 (ps) | τ4 (ps) |
|---|---|---|---|---|---|
| CHX | 0.89 | 0.6 | 3.2 | 99.8 | >1000 |
| TOL | 0.56 | 0.9 | 3.4 | 21.1 | — |
| DOX | 1.17 | 1.0 | 2.6 | 75.0 | — |
| EA | 0.42 | 0.9 | 2.4 | 548.8 | — |
| AC | 0.30 | 0.8 | 18.9 | — | — |
| MeCN | 0.36 | 0.3 | 4.2 | — | — |
| PEG 300 | 80* | 4.7 | 22.8 | 94.1 | — |
However, in highly polar solvents, a sharp decrease in decay time is observed, with lifetimes dropping to ∼18 ps in AC and ∼4 ps in MeCN. This drastic reduction in lifetime in highly polar environments can likely be attributed to ultrafast solvent dynamics that accelerate non-radiative relaxation to the ground state. The relatively longer lifetime of DMABNA in CHX and its significantly shorter lifetime in MeCN can be better understood by comparing it with its structural analog, 4-dimethylamino-4′-nitrostilbene (DMANS). The primary structural difference between DMANS and DMABNA lies in substituting a –CH group with a nitrogen atom (N). Extensive studies on the excited-state dynamics of DMANS have been conducted, including work by Palit and co-workers,15 who demonstrated how the relaxation processes of DMANS drastically change with a change in solvent polarity. DMANS in CHX, at an initial time delay of 0.2 ps, exhibits a weak SE band in the 480–550 nm region, along with an ESA band between 570–650 nm, peaking at 590 nm with a shoulder near 630 nm. At longer time delays, both the SE band and the ESA band below 650 nm decay, while a new weak ESA band emerges in the 650–800 nm range, with a maximum at 720 nm. This band is attributed to the lowest triplet excited state. They further analyzed and quantitatively determined the lifetimes of various relaxation processes using a tri-exponential fitting. Three relaxation processes were identified: (1) vibrational relaxation with a time constant of τ1 = 4.5 ps, (2) isomerization around the olefinic double bond with a time constant of τ2 = 27 ps, as previously reported, and (3) a longer component with τ3 ≈ 600 ps, corresponding to the lifetime of the S1 state.15 Comparable behavior was observed for DMABNA in CHX, which also exhibits similar relaxation processes. However, the best fit was obtained with a four-exponential fitting. The fastest component (0.6 ps) can be ascribed to the Franck–Condon state, the second component (3.2 ps) to the vibrational relaxation, the intermediate component (99.8 ps) to the isomerization involving the CN bond, and the long-lived component (>1 ns) near 700 nm is likely associated with the triplet state.46,47
In contrast, DMANS in a highly polar solvent like MeCN displayed time constants of <0.15 ps, 0.5 ps, and 16 ps, corresponding to vibrational relaxation and/or solvation for the fastest component and decay of the LE state and the S1 state for the other components, respectively. Similarly, DMABNA in MeCN showed comparable dynamics, with a bi-exponential fit revealing time constants of 0.3 ps, likely due to solvation or the LE state, and 4 ps for the S1 state decay. This rapid decay is likely due to strong solute–solvent interactions and ultrafast solvation dynamics, where the excessive stabilization of the CT state may enhance coupling between the excited and ground states, promoting faster non-radiative relaxation pathways. A similar, short lifetime is also seen in highly polar solvent AC, as shown in Fig. S1(d).† Furthermore, we conducted fluorescence up-conversion experiments to characterize the emissive lifetime of DMABNA in various solvents. Additionally, we incorporated the excited state dynamics in a viscous solvent (PEG 300) and a detailed theoretical analysis was performed to investigate the conformational changes of DMABNA in polar solvents.
Time-resolved fluorescence measurements
Time-resolved fluorescence measurements were carried out to investigate the excited-state dynamics of emissive states utilizing the femtosecond fluorescence up-conversion technique with an excitation wavelength of 400 nm. The emission decay kinetics were recorded at the corresponding emission maxima in each solvent, as illustrated in Fig. 2. The decay kinetics were best fitted with bi-exponentials with time constants of τ1 and τ2 that are tabulated in Table 2. In the case of CHX, the τ2 lifetime value is found to be just 1.5 ps, which is much lower as compared to the longest time constant obtained in fs-TA analysis. This is because the relaxation from the excited S1 state is mostly via a non-radiative pathway rather than a radiative manner, as supported by its higher non-radiative rate constant (Table 2). However, a gradual increment in lifetime, i.e., lifetimes of 7.5 ps and ∼33 ps, were observed in TOL and DOX, respectively. In the case of EA, the emissive lifetime is noticed to be of ∼198 ps, as supported by an enhanced fluorescence quantum yield in EA. The Lippert–Mataga's plot clearly depicts that the emissive state lifetime is significantly affected by the solvent polarity. Interestingly, the fluorescence lifetimes in high polarity solvents such as AC and MeCN are found to be ∼15.4 ps and ∼2.1 ps, respectively. Such huge changes in the lifetime in polar solvents reveal that the excited state evolves through a state which may be significantly polar in nature and an effective stabilization by the polar environment (Fig. 6). | ||
| Fig. 6 The fs-fluorescence up-conversion spectra of DMABNA in solvents of varying polarity collected at their emission maxima. The solid lines represent the fitting and the time constants are shown in the inset. | ||
Fig. S3c† represents the fs-TA transient absorption spectra of DMABNA in PEG 300 for different time delays. The fs-TA spectra features positive band (ESA) centered at ∼500 nm and negative band around ∼550 nm. The negative band is attributed to SE as the spectral feature matches with the steady-state emission. As the time delay increases, the ESA band exhibits decay of its amplitude. While in case of SE, it exhibits a red-shift to ∼580 nm along with the decay of its amplitude. The fs-TA data was globally analyzed and the data was best fitted with three time components at τ1 ∼ 4.7 ps, τ2 ∼ 22.8 ps, and τ3 ∼ 94 ps. The species-associated spectra and the kinetics profiles of these species are represented in Fig. S3d and e,† respectively. The time components of 4.7 ps, 22.8 ps, and 94 ps are attributed to the formation of the CT state, structural relaxation of CT state, and the lifetime of the relaxed CT state, respectively. The τ1 and τ2 components are noticed to be longer as compared to those in other solvents, particularly in moderate to high polar solvents. It is a clear indication that the structural relaxation in both the FC state and the CT state is a slow process as a result of viscous environment leading to the restriction of motion of solute molecules. The fs-TA spectral features and the time components values reveal that the excited-state dynamics of DMABNA in PEG 300 is similar to the dynamics observed in moderately polar solvents. It clearly indicates that the excited-state dynamics of DMABNA in PEG is predominantly governed by the polar nature of the solvent, albeit viscosity playing a significance role.
Computational analysis
We performed density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations to elucidate the geometries of both the ground and excited states, as well as to characterize the charge transfer process that may have a significant effect on the conformational relaxation in the excited state. The B3LYP functional, widely regarded as one of the most reliable hybrid functionals in DFT, was used with the 6-311G(d,p) basis set. The ground state geometry optimizations consistently showed a non-planar geometry with a torsion between the dimethylamino-aniline-imine (DMAI) and the nitrophenyl (NP) ring of DMABNA in the gas phase as well as in solvent media. The obtained parameters are summarized in Table 4. It is apparent from the table that solvent has minimal influence on the ground-state conformation. Frontier molecular orbital analysis provided further insights into the charge transfer characteristics of DMABNA. As depicted in Fig. 7, the HOMO is predominantly localized on the DMAI donor group, while the LUMO exhibits a significant charge density shift toward the NP acceptor moiety upon excitation. This redistribution of electron density is characteristic of ICT in donor–acceptor systems, reinforcing the role of DMABNA as a model for studying such phenomena. DMABNA is a prototype of the donor–acceptor stilbenes, and numerous reports favor the twisting conformation in the excited state.15,17,48 Upon TDDFT calculations, we observed that in the excited-state, DMABNA retains a non-planar geometry, such as in its ground state configuration. Furthermore, to understand the conformational changes taking place in the excited state with respect to the ground state, potential energy surface (PES) scans were performed by introducing twists around specific Σ-bonds (dihedral angles indicated as χ1, χ2, χ3, and χ4) to evaluate the possibility of a twisted-intramolecular charge transfer (TICT) state (Fig. 8).| Gas/solvent | μg (D) | μe (D) | Dihedral angle χ (gs) (degree) | Dihedral angle χ (es) (degree) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |||
| Gas | 11.3 | 10.9 | −0.5 | 1.6 | 42.6 | 0.5 | −1.9 | 4.3 | 32.1 | −0.3 |
| CHX | 12.8 | 12.8 | −0.3 | 1.5 | 41.6 | 0.9 | −1.8 | 1.5 | 35.7 | −0.1 |
| TOL | 13.0 | 13.1 | −0.3 | 1.5 | 41.4 | 1.04 | −1.7 | 1.3 | 36.0 | −0.1 |
| AC | 14.8 | 15.6 | −0.1 | 1.7 | 39.5 | 0.9 | −1.6 | 1.0 | 34.4 | 0.0 |
| MeCN | 15.0 | 15.8 | −0.1 | 1.7 | 39.4 | 0.8 | −1.6 | 1.1 | 34.0 | 0.0 |
 | ||
| Fig. 7 (a) Ground state and (b) excited state optimized geometries of DMABNA in MeCN and their HOMO–LUMO electron densities. | ||
 | ||
| Fig. 8 The PES of DMABNA in the ground (S0) and excited (S1) state in MeCN with respect to different dihedral angles (a) χ1, (b) χ2, (c) χ3, and (d) χ4 varying in the range 0 to 180°. | ||
It has been observed that the rotational barrier in the S0 state for twisting along the four dihedral angles (χ1, χ2, χ3, and χ4) are 0.372, 0.507, 0.073, and 0.337 eV with respect to the maximum at a twist angle of 90° for χ2, χ3, and χ4 and at 110° for χ1. However, in the S1 state, the maximum barrier for χ1 is 0.326 eV at a twist angle of 70° and the minimum at a twisting angle of 90°. Also, barrier heights of 0.152 and 0.272 eV with respect to χ2 and χ4 within the minimum twist at 80° and 90°, respectively, were observed. However, for χ3, no significant change of PES was noticed for both S1 and S0, suggesting that molecular conformation is stable across both states. This analysis implies that χ1, χ2, and χ4 play a key role in structural response upon photoexcitation of DMABNA, with χ2 particularly suited for conformational changes in the excited state due to its lower barrier height as compared to χ1 and χ4. Such twisting geometries align with experimental observations of low quantum yield and short emission lifetime in polar solvents, supporting the TICT as the likely mechanism for the excited-state relaxation of DMABNA.
Thus, we can conclude that in a polar solvent (MeCN), the lowest excited singlet state (S1) is likely to be a TICT state, characterized by a twisted dimethyl amino aniline unit (χ2). In the case of DMANS in a non-polar solvent, CHX, the longer lifetime is predominantly associated with a higher barrier to achieving the phantom state (or 1P* state) for the ultimate cis–trans isomerization process through the olefinic CC bond.15,49,50 Similarly, in DMABNA, a stilbene prototype twisting around the imine –CN bond is believed to lead to the phantom state. Due to the high activation barrier in CHX, this twisted conformation is generally inaccessible.
Conclusion
Our study investigates the photophysical behavior of the donor–acceptor benzylideneaniline derivative, DMABNA, focusing on effects of solvent polarity. Steady-state absorption and emission measurements reveal a significant solvent-dependent behavior. In non-polar solvents like cyclohexane, DMABNA shows a single emission band at 455 nm, while in highly polar solvents such as acetonitrile, a dual-band emission at 520 nm and 730 nm is observed along with a drastic decrease in fluorescence quantum yield. The pronounced redshift in emission and high Stokes shift with solvent polarity indicate an intramolecular charge transfer (ICT) process in the excited state. The fs-TA spectroscopy reveals longer lifetimes in cyclohexane, with a rise component at higher time delays extending beyond 1 ns, suggesting triplet-state involvement. In contrast, in acetonitrile, the excited-state lifetime reduces drastically to 4.2 ps, likely due to rapid charge separation in the excited state. Interestingly, fluorescence lifetimes are extremely short in both the solvents (1.5 ps in cyclohexane and 2.1 ps in acetonitrile), consistent with the fs-TA findings in polar solvents but highlighting discrepancies in non-polar solvents, possibly due to triplet-state dynamics.The computational analysis provides further insights into the observed behavior, revealing that DMABNA undergoes notable twisting in the S1 state along four dihedrals. The most significant structural change occurs along the dimethylamino-aniline dihedral, promoting charge separation. This conformation-dependent charge separation in polar solvents suggests that solvent interactions play a key role in the excited-state relaxation dynamics of DMABNA. The resulting solvent-induced charge transfer character accounts for the low fluorescence quantum yield, short fluorescence lifetime, and solvent-dependent photophysical properties, offering a refined perspective on DMABNA's excited-state behavior.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Prajoy Kumar Mitra: conceptualization (equal); data curation (lead); formal analysis (lead); methodology (lead); software (lead); writing – original draft (lead); writing – review & editing (supporting). Preetika Verma: methodology (supporting). Yapamanu Adithya Lakshmanna: conceptualization (equal); data curation (supporting); formal analysis (lead); funding acquisition (lead); project administration (lead); supervision (lead); writing – review & editing (lead).Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
YAL acknowledges the financial support of this work through the CRG grant (CRG/2020/000321) of SERB. We acknowledge the high-performance computing facility provided by the Padmanabha cluster at IISER TVM, India. PKM, and PV acknowledge IISER TVM for the fellowships.References
- A. Aviram, Molecular electronics—science and technology, Angew. Chem., 1989, 101(4), 536–537 CrossRef
.
- A. Tomkevičienė and J. V. Gražulevičius, Mater. Sci., 2011, 17, 335–342 CrossRef
- J. N. J. Ni, T. T. T. Tano, Y. I. Y. Ichino, T. H. T. Hanada, T. K. T. Kamata, N. T. N. Takada and K. Y. K. Yase, Jpn. J. Appl. Phys., 2001, 40, L948 CrossRef CAS
- M. Arivazhagan, Spectrochim. Acta, Part A, 2011, 82, 228–234 CrossRef CAS PubMed
- M. El-Nahass, H. Zeyada, M. Aziz and N. El-Ghamaz, Opt. Mater., 2004, 27, 491–498 CrossRef CAS
- J. M. Tour, J. Org. Chem., 2007, 72, 7477–7496 CrossRef CAS PubMed
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, John Wiley & Sons, 2021 Search PubMed
- C. S. Mak, Q. Y. Leung, W. K. Chan and A. B. Djurišić, Nanotechnology, 2008, 19, 424008 CrossRef PubMed
- S. Roquet, A. Cravino, P. Leriche, O. Alévêque, P. Frere and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459–3466 CrossRef CAS PubMed
- B. L. Feringa, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS PubMed
- V. Balzani, P. Ceroni, A. Credi, M. Gómez-López, C. Hamers, J. F. Stoddart and R. Wolf, New J. Chem., 2001, 25, 25–31 RSC
- G. Signore, R. Nifosi, L. Albertazzi, B. Storti and R. Bizzarri, J. Am. Chem. Soc., 2010, 132, 1276–1288 CrossRef CAS PubMed
- S. Qiu, A. T. Frawley, K. G. Leslie and H. L. Anderson, Chem. Sci., 2023, 14, 9123–9135 RSC
- I. Georgieva, A. l. J. Aquino, F. Plasser, N. Trendafilova, A. Köhn and H. Lischka, J. Phys. Chem. A, 2015, 119, 6232–6243 CrossRef CAS PubMed
- C. Singh, R. Ghosh, J. A. Mondal and D. K. Palit, J. Photochem. Photobiol., A, 2013, 263, 50–60 CrossRef CAS
- S. Lee, M. Jen, T. Jang, G. Lee and Y. Pang, Sci. Rep., 2022, 12, 6557 CrossRef CAS
- A. Nandi, R. Ghosh and D. K. Palit, J. Photochem. Photobiol., A, 2016, 321, 171–179 CrossRef CAS
- S. K. Panja, N. Dwivedi and S. Saha, RSC Adv., 2016, 6, 105786–105794 RSC
- M. Ahn, M.-J. Kim, D. W. Cho and K.-R. Wee, J. Org. Chem., 2020, 86, 403–413 CrossRef PubMed
- D. Jana and S. Jana, ACS Omega, 2020, 5, 9944–9956 CrossRef CAS PubMed
- M. Maus, W. Rettig, D. Bonafoux and R. Lapouyade, J. Phys. Chem. A, 1999, 103, 3388–3401 CrossRef CAS
- G. Haberhauer, Chem.–Eur. J., 2017, 23, 9288–9296 CrossRef CAS PubMed
- C. A. Guido, B. Mennucci, D. Jacquemin and C. Adamo, Phys. Chem. Chem. Phys., 2010, 12, 8016–8023 RSC
- C. Zhong, Phys. Chem. Chem. Phys., 2015, 17, 9248–9257 RSC
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS
- M. Akbarimoosavi, E. Rohwer, A. Rondi, J. Hankache, Y. Geng, S. Decurtins, A. Hauser, S.-X. Liu, T. Feurer and A. Cannizzo, J. Phys. Chem. C, 2019, 123, 8500–8511 CrossRef CAS
- L. Liu, S. Li, N. Zhang, Q. Shi, K. Liu, T. Liu, Z. Huang, L. Ding and Y. Fang, J. Phys. Chem. B, 2023, 127, 10171–10178 CrossRef CAS PubMed
- T. Yoshihara, S. I. Druzhinin and K. A. Zachariasse, J. Am. Chem. Soc., 2004, 126, 8535–8539 CrossRef CAS PubMed
- G. Haberhauer, R. Gleiter and C. Burkhart, Chem.–Eur. J., 2016, 22, 971–978 CrossRef CAS PubMed
- Y. Guo, M. Feng, Z. Kuang, C. S. Abeywickrama, Y. Pang and A. Xia, J. Phys. Chem. B, 2023, 127, 7764–7771 CrossRef CAS PubMed
- T. Chen, J. Lou, H. Wu, J. Luo, D. Yang, X. Qiao, H. Zhang, B. Z. Tang and Z. Wang, Adv. Opt. Mater., 2023, 11, 2301053 CrossRef CAS
- Z. N. Scheller, D. Liese, H. Siera, N. Semleit, M. Schmiedtchen, C. Wölper and G. Haberhauer, Chem.–Eur. J., 2024, 30, e202304143 CrossRef CAS PubMed
- X. Zhao, J. Cheng, L. Zhang, B. Yun, K. Yan, B. Wu, X. Liu, F. Zhang and Z. Lei, Cell Rep. Phys. Sci., 2023, 4, 101691 CrossRef CAS
- T. Staněk, M. Dvořák, N. Almonasy, M. Nepraš, I. Šloufová and M. Michl, Dyes Pigm., 2017, 141, 121–127 CrossRef
- K. A. Zachariasse, S. I. Druzhinin, W. Bosch and R. Machinek, J. Am. Chem. Soc., 2004, 126, 1705–1715 CrossRef CAS PubMed
- C. A. van Walree, O. Franssen, A. W. Marsman, M. C. Flipse and L. W. Jenneskens, J. Chem. Soc., Perkin Trans. 2, 1997, 799–808 RSC
- C. A. Van Walree, A. W. Maarsman, A. W. Marsman, M. C. Flipse, L. W. Jenneskens, W. J. Smeets and A. L. Spek, J. Chem. Soc., Perkin Trans. 2, 1997, 809–820 RSC
- E. El-Menyawy, H. Zeyada and M. El-Nahass, Int. J. Nanoelectron. Mater., 2011, 4, 121–134 Search PubMed
- M. El-Mansy, M. El-Nahass, N. M. Khusayfan and E. El-Menyawy, Spectrochim. Acta, Part A, 2013, 111, 217–222 CrossRef CAS PubMed
- H. Nakai, M. Shiro, K. Emuzi, S. Sakata and T. Kubota, Acta Crystallogr., Sect. B, 1976, 32, 1827–1833 CrossRef
- H. Nakai, K. Ezumi and M. Shiro, Acta Crystallogr., Sect. B, 1981, 37, 193–197 CrossRef
- H. Xu, K. Sohlberg and Y. Wei, J. Mol. Struct.: THEOCHEM, 2003, 634, 311–314 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- M. Horng, J. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS
- A. Jiménez-Sánchez and R. Santillan, Analyst, 2016, 141, 4108–4120 RSC
- H. Görner and E. Fischer, J. Photochem. Photobiol., A, 1991, 57, 235–246 CrossRef
- T. Suzuki, Y. Kaneko and T. Arai, Chem. Lett., 2000, 29, 756–757 CrossRef
- R. Ghosh, A. Nandi and D. K. Palit, Phys. Chem. Chem. Phys., 2016, 18, 7661–7671 RSC
- H. Gruen and H. Goerner, J. Phys. Chem., 1989, 93, 7144–7152 CrossRef CAS
- J. Oberlé, G. Jonusauskas, E. Abraham, R. Lapouyade and C. Rullière, Bull. Chem. Soc. Jpn., 2002, 75, 1041–1047 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01814e |
| This journal is © The Royal Society of Chemistry 2025 |