
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe stereo-divergent functionalization of alkynes: a comprehensive review
Ranjay Shaw
 a,
Ashmita Singh
b,
Ismail Althagafi
c,
Ramendra Pratap
*b and
Dharmendra Kumar Yadav
*d
a,
Ashmita Singh
b,
Ismail Althagafi
c,
Ramendra Pratap
*b and
Dharmendra Kumar Yadav
*d
aDepartment of Chemistry, GLA University, Mathura, 281406, India
bDepartment of Chemistry, University of Delhi, Delhi, India 110007. E-mail: rpratap@chemistry.du.ac.in
cDepartment of Chemistry, Faculty of Science, Umm Al-Qura University, Makkah, 21955, Saudi Arabia
dDepartment of Biologics, College of Pharmacy, Gachon University, Hambakmoeiro 191, Yeonsu-gu, Incheon 21924, Republic of Korea. E-mail: dharmendra30oct@gmail.com
First published on 17th April 2025
Abstract
Alkynes are central in crafting pharmaceuticals, agrochemicals, and materials owing to their reactivity and linear geometry. This review unveils cutting-edge advancements in the stereo-divergent functionalization of alkynes, transforming them into invaluable tools for synthesizing stereochemically defined alkenes and alkanes. The review highlights ground-breaking methodologies that achieve exceptional E- and Z-selectivity using innovative catalysts like cobalt, nickel, and palladium through hydrogenation, hydroboration, and hydrosilylation. Recent breakthroughs such as dual-catalytic systems and energy transfer catalysis enable unprecedented stereocontrol. Sustainable strategies including water as a hydrogen source and recyclable catalysts align with green chemistry principles, paving the way for eco-friendly synthesis. This synthesis of cutting-edge techniques and their applications inspire new avenues in synthetic chemistry, offering transformative tools for creating complex molecular architectures with precision and sustainability.
 Ranjay Shaw | Ranjay Shaw completed his master's degree in chemistry from Vinoba Bhave University, Hazaribag, India. He earned his PhD in 2021 from the University of Delhi, followed by postdoctoral research at the Indian Institute of Technology (IIT), Delhi, from 2021 to 2023. In 2023, he joined GLA University, Mathura, as an Assistant Professor. His research interests lie in organic synthesis, medicinal chemistry, and drug discovery. Dr Shaw focuses on the design and synthesis of heterocycles and carbocycles, exploring their potential therapeutic applications. His work aims to contribute to the development of novel and sustainable methodology for biologically benign organic scaffolds. |
 Ashmita Singh | Ashmita Singh was born in New Delhi, India, in 1992. She received her BSc (2013) and MSc (2015) degrees in chemistry. In 2022, she received her PhD degree in chemistry from Guru Gobind Singh Indraprastha University, New Delhi, India, under the supervision of Professor A. K. Narula, where her research focused on the exploration of N-heterocyclic carbenes (NHCs) in the synthesis of drugs and drug-like molecules. She is a highly skilled researcher specializing in organic synthesis and synthetic chemistry with peer-reviewed research publications in esteemed international journals. Currently, she is working as a Women Scientist in a project awarded by the DST WISE PDF scheme at the University of Delhi, under the mentorship of Professor Ramendra Pratap. Through her ongoing research, she continues to contribute to advancements in catalysis and synthetic methodologies. |
 Ismail Althagafi | Ismail Althagafi is a Professor at the Department of Chemistry, Faculty of Sciences, Umm Al-Qura University, Makkah (Saudi Arabia). He obtained his bachelor's degree from Umm Al-Qura University and his master's and PhD degrees from Cardiff University (United Kingdom). His current research interest includes physical organic chemistry, synthetic chemistry, and bio-organic chemistry. He has published more than 100 research publications in different journals. |
 Ramendra Pratap | Prof. Ramendra Pratap completed his master's degree from the University of Gorakhpur in 2001, and then, he moved to Central Drug Research Institute, Lucknow, Uttar Pradesh, India, for doctoral research. He worked with Dr Vishnu Ji Ram for four and a half years on ring transformation reactions of 2H-pyran-2-ones. He worked on nucleoside modification chemistry as a postdoctoral fellow at the City University of New York, USA, for two years. He received a renowned Humboldt fellowship and shifted to the University of Saarland, Germany. In Saarbrucken, he was involved in Mo-catalyzed reactions. In September 2010, he joined the Department of Chemistry, University of Delhi, India, as an Assistant Professor. Currently, he is working as a full Professor. To date, he has published more than 100 research papers in various international journals. He has also received a JSPS invitation fellowship during 2016–17. His research focuses on developing various heterocycles, carbocycles, materials, and metal-catalyzed bond formation reactions. |
 Dharmendra Kumar Yadav | Dr Dharmendra Kumar Yadav has completed his PhD degree in Biological Science from CSIR-Central Institute of Medicinal and Aromatic Plants, Lucknow, India. He completed his postdoctoral studies at Hanyang University, Korea, and University of Delhi, India. Presently, he is working as an Assistant Professor and Principal Investigator of an NRF project (Korea Government) in the College of Pharmacy in Gachon University, Korea. He received the Young Scientist award from the Science and Engineering Research Board, New Delhi. He has worked as a Young Scientist at All India Institute of Medical Science Jodhpur, India. He has published more than 150 papers in reputed journals, 3 book chapters and a patent. He is continuing his research in atomic level molecular simulation of plasma medicine, computer-aided drug design and molecular modelling of biological networks. He is also a member of several scientific societies and academic bodies. |
1. Introduction
In the realm of organic chemistry, the strategic manipulation of functional groups and the selective control of stereochemistry play pivotal roles in the synthesis of complex molecular architectures. In asymmetric synthesis, stereo-divergent functionalization represents a paradigm shift in this endeavor, emphasizing the selective manipulation of stereochemical outcomes to generate a diverse array of stereochemically distinct products. The concept of stereo-divergent functionalization emerges as a compelling strategy, offering chemists a nuanced approach in accessing a plethora of stereoisomeric products from a single starting material.1–3 This approach to synthesis enhances the synthetic efficiency and offers insights into the underlying mechanistic intricacies governing chemical transformations. Moreover, stereo-divergent strategies pave the way for the creation of molecular libraries with enriched structural complexity and biological relevance by embracing the concept of stereochemical diversity.4,5 Stereo-divergent routes may offer efficient approaches in obtaining various naturally occurring or biologically active enantiomers and diastereomers from the same starting material.6–8Among the myriad of functional groups, alkynes have garnered significant attention owing to their inherent versatility and reactivity, making them indispensable building blocks in the construction of diverse organic compounds.9–11 The synthetic landscape of alkynes is characterized by their unique electronic properties and distinctive linear geometry, which lend them to a wide array of transformations.12,13 Exploiting the potential of alkynes as synthetic precursors, researchers have attempted to functionalize these compounds and achieve precise stereochemical control, thereby expanding the repertoire of available stereoisomeric motifs.14 Recently, significant progress has been made in the stereo-divergent functionalization of alkynes, resulting in a generation of diverse E/Z alkenes or directly chiral alkanes.15 Despite their potential in synthetic chemistry, literature reviews specifically addressing the stereo-divergent functionalization of alkynes are scarce, underscoring the necessity for a comprehensive compilation of literature on the synthesis of alkynes.
This comprehensive review delves into the recent updates on alkyne synthesis from non-alkyne sources and their stereo-divergent functionalization, examining the fundamental principles, synthetic methodologies, and transformative applications that characterize this growing field. We thoroughly investigate catalytic transformations and innovative stereochemical control strategies, elucidating the complexities and challenges inherent in this dynamic synthetic paradigm. Additionally, this review highlights the broader implications of stereo-divergent functionalization beyond synthetic chemistry, emphasizing its potential to inspire new paradigms in molecular design, drug discovery, and materials science. By elucidating the fundamental concepts and presenting exemplary case studies, we aim to stimulate further research and innovation in this rapidly evolving field, ultimately advancing chemical synthesis and molecular engineering.
In short, the study of stereo-divergent functionalization of alkynes represents a blend of creativity, precision, and scientific rigor, continuously pushing the boundaries of synthetic possibility and revealing new aspects of molecular complexity. This article guides readers through the complex realm of stereochemical manipulation, where innovative thinking intersects with molecular precision in advancing the future of organic synthesis.
2. Recent updates in alkyne synthesis from non-alkyne sources
Metal-catalyzed coupling of terminal alkynes is a prevalent method for synthesizing desired internal alkyne products. However, constructing C–C triple bonds from non-alkyne precursors typically involves α,β-elimination, carbene rearrangement, or fragmentation reactions. In 2020, our group published a comprehensive review on the synthesis of alkynes from non-alkyne sources.16 Therefore, this article focuses exclusively on recent advancements in the construction of C–C triple bonds from non-alkyne sources.Terminal alkynes can be synthesized through either a carbene pathway or β-elimination from 1,1-dibromoalkenes.16 Some of these reactions require the use of strong and air-sensitive bases like n-BuLi, LDA, Grignard reagents, or inorganic bases in aqueous conditions. Recently, Rao et al. developed a straightforward synthetic approach for producing terminal alkynes (2) from 1,1-dibromoalkenes (1) under dry reaction conditions, utilizing succinimide and K2CO3 in DMSO (Scheme 1).17 This method demonstrated wide applicability in the synthesis of a broad spectrum of aromatic alkyne (2). Substitution of the electron-rich aromatic ring on substrate delivered a higher yield (64–86%) of product compared to substrates with an electron-deficient aromatic ring. However polycyclic aromatic substituted 1,1-dibromoalkenes afforded corresponding terminal alkyne in 75–96%. The reaction initiates with the base-mediated dehydrobromination of substrate 1 to 1-bromoalkyne (A). Succinimide plays a dual role in the reaction. The in situ generated succinimide anion acts as a good nucleophile in DMSO, facilitating nucleophilic substitution in 1-bromoalkyne (A) to form the acetylide anion (B). Concurrently, succinimide functions as a proton donor during the reaction, protonating the anions (B) to yield the alkyne product (2).
 | ||
| Scheme 1 Dehydrohalogenation of 1,1-dibromoalkenes to alkyne. | ||
Chen et al. developed a one-pot approach for the synthesis of diarylacetylene (10) from arylaldehydes (3) and 1-(arylmethyl) benzotriazoles (4) in the presence of LiHMDS (Scheme 2).18 Both the electron-deficient and donating substituents on the aromatic ring of aryl aldehydes (3) and 1-(aryl methyl) benzotriazoles (4) were well tolerated and results in moderate to good yield (48–98%) of alkyne products (10) without any specific trend. However, the halide substitution at the meta-position of the aromatic ring of substrate 4 results in a very low yield compared to substrates with halide substitution at the para-position. The reaction was supposed to proceed through imine formation (6), followed by Mannich-type addition of benzotriazoles 4 to imine intermediate 6 and double elimination of LiNHSiMe3 and benzotriazoles.
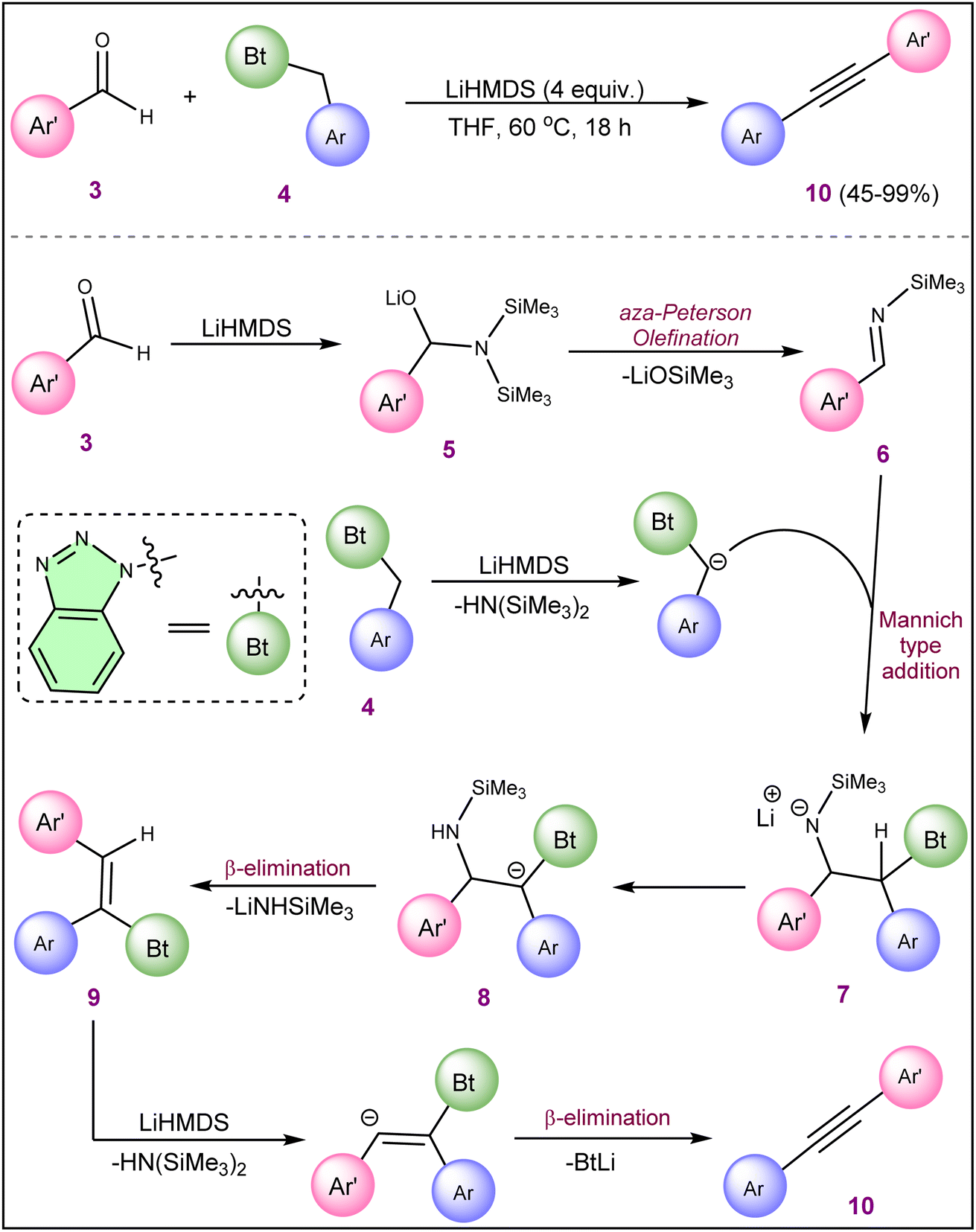 | ||
| Scheme 2 Synthesis of diaryl alkyne from aryl aldehydes and 1-(aryl methyl) benzotriazoles. | ||
In this context, Kimura et al. reported a Fritsch–Buttenberg–Wiechell (FBW) rearrangement of magnesium alkylidene carbenoids (13) to internal alkynes (14) (Scheme 3).19 Magnesium alkylidene carbenoids (13) are reactive intermediates produced from isopropyl magnesium chloride and 1-chlorovinyl p-tolyl sulfoxides. These 1-chlorovinyl p-tolyl sulfoxides are synthesized from carbonyl compounds and chloromethyl p-tolyl sulfoxide through a sulfoxide/magnesium exchange reaction. Several alkyne products (14) were synthesized in good to excellent yield using this method.
 | ||
| Scheme 3 FBW rearrangement of magnesium alkylidene carbenoids to internal alkynes. | ||
There is no significant difference in reactivity between the geometric isomers of 2-alkynyl-2-phenyl-substituted sulfoxide (11, R1 & R2 = aryl and alkynyl). However, 2-methyl-substituted sulfoxides (11, R1 & R2 = aryl and methyl) show reactivity differences between their isomers. The (Z)-sulfoxides (11, R1 = aryl and R2 = methyl) with methyl and chloro-groups trans to each other produce alkynes with low efficiency. As the migratory aptitude (aryl, alkynyl ≫ alkyl) matches carbanion stability trends, the FBW rearrangement of magnesium alkylidene carbenoids (13) appears to be an anionotropic rearrangement. The trans geometry of the chloro-group and the migrating substituent is crucial for a successful 1,2-rearrangement, explaining the observed reactivity differences in geometric isomers of 2-methyl-substituted sulfoxides (11, R1 and R2 = aryl and methyl, respectively).
Sun et al. discovered a modular method for synthesizing alkynes 20 by reacting carboxylic esters 16 with lithiated gem-diborylalkanes 15′ and aryl triflimides 18 (Scheme 4).20 The process involves forming an intermediate α-boryl lithium enolate 17, which is then triflated using triflimide 18 to a borylated vinyl triflate intermediate 19 and subsequently quenched with water to yield the alkyne product 20. This innovative approach allows for the efficient conversion of various aliphatic and aromatic carboxylic acid esters (16) into both internal and terminal alkyne products (20) in a short reaction period. The process accommodates a broad spectrum of functional groups, including halides, amides, amines, carbamate, –OMe, and –CF3, positioned on both esters (16) and gem-diborylalkane (15) substrates. Additionally, it enables the transformation of chiral α-substituted esters into chiral propargyl compounds without causing racemization.
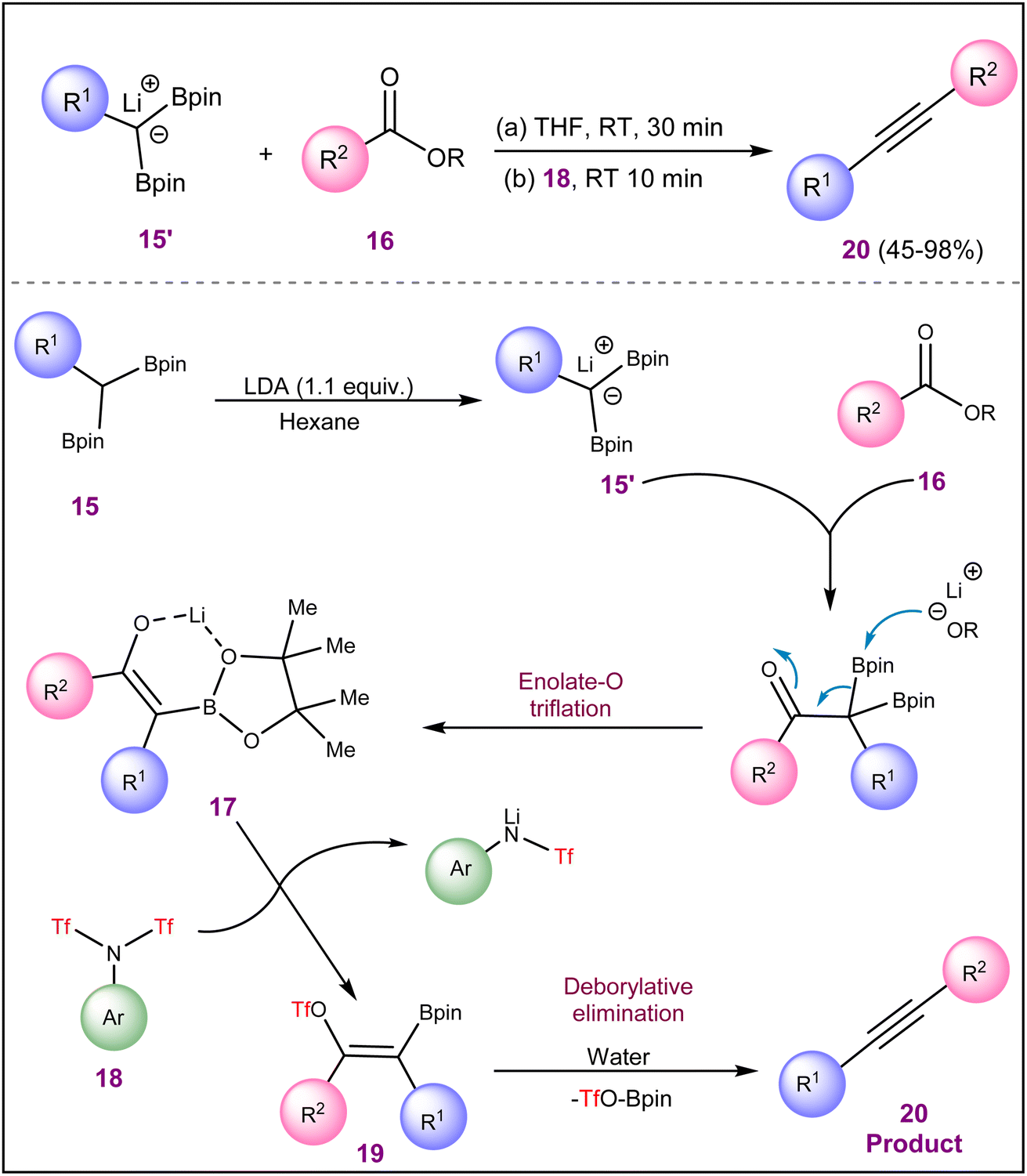 | ||
| Scheme 4 Synthesis of alkynes using esters and lithiated gem-diborylalkanes. | ||
On the other hand, Chen et al. discovered a dual-function Cytochrome P450 monooxygenase involved in cyclohexanoid terpenoid biosynthesis that facilitates enyne formation from a prenyl chain.21 Initially, this P450 enzyme catalyzes the dehydrogenation of the prenyl chain (21) to produce a cis-diene intermediate 24. Subsequently, the enzyme functions as an acetylenase, forming an alkyne group, resulting in the synthesis of a 1,3-enyne (26). Microsome extracts from Saccharomyces cerevisiae expressing the AtyI gene were prepared and incubated with 4-hydroxy-3-prenylbenzoic acid (21) and NADPH, which led to the production of eutypinic acid (26). These findings demonstrate that AtyI catalyzes four-electron oxidation and is the key enzyme responsible for incorporating the enyne group. Similarly, the Gao group reported the creation of an alkyne moiety using a novel cytochrome P450 enzyme named BisI.22 This enzyme exhibits versatile activity toward both C5 and C15 prenyl chains, following a similar reaction pathway demonstrated in Scheme 5.
 | ||
| Scheme 5 P450 catalyzed dehydrogenation of 4-hydroxy-3-prenylbenzoic acid to eutypinic acid. | ||
3. Stereo-divergent functionalization of alkynes
A broad spectrum of functionalized alkenes and alkanes has been synthesized through the stereo-divergent functionalization of alkynes. Among these reactions, semi-hydrogenation and hydrosilylation of alkynes are the most extensively studied. In this review, we categorize and analyze these methodologies based on the type of functionalization achieved with alkynes, providing a comprehensive understanding of the processes involved.3.1 Stereo-divergent hydrogenation
Stereodivergent semi-hydrogenation of alkynes has emerged as a transformative process in modern organic chemistry, enabling the selective synthesis of E- or Z-alkenes as valuable intermediates for pharmaceuticals, agrochemicals, and other fine chemicals.23–25 Recent advances in catalytic systems, particularly those involving transition metals such as nickel, cobalt, ruthenium, palladium, and chromium, have significantly improved our ability to control stereoselectivity under mild and environmentally friendly conditions.26–31 Here, the authors, consolidate findings from recent studies that illustrate the latest developments and mechanistic insights in this field, providing a thorough overview of the strategies employed to achieve stereo-divergent semi-hydrogenation of alkynes.In 2016, one of the cornerstone studies in this area focuses on cobalt-catalyzed semi-hydrogenation using NNP and PNP-type pincer ligands. In this work, Fu et al. reported the cobalt-catalyzed transfer hydrogenation of alkynes (27), with methanol serving as the hydrogen source. The system achieves effective stereocontrol through carefully designed catalysts 28 and 29 to afford the trans-alkene (30) and cis-alkene (31) products, respectively (Scheme 6).32 Operating under mild conditions, the reaction tolerates a wide variety of functional groups and provides good yields with catalyst loadings as low as 0.2 mol%. The broad applicability of this method is demonstrated by the successful synthesis of over 50 alkenes with high chemo- and stereoselectivity. The findings highlight the potential of methanol as a sustainable hydrogen source in semi-hydrogenation reactions and demonstrate cobalt's versatility in catalytic transformations.
 | ||
| Scheme 6 Cobalt-catalyzed transfer hydrogenation of alkynes, with methanol serving as the hydrogen source. | ||
The underlying mechanisms of selectivity control were evaluated by combined density functional theory (DFT) calculations with experimental validation (Scheme 7).33 They discovered that cobalt(I) hydride intermediates play a pivotal role in steering the reaction towards either E- or Z-alkenes, depending on the reaction conditions. Both pre-catalysts (28) and (32) predominantly yield the E-alkene (30) as the main product during the semi-hydrogenation of alkynes with ammonia boranes. However, the stereoselectivity of the reaction shifts when the isopropyl group in pre-catalyst (32) is replaced by a bulkier tertiary butyl group (29), favoring the formation of the Z-alkene (31). The catalytic cycle begins with the insertion of the alkyne into the Co–H bond, followed by protonation of the resulting stilbenyl cobalt(I) intermediate to produce Z-stilbene. Regeneration of the active cobalt catalyst occurs in the presence of ammonia borane and methanol. Z-stilbene undergoes further transformation, with cobalt-catalyzed β-hydride elimination leading to the final E-stilbene product. This alteration in stereoselectivity suggests that the steric hindrance introduced by the larger substituent plays a crucial role in controlling the product outcome, potentially influencing the transition state or intermediate stability during the catalytic cycle. The suppression of unwanted over-reduction to alkanes was achieved by controlling the methanol-mediated protonation steps, offering a robust strategy for designing more selective catalysts.
 | ||
| Scheme 7 Cobalt-catalyzed semi-hydrogenation of alkynes using NNP- and PNP-type pincer ligands. | ||
In 2011, Li and Hua presented a distinct approach in their work on ruthenium-catalyzed semi-hydrogenation of functionalized diaryl alkynes (33) using DMF and water as the hydrogen source, where the choice of acid (acetic acid or trifluoroacetic acid) significantly impacts the stereoselectivity, yielding either cis- or trans-stilbenes.34 The selectivity of the reaction was controlled by the choice of additives, with trifluoroacetic acid (TFA) leading to the E-isomer (34) and acetic acid (HOAc) favoring the Z-isomer (35) (Scheme 8). This research underscores the importance of simple additives in fine-tuning the reaction pathway and achieving the desired product configuration, thus providing a straightforward yet powerful tool for chemists.
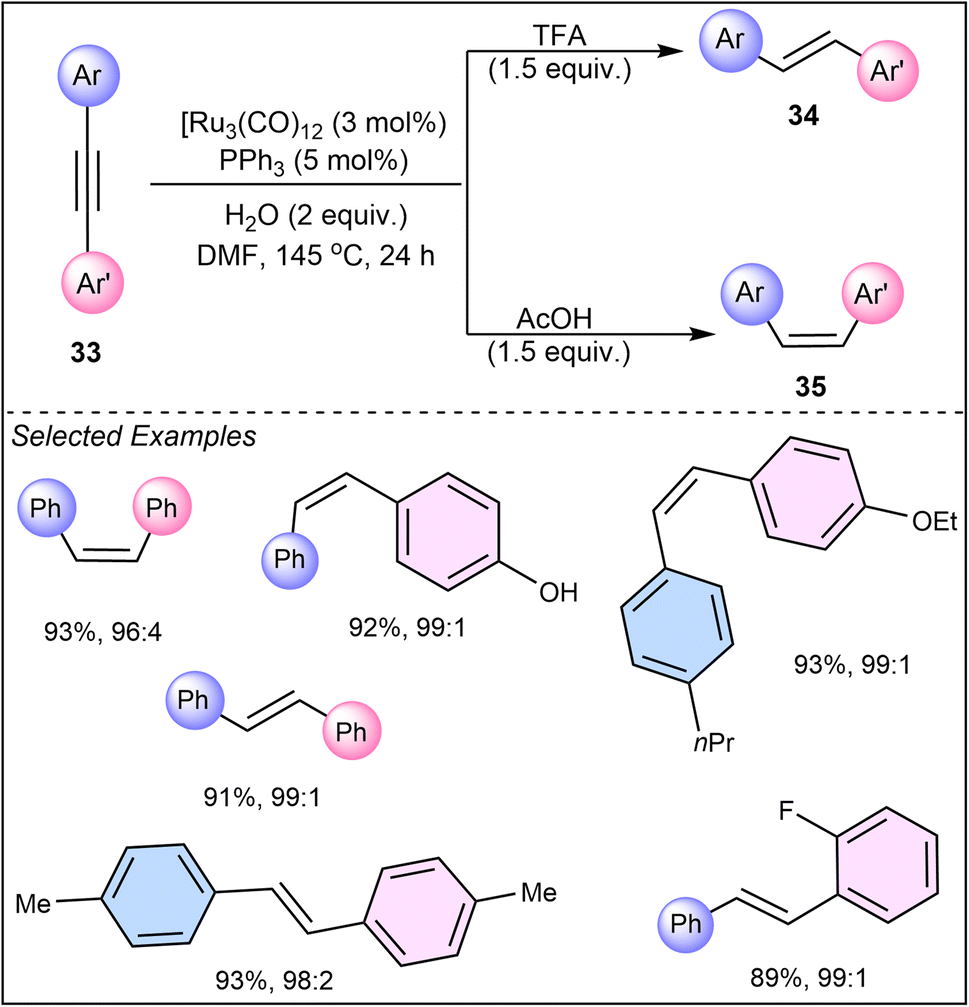 | ||
| Scheme 8 Ruthenium-catalyzed semi-hydrogenation of diaryl alkynes using DMF and water as the hydrogen source. | ||
In 2021 Wu et al. presented an innovative approach involving Ni-catalyzed semi-hydrogenation of alkyne (36), with water as a hydrogen source (Scheme 9).35 By carefully modulating the metal species during the early stages of the reaction, the researchers demonstrated precise control over the stereoselectivity of the resulting alkenes. Unlike many previous reactions where E-alkenes are generated via isomerization of Z-alkenes, this method achieves stereoselectivity through parallel catalytic pathways, forming the isomers independently. Mechanistic studies indicate that the choice of base plays a pivotal role in modulating the reaction pathways. By introducing different bases, nickel species in distinct valence states can be accessed, initiating two separate catalytic cycles that selectively lead to either the E- or Z-isomers. Ph2CONa base selectively yields Z-isomer (39) in the presence of ligand (37) whereas CF3CONa base, along with ligand (38), yields E-alkene (40). This strategy has been successfully applied to nearly 70 substrates, including internal and terminal alkynes, enynes, and diynes, producing semi-hydrogenated products with high yields and selectivity. This study stands out for its commitment to sustainability, highlighting the potential of using water—a green and non-toxic hydrogen donor—as a viable alternative to traditional reductants.
 | ||
| Scheme 9 Ni-catalyzed semi-hydrogenation with water as a hydrogen source. | ||
In 2023, Hu et al. further expanded the toolkit, exploring a photoinduced dual-catalytic system combining nickel hydride catalysis with photoredox catalysis for stereodivergent hydrogenation of alkyne 41 (Scheme 10).36 Utilizing the dual catalytic system, the methodology offers a versatile strategy for controlling stereoselectivity between alkene products 43 and 45 with triethylamine acting as a sacrificial reductant and a source of hydrogen atoms. This system operates under mild conditions and relies on the pKa of alcohol additives to control the stereoselectivity. This approach reduces the need for external hydrogen sources and represents a versatile, green chemistry method that could be scaled up for industrial applications. The mechanistic pathway involves several key steps: the photo-induced formation of a nickel hydride species followed by syn-hydro-nickelization of alkyne I, and subsequent alkenyl-nickel isomerization II. Unlike many traditional methods where stereoselectivity is achieved through post-reduction photoisomerization, this process controls stereochemistry at an earlier stage. The final stereoselective outcome is determined by the rate of protonolysis step III, which can be modulated by varying the pKa of the alcohol additive used. It provides a practical, efficient means for achieving high stereoselectivity without relying on secondary photoisomerization processes.
 | ||
| Scheme 10 Stereodivergent hydrogenation through a photoinduced nickel hydride catalysis. | ||
Another notable study on anion-controlled semi-hydrogenation features a cobalt-catalyzed method using water/methanol as the hydrogen source. In 2019, Li et al. demonstrated how varying the solvent or including bidentate phosphine ligand dppe can steer the reaction towards either E- or Z-alkenes (Scheme 11).37 In the presence of CoI2 catalyst, reductant zinc, water, and methanol, the internal alkyne substrates (46) were converted into Z-alkenes (47). The addition of the dppe ligand in this reaction leads to stereoselective formation of E-alkenes (48). The developed methodology was applicable to an array of internal alkynes. This method showcases the potential of using inexpensive and readily available base metals in achieving sustainable and selective hydrogenation reactions. The plausible mechanism for the formation of both isomers was supported by different catalytic cycles. Cobalt catalyst was first reduced by Zn, which then coordinates with water to form a complex. This complex binds with the alkyne substrate which becomes protonated by methanol and leads to the formation of Z-alkene (47) (catalytic cycle I). The formation of E-alkene (48) follows another pathway (catalytic cycle II) as it includes the dppe ligand. In this, the cobalt catalyst gets reduced by Zn after combining with ligand dppe. The generated catalyst further binds with the alkyne moiety and water molecules followed by the migratory insertion. To circumvent the steric repulsion caused by bulky dppe ligand, the resultant product was formed with the anti-selective stereoisomer 48.
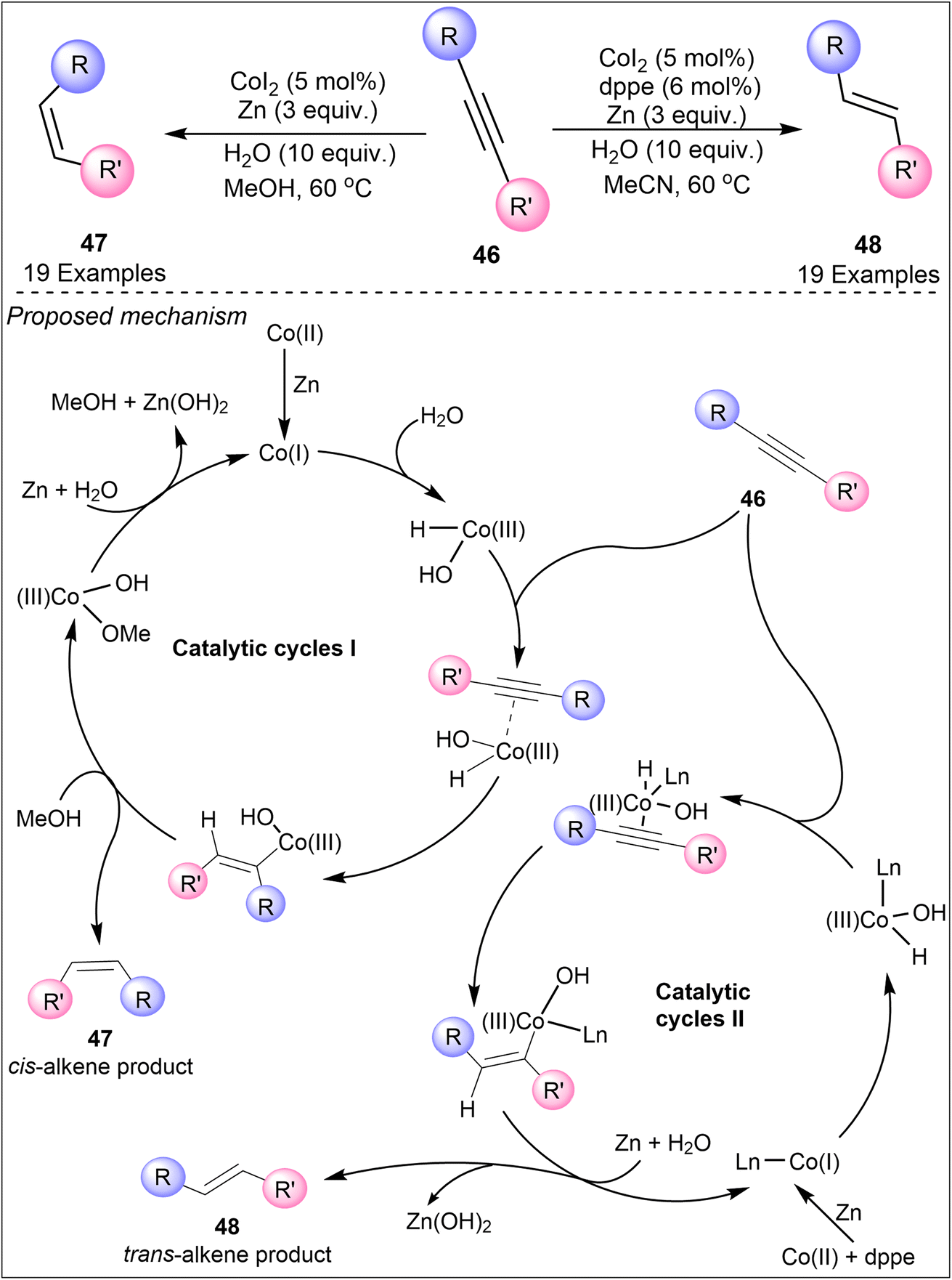 | ||
| Scheme 11 Anion-controlled semi-hydrogenation through cobalt-catalysis using water/MeOH as the H-source. | ||
The field of stereodivergent semi-hydrogenation also benefits from innovations in palladium catalysis, where the interplay of ligands and acidic co-catalysts can direct the reaction toward either hydroformylation or semi-hydrogenation. In 2020, Liu et al. highlighted how specific ligand environments and co-catalyst choices can substantially influence reaction outcomes, allowing for a high degree of control over product formation.38 This study introduces a palladium-catalyzed system designed for the chemo-divergent functionalization of alkynes 49 using syngas (Scheme 12). The key to this selectivity is an advanced ligand, 50, featuring a 2-pyridyl substituent that acts as an internal base. Depending on the reaction conditions, this system allows for either hydroformylation or semi-hydrogenation of a broad range of alkynes with high chemo- and stereoselectivity. The reaction in the presence of CF3SO3H demonstrated syn-hydroformylation of substrate 49 to afford the alkene products 51. Meanwhile, PTSA·H2O shows simple anti-semi-hydrogenation of 49 to result in E-alkenes (52) as products. Mechanistic investigations, including density functional theory (DFT) calculations, kinetic studies, and control experiments, revealed that the strength and concentration of acidic cocatalysts are critical factors in determining chemoselectivity. The DFT analysis showed that ligand 50 plays a dual role: it facilitates heterolytic hydrogen activation, similar to frustrated Lewis pair (FLP) systems, during the hydrogenolysis step in hydroformylation, while simultaneously preventing CO coordination under strong acidic conditions to promote semi-hydrogenation. This work provides new strategies for palladium-catalyzed transformations and opens up avenues for further catalyst development.
 | ||
| Scheme 12 Palladium catalyzed stereodivergent semi-hydrogenation. | ||
Chromium catalysts have also proven effective in stereodivergent hydrogenation, particularly those employing cyclic (alkyl)(amino)carbene ligands. In, 2023 Ling et al. presented a chromium-catalyzed system for E- and Z-selective olefin synthesis through hydrogenation of alkyne (53) that demonstrates the critical role of carbene ligands design in determining reaction selectivity (Scheme 13).39 Using a cyclic (alkyl)(amino)carbene ligand with a phosphine anchor 54, the hydrogenation proceeds via a trans addition, yielding E-olefins (56) with high selectivity. In contrast, switching to a carbene ligand containing an imino anchor 55 enables the stereoselective formation of predominantly Z-isomers (57). This ligand-driven stereo inversion strategy allows for selective control over E and Z geometry using a single metal catalyst, overcoming the typical requirement of employing different metals for each isomer. Their work contributes valuable insights into the mechanistic pathways involved in metal-catalyzed hydrogenation, laying the groundwork for future advancements in catalyst design.
 | ||
| Scheme 13 Chromium-catalyzed E- and Z-selective olefin synthesis through alkyne hydrogenation. | ||
An intriguing study on reversible catalyst inhibition using ruthenium introduces a novel strategy to switch between E- and Z-selectivity in the hydrogenation of alkyne 58. In 2022, Luo et al. showed that a catalytic thiol can act as a reversible inhibitor, allowing for fine-tuning of the product configuration of a ruthenium (Ru-59) catalyzed semi-hydrogenation process by simply adjusting the inhibitor's concentration (Scheme 14).40 Mechanistic investigations revealed that the Z-alkene (60) serves as an intermediate in the formation of the E-alkene (61). The addition of a catalytic amount of bidentate thiol (NACET) effectively blocks the Z/E isomerization step by forming stable ruthenium-thiol(ate) complexes, while still permitting the primary hydrogenation to proceed. As a result, the absence or presence of the catalytic thiol dictates the stereoselectivity of the reaction: the reaction proceeds to the E-alkene 61 without the thiol, while the process halts at the Z-alkene 60 intermediate with the thiol present. This innovative approach offers a new dimension of control in stereodivergent hydrogenation chemistry.
 | ||
| Scheme 14 Ruthenium catalyzed E- and Z-selective hydrogenation using catalytic thiol as a reversible inhibitor. | ||
An iterative and stereodivergent approach for synthesizing unbranched polyenes and polyynes was explored in the early study by Adrian and Stark in 2016, offering a method to achieve complete stereocontrol in the formation of both E- and Z-olefins from terminal alkynes 62 (Scheme 15).41 The process involves a series of high-yielding C–C bond couplings, followed by stereospecific alkyne reductions. The synthetic cycle includes a C-3 chain extension to 63 via allylation, followed by chemoselective hydroboration to 64 and reduction of the alkyne. The geometric control of the double bonds is achieved through stereoselective alkyne reduction, using Lindlar hydrogenation for Z-alkenes 65 and aluminium hydride reduction for E-alkenes 66. Notably, the total synthesis of membranacin (Annonaceous acetogenin) precursor chatenaytrienin-4 was achieved without the need for protecting groups, showcasing the methodology's efficiency. Furthermore, this approach has significant implications for the synthesis of complex natural products, highlighting its versatility and practicality in organic synthesis.
 | ||
| Scheme 15 Iterative and stereodivergent approach for synthesizing unbranched polyenes from polyynes. | ||
In 2021, Chen et al. demonstrated the catalytic efficiency of a bis-pyridyl diamine (PDI) di-Co(III) complex 68 for the semi-hydrogenation of alkynes 67 (Scheme 16).42 The system employs Ph2SiH2 and H2O as hydride sources and is able to switch stereoselectivity between E- and Z-alkenes by modulating the water concentration. This stereodivergent system was highly tolerant to various functional groups, making it a robust and versatile tool for alkyne reduction. The ability to fine-tune the reaction conditions to achieve the desired stereochemistry marks a significant advancement in stereoselective alkyne transformations. In the presence of about 2 equivalents of H2O, the reaction affords the Z-alkene product (69), whereas 5 equivalents of H2O lead to the isomerized product, E-alkene (70).
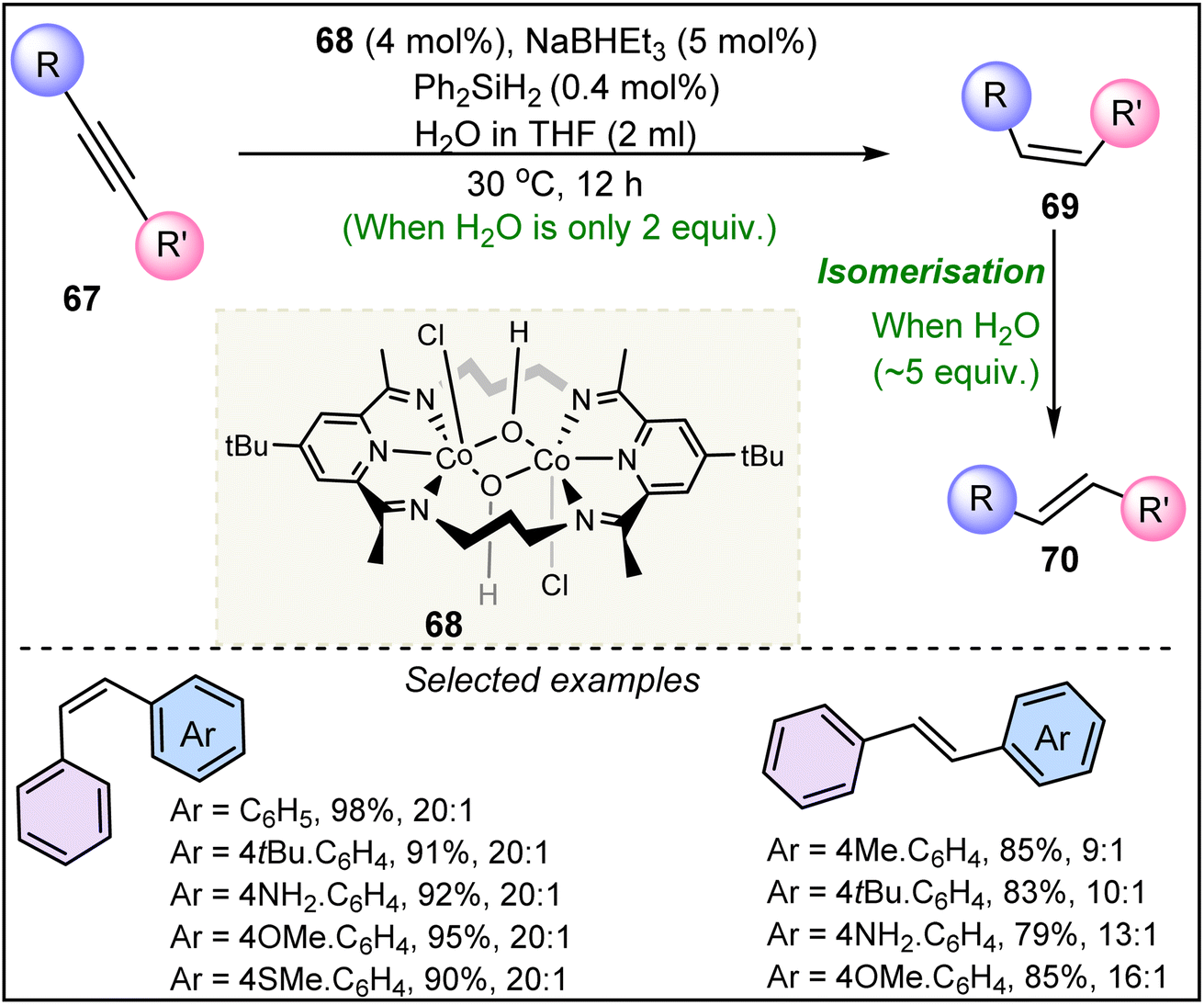 | ||
| Scheme 16 Bis-pyridyl diamine (PDI) di-Co(III) complex catalyzed semi-hydrogenation of alkynes. | ||
In 2018, Rao and Prabhu successfully detailed a palladium-catalyzed system for Z/E stereodivergent olefin synthesis from alkyne (71). The process utilizes H2O as the hydrogen source, with diboron compounds as mediators (Scheme 17).43 The choice of ligands, such as PCy3 (72) for E-olefins (74) and P(o-Tol)3 (73) for Z-olefins (75), plays a crucial role in governing the reaction's stereoselectivity. Additionally, deuterium oxide (D2O) can be used in place of water to produce deuterated alkenes, broadening the utility of this method. Their method demonstrates excellent scalability, enabling the production of stereo-defined olefins in high yields.
 | ||
| Scheme 17 Palladium-catalyzed system for Z/E stereodivergent olefin synthesis from alkynes. | ||
In 2021, Li et al. published a ligand-guided stereoselective semi-hydrogenation of alkynes (76), focusing on how different ligands direct the formation of Z- or E-olefins. The process utilizes cost-effective and stable nickel(II)-salts, with water serving as a sustainable hydrogen source and zinc powder as a reductant (Scheme 18).44 Remarkably, the stereoselectivity—whether producing trans-alkenes (77) or cis-alkenes (78)—is dictated by the specific anion in the nickel salt. This approach was particularly effective for internal alkynes, demonstrating high yields across various substrates, including those with sensitive functional groups such as carbonyls. The role of ligand design in controlling stereochemistry is a significant aspect of this work, providing insights for future applications in catalytic hydrogenation.
 | ||
| Scheme 18 Nickel-catalyzed ligand guided stereoselective hydrogenation. | ||
In 2019, Murugesan et al. developed yet another methodology using Ni(II)-catalyzed hydrogenation of alkynes 79 to E- and Z-alkenes. The study outlines a two-step process, where Z-alkenes 80 initially form, followed by isomerization to E-alkenes 81 (Scheme 19).45 Ni(NO3)2·6H2O used as a catalyst precursor forms active nanoparticles that exhibit excellent selectivity for Z-isomer 80 (Z/E > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). By incorporating multidentate ligands such as triphos or tetraphos, the system becomes E-selective for alkene 81 (E/Z > 99:1). Mechanistic insights suggest that the catalyst favouring Z-alkene formation operates via a heterogeneous pathway, while the E-selective catalyst follows a homogeneous process. In the latter case, alkyne 79 is initially reduced to Z-alkene 80 (Scheme 19, cycle I), which subsequently undergoes isomerization to form E-alkene 81 (Scheme 19, cycle II). Their findings offer a deeper understanding of nickel–hydride complexes and their role in chemoselective hydrogenation. This technique was successfully applied to over 40 substrates and scaled up for multigram synthesis, showcasing its versatility and practicality for larger-scale applications. Their work contributed valuable knowledge to the field of nickel catalysis, enhancing the development of new catalytic systems for stereoselective alkyne transformations.
:1). By incorporating multidentate ligands such as triphos or tetraphos, the system becomes E-selective for alkene 81 (E/Z > 99:1). Mechanistic insights suggest that the catalyst favouring Z-alkene formation operates via a heterogeneous pathway, while the E-selective catalyst follows a homogeneous process. In the latter case, alkyne 79 is initially reduced to Z-alkene 80 (Scheme 19, cycle I), which subsequently undergoes isomerization to form E-alkene 81 (Scheme 19, cycle II). Their findings offer a deeper understanding of nickel–hydride complexes and their role in chemoselective hydrogenation. This technique was successfully applied to over 40 substrates and scaled up for multigram synthesis, showcasing its versatility and practicality for larger-scale applications. Their work contributed valuable knowledge to the field of nickel catalysis, enhancing the development of new catalytic systems for stereoselective alkyne transformations.
 | ||
| Scheme 19 Ni(III)-catalyzed hydrogenation of alkynes to E- and Z-alkenes. | ||
Additionally, Huang et al. reported a novel methodology for Z-selective semi-hydrogenation of alkynes 82, employing Cu(OAc)2 and nitrogen-based ligands like 4,4′-bipyridine (Scheme 20).46 The method is highly effective, offering high yields and stereoselectivity across a broad range of substrates, whether with internal or external alkynes. The optimization of reaction conditions and expanded substrate scope highlight the utility of this approach in producing Z-alkenes 84 with high precision. However, E-isomers 83 were formed predominately in place of Z-isomers when substrates of internal alkynes bearing carboxylate or amide functionality were taken. This work underscores the importance of copper catalysis in achieving stereoselectivity in semi-hydrogenation reactions.
 | ||
| Scheme 20 Cu(OAc)2 and nitrogen-based ligand catalyzed semi-hydrogenation of alkynes. | ||
In 2019, Zhao et al. reported a Pd-assisted semi-hydrogenation of alkynes 85 utilizing water as a hydrogen source and Mn as an electron donor (Scheme 21).47 The developed process was effective against a wider range of alkynes, selectively yielding E- or Z-alkenes in good yield. Initially, Z-alkenes 86 were formed under the developed reaction condition, which could isomerize to E-alkenes 87 at elevated temperatures. In the proposed mechanistic insight, the Pd(0) catalyst plays a pivotal role to form palladium hydrides, engaging in an intricate interaction with water, enhanced by manganese (Mn). This dynamic process leads to the formation of molecular hydrogen through reductive elimination from the palladium. Once the palladium hydrides are generated, they eagerly combine with an alkyne, which leads to a migratory insertion. Further, the reductive elimination step releases cis-alkene 86, while regenerating the versatile Pd(0) catalyst. At elevated temperatures, cis-alkene may continue to react with palladium hydride and could undergo β-hydride elimination, transforming it into trans-alkene 87.
 | ||
| Scheme 21 Pd-assisted semi-hydrogenation utilizing water as the hydrogen source and Mn as an electron donor. | ||
3.2 Stereo-divergent boration
Tetrasubstituted olefins are important structural motifs in many organic compounds and play a crucial role as intermediates in synthetic chemistry.48–50 The alkyne's hydroboration reaction is fundamental in synthetic chemistry, crucial for the construction of alkenyl boron substrates, which serve as key intermediates in synthesizing a diverse range of valuable chemicals. These compounds play a critical role in distinct sectors, particularly in pharmaceuticals and advanced materials.51–56 However, the development of versatile methods to achieve stereodivergent access to both E- and Z-isomers from a single precursor remains a significant challenge.57,58 The development of stereodivergent hydroboration reactions has significantly transformed this domain, enabling the selective production of both Z- and E-alkenyl boron isomers from a single alkyne precursor. This ability to control stereochemistry expands the practical applications of hydroboration reactions and facilitates the design and production of complex substrates with highly specific stereochemical arrangements.59,60 Recent advancements in stereodivergent hydroboration of alkynes have been significantly propelled by several key publications, each contributing a deeper understanding of this critical reaction.In this context, Lu (2012) reported a ruthenium-catalyzed system that enables hydroboration of alkynes (88) with excellent control over stereoselectivity (Scheme 22).61 This method utilizes ruthenium acriphos complexes, which were systematically synthesized and evaluated for their catalytic properties. The key finding was the ability to selectively produce E- or Z-stereoisomers by modifying the ligands and reaction conditions. The potential for catalyzing stereodivergent hydroboration reactions using different activation methods with various Ru catalysts was explored. Complex 89 was highly effective in catalyzing the stereodivergent hydroboration of terminal alkynes 88, selectively yielding E- or Z-configured products based on the activation approach. When treated with pinacolborane, complex 89 facilitates efficient hydroboration reactions, predominantly forming E-vinyl boranes 90. However, the system switched to favour Z-selectivity 91 when activated with potassium tert-butoxide (KOtBu). The developed ruthenium-based catalytic system shows a high degree of stereocontrol under mild reaction conditions and demonstrates compatibility with a variety of terminal alkynes carrying diverse functional groups. The findings underscore the versatility of Ru-complex 89 in enabling efficient, stereodivergent hydroboration reactions by simply adjusting the activation method.
 | ||
| Scheme 22 Ruthenium-catalyzed system that enables the hydroboration of alkynes. | ||
In another study, Jang et al. in 2016 contributed a versatile copper-catalyzed hydroboration method using 1,8-naphthalenediaminatoborane HB(dan) that is applicable to various terminal and internal alkynes 92, achieving high E/Z selectivity through meticulous optimization of ligands and solvents (Scheme 23).62 In their process, Cu catalyst along with ligand DPEphos (bis[(2-diphenylphosphino)phenyl]ether) exclusively yields Z-stereoselective alkenyl borons 93. On the other hand, employing an NHC–Cu complex SIPr–CuCl as a catalyst predominantly leads to E-hydroboration products 94 under mild conditions. The provided mechanistic details suggest that in situ generated copper–hydride intermediates are crucial for stereocontrol. The Z-selectivity was explained by HB(dan) favoring an intermediate conformation in which the phenyl group is positioned cis to the Cu center. This alignment minimizes steric clashes between the incoming dan group and the phenyl group during the σ-bond metathesis step. On the contrary, the bulky NHC–Cu complex discourages this specific intermediate orientation due to steric hindrance, leading to an E-stereochemical outcome 94.
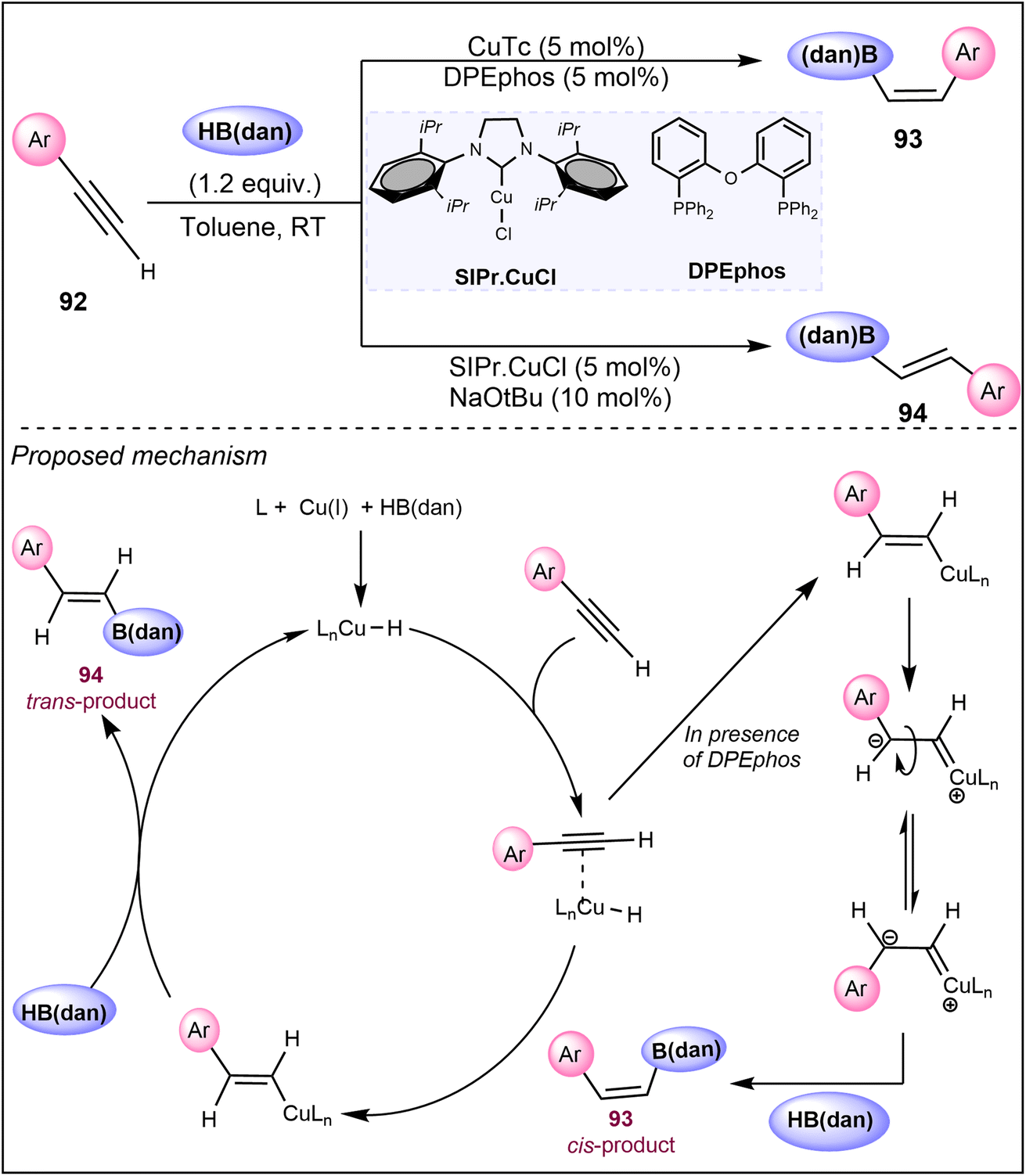 | ||
| Scheme 23 Copper-catalyzed E-/Z-selective hydroboration of terminal and internal alkynes using HB(dan). | ||
Another noticeable contribution was made by Kuang et al. in 2020, where a copper-catalyzed diborylation of CF3-containing 1,3-enynes was developed (95), offering a novel approach for the selective formation of stereochemically defined alkenylboronates (Scheme 24).63 This copper-catalyzed strategy utilizes the distinct reactivity of CF3-enynes (95) to facilitate the regio- and stereoselective addition of boron. Cu catalyst in the presence of the ligand PCy3, base sodium-tert-butoxide, methanol, and THF solvent at 20 °C selectively yields Z-isomeric product 1,3-diborylation (96). Meanwhile, 1,4-diboryl Z-selective product (97) is obtained if the ligand is interchanged with P(OEt)3. Ligand Ph2PCy along with base lithium carbonate and solvent 1,4-dioxane was used for the E-selective product (98). The study demonstrates that E- and Z-isomers could be produced with high selectivity by varying the ligands, bases, and solvents. This copper-catalyzed system enables diborylation without defluorination, expanding the substrate scope to include multi-borylated products. The method is significant for drug discovery and materials science, where trifluoromethylated compounds are highly sought after due to their unique electronic properties. Mechanistic insights into this process were obtained through deuterium labelling and quantum-chemical calculations, which revealed that the Z-selective hydroboration proceeds via trans-addition of the boron reagent.
 | ||
| Scheme 24 Copper-catalyzed stereoselective diborylation of CF3-containing 1,3-enynes. | ||
In 2023, Corpas et al. introduced a groundbreaking iterative dual-metal and energy transfer catalysis approach, allowing for the precise stereochemical control of hydroboration across a broad range of alkynes (Scheme 25).64 The methodology is effective for the synthesis of stereoisomers of tetrasubstituted β-boryl acrylates from internal alkynoates 99 with excellent stereocontrol. Mechanistic insights into these reactions were gained through advanced techniques, including quantum-chemical calculations, quenching experiments, and transient absorption spectroscopy. These studies shed light on the intricate details of both the carboboration and photoisomerization processes, offering a deeper understanding of how stereoselectivity is controlled in this system. The process involves a two-step reaction sequence: first, a syn-carboboration reaction of 99 using B2Pin2 and an electrophile produce a Z-alkene product 100, followed by its E-selective photoisomerization to product 101. The challenge of overcoming the inherent reluctance of electron-deficient internal alkynes to undergo catalytic carboboration was addressed through the cooperative action of copper and palladium catalysis. Additionally, an iridium complex was employed as an effective sensitizer to facilitate the photoisomerization of the sterically hindered alkenes (100). Hence, this work offers a novel dual-metal system that combines iridium and nickel catalysts, with mechanistic studies revealing the importance of energy transfer in achieving stereodivergence.
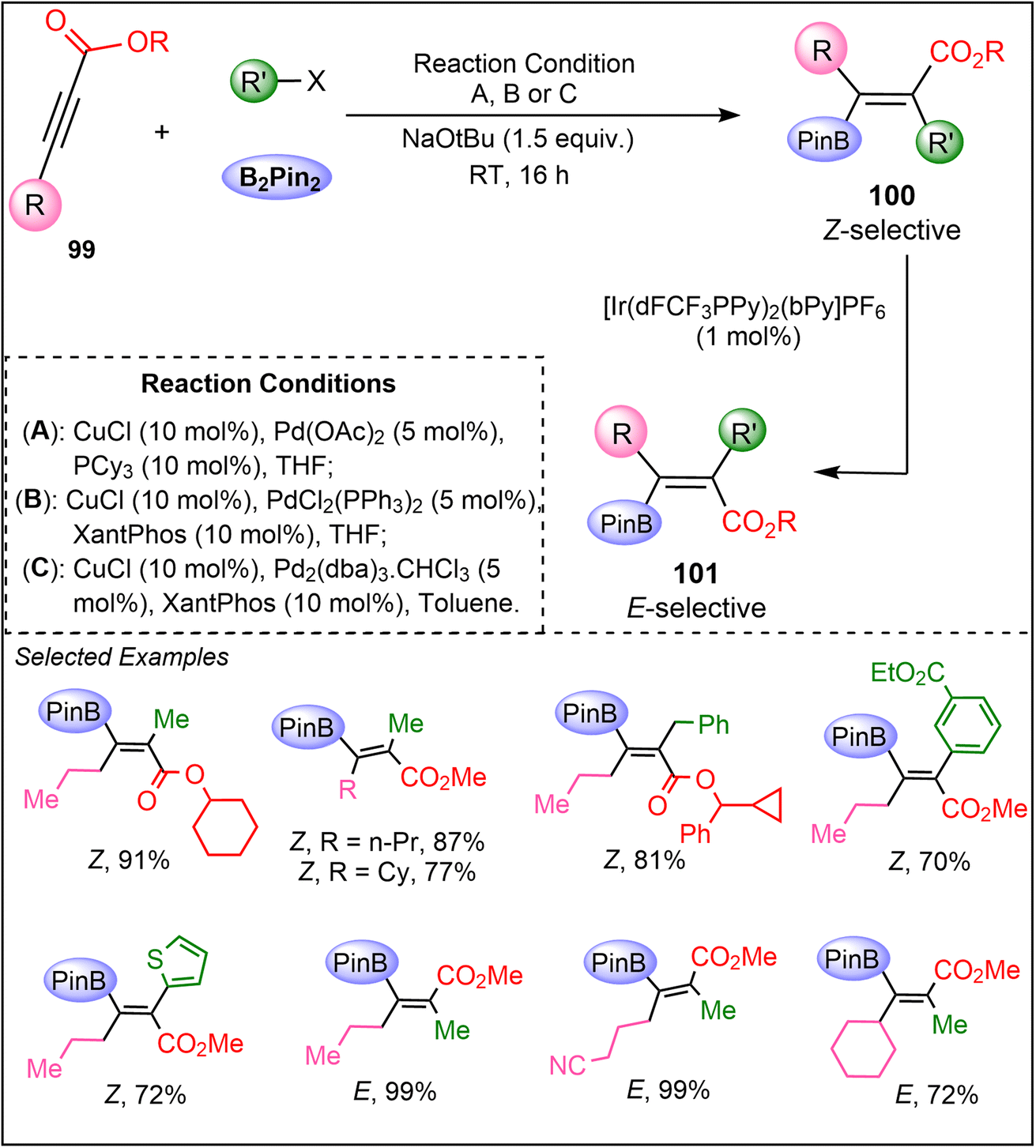 | ||
| Scheme 25 Iterative dual metal (Cu, Ir) catalyzed stereoselective hydroboration of alkynes. | ||
3.3 Stereo-divergent arylation
Recent advancements in the arylation of alkynes have been propelled by innovative research that explores new catalytic strategies, expands substrate scope, and provides deeper mechanistic insights.65–68 Trisubstituted alkenes serve as important synthetic intermediates in organic chemistry, with extensive uses in the production of pharmaceuticals, materials, and fine chemicals.69–72 One of the most efficient methods for synthesizing these compounds is through multicomponent reactions (MCRs). These reactions enable the one-pot difunctionalization of alkynes, where two chemical bonds are formed across a triple bond in a single step. This approach is direct and useful for creating valuable trisubstituted alkenes, offering a simplified and effective route to complex molecule synthesis by minimizing reaction steps and increasing overall efficiency.73–75In this context, Armstrong et al. (2018) made a significant contribution by developing a novel approach for achieving diastereodivergent hydroarylation of terminal alkynes (102) using a tandem catalysis system via palladium and copper co-catalysis (Scheme 26).76 The method allows for selective formation of either E- or Z-isomers of aryl alkenes from the same starting materials 102 by adjusting the stoichiometry of an alcohol additive. This control over stereoselectivity provides access to both isomers with high precision, demonstrating broad functional group tolerance, including compatibility with esters, nitriles, halides, and other functionalities. Z-selective hydroarylation to product 104 was accomplished via a tandem Sonogashira coupling followed by a catalytic semireduction, whereas E-selective hydroarylation product 105 was achieved through an additional catalytic isomerization step of the Z-alkene in the presence of an excess of MeOH in strong alkaline medium.
 | ||
| Scheme 26 Diastereodivergent hydroarylation of terminal alkynes via palladium and copper co-catalysis. | ||
In 2019, Corpas et al. further advanced the field by introducing a dual catalytic system that synergistically combines Pd-catalysis with photocatalysis to achieve regio- and stereocontrol in hydroarylation reactions of unsymmetrical dialkyl alkynes (106) with arylboronic acids, enabling precise access to either the E- or Z-isomer of trisubstituted alkynes (Scheme 27).77 The novelty of this approach lies in the integration of photocatalysis, which enables energy transfer processes that influence the regioselectivity and stereoselectivity of the products. This work introduces a regioselective and stereodivergent catalytic hydroarylation of unsymmetrical dialkyl alkynes 106 with arylboronic acids, enabling precise access to either the E or Z isomer of trisubstituted alkenes. The E-selective product 107 was achieved through a syn-carbopalladation mechanism involving an Ar–Pd species, followed by protodepalladation. The regioselectivity is tightly controlled by a 2-pyridyl sulfonyl (SO2Py) directing group, which ensures accurate positional selectivity during the reaction. The reaction utilizes a Pd/Ir tandem catalytic approach to obtain the Z-isomer 108. This involves a hydroarylation step followed by an E-to-Z photoisomerization, enabling access to the complementary stereochemistry with precision. This versatility allows for the selective formation of stereo-defined olefins and dienes, providing an efficient approach to controlling the stereochemistry in olefin synthesis. The methodology was demonstrated to be versatile, accommodating a wide range of alkynes and aryl halides with various functional groups.
 | ||
| Scheme 27 Photocatalytic approach for regio- and stereocontrol hydroarylation of dialkyl alkynes. | ||
Zhu et al. in 2022 contributed to the field by developing a nickel-catalyzed multicomponent reductive cascade cross-coupling reaction for the arylalkylation of alkynes that integrates electrochemistry and photocatalysis for the stereodivergent synthesis of trisubstituted olefins (Scheme 28).78 This method allows for the selective synthesis of E- or Z-isomers of trisubstituted alkenes by switching between electrochemical and photocatalytic conditions. When the reaction was conducted electrochemically using nickel catalysis, the E-isomer of the trisubstituted alkene (111) was formed exclusively. In contrast, the iridium and nickel-catalyzed photocatalytic transformation under 440 nm blue light results in the Z-selective product (112). Similarly, employing a combination of nickel-catalyzed photocatalysis and electrocatalysis also enables the selective formation of Z-isomeric product (113) with high stereocontrol. The study revealed the complementary roles of Ir-catalyst (110) single-electron transfer (SET) and energy transfer (ET) in controlling stereochemistry, with a broad substrate scope that includes a variety of alkynes (109), along with aryl halides and alkyl bromides. The novelty of this approach lies in its ability to combine electrochemistry with photocatalysis to achieve complex transformations with high stereoselectivity.
 | ||
| Scheme 28 Nickel-catalyzed arylalkylation of alkynes for the stereodivergent synthesis of trisubstituted olefins. | ||
3.4 Stereo-divergent cyanation
Hydrocyanation of alkynes is a vital transformation in organic chemistry, enabling the construction of nitrile-containing compounds, which are highly valuable synthetic intermediates in pharmaceuticals, agrochemicals, and materials science.79–82 The nitrile functional group is versatile, serving as a precursor to amines, amides, carboxylic acids, and other key functional groups.83–85 Moreover, the ability to control the stereochemistry of the resulting alkenes is crucial because the configuration of alkenes (E or Z) can significantly influence the biological and physical properties of target molecules.86,87 Hence, developing efficient and stereoselective methodologies for the hydrocyanation of alkynes has always been an area of significant interest. With recent advancements, there is a growing emphasis on sustainable and cost-effective synthetic catalytic systems that use earth-abundant metals. Here, we focus on the stereodivergent hydrocyanation of alkynes, exploring the unique methodologies developed for E- and Z-alkene synthesis.In this context, Long et al. (2023) presented a detailed study on the palladium-catalyzed stereodivergent hydrocyanation of alkynes (114) to selectively produce E- and Z-acrylonitriles.88 Palladium-based systems have long been favoured for their high catalytic activity and versatility in functionalizing alkynes (Scheme 29).88 In this methodology, ligand selection was key to controlling the stereochemistry of the product, where the monodentate ligands 115 direct the formation of E-isomeric product 117. In contrast, the alkene 117 system facilitates cyclometallation with Pd-catalyst in the presence of bidentate ligands 116, which allows for the isomerization of 117 to Z-isomeric product 118. One of the standout features of their method is its wide substrate compatibility. The study demonstrates that various functionalized propiolamides, including primary, secondary, and tertiary amides, undergo hydrocyanation with high stereoselectivity. This versatility is crucial for practical applications in organic synthesis where functional group tolerance is often a limiting factor. Additionally, the methodology's scalability is demonstrated through large-scale reactions, which are essential for industrial applications. Mechanistic investigations using density functional theory (DFT) calculations reveal that the palladium-catalyzed system operates through a well-defined pathway involving activation of the alkyne, followed by selective hydrogenation and isomerization steps. Their method is noteworthy for its use of water as a hydrogen source, aligning with green chemistry principles.
 | ||
| Scheme 29 Palladium-catalyzed stereodivergent hydrocyanation of alkynes. | ||
Meanwhile, Wang et al. (2022) introduced a cobalt-catalyzed system for the arylcyanation of alkynes, marking an important shift toward the use of earth-abundant and less toxic metals.89 Cobalt is a relatively inexpensive and sustainable alternative to precious metals that exhibits remarkable potential in the stereoselective transformation of alkynes into E- and Z-alkenes (Scheme 30).89 Their system is unique in its ability to switch between cis and trans selectivity by adding Lewis acid cocatalysts, such as Zn(OTf)2. The study highlights the use of a cobalt(II) catalyst with a 4-tBu-dppp (121) ligand system in the presence of Zn(OTf)2 that offers high efficiency and excellent Z-selectivity of product 123. Mechanistic studies suggest that the cobalt catalyst with ligand 120 proceeds via metallacyclopropene intermediates II, which enable facile isomerization of intermediate I to III and control over the stereochemical outcome of product 122. Its compatibility with a variety of functional groups, along with its scalability to multigram synthesis, positions it as a practical and efficient methodology for stereoselective alkyne functionalization. The cobalt-catalyzed process tolerates various aryl nitriles and cyanides, demonstrating its broad applicability across diverse substrates. One of the main advantages of this approach is the ease of catalyst handling and its cost-effectiveness.
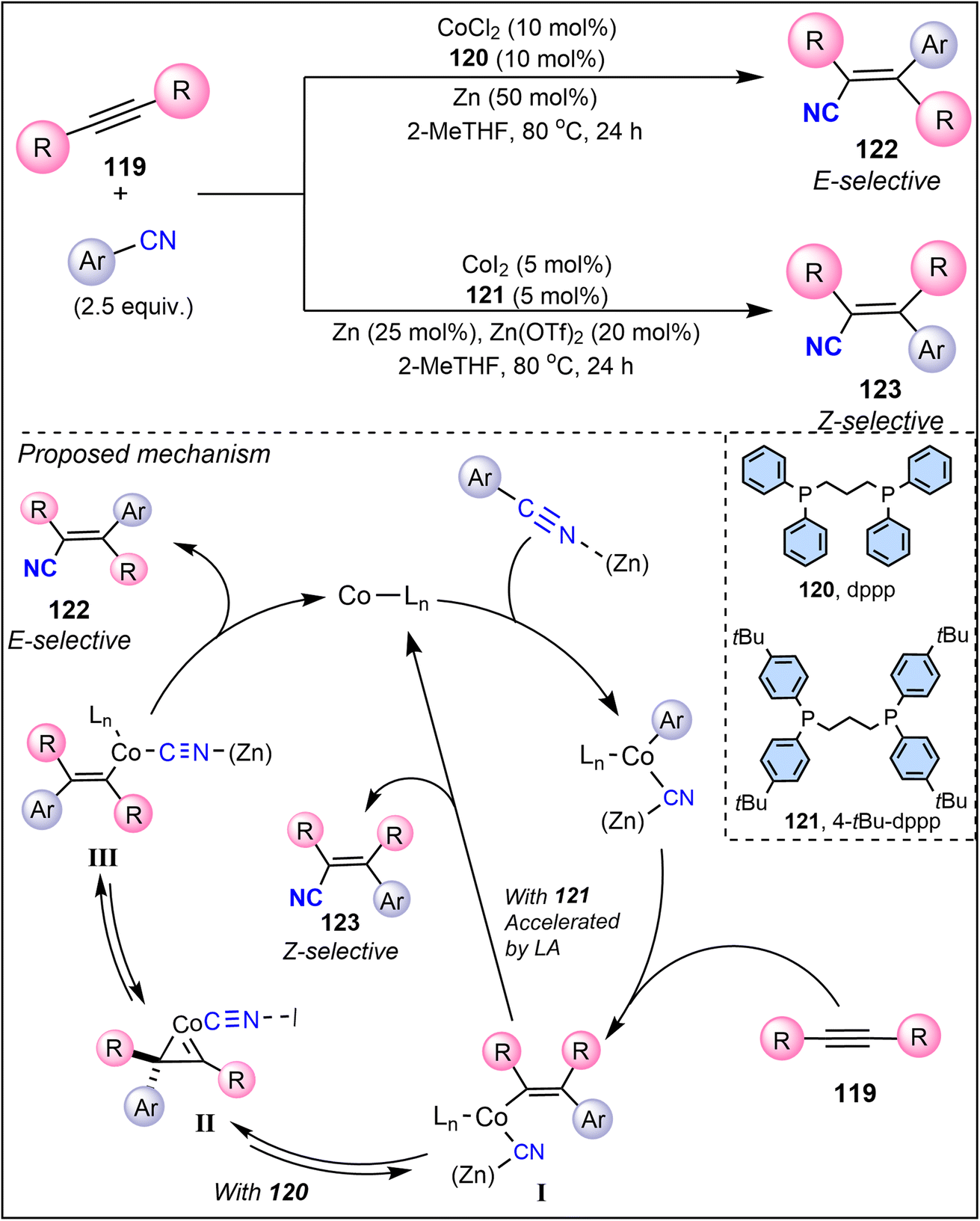 | ||
| Scheme 30 Cobalt-catalyzed system for the arylcyanation of alkynes. | ||
3.5 Stereo-divergent silylation
Vinylsilanes are versatile reagents in synthetic chemistry due to their low toxicity and high stability, facilitating applications in material and medicinal chemistry.90–93 They serve as multifunctional precursor or intermediates in various organic transformations including Hiyama–Denmark coupling94,95 and Tamao–Fleming oxidation.96,97 Hydrosilylation of alkyne is one of the most efficient methods for the synthesis of vinylsilanes.98 Herein, only stereodivergent hydrosilylation approaches were summarized in this section.Mori et al. demonstrated the hydrosilylation of terminal alkynes (124) using RhI(PPh3)3 as a catalyst, revealing that Z- and E-alkenylsilanes can be produced in a stereodivergent way by altering the reaction conditions and the sequence in which the reagents are added (Scheme 31).99 Z-Alkenylsilanes 126 were produced with a high yield (99%) and excellent stereoselectivity (99:1) by adding an alkyne 124 to a pretreated mixture of organosilane and rhodium catalyst at room temperature for 2 hours. In contrast, conducting the reaction with a mixture of an organosilane, an alkyne 124, and the catalyst at 60 °C results in E-products 125, also with a 99% yield and 99% selectivity. Meanwhile, the corresponding rhodium chloride catalyst shows relatively lower reactivity, which could be enhanced by the addition of 5% of NaI in the reaction medium.100
 | ||
| Scheme 31 Hydrosilylation of terminal alkynes using RhI(PPh3)3 catalyst for Z- and E-alkenylsilanes. | ||
In a similar study, the Ozawa group reported a ruthenium catalyst-dependent stereodivergent hydrosilylation of terminal alkynes 127 using various organosilanes (Scheme 32).101 Utilizing RuHCl(CO)(PPh3)3 (A) as the catalyst resulted in the formation of E-selective vinylsilanes 128 with over 99% selectivity and excellent yield. In contrast, Ru(SiMe2Ph)Cl(CO)(i-Pr3)2 (B) as the catalyst produces Z-selective vinylsilanes 129 with 91–99% selectivity. The catalytic pathway for synthesizing E-selective 128 involves alkyne insertion into catalyst A, generating an alkenyl ruthenium complex, followed by insertion of organosilanes through a four-membered transition state (I) to yield 128. The synthesis of Z-selective 129 follows a similar reaction pathway with catalyst B, proceeding through a transition state (II).
 | ||
| Scheme 32 Stereodivergent hydrosilylation of terminal alkynes utilizing RuHCl(CO)(PPh3)3 as the catalyst. | ||
Later on, Yong et al. reported a cobalt(I) catalyzed silane source-dependent stereoselective hydrosilylation of internal alkynes 130 (Scheme 33).102 When an internal alkyne 130 reacts with triethylsilane in the presence of a hemilabile phosphane-tethered cobalt(I) catalyst 131, E-alkenyl silane 133 is formed via syn-addition. A key step in this hydrosilylation process is the oxidation of silane at the vacant coordination sites of the cobalt complex. Interestingly, triethoxysilane instead of triethylsilane reverses the regioselectivity of the reaction, predominantly forming anti-adducts Z-alkenyl silane 134. For unsymmetrical internal alkynes, the yields and regioselectivity of hydrosilylation products are moderate, but syn-hydrosilylation product 133 is exclusively observed in all cases. This indicates that the process is not sterically controlled. Although the reaction mechanism has not been extensively studied, it is speculated to involve syn-hydrosilylation, followed by isomerization through intermediate A.103
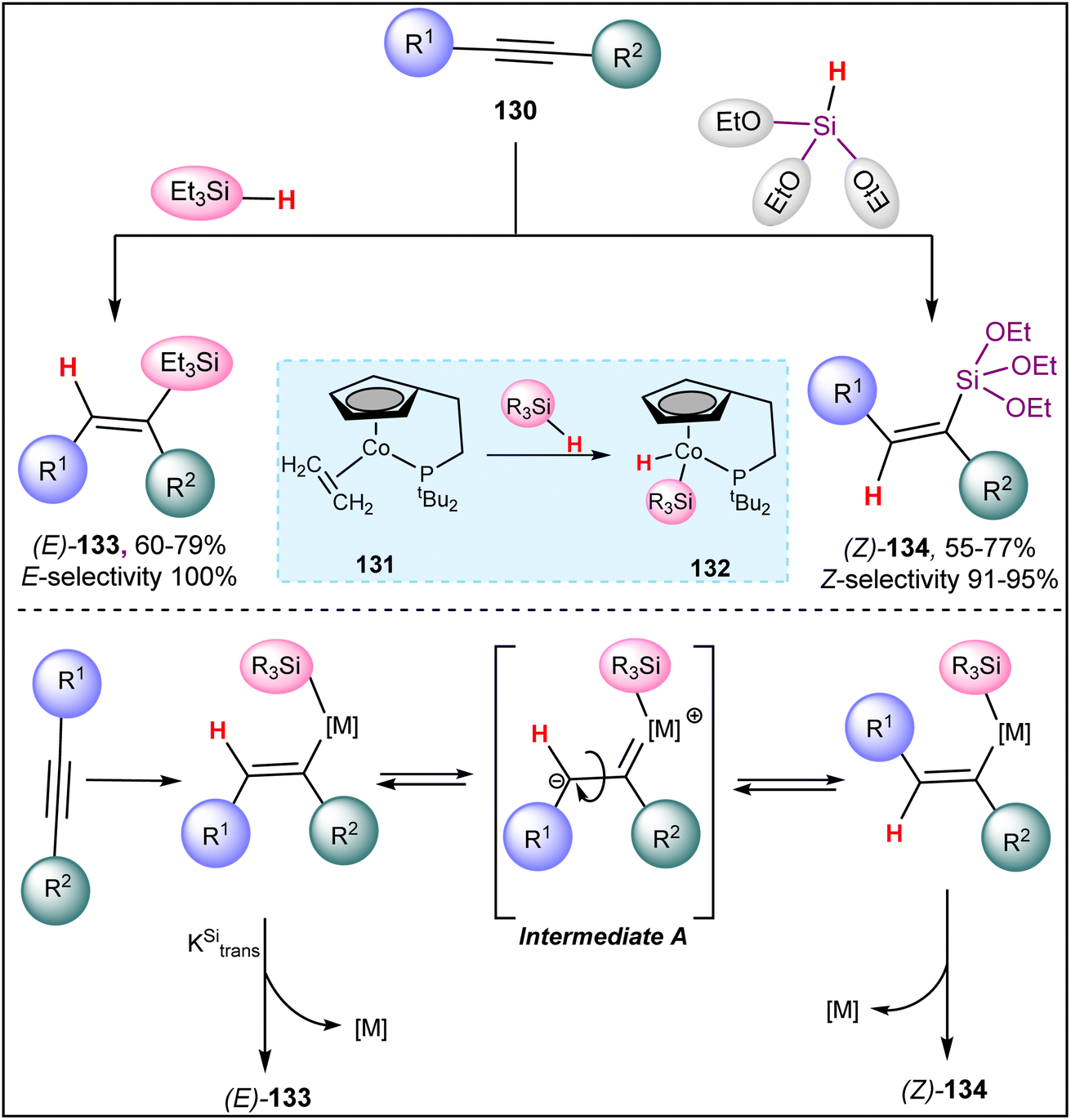 | ||
| Scheme 33 Cobalt(I) catalyzed silane source-dependent stereoselective hydrosilylation of internal alkynes. | ||
In this context, the Plietker group introduced an iron nitrosyl hydride complex 136 catalyzed stereodivergent hydrosilylation of diaryl alkynes 135 (Scheme 34).104 The stereoselectivity of the process is controlled by employing different silane reagents, and a variety of alkynes 135 are converted into their respective E- or Z-configured vinylsilanes in excellent yields. The iron nitrosyl hydride complex FeH(CO)(NO)(PPh3)2 (136) was prepared from protonation of iron tricarbonyl nitrosyl anion [Fe(CO)3(NO)−] with trifluoroacetic acid in the presence of an excess of triphenylphosphine in ether.105 Hydrosilylation of diaryl alkyne 135 using PhSiH3 in the presence of 1 mol% of the Fe-hydride catalyst 136 and 0.5 equivalents of NEt3 in THF at 40 °C afford Z-isomer (trans) of vinyl silane product 138 through a formal trans-addition of the Si–H bond across the C–C triple bond. Instead of PhSiH3, the application of sterically hindered PhMe(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)SiH as the silane reagent inverses the stereoselectivity of the reaction and E-isomer (cis) of vinyl silane 137 is formed as a product.
CH)SiH as the silane reagent inverses the stereoselectivity of the reaction and E-isomer (cis) of vinyl silane 137 is formed as a product.
 | ||
| Scheme 34 Iron nitrosyl hydride complex catalyzed stereodivergent hydrosilylation of diaryl alkynes. | ||
Later on, Ding et al. reported ruthenium-catalyzed ligand-controlled regio- and stereodivergent hydrosilylation of internal silyl alkynes 139 to generate vinyl silanes (Scheme 35).106 The reaction of 1-trimethylsilyl-1-hexyne with triethoxysilane 140 in the presence of a catalytic amount of [cp*Ru(MeCN)3]PF6 (141′) in DCM affords vinyl disilanes 143 in quantitative yield with exclusive α anti addition of silane (α/β > 50:1, Z/E > 50:1) in 24 hours. Remarkably, substituting the cp* ligand with cp in the catalyst 141 completely alters the stereo- and regioselectivity of the reaction, resulting in a β syn-addition product 142 with a quantitative yield (β/α > 50:1, E/Z > 50:1). Notably, this study was one of the few examples of Ru-catalyzed syn-selective alkyne hydrosilylations.107
 | ||
| Scheme 35 [cp*Ru(MeCN)3]PF6 catalyzed ligand-controlled regio- and stereodivergent hydrosilylation. | ||
A wide range of silyl alkynes 139 with different functional groups including esters, mesylates, acetals, and protected alcohols and amines are well tolerated in the mild reaction condition; however, aryl-substituted alkynes are unreactive in the reaction condition. Changing the silyl group on the alkyne substrates does not affect the selectivity and yield of the β syn-addition product 142. The bulky silyl group on the alkyne substrates does not affect the selectivity of the α anti-addition product 143 but significantly reduces the yield. Similarly, the alkoxysilane and chlorosilanes efficiently participate in the hydrosilylation process, but electron-rich trialkyl silanes are unreactive.
Two different catalytic cycles were proposed for the formation of α-anti and β-syn products. In both cases, the reaction starts with the co-ordination of alkyne and silane on the ruthenium catalyst (A and A′), which further undergoes oxidative hydrometalation to form the planar σ-vinyl intermediate (B and B′). An electronic rotation in the σ-vinyl intermediate leads to the formation of metallacyclopropene-like intermediate (C and C′). The preferred rotation causes the β-substituent to position itself opposite to the Cp (or Cp*) to minimize steric hindrance, thereby determining the stereoselectivity of the reaction. Subsequently, the reductive silyl migration from intermediate C or C′ to the carbene center leads to the formation of intermediate D or D′, respectively, which produces the final product through reductive elimination in the presence of precursor 139 and 140.
Similarly, a ligand-dependent palladium-catalyzed stereodivergent approach was reported for the synthesis of E- and Z-enynes from terminal alkyne 144 and silyl iodides (Scheme 36).108 Upon optimizing several ligands, tris(3,5-di-tert-butylphenyl)phosphane (L1) as a ligand in the reaction condition afford Z-conjugated enynes 145 (29–96%) with excellent stereoselectivity up to Z:E > 19:1. Meanwhile, the presence of α,α,α′,α′-tetraaryl-2,2-disubstituted 1,3-dioxolane-4,5-dimethanol (TADDOL) derived phosphoramidite ligand (L2) delivers E-conjugated enyne product 146 in good yield (26–90%) and selectivity (E:Z > 19:1). A broad variety of aryl-substituted alkynes (144) with different electronic properties are successfully tolerated under the reaction conditions; however, alkyl alkynes did not exhibit reactivity. Similarly, various silylated enynes, both E- and Z-selective, could be produced in high yields using different iodosilanes. Even sterically hindered triethylsilyl iodide was reactive, although it yielded a lower product amount. The catalytic cycle of the process starts with the oxidative addition of silyl iodide to Pd-catalyst to generate silylpalladium iodide (A), which reacts with alkyne 144 to yield vinylpalladium species C through intermediate B. In the presence of triaryl phosphine ligands L1, vinylpalladium C undergoes subsequent coupling with another alkyne 144 to afford the Z-selective enynes 145. In contrast, phosphoramidites L2 that are less electron-rich may cause isomerization of vinylpalladium C to the intermediate D, which then leads to the formation of E-enynes 146.
 | ||
| Scheme 36 Ligand-dependent Pd-catalyzed stereodivergent synthesis of E- and Z-enynes using silyl iodides. | ||
Recently, Zhao et al. described regio- and stereodivergent bis silylation reactions of alkynoates 147 using disilane reagents, catalyzed by palladium and Lewis acids (Scheme 37).109 An air-stable disilane reagent, 8-(2-substituted-1,1,2,2-tetramethyldisilanyl)quinoline (TMDQ; 148) was synthesised, which plays a crucial role in controlling selectivity within this catalytic system, enabling the divergent synthesis of 1,2-bissilyl alkenes 149–151. The reaction of alkynoates 147 with an asymmetrical disilane 148 in the presence of a catalytic amount of Pd(dba)2 (5 mol%) in toluene at 120 °C yields the cis-product 149 with over 97% selectivity. Switching the catalytic system to Pd(acac)2 and using 50 mol% of methylaluminum bis(2,6-di-t-butyl-4-methylphenoxide) (MAD) as an additive reverses the reactivity of the silane reagent with alkyne 147, leading to the formation of cis-product 150 in excellent yield and with greater than 97% selectivity. Meanwhile, the trans-product 151 was obtained when the reaction was conducted using a catalytic system of Pd(acac)2 and 40 mol% of tris(pentafluorophenyl)borane (BCF). Increasing the BCF concentration improves selectivity but reduces the overall yield of 151. A wide variety of alkyl and aryl-substituted alkynoates 147 were efficiently converted into corresponding alkenyl silicon derivatives. In the TMS group, silicon is more positively charged than the silicon connected at the C8 position of quinoline of 148, and the α-carbon in alkynoates 147 is more electronegative than the β-carbon. This difference creates a thermodynamic driving force that facilitates the migration of TMS to the α-carbon of the C–C triple bond of 147. The alkyne migration insertion step has the highest activation energy (16.7 kcal mol−1) in the reaction pathway, making it the rate-limiting step and it determines the regioselectivity of the reaction. Adding MAD as an additive completely reverses the regioselectivity of this cis-bis-silylation. MAD acts as a bulky coordinating group, altering regioselectivity by making electronically favoured migratory insertion more difficult due to steric effects. The introduction of BCF changes the reaction mechanism, making reductive elimination the rate-determining step, which leads to E-selectivity in the final product 151.
 | ||
| Scheme 37 Pd and Lewis acid catalyzed regio- and stereodivergent bis silylation of alkynoates. | ||
Yang and Wang envisioned the manganese-catalyzed stereodivergent hydrosilylation of internal alkynes using a wide variety of silanes.110 The reaction of alkyne 152 with different silane 153 in the presence of the catalytic amount of mononuclear MnBr(CO)5 and arsenic ligand, AsPh3 in toluene at 150 °C affords the E-selective vinyl silane product 154 (Scheme 38). The reaction starts with the formation of Mn–Si complex A from Mn-catalyst, ligand, and silane. Complex A produces intermediate C through CO alkyne exchange followed by syn-addition of the Mn–Si bond to alkyne 152. The interaction of complex C with silane 153 followed by σ-bond metathesis via intermediate D leads to the formation of product 154 and Mn–Si complex A. A diverse array of mono-, di-, and tri-substituted silanes with both electron-donating and withdrawing properties are well tolerated in the reaction condition and result in E-configured products 154 in high yields with good to excellent stereoselectivity. Similarly, various aryl and alkyl-substituted alkynes are effectively utilized in this protocol. Surprisingly, the stereoselectivity was reversed in the reaction of alkyne 152 and silane 155 using the dinuclear manganese catalyst Mn2(CO)10 along with dilauroyl peroxide (LPO), resulting in the formation of the Z-isomeric vinyl silane product 156 (Scheme 39). Unlike E-selective hydrosilylation, this Z-selective hydrosilylation protocol is also effective with various terminal aryl alkynes, including those with sensitive halogens. This reaction proceeds through a radical mechanism and initiates from LPO-initiated homolysis of Mn2(CO)10 to an Mn-radical complex A. Silane readily reacts with Mn-complex A to produce HMn(CO)5 and silyl radical. The silyl radical forms an adduct with alkyne to produce E-alkenyl radicals B, which isomerize to sterically preferred Z-alkenyl radicals C.111 Finally, the hydrogenolysis of C with HMn(CO)5 yields the desired product 156 and regenerates the radical Mn-complex A.
 | ||
| Scheme 38 Mn-catalyzed stereodivergent E-hydrosilylation of internal alkynes using a wide variety of silanes. | ||
 | ||
| Scheme 39 Mn-catalyzed stereodivergent Z-hydrosilylation of internal alkynes using a wide variety of silanes. | ||
However, this proposed organometallic catalytic cycle does not fully explain why the Z-isomer is not formed with Mn(CO)5Br. Li's computational study on Mn-catalyzed hydrosilylation provides additional insight into the mechanism, attributing the lack of Z-isomer formation to the large steric hindrance and high energy barrier of isomerization from Mn-complex C to E in Scheme 38.112 In both mononuclear Mn(CO)5Br and binuclear Mn2(CO)10-catalyzed cycles, the rate-determining step is the addition process of the substituted alkyne.
The Aymonier group synthesized onium salt (OS) stabilized metal nanocatalysts (M(0)NCs) in supercritical CO2 (scCO2) and employed them in stereodivergent hydrosilylation of alkynes 157 with triethoxysilane 158 (Scheme 40).113 Hydrosilylation reactions can be catalyzed by various metals such as Pt, Ir, Rh, and Ru, with Pt(0)NCs the most effective. After optimizing these metals with three different OS stabilizers [cetyltrimethylammonium bromide (CTAB), tetrabutylammonium bromide (TBAB) and calcium bis(trifluoromethanesulfonimide) (CTANTf2)], Pt@TBAB exhibits quantitative conversion of alkyne to corresponding vinylsilane (159–161) with up to 71% selectivity of β-E isomer 161. Although the selectivity of the product was less than the previously reported homogeneous ruthenium-catalyzed reaction, this is one of the milestone observations in the field of hydrosilylation through heterogeneous catalysis. Also, a significant loss in β-E-selectivity was observed with increasing concentration of NC from 100 ppm to 10000 ppm.
 | ||
| Scheme 40 Onium salt (OS) stabilized metal nanocatalysts in supercritical CO2 for the E/Z hydrosilylation of alkynes. | ||
To advance hydrosilylation, Fopp et al. described a catalyst-free cis- and trans-selective silylzincation of various α-heteroatom-substituted terminal alkynes 162 using (Me2PhSi)2Zn and [(Me3Si)3Si]2Zn, respectively (Scheme 41).114,115 Both reagents exhibit highly regio- and stereoselective addition across the C–C triple bond of alkynes 162 substituted with nitrogen, sulfur, oxygen, and phosphorus, exclusively yielding β-silyl isomers. The reaction of 162 with (Me2PhSi)2Zn in ether at 0 °C leads to the formation of Z-2-(silyl)vinyl zinc isomer 163. Quenching of this organozinc intermediate 163 with NH4Cl/NH3 produces α-heteroatom-substituted vinyl silanes 164 in moderate to good yields with excellent stereoselectivity. A copper-mediate electrophilic substitution of zinc in 163 proceeds efficiently with complete retention of the geometry across the double bond and further extends the scope of the reaction to product 165. On the other hand, the reaction of 162 with [(Me3Si)3Si]2Zn in n-hexane at lower temperature leads to the formation of E-2-(silyl)vinyl zinc isomer 166, which shows similar reactivity with electrophiles with retention of geometry. The syn-addition of (Me2PhSi)2Zn to the triple bond through a polar mechanism was supposed for the formation of 163.116 Meanwhile, a radical-chain mechanism was supposed for the reversal of stereoselectivity in 166 with [(Me3Si)3Si]2Zn.117
 | ||
| Scheme 41 E-/Z-Selective silylzincation of α-heteroatom-substituted terminal alkynes using organo-zinc. | ||
3.6 Stereo-divergent fluorination
Fluoroalkene are isosteres of peptide amide bonds and have become increasingly prevalent as structural and mechanistic probes in biological118,119 and chemical studies.120,121 These analogs are valuable because they can mimic the electronic and geometric properties of peptide bonds while offering enhanced stability and unique interactions. Given the importance of fluoroalkenes, significant efforts have been made to develop efficient regio- and stereoselective syntheses of these compounds.122–124 In particular, stereodivergent hydrofluorination allows for precise control over the synthesis of specific fluoroalkene isomers with desired configurations.In this context, Guo et al. reported a metal-free hydrofluorination of alkyne 169 using protic tetrafluoroborate salts as tunable hydrofluorinating reagents to control the regio- and stereoselectivity of the reaction (Scheme 42).125 A wide variety of internal alkyne 169 were treated with different tetrafluoroborate salts in DCM or DCE. The reported conditions are compatible with a wide range of functional groups and successfully employed for the late-stage functionalization of drug derivatives and for synthesizing fluorinated drug analogues. The reaction of alkyne 169 with HBF4·Et2O results in the E-isomer of fluoroalkenes 170 as a major product through intermediate A and B, whereas 2,6-dichloropyridinium tetrafluoroborate as tetrafluoroborate salt along with LiBF4 afforded the Z-isomer of fluoroalkenes 171. The presence of LiBF4 as an additive enhances the concentration of tetrafluoroborate anion, promotes the anti-attack of fluoride on intermediate D, suppresses the formation of this side product, and further enhances the yield of Z-alkene product 171. At elevated temperatures, in situ generated BF3 leads to fluoride elimination from B to produce the vinyl cation C, which undergoes isomerization to C′ and reduces the selectivity of the final product 170.
 | ||
| Scheme 42 Metal-free hydrofluorination of alkyne using protic tetrafluoroborate salts as tunable hydrofluorinating reagents. | ||
3.7 Miscellaneous
Halogenated alkenes play a crucial role in various scientific fields, notably in pharmaceuticals, agrochemicals, and chemical industries.126 They serve as key starting materials for metal-mediated cross-coupling reactions,127 which are widely utilized in scientific research for small molecule synthesis and in the industrial production of numerous compounds. In this context, Lauten's group realized a Pd-catalyzed intramolecular alkyne carbohalogenation for stereodivergent addition of aromatic halide across alkyne to prepare vinylic halides.128 1-Halo-2-((prop-2-yn-1-yl)oxy)benzene type substrates 172 undergo intramolecular carbohalogenation in the presence of Pd(Q-Phos)2 catalyst in toluene at 50 °C to produce cis-3-(halomethylene)-2,3-dihydrobenzofurans 175 (Scheme 43). Raising the reaction temperature to 100 °C did not significantly affect the product yield, but it completely reversed the stereoselectivity, resulting in the trans-isomer 177. In situ NMR studies indicate that the thermodynamic trans-product 177 primarily forms through the isomerization of the intermediate 174 rather than directly from the substrate 172. To better understand the mechanism of olefin isomerization, isomerically pure cis-isomers 175 were re-exposed to standard reaction conditions at 100 °C, leading to their conversion into the trans-isomer 177. Similarly, trans-isomer 177 was converted to cis-isomer 175 when treated at 50 °C. The reaction effectively tolerates electron-withdrawing and electron-donating substituents on the aryl bromide substrate 172. | ||
| Scheme 43 Pd-catalyzed intramolecular alkyne carbohalogenation for the stereodivergent addition of aromatic halide across alkyne to yield vinylic halides. | ||
Similarly, Liu et al. reported a catalyst-controlled stereodivergent synthesis of five-membered N-heterocycles (180–182) via tandem annulation of amino alkynes 178 with diazo compounds 179 (Scheme 44).129 The rhodium-catalyzed method involves carbenoid insertion into the N–H bond to generate ylide intermediate A, followed by Conia-ene 5-exo-dig cyclization, resulting in 3-methylene pyrrolidines 180. In contrast, the copper-catalyzed reaction begins with the cross-coupling of an alkyne 178 with diazo compounds 179, producing allenoate intermediates B that may undergo either 5-exo-dig or 5-endo-dig intramolecular hydroamination, yielding the respective cycloaddition products 181 and 182. When this copper-catalyzed transformation was carried out with N-tosyl amino alkynes in acetonitrile, it led to the formation of dihydropyrroles 181 via 5-exo-dig cyclization. However, using sterically hindered N-benzyl amino alkynes in chloroform produces E-selective pyrrolidines 182. Generally, the chiral propargyls 178 substrates containing either electron-rich or electron-deficient aryl or alkyl substituents adjacent to the N-atom gave the corresponding products in moderate to high yields and with good stereoselectivity.
 | ||
| Scheme 44 Catalyst-controlled stereodivergent synthesis of five-membered N-heterocycles via the tandem annulation of amino alkynes with diazo compounds. | ||
Like halogenated alkene, CF3-alkenes are excellent precursors in various organic transformations and are of great importance in pharmaceutical, agricultural, and materials science.130,131 Hydrometallation of α-CF3-alkynes is one of the most efficient approaches for producing trifluoromethylated alkene precursors.132 In this context a stereoselectivity tunable hydrogermylation reaction was developed for the synthesis of α-CF3-vinylgermanes 185 and 186, which could be further utilized as precursors in cross-coupling reactions (Scheme 45).133,134 Hydrogermylation of aryl and alkyl α-CF3-alkynes 183 with organogermanium hydrides 184 selectively produces Z-isomer of α-CF3-vinylgermane 185 in 71–87% isolated yield in the presence of a radical initiator peroxydisulfate. Radical initiator (NH4)2S2O8 efficiently oxidizes organogermanium species 184 to the corresponding organogermyl radical, which leads to the trans-addition of radicals and forms Z-selective vinylgermanes 185. Meanwhile, exploration of Pd(PPh3)4-catalyzed hydrogermylation shows a regio- and stereoselective cis-addition of reagent 184 to α-CF3-alkyne substrate 183 to form E-isomer of α-CF3-vinylgermane 186 in moderate to good yield (43–91%). The synthesized vinylgermanes were further investigated in Pd-catalyzed coupling reactions with aryl halides to expand the scope of the reaction and prepare α-CF3-styrenes 187 and 188.
 | ||
| Scheme 45 Stereoselectivity tunable hydrogermylation reaction for the synthesis of α-CF3-vinylgermanes. | ||
Homoallylic ketones, also known as γ,δ-unsaturated ketones are widely employed in the synthesis of heterocycles, natural products, electronic optical organic materials, peptidomimetics, and much more.135 Cruz and Dong demonstrated a chiral Jacobsen's amine (L1 and L2) dependent stereodivergent synthesis of α-β-chiral homoallylic ketones 191 and 192 through Rh-hydride catalyzed coupling of α-branched aldehydes 189 with alkynes 190 (Scheme 46).136 Electron-rich aromatic aldehydes (189) show good performance under the reaction conditions, while the electronic properties of the substituents on alkynes 190 did not significantly affect the reactivity. The reaction achieved excellent diastereoselectivity and reactivity (68–95%, up to >20:1 dr and >99% ee) for the anti-diastereomer product (S,S)-191 when the chiral organocatalyst amine (S,S)-L1 was used along with a Rh-(R)-DTBM-BINAP catalyst. By switching to the ligand (R,S)-L2, the diastereoselectivity was reversed, allowing for the formation of the syn-diastereomer (R,S)-192 (81–93%, up to 8:1 dr and >99% ee). A detailed computational study on the mechanism of the coupling of aldehydes and alkynes catalyzed synergistically by rhodium and amine revealed the reaction pathway.137 Rh-hydride species A was generated in situ from [Rh(cod)Cl]2 and phosphoric acid. The Rh-hydride reacts with alkyne and generates a Rh-π-allyl complex C via the formation of allene intermediate B. At the same time, the amine organocatalyst generates an enamine D with aldehyde substrate 189. The regioselective C–C bond formation between Rh-π-allyl complex C and enamine D (at the more substituted carbon of complex) forms the intermediate E which undergoes hydrolysis to result in the desired homoallylic ketones 191 and 192. The diastereoselectivity was determined by the syn- or anti-configuration of intermediate C and D during C–C bond formation, which was influenced by the steric hindrance created by the chiral Jacobsen's amine (L1 and L2) and the chiral Rh-(R)-DTBM-BINAP catalyst.
 | ||
| Scheme 46 Chiral Jacobsen's amine-dependent stereodivergent synthesis of α-β-chiral homoallylic ketones and through Rh-hydride catalyzed coupling of α-branched aldehydes with alkynes. | ||
Vinyl sulfone serves as a versatile building block in organic transformations and is also a core structural component in many drug candidates and bioactive compounds.138 It has been recognized as a substitute for α,β-unsaturated carbonyl groups, readily participating in 1,4-addition and cycloaddition reactions.139,140 In this context, Long et al. demonstrated a ligand-controlled stereodivergent difunctionalization of terminal alkynes 193 using sodium sulfinates 194 and vinyl triflates 193 (Scheme 47).141 This approach employs dual photoredox and nickel catalysis to efficiently produce synthetically valuable cis- and trans-sulfonyl-1,3-dienes in moderate to good yield. The reaction of alkyne substrate 193 with 194 and 195 in the presence of a catalytic amount of Ru(dtbbpy)3(PF6)2 and Ni(OAc)2·4H2O, along with terpyridine as the ligand, results in an anti-addition process, producing highly E-selective 1,3-dienes 196 (E/Z > 99:1) as a product. In contrast, when 1,10-phenanthroline was used as the ligand with the NiCl2·dppf catalyst, the reaction favoured syn-addition, yielding Z-selective 1,3-dienes 197 (Z/E > 99:1). Aromatic and aliphatic alkynes 193 and sulfinates 194 serve as suitable substrates for both reactions. The reaction conditions are compatible with a broad range of functional groups in sulfinates 194 and alkynes 193, including halides and esters. Additionally, cyclic vinyl triflates 195 provide good yields of 1,3-dienes (196 and 197), while acyclic vinyl triflates result in lower diene yields, albeit with excellent selectivity.
 | ||
| Scheme 47 Ligand-controlled stereodivergent functionalization of terminal alkynes using sodium sulfinates and vinyl triflates. | ||
Upon light excitation, the photoexcited *Ru(dtbbpy)32+  interacts with 194 (Ered ≈ +0.45 V vs. SCE in CH3CN), releasing a sulfonyl radical and generating Ru(I). When terpyridine is used as the ligand, the sulfonyl radical is trapped by Ni(I) to form Ni(II)–SO2R (A), which is further reduced by Ru(I) to produce Ni(I)–SO2R complex (B). The alkyne 193 coordination to intermediate B and successive regioselective migratory insertion yields the cis-alkenyl-Ni(I) species C, which undergoes anti/syn-isomerization (activation energy = 25.1 kcal mol−1), leading to the intermediate trans-alkenyl-Ni(I) (D). The oxidative addition of D with vinyl triflate 195 via an SN-Ar type mechanism forms trans-Ni(III) (E), which produces anti-addition dienes 196 through reductive elimination.
interacts with 194 (Ered ≈ +0.45 V vs. SCE in CH3CN), releasing a sulfonyl radical and generating Ru(I). When terpyridine is used as the ligand, the sulfonyl radical is trapped by Ni(I) to form Ni(II)–SO2R (A), which is further reduced by Ru(I) to produce Ni(I)–SO2R complex (B). The alkyne 193 coordination to intermediate B and successive regioselective migratory insertion yields the cis-alkenyl-Ni(I) species C, which undergoes anti/syn-isomerization (activation energy = 25.1 kcal mol−1), leading to the intermediate trans-alkenyl-Ni(I) (D). The oxidative addition of D with vinyl triflate 195 via an SN-Ar type mechanism forms trans-Ni(III) (E), which produces anti-addition dienes 196 through reductive elimination.
On the other hand, dppf-ligated Ni(I) (F) is a relatively more electron-rich and sterically less hindered catalytic system, generating a Ni(0) species (G) through single-electron transfer (SET) reduction by Ru(I) species, which binds with vinyl triflate 195, and undergoes SN-Ar type oxidative addition to form the dppf-ligated alkenyl-Ni(II) intermediate H. A subsequent ligand exchange with 1,10-phenanthroline produces phenanthroline-ligated alkenyl-Ni(II) complex I. Simultaneously, the sulfonyl radical adds to alkyne 193, forming vinyl radical J, which is captured by complex I, resulting in the more stable cis-Ni(III) species K. Reductive elimination of Ni(III) complex K produces the syn-selective product 197 and regenerates Ni(I)-species, which undergoes ligand exchange with dppf to reform the catalyst F. Finally, Ru(dtbbpy)3+, (EII/I1/2 = −1.45 V vs. SCE), reduce (dppf)Ni(I) F or (terpy)Ni(II) A,  regenerating the ground-state Ru(dtbbpy)32+ and completing the catalytic cycles.
regenerating the ground-state Ru(dtbbpy)32+ and completing the catalytic cycles.
Concurrently, the Srivastava group reported stereodivergent difunctionalizations of terminal and internal alkynes 198 using various sulfinates 199 and isocyanides 200 to synthesize Z- and E-β-sulfonylacrylamides 203 and 204 (Scheme 48).142 The Z-β-sulfonylacrylamides 203 can be synthesized in a one-pot process by sequentially adding sulfonates 199 and isocyanides 200 to terminal alkynes 198 in ethanol. In contrast, a two-step approach is used to produce E-sulfonylacrylamides 204, which involves the preparation of E-β-iodovinylsulfones 202 from sulfinates 199 and terminal or internal alkynes 198 in water, followed by a palladium-catalyzed isocyanide (200) addition reaction.
 | ||
| Scheme 48 Stereodivergent difunctionalization of terminal and internal alkynes using various sulfinates and isocyanides to synthesize Z- and E-β-sulfonylacrylamides. | ||
Both reactions proceed smoothly with aliphatic isocyanides (200), but aromatic isocyanides are unsuitable. Appreciatively, both electron-rich and electron-poor acetylenes (198) with aromatic or aliphatic substitution react smoothly, providing the corresponding sulfonylacrylamides 203 and 204 products in good to excellent yields. Similarly, both aromatic and aliphatic sulfonates 199 afford the products 203 and 204, but the yields are slightly lower with aliphatic sulfinates. A reaction mechanism is proposed for the metal-free addition of isocyanide to the in situ-generated E-intermediate 201, leading to the formation of Z-β-sulfonylacrylamide 203 through a Michael-type addition–elimination sequence. It is presumed that Z-selectivity is preferred due to the charge stabilization after Michael-type addition (Scheme 49(1)). The formation of E-β-sulfonylacrylamide 204 occurs through Pd-catalyzed isocyanide addition to E-intermediate 202 (Scheme 49(2)). This reaction initiates with the generation of Pd(0) species from Pd(OAc)2 and DPPF in the presence of DBU, which undergoes oxidative addition with 202 to generate organopalladium complex A, followed by coordinates with isocyanide 200 to form complex B. Subsequent migratory insertion of isocyanide gives rise to complex C, which takes a water molecule to furnish complex D and undergoes successive reductive elimination and then tautomerization to afford E-β-sulfonylacrylamide 204.
 | ||
| Scheme 49 Stereodivergent difunctionalisation of alkyne to (1): Z-sulfonylacrylamides and (2): E-sulfonylacrylamides. | ||
In the realm of alkyne functionalization, Qin et al. introduced a metallaphotoredox approach for stereodivergent three-component carboallylation of terminal alkynes 205 with allylic carbonates 206 and alkyl trifluoroborates 207 (Scheme 50).143 The reaction of terminal alkyne 205, allylic carbonate 206, and tert-butyl trifluoroborate 207 in the presence of a catalytic amount of Ni(phen)Cl2, 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) as the ligand, 4CzIPN as the photocatalyst, and pyrene as an additive was carried out in a DMAc/MeCN solvent mixture under blue LED irradiation (λmax = 467 nm) at 35 °C, yielding the desired (E,Z)-1,4-diene product 208 with an 84% yield and high stereoselectivity (E/Z = 92:8). Interestingly, the stereoisomer (Z,Z)-1,4-diene 209 was obtained with a 79% yield and an opposite stereoselectivity (E/Z = 16:84) when the reaction was conducted without pyrene in the DME solvent.
 | ||
| Scheme 50 Metallaphotoredox approach for the stereodivergent carboallylation of terminal alkynes with allylic carbonates and alkyl trifluoroborates. | ||
This redox-neutral dual catalytic method employs commercially available organic photocatalyst 4CzIPN and nickel catalysts to initiate a radical addition/alkenyl–allyl coupling sequence, providing a straightforward route to functionalized 1,4-dienes 208 and 209 with high chemo-, regio-, and stereoselectivity. The process begins with the single-electron oxidation of alkyl trifluoroborate (Ered1/2 = +1.26 V vs. SCE) by photoexcited [4CzIPN]* (E1/2 = +1.35 V vs. SCE), generating an alkyl radical A, which then adds to an alkyne 205 to form alkenyl radical B. The catalytic cycle continues with the oxidative addition of allylic ester 206 to a Ni0 catalyst, forming allylnickel intermediate C. This intermediate captures alkenyl radical B to generate a trans-(alkenyl)(allyl)NiIII-species D, which yields the (E,Z)-skipped diene product 208 and NiI-species E upon reductive elimination. A final single-electron transfer between the reducing photocatalyst and NiI-species E regenerates the ground-state photocatalyst and Ni0 catalyst, closing both catalytic cycles. The (E,Z)-skipped diene 208 remains stable in the presence of pyrene but undergoes photoinduced *4CzIPN-enabled E-to-Z isomerization to form 209 in the absence of pyrene, providing modular access to both trans- and cis-1,4-dienes.
4. Conclusion
The collective research presented in this review reflects significant progress in the field of stereodivergent semi-hydrogenation of alkynes. By leveraging different metals, ligand architectures, and sustainable hydrogen sources, researchers have developed a diverse array of catalytic systems capable of achieving high levels of stereoselectivity. The emphasis on green chemistry principles, such as the use of water as a hydrogen source and the avoidance of toxic reagents, further underscores the drive toward more sustainable chemical processes. Moving forward, the field is poised to benefit from continued exploration into catalyst stability, reaction scalability, and expanded substrate scope, which will undoubtedly lead to new discoveries and applications in selective alkyne hydrogenation.This comprehensive overview provides a solid foundation for understanding the current state of research in stereodivergent semi-hydrogenation, highlighting the challenges and opportunities. By integrating innovative mechanistic insights with environmentally conscious methodologies, the field will continue to evolve, driving future breakthroughs in selective alkene synthesis for pharmaceuticals, materials science, and beyond.
Data availability
The datasets generated during the current study are compilations of publicly available literature.Conflicts of interest
There are no conflics of interest to declare.Acknowledgements
RP and AS thank the University of Delhi for providing suitable infrastructure for the literature search. AS thanks DST for providing a salary under the WISE-PDF scheme (WISE-PDF/CS-39/2024) which helped her to focus on this work. RS thanks GLA University, IA thanks Umm Al-Qura University and DKY thanks Gachon University for their support.References
- I. P. Beletskaya, C. Najera and M. Yus, Stereodivergent catalysis, Chem. Rev., 2018, 118, 5080–5200 CrossRef CAS PubMed.
- A. R. Healy, Stereodivergent Carbon–Carbon Bond-Forming Reactions, Synthesis, 2024, 56(14), 2223–2233 CrossRef CAS.
- H. Wang, Q. Zhang and W. Zi, Synergistic Catalysis Involving Palladium for Stereodivergent Csp3–Csp3 Coupling Reactions, Acc. Chem. Res., 2024, 57, 468–488 CAS.
- V. Srinivasulu, G. Srikanth, M. A. Khanfar, I. A. Abu-Yousef, A. F. Majdalawieh, R. Mazitschek, S. C. Setty, A. Sebastian and T. H. Al-Tel, Stereodivergent complexity-to-diversity strategy en route to the synthesis of nature-inspired Skeleta, J. Org. Chem., 2022, 87, 1377–1397 CrossRef CAS PubMed.
- S.-M. Xu, L. Wei, C. Shen, L. Xiao, H.-Y. Tao and C.-J. Wang, Stereodivergent assembly of tetrahydro-γ-carbolines via synergistic catalytic asymmetric cascade reaction, Nat. Commun., 2019, 10, 5553 CrossRef PubMed.
- C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Stereodivergent routes in organic synthesis: carbohydrates, amino acids, alkaloids and terpenes, Org. Biomol. Chem., 2020, 18, 1232–1278 RSC.
- C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Stereodivergent routes in organic synthesis: marine natural products, lactones, other natural products, heterocycles and unnatural compounds, Org. Biomol. Chem., 2020, 18, 1279–1336 RSC.
- H. Takamura, Recent topics of the stereodivergent synthesis of natural products, Tetrahedron Lett., 2018, 59, 955–966 CrossRef CAS.
- M. Bortolami, R. Petrucci, D. Rocco, V. Scarano and I. Chiarotto, Alkynes as Building Blocks, Intermediates and Products in the Electrochemical Procedures Since 2000, ChemElectroChem, 2021, 8, 3604–3613 CrossRef CAS.
- N. Chalotra, J. Kumar, T. Naqvi and B. A. Shah, Photocatalytic functionalizations of alkynes, Chem. Commun., 2021, 57, 11285–11300 RSC.
- A. Ramani, B. Desai, M. Patel and T. Naveen, Recent advances in the functionalization of the terminal and internal alkynes, Asian J. Org. Chem., 2022, 11, e202200047 CrossRef CAS.
- F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, John Wiley & Sons, 2006 Search PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A stepwise huisgen cycloaddition process: copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes, Angew. Chem., 2002, 114, 2708–2711 CrossRef.
- J. Corpas, P. Mauleón, R. Gomez Arrayas and J. C. Carretero, E/Z Photoisomerization of Olefins as an Emergent Strategy for the Control of Stereodivergence in Catalysis, Adv. Synth. Catal., 2022, 364, 1348–1370 CrossRef CAS.
- Y. Ke, W. Li, W. Liu and W. Kong, Ni-catalyzed ligand-controlled divergent and selective synthesis, Sci. China:Chem., 2023, 66, 2951–2976 CrossRef CAS.
- R. Shaw, A. Elagamy, I. Althagafi and R. Pratap, Synthesis of alkynes from non-alkyne sources, Org. Biomol. Chem., 2020, 18, 3797–3817 RSC.
- M. L. N. Rao and S. S. Islam, Synthesis of alkynes under dry reaction conditions, Tetrahedron Lett., 2021, 71, 153051 CrossRef CAS.
- J. Chen, X. Zhang, J. Wu, R. Wang, C. Lei and Y. An, Facile one-pot synthesis of diarylacetylenes from arylaldehydes via an addition-double elimination process, Org. Biomol. Chem., 2021, 19, 4701–4705 RSC.
- T. Kimura, K. Sekiguchi, A. Ando and A. Imafuji, Fritsch–Buttenberg–Wiechell rearrangement of magnesium alkylidene carbenoids leading to the formation of alkynes, Beilstein J. Org. Chem., 2021, 17, 1352–1359 CrossRef CAS PubMed.
- W. Sun, L. Xu, Y. Qin and C. Liu, Alkyne synthesis through coupling of gem-diborylalkanes with carboxylic acid esters, Nat. Synth., 2023, 2, 413–422 CrossRef CAS.
- Y. R. Chen, A. Naresh, S. Y. Liang, C. H. Lin, R. J. Chein and H. C. Lin, Discovery of a dual function cytochrome P450 that catalyzes enyne formation in cyclohexanoid terpenoid biosynthesis, Angew. Chem., Int. Ed., 2020, 59, 13537–13541 CrossRef CAS PubMed.
- J. M. Lv, Y. H. Gao, H. Zhao, T. Awakawa, L. Liu, G. D. Chen, X. S. Yao, D. Hu, I. Abe and H. Gao, Biosynthesis of biscognienyne B Involving a cytochrome P450-dependent alkynylation, Angew. Chem., 2020, 132, 13633–13638 CrossRef.
- R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, 2nd edn, 1999 Search PubMed.
- A. M. Kluwer, and C. J. Elsevier, Homogeneous Hydrogenation of Alkynes and Dienes, VCH, Weinheim, Germany, 2007, pp. 374−411 Search PubMed.
- S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Catalytic Z-selective olefin cross-metathesis for natural product synthesis, Nature, 2011, 471, 461–466 CrossRef CAS PubMed.
- E. Richmond and J. Moran, Ligand control of E/Z selectivity in nickel-catalyzed transfer hydrogenative alkyne semireduction, J. Org. Chem., 2015, 80, 6922–6929 CrossRef CAS PubMed.
- M. S. Holzwarth and B. Plietker, Biorelevant metals in sustainable metal catalysis—a survey, ChemCatChem, 2013, 5(7), 1650–1679 CrossRef CAS.
- C. Q. Zhao, Y. G. Chen, H. Qiu, L. Wei, P. Fang and T. S. Mei, Water as a hydrogenating agent: stereodivergent Pd-catalyzed semi-hydrogenation of alkynes, Org. Lett., 2019, 21, 1412–1416 CrossRef CAS PubMed.
- J. Li and R. Hua, Stereodivergent Ruthenium-Catalyzed Transfer Semi-hydrogenation of Diaryl Alkynes, Eur. J. Org Chem., 2011, 17, 8462–8465 CrossRef CAS PubMed.
- T. Chen, J. Xiao, Y. Zhou, S. Yin and L. B. Han, Nickel-catalyzed (E)-selective semi-hydrogenation of internal alkynes with hypophosphorous acid, Organomet. Chem., 2014, 749, 51–54 CrossRef CAS.
- A. F. Trindade, P. M. Gois and C. A. Afonso, Recyclable stereoselective catalysts, Chem. Rev., 2009, 109(2), 418–514 CrossRef CAS PubMed.
- S. Fu, N. Y. Chen, X. Liu, Z. Shao, S. P. Luo and Q. Liu, Ligand-controlled cobalt-catalyzed transfer hydrogenation of alkynes: stereodivergent synthesis of Z-and E-alkenes, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS PubMed.
- X. Qi, X. Liu, L. Qu, Q. Liu and Y. Lan, Mechanistic insight into cobalt-catalyzed stereodivergent semi-hydrogenation of alkynes: The story of selectivity control, J. Catal., 2018, 362, 25–34 CrossRef CAS.
- J. Li and R. Hua, Stereodivergent Ruthenium-Catalyzed Transfer Semi-hydrogenation of Diaryl Alkynes, Chem. - Eur. J., 2011, 17, 8462–8465 CrossRef CAS PubMed.
- Y. Wu, Y. Ao, Z. Li, C. Liu, J. Zhao, W. Gao, X. Li, H. Wang, Y. Liu and Y. Liu, Modulation of metal species as control point for Ni-catalyzed stereodivergent semi-hydrogenation of alkynes with water, Nat. Commun., 2023, 14, 1655–1665 CrossRef CAS PubMed.
- T. Hu, M. Jaber and A. Amgoune, Photoinduced NiH Catalysis with Trialkylamines for the Stereodivergent Transfer Semi-Hydrogenation of Alkynes, Chem. - Eur. J., 2023, 29, e202301636 CrossRef CAS PubMed.
- K. Li, R. Khan, X. Zhang, Y. Gao, Y. Zhou, H. Tan, J. Chena and B. Fan, Cobalt catalyzed stereodivergent semi-hydrogenation of alkynes using H2O as the hydrogen source, Chem. Commun., 2019, 55, 5663–5666 RSC.
- J. Liu, Z. Wei, J. Yang, Y. Ge, D. Wei, R. Jackstell, H. Jiao and M. Beller, Tuning the Selectivity of Palladium Catalysts for Hydroformylation and Semi-hydrogenation of Alkynes: Experimental and Mechanistic Studies, ACS Catal., 2020, 10, 12167–12181 CrossRef CAS.
- L. Ling, L. Leo, C. Hu, H. Chen, L. Long, X. Zhang, M. Luo, L. Zhao and X. Zeng, Chromium-catalyzed stereodivergent E-and Z-selective alkyne hydrogenation controlled by cyclic (alkyl)(amino) carbene ligands, Nat. Commun., 2023, 14, 190–199 CrossRef PubMed.
- J. Luo, Y. Liang, M. Montag, Y. Diskin, L. Avram and D. Milstein, Controlled selectivity through reversible inhibition of the catalyst: stereodivergent semi-hydrogenation of alkynes, J. Am. Chem. Soc., 2022, 144, 13266–13275 CrossRef CAS PubMed.
- J. Adrian and C. B. W. Stark, Modular and stereodivergent approach to unbranched 1, 5, 9, n-Polyenes: total synthesis of Chatenaytrienin-4, J. Org. Chem., 2016, 81, 8175–8186 CrossRef CAS PubMed.
- K. Chen, H. Zhu, Y. Li, Q. Peng, Y. Guo and X. Wang, Dinuclear cobalt complex-catalyzed stereodivergent semireduction of alkynes: switchable selectivities controlled by H2O, ACS Catal., 2021, 11, 13696–13705 CrossRef CAS.
- S. Rao and K. R. Prabhu, Stereodivergent alkyne reduction by using water as the hydrogen source, Chem. - Eur. J., 2018, 24, 13954–13962 CrossRef CAS PubMed.
- K. Li, C. Yang, J. Chen, C. Pan, R. Fan, Y. Zhou, Y. Luo, D. Yang and B. Fan, Anion Controlled Stereodivergent Semi-Hydrogenation of Alkynes using Water as Hydrogen Source, Asian J. Org. Chem., 2021, 10, 2143–2146 CrossRef CAS.
- K. Murugesan, C. B. Bheeter, P. R. Linnebank, A. Spannenberg, J. N. H. Reek, R. V. Jagadeesh and M. Beller, Nickel-Catalyzed Stereodivergent Synthesis of E-and Z-Alkenes by Hydrogenation of Alkynes, ChemSusChem, 2019, 12, 3363–3369 CrossRef CAS PubMed.
- J. Huang, X. Li, H. Wen, L. Ouyang, N. Luo, J. Liao and R. Luo, Substrate-controlled Cu (OAc) 2-catalyzed stereoselective semi-reduction of alkynes with MeOH as the hydrogen source, ACS Omega, 2021, 6, 11740–11749 CrossRef CAS PubMed.
- C. Q. Zhao, Y.-G. Chen, H. Qiu, L. Wei, P. Fang and T.-S. Mei, Water as a hydrogenating agent: stereodivergent Pd-catalyzed semi-hydrogenation of alkynes, Org. Lett., 2019, 21, 1412–1416 CrossRef CAS PubMed.
- A. B. Flynn and W. W. Ogilvie, Stereocontrolled synthesis of tetrasubstituted olefins, Chem. Rev., 2007, 107, 4698–4745 CrossRef CAS PubMed.
- F. Buttard, J. Sharma and P. Champagne, Recent advances in the stereoselective synthesis of acyclic all-carbon tetrasubstituted alkenes, Chem. Commun., 2021, 57, 4071–4088 RSC.
- S. Kraft, K. Ryan and R. Kargbo, Recent advances in asymmetric hydrogenation of tetrasubstituted olefins, J. Am. Chem. Soc., 2017, 139, 11630–11641 CrossRef CAS PubMed.
- M. G. Davidson, A. K. Hughes, T. B. Marder and K. Wade, Contemporary Boron Chemistry, Royal Society of Chemistry: Cambridge, 2000 Search PubMed.
- P. V. Ramachandran and H. Brown, Organoboranes for Syntheses, ACS Symposium Series American Chemical Society, Washington, DC, 2001 Search PubMed.
- W. G. Woods, I. S. Bengelsdorf and D. L. Hunter, 2-Vinyl-4, 4, 6-trimethyl-1, 3, 2-dioxaborinane. I. Synthesis and Properties1, 2, J. Org. Chem., 1966, 31, 2766–2768 CrossRef CAS.
- I. Beletskaya and A. Pelter, Hydroborations catalyzed by transition metal complexes, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS.
- J. Yun, Copper (I)-Catalyzed Boron Addition Reactions of Alkynes with Diboron Reagents, Asian J. Org. Chem., 2013, 2, 1016–1025 CrossRef CAS.
- K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Copper-Catalyzed Highly Regio-and Stereoselective Directed Hydroboration of Unsymmetrical Internal Alkynes: Controlling Regioselectivity by Choice of Catalytic Species, Chem. - Eur. J., 2012, 18, 4179–4184 CrossRef CAS PubMed.
- H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita and K. Takaki, Copper-Catalyzed Borylation Reactions of Alkynes and Arynes, Angew. Chem., 2011, 1(124), 239–242 Search PubMed.
- D. Yukimori, Y. Nagashima, C. Wang, A. Muranaka and M. Uchiyama, Quadruple borylation of terminal alkynes, J. Am. Chem. Soc., 2019, 141, 9819–9822 CrossRef CAS PubMed.
- S. L. Schreiber, Molecular diversity by design, Nature, 2009, 457, 153–154 CrossRef CAS PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules, Nat. Commun., 2010, 1, 80 CrossRef PubMed.
- L. Lu, Ruthenium Acriphos Complexes as Catalysts for the Functionalization of Alkynes, Alkenes and Arenes, PhD thesis, RWTH Aachen University, 2019, p. 2020.
- W. J. Jang, W. L. Lee, J. H. Moon, J. Y. Lee and J. Yun, Copper-catalyzed trans-hydroboration of terminal aryl alkynes: stereodivergent synthesis of alkenylboron compounds, Org. Lett., 2016, 18, 1390–1393 CrossRef CAS PubMed.
- Z. Kuang, H. Chen, J. Qiu, Z. Ou, Y. Lan and Q. Song, Cu-catalyzed regio-and stereodivergent chemoselective sp2/sp3 1, 3-and 1, 4-diborylations of CF3-containing 1, 3-enynes, Chem, 2020, 6, 2347–2363 CAS.
- J. Corpas, M. Gomez-Mendoza, E. M. Arpa, V. A. de la Peña O'Shea, B. Durbeej, J. C. Carretero, P. Mauleón and R. G. Arrayás, Iterative Dual-Metal and Energy Transfer Catalysis Enables Stereodivergence in Alkyne Difunctionalization: Carboboration as Case Study, ACS Catal., 2023, 13, 14914–14927 CrossRef CAS PubMed.
- X. Zeng, Recent advances in catalytic sequential reactions involving hydrocelement addition to carbon–carbon multiple bonds, Chem. Rev., 2013, 113, 6864–6900 CrossRef CAS PubMed.
- J. Chen, J. Guo and Z. Lu, Recent advances in hydrometallation of alkenes and alkynes via the first row transition metal catalysis, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS.
- C. Lin and L. Shen, Recent Progress in Transition Metal-Catalyzed Regioselective Functionalization of Unactivated Alkenes/Alkynes Assisted by Bidentate Directing Groups, ChemCatChem, 2019, 11, 961–968 CrossRef CAS.
- Y. Kawasaki, Y. Ishikawa, K. Igawa and K. J. Tomooka, Directing group-controlled hydrosilylation: regioselective functionalization of alkyne, J. Am. Chem. Soc., 2011, 133, 20712–20715 CrossRef CAS PubMed.
- G. Rossino, G. Marrubini, M. Brindisi, M. Granje, P. Linciano, D. Rossi and S. Collina, A green Heck reaction protocol towards trisubstituted alkenes, versatile pharmaceutical intermediates, Front. Chem., 2024, 12, 1431382 CrossRef CAS PubMed.
- M. J. Hesse, C. P. Butts, C. L. Willis and V. K. Aggarwal, Diastereodivergent synthesis of trisubstituted alkenes through protodeboronation of allylic boronic esters: application to the synthesis of the Californian red scale beetle pheromone, Angew. Chem., Int. Ed., 2012, 12, 12444–12448 CrossRef PubMed.
- M. R. Wilson and R. E. Taylor, Strained alkenes in natural product synthesis, Angew. Chem., Int. Ed., 2013, 52, 4078–4087 CrossRef CAS PubMed.
- C. Lecourt, S. Dhambri, L. Allievi, Y. Sanogo, N. Zeghbib, R. B. Othman, M. I. Lannou, G. Sorin and J. Ardisson, Natural products and ring-closing metathesis: synthesis of sterically congested olefins, Nat. Prod. Rep., 2018, 35, 105–124 RSC.
- T. J. Müller, and K. Deilhof, Alkynes in Multicomponent Synthesis of Heterocycles. Multicomponent Reactions in Organic Synthesis, 2014, 333–378 Search PubMed.
- L. F. Tietze and A. Modi, Multicomponent domino reactions for the synthesis of biologically active natural products and drugs, Med. Res. Rev., 2000, 20, 304–322 CrossRef CAS PubMed.
- B. E. Maryanoff and A. B. Reitz, The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- M. K. Armstrong, M. B. Goodstein and G. Lalic, Diastereodivergent reductive cross coupling of alkynes through tandem catalysis: Z-and E-selective hydroarylation of terminal alkynes, J. Am. Chem. Soc., 2018, 140, 10233–10241 CrossRef CAS PubMed.
- J. Corpas, M. T. Quirós, P. Mauleon, R. G. Arrayas and J. C. Carretero, Metal-and photocatalysis to gain regiocontrol and stereodivergence in hydroarylations of unsymmetrical dialkyl alkynes, ACS Catal., 2019, 9, 10567–10574 CrossRef CAS.
- C. Zhu, H. Yue and M. Rueping, Nickel catalyzed multicomponent stereodivergent synthesis of olefins enabled by electrochemistry, photocatalysis and photo-electrochemistry, Nat. Commun., 2022, 13, 3240–3249 CrossRef CAS PubMed.
- Y. Nakao, Metal-mediated C–CN bond activation in organic synthesis, Chem. Rev., 2020, 121, 327–344 CrossRef PubMed.
- D. H. Gerlach, A. R. Kane, G. W. Parshall, J. P. Jesson and E. L. Muetterties, Reactivity of trialkylphosphine complexes of platinum (O), J. Am. Chem. Soc., 1971, 93, 3543–3544 CrossRef.
- G. W. Parshall, sigma.-Aryl compounds of nickel, palladium, and platinum. Synthesis and bonding studies, J. Am. Chem. Soc., 1974, 96, 2360–2366 CrossRef CAS.
- E. I. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Recent advances in efficient and selective synthesis of di-, tri-, and tetrasubstituted alkenes via Pd-catalyzed alkenylation− carbonyl olefination synergy, Acc. Chem. Res., 2008, 41, 1474–1485 CrossRef CAS PubMed.
- T. T. Nguyen, M. J. Koh, T. J. Mann, R. R. Schrock and A. H. Hoveyda, Synthesis of E-and Z-trisubstituted alkenes by catalytic cross-metathesis, Nature, 2017, 552, 347–354 CrossRef CAS PubMed.
- C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, A multicomponent synthesis of stereodefined olefins via nickel catalysis and single electron/triplet energy transfer, Nat. Catal., 2019, 2, 678–687 CrossRef CAS.
- X. Fang, P. Yu and B. Morandi, Catalytic reversible alkene-nitrile interconversion through controllable transfer hydrocyanation, Science, 2016, 351, 832–837 CrossRef CAS PubMed.
- F. L. Taw, P. S. White, R. G. Bergman and M. Brookhart, Carbon− Carbon Bond Activation of R− CN (R= Me, Ar, iPr, tBu) Using a Cationic Rh (III) Complex, J. Am. Chem. Soc., 2002, 124, 4192–4193 CrossRef CAS PubMed.
- F. L. Taw, A. H. Mueller, R. G. Bergman and M. Brookhart, A mechanistic investigation of the carbon− carbon bond cleavage of aryl and alkyl cyanides using a cationic Rh (III) silyl complex, J. Am. Chem. Soc., 2003, 125, 9808–9813 CrossRef CAS PubMed.
- J. Long, R. Zhao, G. J. Cheng and X. Fang, Palladium-Catalyzed Alkyne Hydrocyanation toward Ligand-Controlled Stereodivergent Synthesis of (E)-and (Z)-Trisubstituted Acrylonitriles, Angew. Chem., Int. Ed., 2023, 62, e202304543 CrossRef CAS PubMed.
- C. S. Wang, Y. Yu, Y. Sunada, C. Wang and N. Yoshikai, Cobalt-Catalyzed Carbo-and Hydrocyanation of Alkynes via C–CN Bond Activation, ACS Catal., 2022, 12, 4054–4066 CrossRef CAS.
- T. Y. Luh and S. T. Liu, Synthetic applications of allylsilanes and vinylsilanes, Chem. Org. Silicon Compd., 1998, 2, 1793–1868 CAS.
- Z. H. Ye, F. H. Gou, Y. Wu, C. Y. Li and P. Wang, Diverse Synthesis of Alkenylsilanes via Pd-Catalyzed Alkenyl C–H Silylation, Org. Lett., 2023, 25, 2145–2150 CrossRef CAS PubMed.
- A. Barbero and F. J. Pulido, Allylsilanes and Vinylsilanes from Silylcupration of Carbon− Carbon Multiple Bonds: Scope and Synthetic Applications, Acc. Chem. Res., 2004, 37, 817–825 CrossRef CAS PubMed.
- K. Indukuri, L. Cornelissen and O. Riant, Recent developments in the chemistry of vinylsiloxanes, Synthesis, 2016, 48, 4400–4422 CrossRef CAS.
- Y. Nakao and T. Hiyama, Silicon-based cross-coupling reaction: an environmentally benign version, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC.
- S. E. Denmark and C. S. Regens, Palladium-catalyzed cross-coupling reactions of organosilanols and their salts: practical alternatives to boron-and tin-based methods, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef CAS PubMed.
- I. Fleming, A. Barbero and D. Walter, Stereochemical control in organic synthesis using silicon-containing compounds, Chem. Rev., 1997, 97, 2063–2192 CrossRef CAS PubMed.
- G. R. Jones and Y. Landais, The oxidation of the carbon-silicon bond, Tetrahedron, 1996, 52, 7599–7662 CrossRef CAS.
- E. A. Peterson, Recent Advances in the Hydrosilylation of Alkynes, 2005, https://www.scientificspectator.com/documents/Olenick%20Compilation/CH%2034%20Hydrosilylation%20Article.pdf, Accessed on 14th April 2025 Search PubMed.
- A. Mori, E. Takahisa, H. Kajiro, Y. Nishihara and T. Hiyama, Stereodivergent hydrosilylation of 1-alkynes catalyzed by RhI (PPh3)3 leading to (E)-and (Z)-alkenylsilanes and the application to polymer synthesis, Polyhedron, 2000, 19, 567–568 CrossRef CAS.
- A. Mori, E. Takahisa, Y. Yamamura, T. Kato, A. P. Mudalige, H. Kajiro, K. Hirabayashi, Y. Nishihara and T. Hiyama, Stereodivergent syntheses of (Z)-and (E)-alkenylsilanes via hydrosilylation of terminal alkynes catalyzed by rhodium (I) iodide complexes and application to silicon-containing polymer syntheses, Organometallics, 2004, 23, 1755–1765 CrossRef CAS.
- H. Katayama, K. Taniguchi, M. Kobayashi, T. Sagawa, T. Minami and F. Ozawa, Ruthenium-catalyzed hydrosilylation of terminal alkynes: stereodivergent synthesis of (E)-and (Z)-alkenylsilanes, J. Organomet. Chem., 2002, 645, 192–200 CrossRef CAS.
- L. Yong, K. Kirleis and H. Butenschön, Stereodivergent formation of alkenylsilanes: syn or anti hydrosilylation of alkynes catalyzed by a cyclopentadienylcobalt (I) chelate bearing a pendant phosphane tether, Adv. Synth. Catal., 2006, 348, 833–836 CrossRef CAS.
- I. Ojima, N. Clos, R. J. Donovan and P. Ingallina, Hydrosilylation of 1-hexyne catalyzed by rhodium and cobalt-rhodium mixed-metal complexes. Mechanism of apparent trans addition, Organometallics, 1990, 9, 3127–3133 CrossRef CAS.
- C. Belger and B. Plietker, Hydrosilylation of 1-hexyne catalyzed by rhodium and cobalt-rhodium mixed-metal complexes. Mechanism of apparent trans addition, Chem. Commun., 2012, 48, 5419–5421 RSC.
- M. Cygler, F. R. Ahmed, A. Forgues and J. L. A. Roustan, Synthesis and crystal structure of an iron nitrosyl carbonyl hydride, Inorg. Chem., 1983, 22, 1026–1030 CrossRef CAS.
- S. Ding, L. J. Song, L. W. Chung, X. Zhang, J. Sun and Y. D. Wu, Ligand-controlled remarkable regio-and stereodivergence in intermolecular hydrosilylation of internal alkynes: experimental and theoretical studies, J. Am. Chem. Soc., 2013, 135, 13835–13842 CrossRef CAS PubMed.
- S. V. Maifeld, M. N. Tran and D. Lee, Hydrosilylation of alkynes catalyzed by ruthenium carbene complexes, Tetrahedron Lett., 2005, 46, 105–108 CrossRef CAS.
- K. L. Song, B. Wu, W. E. Gan, Y. Zeng, Y. J. Zhang, J. Cao and L. W. Xu, Stereo-divergent synthesis of silyl-enyne via palladium-catalyzed coupling of alkynes and iodosilanes, Org. Chem. Front., 2022, 9, 3718–3722 RSC.
- S. Zhao, L. Ding, Y. Sun, M. Wang and D. Zhao, Synergistic Palladium/Lewis Acid-Catalyzed Regio-and Stereo-divergent Bissilylation of Alkynoates: Scope, Mechanism, and Origin of Selectivity, Angew. Chem., 2023, 135, e202309169 CrossRef.
- X. Yang and C. Wang, Dichotomy of manganese catalysis via organometallic or radical mechanism: stereodivergent hydrosilylation of alkynes, Angew. Chem., 2018, 130, 935–940 CrossRef.
- B. Kopping, C. Chatgilialoglu, M. Zehnder and B. Giese, Tris (trimethylsilyl) silane: an efficient hydrosilylating agent of alkenes and alkynes, J. Org. Chem., 1992, 57, 3994–4000 CrossRef CAS.
- Q. Li, S. Huo, L. Meng and X. Li, Mechanism and origin of the stereoselectivity of manganese-catalyzed hydrosilylation of alkynes: a DFT study, Catal. Sci. Technol., 2022, 12, 2649–2658 RSC.
- O. Pascu, V. Liautard, M. Vaultier, M. Pucheault and C. Aymonier, Catalyzed stereodivergent hydrosilylation with Onium Salts stabilised M (0) nanocatalysts prepared in scCO2, RSC Adv., 2014, 4, 59953–59960 RSC.
- C. Fopp, E. Romain, K. Isaac, F. Chemla, F. Ferreira, O. Jackowski, M. Oestreich and A. Perez-Luna, Stereodivergent silylzincation of α-heteroatom-substituted alkynes, Org. Lett., 2016, 18, 2054–2057 CrossRef CAS PubMed.
- C. Fopp, K. Isaac, E. Romain, F. Chemla, F. Ferreira, O. Jackowski, M. Oestreich and A. Perez-Luna, Stereodivergent synthesis of β-heteroatom-substituted vinylsilanes by sequential silylzincation–copper (I)-mediated electrophilic substitution, Synthesis, 2017, 49, 724–735 CAS.
- S. Nakamura, M. Uchiyama and T. Ohwada, Chemoselective silylzincation of functionalized terminal alkynes using dianion-type zincate (SiBNOL-Zn-ate): regiocontrolled synthesis of vinylsilanes, J. Am. Chem. Soc., 2004, 126, 11146–11147 CrossRef CAS PubMed.
- J. Maury, L. Feray and M. P. Bertrand, Unprecedented noncatalyzed anti-carbozincation of diethyl acetylenedicarboxylate through alkylzinc group radical transfer, Org. Lett., 2011, 13, 1884–1887 CrossRef CAS PubMed.
- P. A. Bartlett and A. Otake, Fluoroalkenes as peptide isosteres: Ground state analog inhibitors of thermolysin, J. Org. Chem., 1995, 60, 3107–3111 CrossRef CAS.
- A. Otaka, E. Mitsuyama, J. Watanabe, H. Watanabe and N. Fujii, Synthesis of fluorine-containing bioisosteres corresponding to phosphoamino acids and dipeptide units, Biomolecules, 2004, 76, 140–149 CAS.
- J. J. Urban, B. G. Tillman and W. A. Cronin, Fluoroolefins as peptide mimetics: A computational study of structure, charge distribution, hydration, and hydrogen bonding, J. Phys. Chem. A, 2006, 110, 11120–11129 CrossRef CAS PubMed.
- C. E. Jakobsche, G. Peris and S. J. Miller, Functional analysis of an aspartate-based epoxidation catalyst with amide-to-alkene peptidomimetic catalyst analogues, Angew. Chem., Int. Ed., 2008, 47, 6707 CrossRef CAS PubMed.
- S. Couve-Bonnaire, D. Cahard and X. Pannecoucke, Chiral dipeptide mimics possessing a fluoroolefin moiety: a relevant tool for conformational and medicinal studies, Org. Biomol. Chem., 2007, 5, 1151–1157 RSC.
- S. Sano, Y. Kuroda, K. Saito, Y. Ose and Y. Nagao, Tandem reduction–olefination of triethyl 2-acyl-2-fluoro-2-phosphonoacetates and a synthetic approach to Cbz-Gly-Ψ [(Z)-CFC]-Gly dipeptide isostere, Tetrahedron, 2006, 62, 11881–11890 CrossRef CAS.
- T. Narumi, K. Tomita, E. Inokuchi, K. Kobayashi, S. Oishi, H. Ohno and N. Fujii, Diastereoselective synthesis of highly functionalized fluoroalkene dipeptide isosteres and its application to Fmoc-based solid-phase synthesis of a cyclic pentapeptide mimetic, Tetrahedron, 2008, 64, 4332–4346 CrossRef CAS.
- R. Guo, X. Qi, H. Xiang, P. Geaneotes, R. Wang, P. Liu and Y. M. Wang, Stereodivergent alkyne hydrofluorination using protic tetrafluoroborates as tunable reagents, Angew. Chem., Int. Ed., 2020, 59, 16651–16660 CrossRef CAS PubMed.
- D. S. Bae, M. E. Andersen and H. J. Clewell III, Halogenated Alkenes. Physiologically Based Pharmacokinetic Modeling: Science and Applications, 2005, pp. 55–78 Search PubMed.
- T. Kohei and N. Miyaura, Introduction to cross-coupling reactions, Cross-Coupling Reactions: A Practical Guide, 2002, pp. 1–9 Search PubMed.
- C. M. Le, P. J. C. Menzies, D. A. Petrone and M. Lautens, Synergistic Steric Effects in the Development of a Palladium-Catalyzed Alkyne Carbohalogenation: Stereodivergent Synthesis of Vinyl Halides, Angew. Chem., 2015, 127, 256–259 CrossRef.
- K. Liu, C. Zhu, J. Min, S. Peng, G. Xu and J. Sun, Stereodivergent Synthesis of N-Heterocycles by Catalyst-Controlled, Activity-Directed Tandem Annulation of Diazo Compounds with Amino Alkynes, Angew. Chem., Int. Ed., 2015, 54, 12962–12967 CrossRef CAS PubMed.
- F. Tian, G. Yan and J. Yu, Recent advances in the synthesis and applications of α-(trifluoromethyl) styrenes in organic synthesis, Chem. Commun., 2019, 55, 13486–13505 RSC.
- Y. Yamamoto, Recent Advances in the Synthesis of Fluoroalkylated Alkenes and Alkynes via Fluoroalkyl Radical Substitution Reactions, Adv. Synth. Catal., 2023, 365, 3572–3585 CrossRef CAS.
- T. Konno, Trifluoromethylated internal alkynes: Versatile building blocks for the preparation of various fluorine-containing molecules, Synlett, 2014, 25, 1350–1370 CrossRef.
- S. Schweizer, C. Tresse, P. Bisseret, J. Lalevee, G. Evano and N. Blanchard, Stereodivergent hydrogermylations of α-trifluoromethylated alkynes and their applications in cross-coupling reactions, Org. Lett., 2015, 17, 1794–1797 CrossRef CAS PubMed.
- C. Tresse, S. Schweizer, P. Bisseret, J. Lalevee, G. Evano and N. Blanchard, Stereodivergent hydrosilylation, hydrostannylation, and hydrogermylation of α-trifluoromethylated alkynes and their synthetic applications, Synthesis, 2016, 48, 3317–3330 CrossRef CAS.
- A. Douchez, A. Geranurimi and W. D. Lubell, Applications of γ, δ-unsaturated ketones synthesized by copper-catalyzed cascade addition of vinyl Grignard reagents to esters, Acc. Chem. Res., 2018, 51, 2574–2588 CrossRef CAS PubMed.
- F. A. Cruz and V. M. Dong, Stereodivergent coupling of aldehydes and alkynes via synergistic catalysis using Rh and Jacobsen's amine, J. Am. Chem. Soc., 2017, 139, 1029–1032 CrossRef CAS PubMed.
- H.-R. Wei, Y.-Y. Xing, J.-B. Liu, W.-J. Wang, F. Huang, C.-Z. Sun and D.-Z. Chen, A mechanism exploration of stereodivergent coupling of aldehydes and alkynes catalyzed synergistically by rhodium and amine, Org. Chem. Front., 2019, 6, 3282–3291 RSC.
- A. N. R. Alba, X. Companyó and R. Rios, Sulfones: new reagents in organocatalysis, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC.
- R. Ahmadi and S. Emami, Recent applications of vinyl sulfone motif in drug design and discovery, Eur. J. Med. Chem., 2022, 234, 114255 CrossRef CAS PubMed.
- D. C. Meadows and J. Gervay-Hague, Vinyl sulfones: Synthetic preparations and medicinal chemistry applications, Med. Res. Rev., 2006, 26, 793–814 CrossRef CAS PubMed.
- T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Ligand-controlled stereodivergent alkenylation of alkynes to access functionalized trans-and cis-1, 3-dienes, Nat. Commun., 2023, 14, 55 CrossRef CAS PubMed.
- S. Tripathi, M. Kumar, M. D. Ambule, A. Saxena, R. Kant, S. K. Shukla and A. K. Srivastava, Stereodivergent Synthesis of (Z)-/(E)-β-Sulfonylacrylamides via Tandem Difunctionalization of Alkynes with Sulfinates and Isocyanides, Org. Lett., 2022, 24, 7632–7636 CrossRef CAS PubMed.
- J. Qin, Z. Zhang, Y. Lu, S. Zhu and L. Chu, Divergent 1, 2-carboallylation of terminal alkynes enabled by metallaphotoredox catalysis with switchable triplet energy transfer, Chem. Sci., 2023, 14, 12143–12151 RSC.
| This journal is © The Royal Society of Chemistry 2025 |