
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent catalyst-free regioselective synthesis and binding studies of 3-aroyl-2-methylimidazo[1,2-a]pyrimidines with BSA using biophysical and computational techniques†
Ranjana Aggarwal *ab,
Manisha Sharmaa,
Garima Sumranc and
Parvin Kumara
*ab,
Manisha Sharmaa,
Garima Sumranc and
Parvin Kumara
aDepartment of Chemistry, Kurukshetra University, Kurukshetra-136119, Haryana, India
bCouncil of Scientific and Industrial Research-National Institute of Science Communication and Policy Research, New Delhi 110012, India. E-mail: ranjana67in@yahoo.com; ranjanaaggarwal67@gmail.com; Tel: +91-9896740740
cDepartment of Chemistry, D. A. V. College (Lahore), Ambala City, Haryana 134 003, India
First published on 14th May 2025
Abstract
A facile and environmentally benign protocol for regioselective synthesis of diversely substituted imidazo[1,2-a]pyrimidines 5a–h has been described via multicomponent reaction of unsymmetrical β-diketones 1, N-bromosuccinimide 2 and 2-aminopyrimidine 4 in DCM. The reaction proceeds through in situ formation of α-bromo-β-diketones 3 and their ensuing condensation with 2-aminopyrimidine without the need of any organic or inorganic catalyst. The structure of the regioisomeric product was characterized by 1H, 13C NMR, heteronuclear 2D NMR and HRMS studies. The present protocol offers several advantages such as avoidance of metal-based and toxic catalysts, broad substrate scope with respect to substitutions on β-diketones, operational simplicity, easy work-up and high yields. Computational molecular docking studies were carried out to examine the interaction of imidazo[1,2-a]pyrimidines with bovine serum albumin (BSA). Moreover, different spectroscopic approaches viz. UV-visible, steady-state fluorescence and competitive displacement assays were carried out to investigate the binding mechanisms of imidazo[1,2-a]pyrimidines (5c, 5e and 5h) with BSA. The results thus obtained revealed that imidazo[1,2-a]pyrimidines showed moderate binding with BSA through a static quenching mechanism and compound 5e had more affinity to bind in site I of BSA.
1. Introduction
Nitrogen containing heterocyclic compounds are constituents of many biologically important molecules, including several endogenous metabolites, natural products and pharmaceuticals.1,2 Imidazopyrimidines containing a bridgehead nitrogen atom are an important class of fused heterocycles due to their structural resemblance to purine bases present in DNA and RNA.3 In particular, imidazo[1,2-a]pyrimidines are considered as important scaffolds in medicinal and pharmaceutical science, because of their broad spectrum of biological activities such as cytotoxic,4 antimicrobial,5 anti-inflammatory,6 antiviral,7 anti-tubercular,8 antileishmanial,9 and antimalarial activity.10 Imidazo[1,2-a]pyrimidine derivatives also acted as acetylcholinesterase inhibitors11 for Alzheimer's disease treatment, α-glucosidase inhibitors to treat diabetes mellitus,12 dual inhibitors of both ACE2 and spike protein to prevent SARS-CoV-2 infection,13 GABA receptor ligands for treatment of anxiety disorders,14 and p38 MAP kinase inhibitors for treatment of inflammatory conditions such as rheumatoid arthritis and Crohn's disease.15 Additionally, a photophysical study of imidazo[1,2-a]pyrimidines demonstrated that these derivatives serve as photosensitizers in intracellular environments that generate singlet oxygen upon irradiation for killing cancer cells,16 and act as a fluorescent chemo sensor to detect zinc ions,17 and an electrochemical study showed that imidazo[1,2-a]pyrimidines are proven effective corrosion inhibitors for copper.18 Moreover, many drugs containing imidazo[1,2-a]pyrimidine scaffold as a core unit such as fasiplon, taniplon, and divaplon are available in the market (Fig. 1). | ||
| Fig. 1 Representative molecules containing imidazo[1,2-a]pyrimidine scaffold. | ||
Serum albumins, major constituent proteins of blood, are a type of biological receptor that are intricate in transport and distribution of endogenous and exogenous chemicals including thyroid hormones, metals, fatty acids, steroids, and other molecules throughout the bloodstream.19 Serum albumins also play a crucial role in maintaining the extracellular fluid by influencing the plasma oncotic pressure.20 The drug–protein interaction significantly influences pharmacokinetic properties such as absorption, delivery, metabolism, and excretion (ADME) and pharmacodynamics properties of drugs.21 In order to achieve the desired therapeutic effect, drugs must bind to serum albumin with optimum binding affinity. Therefore, investigation of the molecular interactions between proteins and small organic molecules and drugs is very crucial to design and develop novel and more efficient drugs with improved plasma solubility/stability.
Bovine serum albumin (BSA) is one of the most extensively researched proteins as primary molecular targets due to its capability to reversibly bind to a wide variety of drugs.22,23 BSA is more widely used as a crucial model protein to study the interaction of small molecules with serum albumins due to its cost effectiveness, wide availability, good stability and 76% structural homology with Human serum albumin (HSA).24 BSA's primary structure contains 583 amino acids with 17 disulfide bonds and a free cysteine residue.25 It has three homologous α-helical domains (I–III), each containing two subdomains A and B.26 BSA exhibits intrinsic fluorescence specifically due to Trp134 and Trp213 residues which are located on the surface of subdomain IB and in the hydrophobic cavity of subdomain IIA, respectively27 and the quenching of this fluorescence by small molecules is used in BSA-ligand interaction studies.
In view of their biological potential, to date different synthetic strategies have been developed for the construction of functionalized imidazo[1,2-a]pyrimidine derivatives.28 The traditional and most commonly used method for the synthesis of imidazo[1,2-a]pyrimidines is Hantzsch-type synthesis which involves the cyclo-condensation of α-functionalized carbonyl compounds with 2-aminopyrimidine in the presence of various catalysts such as neutral Al2O3,29 KI, K2CO3,30 PTSA,31 NaHCO3,32 etc. For α-bromination of 1,3-diketones, N-bromosuccinimide (NBS) was utilized over other reagents e.g. molecular bromine, sodium hypobromite, tetraalkylammonium tribromide, CuBr2 with Koser's reagent, HBr–H2O2, due to its easily accessible, easy to handle, inexpensive, and selective brominating property.33 In recent years, three component reactions of aldehydes, 2-aminopyrimidine and nitriles/isonitriles/alkyne or their derivatives have been reported to produce imidazo[1,2-a]pyrimidines.17,28 Other method involves the reaction between 2-aminopyrimidine, ketone and [hydroxy(tosyloxy)iodo]benzene (HTIB) in one pot.34 More recently, Chanda et al.35 described water and isopropanol mediated catalyst-free microwave assisted protocol for the synthesis of imidazo[1,2-a]pyrimidines which shows progress towards green synthetic methods. However, most of these methods often suffer from a drawback of harsh reaction conditions calling for high temperature, hazardous high boiling organic solvents, long reaction times, decomposition of substrates during overheating and lower yields of the products. The necessity of thermal activation for the imidazo[1,2-a]pyrimidines ring closure is one of the main and most disappointing limitations on the way to medicinal utilization of this heterocyclic motif. Therefore, the development of energy and environment-efficient greener synthetic methodologies for the construction of these versatile bicyclic heterocycles is always in demand.
Synthetic strategy for functionalized imidazo[1,2-a]pyrimidine through a classical Hanztsch reaction between the α-bromo-1,3-diketones and 2-aminopyrimidine is shown in Scheme 1. The reaction of unsymmetrical diketones 1 with 2-aminopyrimidine 4 in the presence of NBS may lead to the formation of two possible regioisomers: 3-aroyl-2-methyl-imidazo[1,2-a]pyrimidines 5 and 3-acetyl-2-aryl-imidazo[1,2-a]pyrimidines 6, by different permutations, as depicted in Scheme 1. Differently substituted acetophenones on reaction with ethyl acetate in the presence of sodium ethoxide were converted into corresponding unsymmetrical 1,3-diketones through Claisen condensation following the literature protocol.
 | ||
| Scheme 1 Retrosynthetic approach for the construction of imidazo[1,2-a]pyrimidine. | ||
Recently, multicomponent reactions (MCRs) have gained considerable interest as a sustainable approach offering several advantages such as efficiency, selectivity, high atom economy, reduced waste generation and short reaction time.36 Currently our research group is exploring regioselective reaction of substituted unsymmetrical α-bromo-1,3-diketones with various binucleophiles,37 therefore, it was planned to examine study of regioselectivity pattern of the reaction between α-bromo-1,3-diketones and 2-aminopyrimidine to construct the functionalized imidazo[1,2-a]pyrimidine scaffold using MCR approach. To the best of our knowledge, the reaction between 2-aminopyrimidine and unsymmetrical α-bromo-1,3-diketones has not been proposed yet. Intrigued by these findings and in continuation of our ongoing efforts towards the synthesis of fused nitrogen-bridged heterocyclic compounds using green synthetic protocols,37–39 herein we describe a highly efficient, catalyst-free regioselective approach for the synthesis of functionalized imidazo[1,2-a]pyrimidine derivatives involving in situ bromination of unsymmetrical 1,3-diketones by NBS and condensation with 2-aminopyrimidine under mild reaction condition and characterization of regioisomer on the basis of heteronuclear 2D NMR [(1H–13C) heteronuclear multiple bond correlation, HMBC, (1H–13C) heteronuclear multiple quantum coherence, HSQC, (1H–15N) HMBC] spectroscopic studies. The molecular docking studies have been executed to determine interaction mode of imidazo[1,2-a]pyrimidine derivatives with BSA. Binding studies of representative compounds with BSA have been investigated using UV-visible absorption spectroscopy and fluorescence emission spectroscopy.
2. Result and discussion
2.1 Chemistry
In order to synthesize imidazo[1,2-a]pyrimidines, a model reaction was carried out by stirring an equimolar amount of 1-phenylbutane-1,3-dione 1 with NBS 2 to form 2-bromo-1-phenylbutane-1,3-dione 3 in situ and then followed by the subsequent addition of 2-aminopyrimidine 4 in acetone at room temperature. Thin layer chromatography (TLC) examination of the reaction mixture denoted that the reaction was complete within 24 h, resulting in the formation of a single regioisomer out of two possible regioisomers with only 25% yield (entry 1, Table 1).| Entry | Solventa | Temperaturea | Time | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: unsymmetrical β-diketone (1a, 1 mmol), NBS (2, 1 mmol), 2-aminopyrimidine (4, 1 mmol) and an appropriate solvent (10.0 mL).b Isolated yield.c NR, no reaction.d Domino reaction. | ||||
| 1 | Acetone | r.t. | 24 h | 25 |
| 2 | MeOH | Reflux | 6 h | 45 |
| 3 | EtOH | Reflux | 6 h | 52 |
| 4 | AcOH | Reflux | 4 h | 48 |
| 5 | DCM | r.t. | 6 h 45 min | 75 |
| 6 | DCM | Reflux | 2.5 h | 65 |
| 7 | MeCN | Reflux | 6 h | 55 |
| 8 | Toluene | Reflux | 5 h | NRc |
| 9 | Solvent-free | r.t. | 3 h | Trace |
| 10 | Solvent-free | 90 °C | 1.5 h | 60 |
| 11 | DCMd | r.t. | 4 h | 80 |
Further to optimize the reaction condition, we explored the model reaction in different polar protic (MeOH, EtOH, AcOH); polar aprotic (DCM, chloroform, acetone, MeCN) and nonpolar aprotic (toluene) solvents at different temperatures (room temperature up to reflux) to determine the most appropriate reaction conditions (entry 2–8, Table 1). Among the solvents screened, DCM improved the yield significantly to 75% at room temperature (entry 5, Table 1). To further improve the yield, reaction was also carried out in DCM by the gradual increase of temperature from r.t. to refluxing temperature. However, there was no further improvisation of the product yield. In addition, the reaction was performed at r.t. under solvent-free conditions which furnished the desired product in trace amount (entry 9, Table 1). However, on heating under solvent-free conditions, the desired product was obtained but only with 60% yield (entry 10, Table 1).
Having optimized conditions identified for sequential synthesis, we explored further one-pot multicomponent domino process by stirring an equimolar mixture of 2-aminopyrimidine 4, 1-phenylbutane-1,3-dione 1 and NBS 2 in DCM at r.t. The reaction proceeded smoothly to yield the single product in 4 h as indicated by TLC out of two possible regioisomers. It is noteworthy to mention that this domino approach increased the yield (entry 11 vs. 5) and the product was isolated in 80% yield in short reaction time (4 h vs. 6 h 45, entry 11 vs. 5) than the sequential synthesis protocol. Hence, this one-pot MCRs approach was chosen as the model method (Scheme 2).
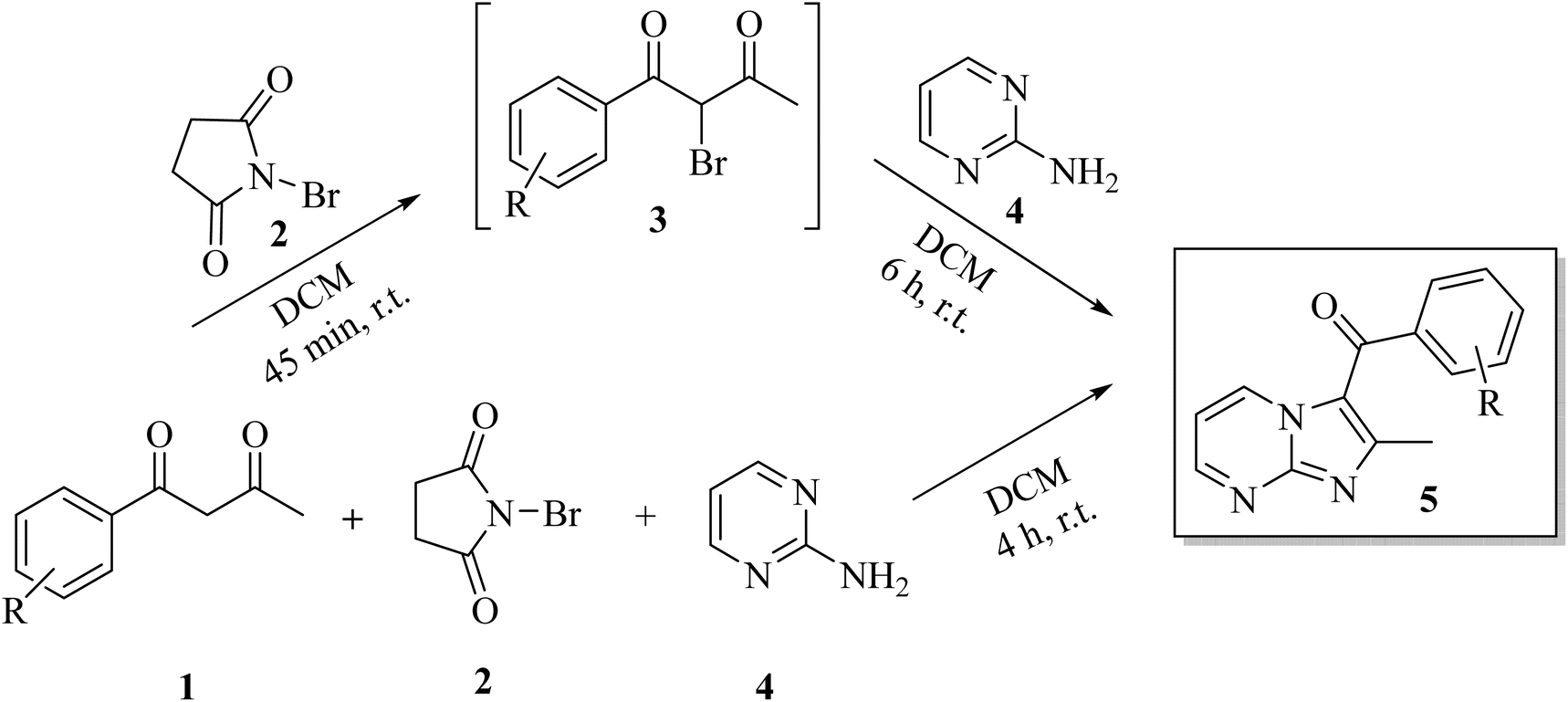 | ||
| Scheme 2 Sequential and multicomponent regioselective synthesis of 3-aroylimidazo[1,2-a]pyrimidines. | ||
To get an insight into the extensibility and efficiency of the developed protocol, differently substituted unsymmetrical β-diketones containing electron donating and electron withdrawing substituents were reacted with 2-aminopyrimidine under optimized conditions. All the reaction combinations underwent successful condensation and yielded the corresponding products in high yields with high regioselectivity. Generally, β-diketones bearing electron-donating groups provided better yields of products than β-diketones bearing electron-withdrawing groups. Differently synthesized imidazo[1,2-a]pyrimidines are outlined in Table 2.

The successful conversion of reactants into products was confirmed through spectroscopic (1H-NMR, 13C-NMR, IR) data analysis. The IR spectrum of the attained product 5a displayed a sharp absorption band at 1648 cm−1 corresponding to carbonyl group stretch indicating the product formation. The 1H NMR spectrum of compound 5a displayed a singlet of three proton intensity at δ 2.21 ppm corresponding to the methyl group, three signals each as doublet of doublet of one proton intensity at 7.62, 9.02 and 9.65 ppm attributable to H-6, H-5, H-7 protons of pyrimidine nucleus, respectively, and three signals for two, one and two proton intensity at 7.62, 7.73, 7.80 ppm corresponding to phenyl protons. 13C NMR spectrum of compound 5a displayed signals at δ 15.44 and 186.20 ppm corresponding to methyl and carbonyl carbon, along with required number of signals for aromatic carbons, thus validating the successful condensation of 2-aminopyrimidine with α-bromo-β-diketones to give the desired product 5a. Additionally, the mass spectra of 5a exhibited a peak at m/z = 238.0890, representing the molecular ion, [M + H]+, which aligns with the expected composition for the title compound (C14H11N3O).
In order to unambiguously assign the correct regioisomeric structure to reaction product as 5 out of 5 and 6, multinuclear 2D NMR experiments [(1H–13C) HMBC, (1H–13C) HSQC and (1H–15N) HMBC] were carried out for compound 5a. The 2D NMR correlation results and 1H, 13C and 15N chemical shifts for compound 5a are shown in Fig. 2 and Table 3.
 | ||
| Fig. 2 1H (in red), 13C (in blue) and 15N (in green) NMR chemical shift values and 2D NMR correlation illustration for compound 5a and 5c. | ||
| Chemical shifts (δ in ppm) | gs-HMQC correlation | gs-HMBC correlation | Assignments |
|---|---|---|---|
| 186.20 | — | 7.80 (H2′/H6′) | CO |
| 156.73 | 9.02 (5H) | 7.62 (H6) | C5 |
| 9.65 (H7) | |||
| 146.82 (C9) | |||
| 147.60 | — | 2.21 (2-CH3) | C2 |
| 146.82 | — | 9.02 (H5) | C9 |
| 9.65 (H7) | |||
| 138.81 | — | 7.62 (H3′/H5′) | C1′ |
| 138.07 | 9.65 (7H) | 7.62 (H6) | C7 |
| 9.02 (H5) | |||
| 146.82 (C9) | |||
| 133.52 | 7.73 (H4′) | 7.62 (H3′/H5′) | C4′ |
| 129.38 | 7.62 (H3′/H5′) | 138.81 (C1′) | C3′/C5′ |
| 129.27 | 7.80 (H2′/H6′) | 7.73 (H4′) | C2′/C6′ |
| 186.20 (CO) | |||
| 119.67 | — | 2.21(2-CH3) | C3 |
| 113.66 | 7.62 (6H) | 9.65 (7H) | C6 |
| 9.02 (5H) | |||
| 15.44 | 2.21 (2-CH3) | C2 (147.60) | 2-CH3 |
| C3 (119.67) |
The (1H–13C) HMBC spectrum of 5a displayed cross peak of carbonyl carbon at δ 186.20 ppm with H2′/H6′ (δ 7.80) protons of aryl ring confirming the presence of CO with aryl/heteroaryl ring. Similarly (1H–13C) HMBC spectrum showed cross peaks of methyl protons (δ 2.21 ppm) with C-2 (δ 147.60 ppm) and C-3 (δ 119.67 ppm) which indicated the presence of methyl substituent at position-2 of imidazo[1,2-a]pyridine nucleus. Further, (1H–15N) HMBC of compound 5a also showed a cross peak of methyl protons (δ 2.21) with N-1 (δ −215.9), thus confirming the presence of methyl substituent at position-2 of imidazo[1,2-a]pyrimidinyl scaffold. Had the structure been 3-acetyl-2-arylimidazo[1,2-a]pyrimidine 6a, then the correlation between the methyl protons with N-1, C-3 and the cross peak of carbonyl carbon with the H2′/H6′ proton of the aryl part would have been absent. Thus, the structure can certainly be assigned as 1-phenyl-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone 5a. Similar correlation results of (1H–13C) HMBC and (1H–15N) HMBC were observed for the compound 5c as shown in Fig. 2.
The plausible mechanism for regioselective synthesis of imidazo[1,2-a]pyrimidine is depicted in Scheme 3. In the first step, ring nitrogen of 2-aminopyrimidine attacks nucleophilically on α-bromo-1,3-diketones to give the intermediate that is cyclized by nucleophilic addition of imine nitrogen to either of the carbonyl carbons to form regioisomer 2-alkyl/arylimidazo[1,2-a]pyrimidine 5 or 6 (Route 1). Due to more electrophilic character and less steric hindrance on carbonyl carbon adjacent to methyl, imine nitrogen underwent nucleophilic addition on this carbonyl carbon (Path-a) followed by removal of water to give 2-alkylimidazo[1,2-a]pyrimidine 5 as the final product.
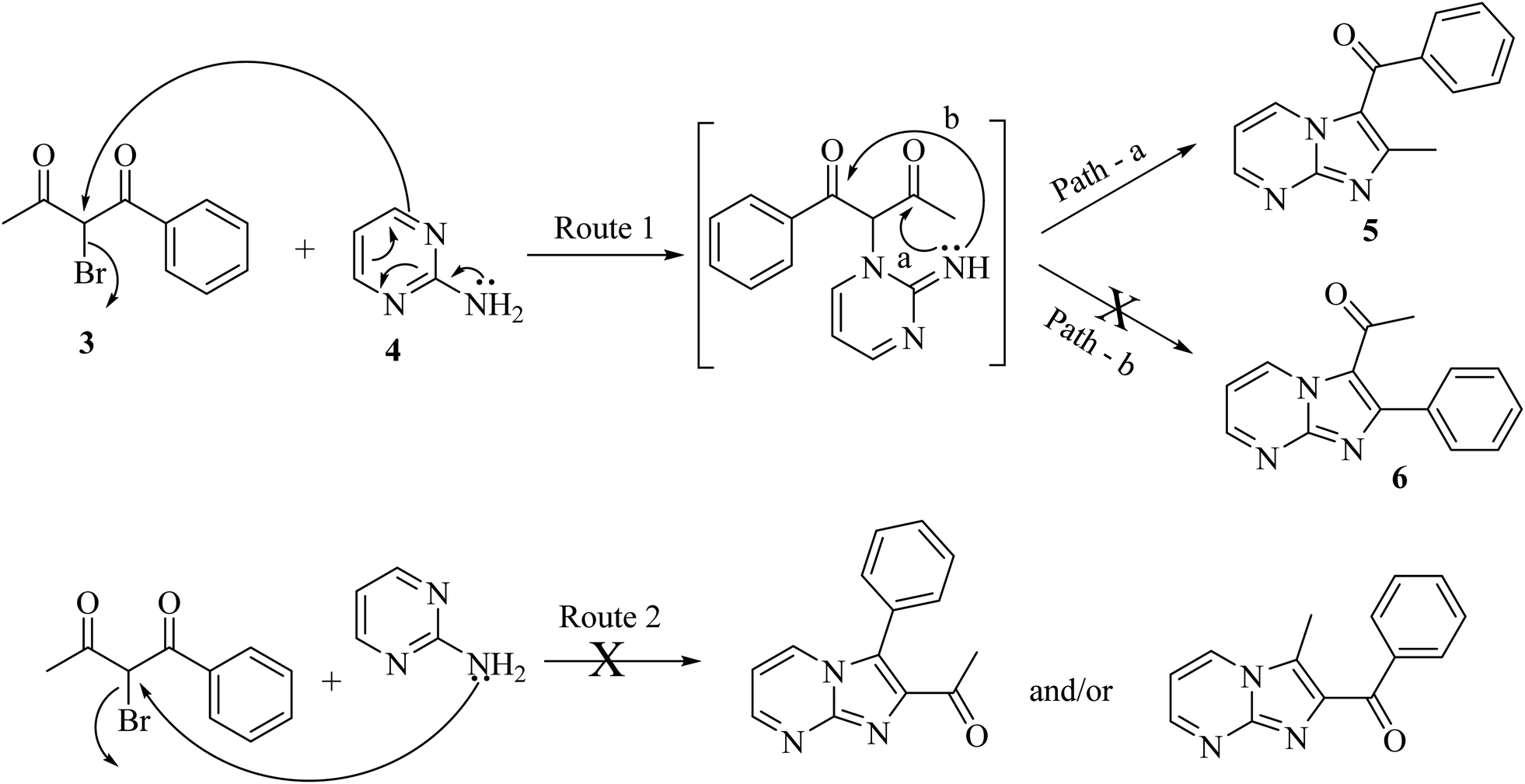 | ||
| Scheme 3 Plausible mechanism for regioselective synthesis of imidazo[1,2-a]pyrimidine. | ||
2.2 Biological studies
Outcomes from molecular docking simulation studies showed that imidazo[1,2-a]pyrimidine derivatives 5a–h exhibited binding interactions with the BSA protein mainly through hydrophobic interaction as well as through hydrogen bonding, electrostatic and van der Waals interaction with binding free energy in the range of −8.25 to −9.04 kcal mol−1 (Table 4). Among the synthesized imidazo[1,2-a]pyrimidine derivatives, compound 5e with 4-methoxy substitution at phenyl ring binds with the albumin protein (BSA, PDB ID: 4f5s) in AutoDock more efficiently than other derivatives with a maximum docking score of −9.04 kcal mol−1 (Table 4). Results of docking analysis using BIOVIA Discovery Studio Visualizer (DSV) have shown that 5e interacts with BSA in the active pocket of chain A. Compound 5e interacts with various amino acids (Tyr137, Lys136, Pro117, Tyr160, Met184, Ile181, Val188, Leu115, Phe133) through hydrogen bonding and various hydrophobic interactions as shown in the 2D and 3D plots (Fig. 3). Also, compound 5e formed a conventional hydrogen bond with Arg185 via its carbonyl group. The lowest binding free energy conformer of compounds 5a–h, PBZ and IBP with BSA is shown in Fig. 4a–j.
| Compounds | Binding energy (kcal mol−1) | Interacted residues |
|---|---|---|
| a π-donor hydrogen bonding.b π–π T-shaped.c π–π stacked.d Alkyl.e π–alkyl.f van der Waals.g hydrogen bonding.h π–sulfur.i π–sigma.j Carbon–hydrogen bond.k π–cation.l Amide π-stacking. | ||
| 5a | −8.69 | Tyr160a,b,c,d, Tyr137b,c, Arg185e, Leu115e, Ile181d,e, Pro117d, Met184e, Val188e, Lys136f, Glu140f, Ile141f, Leu122f |
| 5b | −8.51 | Tyr160a,d, Tyr137b,g, Ile181d, Met184d,h, Arg185d,e, Val188e, Pro117e, Leu115e, Ile141i, Leu189f |
| 5c | −8.50 | Tyr137a,b, Met184c,h, Arg185k,e, Val188e, Ile181e, Leu115e, Pro117e, Lys114e, Leu122f, Tyr160f, Ile141f |
| 5d | −8.95 | Tyr160b,d, Met184d, Arg185d,e,g,i, Ile181d,e, Pro117e, Leu115e, Tyr137f, Ile141f, Val188f, Glu182f, Glu140f, Lys136f |
| 5e | −9.04 | Tyr137a,b, Lys136d, Pro117d, Tyr160b,d, Met184e, Ile181d,e, Arg185e,g, Val188e, Leu115e, Phe133j |
| 5f | −8.76 | Tyr160a,d, Tyr137c, Met184d, Ile181d,e, Arg185d,e,g,k, Pro117e, Glu140j, Lys136j, Leu115d,i, Leu122f, Ile141f, Glu182f |
| 5g | −8.28 | Tyr160a,c,d, Tyr137a,c,d, Val188e, Met184e, Arg185e,g, Ile181d,e, Ile141d, Pro117d,e, Leu115d,i, Leu122f, Glu140f |
| 5h | −8.25 | Tyr160b,g,h,l, Arg185e,g, Ile181d,e, Val188e, Tyr137b,h, Met184h, Leu115e, Pro117d,e |
| PBZ | −8.81 | Tyr137a, Tyr160a, Pro117d, Ile181d, Val188e, Arg185e, Glu140f, Met184f, Leu178f, Glu182f, Leu115i, Ile141i |
| IBP | −7.46 | Leu115d,e, Ile181d, Arg185d,g,k, Lys114d,j, Pro117e, Glu182f, Tyr160f, Tyr137f, Arg144f |
 | ||
| Fig. 3 (a and b) 2D and 3D poses of the interaction of ligand 5e with BSA protein. | ||
 | ||
| Fig. 4 (a–j) 2D diagram showing of the interaction of ligand 5a–h, phenylbutazone (PBZ) and ibuprofen (IBP) with BSA protein. | ||
Docking analysis showed that almost all the derivatives exhibit strong interactions with the A chain of the protein with few exceptions where derivatives 5a and 5b were found to interact with the protein through chain B. Compound 5e was selected for further in vitro analysis with BSA protein using various spectroscopic techniques due to the best docking score. To compare the results, two compounds bearing substituents of different nature, 5c with 4-chlorophenyl moiety and 5h with 2-thienyl group, were chosen.
2.2.2.1 UV-visible absorption spectroscopy studies. Recently UV-visible spectroscopy has been a valuable and imperative tool for studying the conformational changes of proteins during interactions between drugs and proteins due to its good sensitivity, convenience of usage and versatility.40 Furthermore, this technique can also be used to investigate the type of quenching (static/dynamic) between ligand and protein. When BSA interacts with ligands, the biomolecule's electronic environment is perturbed, which results in a wavelength shift or alteration in the intensity in UV-visible spectra.41 During static quenching, a significant change in absorption spectra is observed on subsequent addition of ligand, while the absorption spectra remain unchanged in dynamic quenching.
Among the synthesized compounds, 5e having electron donating 4-OMePh ring, 5h having 2-thienyl ring and 5c having electron withdrawing 4-ClPh ring were selected for UV-visible spectral studies with BSA. Absorption spectra of BSA were recorded in the absence and presence of 5e, 5h, and 5c at a varying concentration ranging from 0 to 40, 0 to 28 and 0 to 36 μM, respectively, in the wavelength range of 250–350 nm at room temperature. The UV-visible absorption spectra of BSA display characteristic peaks at about 278 nm, which could result from π–π* transition due to the presence of three aromatic amino acids (tryptophan, tyrosine and phenylalanine) on the protein chain surface.42 In the presence of increasing concentration of 5e, 5h and 5c, the intensity of the BSA spectra continues to rise due to the changes in the environment surrounding aromatic amino acids Trp, Phe and Tyr (Fig. 5a–c). This perturbation in the absorption spectra of BSA indicates the preferable interactions between BSA and compounds studied. The hyperchromic effect in the absorption spectra of BSA on subsequent addition of 5e, 5h and 5c indicated a possible static interaction between BSA and imidazo[1,2-a]pyrimidine ligands.
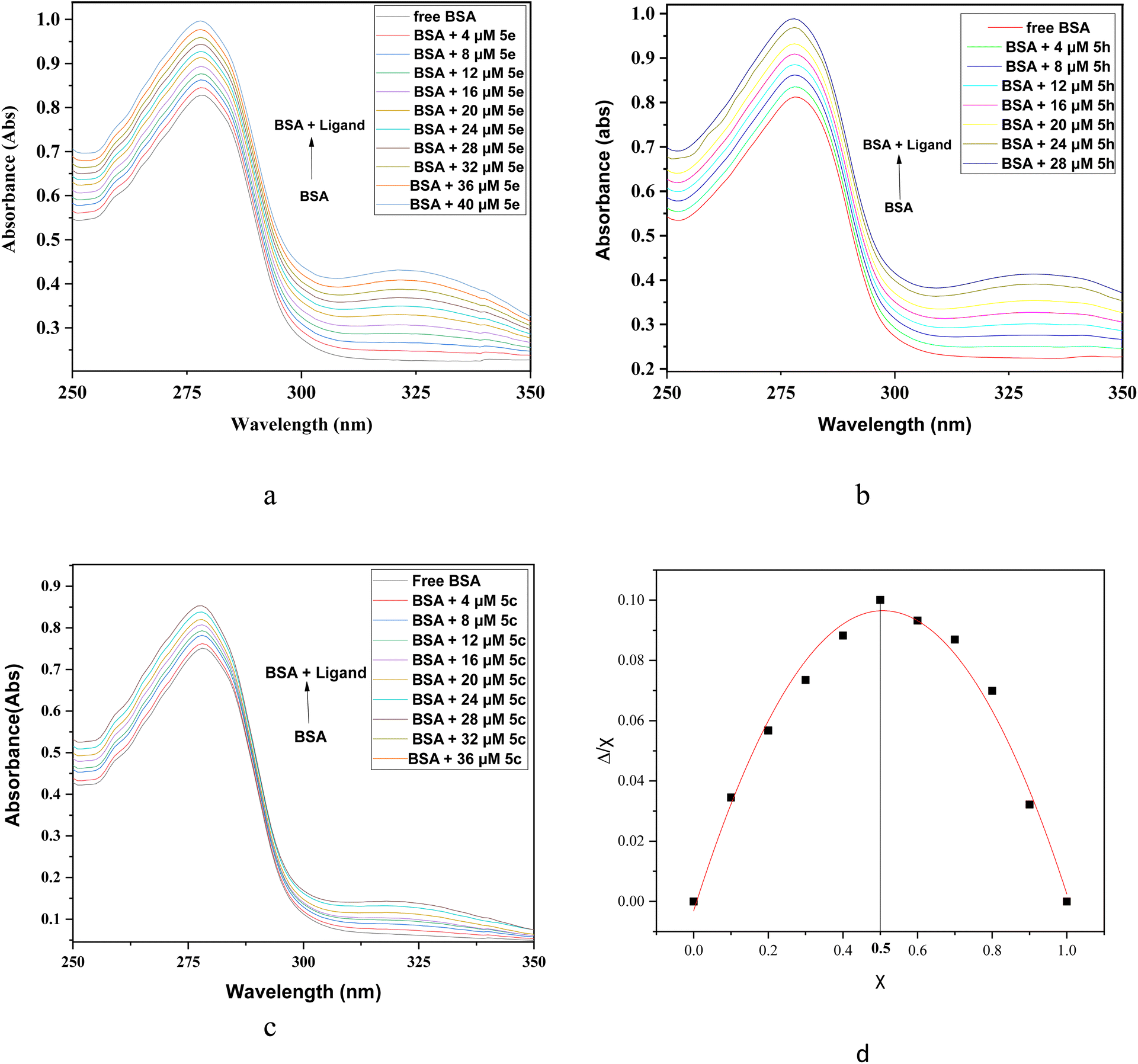 | ||
| Fig. 5 UV-visible spectra of BSA-ligand complex system at increasing concentrations of ligand at constant BSA concentration of 15 μM in physiological pH 7.2 of Tris–HCl buffer at room temperature. (a) BSA spectra at variable concentration of 5e (0–40 μM); (b) BSA spectra at variable concentration of 5h (0–28 μM); (c) BSA spectra at variable concentration of 5c (0–36 μM). (d) Job's plot for BSA-5e complex system. | ||
The stoichiometric ratio in which BSA protein interacts with a ligand was determined employing Job's method of continuous variation. Ten different solutions were prepared with different volumes of BSA and 5e fixed at a constant concentration of 15 μM. In Job's plot, the absorbance at λmax (278 nm) was measured and absorbance versus mole fraction of ligand 5e was plotted (Fig. 5d). The maxima in Job's plot was observed at 0.5 on the axis which indicates 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry between BSA and ligand 5e.43
:1 binding stoichiometry between BSA and ligand 5e.43
 | ||
| Fig. 6 Emission spectra of BSA (15 μM) and in presence of increasing concentration of compounds (a) 5e (0–60 μM), (b) 5h (0–56 μM), (c) 5c (0–56 μM) and (d) emission spectra of ligand 5e (0.1 mM) and in presence of increasing concentration of BSA (0–40 μM). | ||
Further to understand the interaction between ligand 5e and BSA, a fixed concentration of compound 5e (0.1 mM) was titrated with increasing BSA concentration (0–40 μM) at room temperature (25 °C) and the effects on emission profiles was monitored following excitation at 367 nm (Fig. 6d). Free ligand (5e) exhibits maximum in absorption peak at 367 nm. Therefore, the emission spectra were recorded in the spectral range of 387–700 nm by fixing the excitation wavelength at 367 nm. Ligand 5e exhibited a strong fluorescence emission at 454 nm in the absence of BSA. As expected, emission intensity increases at 481 nm as the concentration of BSA is increased (Fig. 6d). The emission increases linearly as a function of BSA concentration until about 28 μM, where it reaches a plateau that may be attributed to a self-quenching phenomenon.44 Red shift in emission maxima was observed in the presence of BSA.
The static and dynamic quenching process can be identified by calculating the quenching constant Kq using the Stern–Volmer equation45 (eqn (1)).
 | (1) |
In order to find the effectiveness of binding of compounds with BSA, fluorescent data were utilized to compute quenching constant (Kq) and Stern–Volmer constant (KSV) by plotting emission spectral data at λmax (343 nm) at room temperature by employing the Stern–Volmer equation (eqn (1)). Based on the linear fit graph of fluorescence intensity (Fo) of BSA in the absence of ligand to the fluorescence intensity (F) of BSA in the presence of ligand at varying concentration was plotted against increasing concentration of ligand [Q] (Fig. 7), quenching constant (Kq) and Stern–Volmer constant (Ksv) were calculated and shown in Table 5. The values of quenching constant Kq are 2.0 × 1012, 4.9 × 1012, and 5.0 × 1012 M−1 s−1 for compounds (5c, 5e and 5h, respectively) which were found to be higher than maximum scatter collision quenching constant46 (2 × 1010 M−1 s−1), indicating the quenching of fluorophore (BSA) by our compounds is initiated by the static mechanism with formation complex at the ground state. According to Kq values, the capability of the compounds to quench the emission intensity of BSA follows the sequence 5h > 5e > 5c.
 | ||
| Fig. 7 (a) Stern–Volmer plot of BSA quenching by compounds 5e, 5h and 5c. (b) Double logarithmic plot employed to determine binding parameters for compounds 5e, 5h and 5c. | ||
| Compd | KSV × 104 (M−1) | Kq × 1012 (M−1 s−1) | LogKb |
Kb (M−1) | n | ΔG° (kcal mol−1) |
|---|---|---|---|---|---|---|
| 5c | 2.0 ± 0.03 | 2.0 ± 0.03 | 4.6 ± 0.1 | 4.7 × 104 | 1.0 | −6.27 |
| 5e | 4.9 ± 0.07 | 4.9 ± 0.07 | 4.8 ± 0.1 | 6.4 × 104 | 1.0 ± 0.02 | −6.54 |
| 5h | 5.0 ± 0.1 | 5.0 ± 0.1 | 6.6 ± 0.1 | 4.2 × 106 | 1.4 ± 0.03 | −9.00 |
2.2.3.1 Identification of binding constant and number of binding sites. Further, Modified Stern–Volmer equation46 (eqn (2)) was used to calculate binding constant Kb and number of binding sites (n).
 | (2) |
From the plot of log(Fo − F)/F against log[Q] (Fig. 8), the binding constant Kb and the number of binding sites n have been obtained from the intercept and slope, respectively. The results are mentioned in Table 5. The value of n unveils the presence of a single binding site in BSA for imidazopyrimidines. The desired value for binding constant is contemplated to be in the range 104–106 M−1. The value of Kb observed for 5c, 5e and 5h are in this range, suggesting that the ligand binding to BSA protein is reversible and allows the transport and release of the ligand to the target site. Results indicate that the compounds bind to BSA in the order of 5h > 5e > 5c. Furthermore, the negative value of standard Gibbs free energy change (ΔG°) from eqn (3) suggests the spontaneity of the binding process that resulted into BSA–imidazopyrimidine complex.
|
G° = −RTlnKb
| (3) |
 | ||
| Fig. 8 Fluorescence spectra of (a) BSA-PBZ and (b) BSA-IBP complex in the absence and presence of increasing concentration of 5e (0–62 μM) at 298 K. (c) Double logarithmic plot employed to determine binding constants for compound 5e the absence (DMSO) and presence of site markers, ibuprofen and phenylbutazone. | ||
In this experiment, the fluorescence spectra of BSA–PBZ and BSA–IBP complexes (1:1) were recorded in the absence and presence of the subsequent increasing amounts of ligand 5e (0–52 μM) (Fig. 8a and b). A decrement in the emission intensity was observed by the increase in the concentration of 5e, and the data thus obtained was utilized to plot a graph of log(Fo − F)/F against log[Q] for complex 5e in the absence and presence of site markers as shown in Fig. 8c. The binding constant Kb for phenylbutazone and ibuprofen was found to be 0.0085 × 104 and 0.374 × 104, respectively (Table 6). It can be observed that the Kb and log Kb values of the compound 5e with BSA significantly decrease in the presence of phenylbutazone and ibuprofen, compared to the Kb value in the absence of any site markers, indicating that there are competitive interactions between compound 5e and the two site makers with BSA.48 Therefore, it can be inferred that the compound 5e may interact with BSA at both site I and site II. Nevertheless, the value of Kb is much lower in the presence of phenylbutazone, suggesting compound 5e mainly binds in Sudlow's Site I of BSA.
| System | LogKb |
Kb (M−1) |
|---|---|---|
| BSA + 5e | 4.790 | 6.2 × 104 |
| BSA + PBZ + 5e | 1.932 | 0.0085 × 104 |
| BSA + IBP + 5e | 2.573 | 0.374 × 104 |
3. Conclusions
In summary, we have developed a simple and efficient approach for the synthesis of 2,3-di substituted imidazo[1,2-a]pyridimines from α-bromo-β-diketones and 2-aminopyrimidine which could serve as promising scaffolds for the development of novel bioactive molecules. The structure of regioisomeric product was characterized by 1H, 13C, HMBC, HMQC, IR spectral, and mass spectrometric studies. The salient features of this approach include catalyst-free mild reaction conditions, inexpensive reagents, broad substrate scope, high yields, and easy workup procedure in a single synthetic operation. Spectroscopic methods and molecular docking studies were used to explore binding interaction of imidazo[1,2-a]pyridimines with BSA. UV-visible study confirmed the interaction and 1:1 binding stoichiometry. Fluorescence analyses revealed that compounds bind moderately with BSA through static quenching mechanism and binding constant revealed that compound 5h exhibits greater binding affinity than 5c and 5e. Negative ΔG° values suggested that the binding process was spontaneous. Furthermore, site-marker competitive binding assay indicated that compound 5e interacts with BSA at both site I and site II and site I is the main binding site. The present study illustrates that the nature of substituents modifies binding properties of imidazo[1,2-a]pyrimidines to BSA and it is expected to influence design, development and synthesis of novel imidazo[1,2-a]pyrimidine derivatives with significant medical applications.
4. Experimental
4.1 General
All the chemicals and solvents used in the present study were purchased from commercial suppliers of Hi-media and Avera, India, and used without further purification. To monitor the progress of the reaction and purity of the products, TLC experiments were performed on 0.2 mm Merck precoated silica gel 60 F254-coated aluminum plates. The mixture of ethyl acetate and petroleum ether was used as the mobile phase and spots were visualized under UV light at 254 nm. Melting points were determined using an electrical digital Melting Point Apparatus (MEPA) in an open capillary tube and were uncorrected. IR spectra were recorded on Buck Scientific IR M-500 instrument in KBr pellets (υmax in cm−1). 1H and 13C NMR spectra were recorded on a Jeol ECZS-400 instrument at 400 and 100 MHz, respectively, using DMSO-d6 as a solvent and tetramethylsilane (TMS) as an internal standard (the chemical shift in δ scale and coupling constants (J) were expressed in parts per million (ppm) and hertz, respectively). High-resolution mass spectra (HRMS) were measured in the ESI+ mode at CIL GJU, Hisar. 2D correlation spectroscopy, (1H–13C) gs-HSQC, (1H–13C) gs-HMBC of the samples were carried out at Kurukshetra University, Kurukshetra. The UV-vis spectra were recorded on a UV-vis spectrophotometer 117 (Systronic, India) with 1 cm path-length cell. Fluorescence spectra were recorded on the Shimadzu-5301pc spectro-fluorophotometer (Kyoto, Japan).:40, v/v). After the completion of reaction (as indicated by TLC), the solvent was evaporated on the rotary evaporator and the resulting mixture was neutralized with a saturated solution of sodium bicarbonate and extracted with ethyl acetate. The gummy mass obtained was triturated with petroleum ether. Then acetone (5 mL) was added, stirred at room temperature for 15–20 minutes and the residue thus obtained was filtered, washed with cold ethanol, dried, and recrystallized from ethanol to obtain the target 3-aroylimidazo[1,2-a]pyrimidine (5a–h).
4.1.1.1 1-Phenyl-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone (5a). White solid; m.p. 187 °C; yield: 0.190 g (80%); IR (KBr) νmax (cm−1): 1648 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).1H NMR (400 MHz, DMSO-d6): δ 9.64–9.66 (d, 1H, J = 6.8 Hz, 7 H), 9.02 (d, 1H, J = 4.0 Hz, 5-H), 7.79–7.81 (d, 2H, J = 7.6 Hz, Ph-2′,6′-H), 7.73 (dd, 1H, Jo = 6.8, Jm = 4.4 Hz, Ph-4′-H), 7.60–7.64 (m, 3H, Ph-3′,5′, 6-H), 2.21 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 186.20, 156.99, 147.59, 146.82, 138.81, 138.08, 133.52, 129.38, 129.27, 119.67, 113.66, 15.44.
HRMS (ESI) m/z: 238.0890 [M + H]+.
Elemental analysis: calcd. for C14H11N3O: C, 70.87; H, 4.67; N, 17.71% found: C, 70.65; H, 4.74; N, 17.48%.
4.1.1.2 1-(4′-Fluorophenyl)-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone (5b). White solid; m.p. 206 °C; yield: 0.202 g (79%); IR (KBr) νmax (cm−1): 1650 (C
O).1H NMR (400 MHz, DMSO-d6): δ 9.60–9.62 (dd, 1H, J = 7.2, J = 2.0 Hz, 7-H), 8.99–9.01 (dd, 1H, J = 4.4, J = 2.0 Hz, 5-H), 7.87–7.91 (dt, 2H, Jo = 8.8, J(m)HF = 5.6 Hz, Ph-2′,6′-H), 7.59–7.62 (dd, 1H, J = 6.8 Hz, J = 4.4 Hz, 6-H), 7.43–7.48 (t, 2H, Jo = 8.8 Hz, Ph-3′,5′-H), 2.24 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 184.85, 156.91, 147.04, 138.05, 135.44, 135.41, 132.48, 132.38, 119.67, 116.68, 116.46, 113.56, 15.65.
HRMS (ESI) m/z: 256.0836 [M + H]+.
Elemental analysis: calcd. for C14H10FN3O: C, 65.88; H, 3.95; N, 16.46% found: C, 65.81; H, 3.81; N, 16.63%.
4.1.1.3 1-(4′-Chlorophenyl)-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone (5c). White solid; m.p. 224 °C; yield: 0.210 g (77%); IR (KBr) νmax (cm−1): 1650 (C
O).1H NMR (400 MHz, DMSO-d6): δ 9.61–9.63 (d, 1H, J = 6.8 Hz, 7-H), 9.01 (d, 1H, J = 3.2 Hz, 5-H), 7.81–7.83 (d, 2H, J = 8.4 Hz, Ph-2′,6′-H), 7.68–7.70 (d, 2H, J = 8.4 Hz, Ph-3′,5′-H), 7.61–7.64 (dd, 1H, J = 6.4, J = 4.4 Hz, 6-H), 2.24 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 184.96, 156.98, 147.98, 146.98, 138.31, 138.05, 137.50, 131.28, 129.53, 119.56, 113.63, 15.63.
HRMS (ESI) m/z: 272.0469 [M + H]+; 274.0463[M + 1 + 2]+; (3:1).
Elemental analysis: calcd. for C14H10ClN3O: C, 61.89; H, 3.71; N, 15.47% found: C, 61.78; H, 3.63; N, 15.25%.
4.1.1.4 1-(4′-Bromophenyl)-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone (5d). White solid; m.p. 248 °C; yield: 0.227 g (72%); IR (KBr) νmax (cm−1): 1650 (C
O).1H NMR (400 MHz, DMSO-d6): δ 9.61–9.63 (dd, 1H, J = 6.8, J = 1.2 Hz, 7-H), 8.97 (dd, 1H, J = 4.0, J = 1.2 Hz, 5-H), 7.82–7.84 (d, 2H, J = 8.4 Hz, Ph-2′,6′-H), 7.71–7.73 (d, 2H, J = 8.4 Hz, Ph-3′,5′-H), 7.56–7.58 (dd, 1H, J = 6.8, J = 4.4 Hz, 6-H), 2.21 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 185.19, 156.39, 149.45, 147.70, 138.11, 137.81, 132.42, 131.27, 127.15, 119.54, 113.15, 16.14.
HRMS (ESI) m/z: 316.9978 [M + H]+; 318.9959[M + 1 + 2]+; (1:1).
Elemental analysis: calcd. for C14H10BrN3O: C, 53.19; H, 3.19; N, 13.29% found: C, 52.98; H, 3.24; N, 13.43%.
4.1.1.5 1-(4′-Methoxyphenyl)-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone (5e). White solid; m.p. 190 °C; yield: 0.230 g (86%); IR (KBr) νmax (cm−1): 1645 (C
O).1H NMR (400 MHz, DMSO-d6): δ 9.52–9.54 (dd, 1H, J = 7.0, J = 2.4 Hz, 7-H), 9.01–9.03 (dd, 1H, J = 4.8, J = 2.0 Hz, 5-H), 7.82–7.84 (d, 2H, J = 8.8 Hz, Ph-2′,6′-H), 7.61–7.64 (dd, 1H, J = 6.0, J = 4.8 Hz, 6-H), 7.14–7.16 (d, 2H, J = 9.2 Hz, Ph-3′,5′-H), 3.90 (s, 3H, Ph-4′-OMe), 2.31 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 184.54, 164.08, 157.17, 146.21, 138.12, 132.26, 130.82, 119.82, 114.78, 113.76, 56.23, 15.07.
HRMS (ESI) m/z: 268.1003 [M + H]+.
Elemental analysis: calcd. for C15H13N3O2: C, 67.40; H, 4.90; N, 15.72% found: C, 67.28; H, 4.74; N, 15.59%.
4.1.1.6 1-(3′-Methoxyphenyl)-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone (5f). White solid; m.p. 215 °C; yield: 0.220 g (82%); IR (KBr) νmax (cm−1): 1645 (C
O).1H NMR (400 MHz, DMSO-d6): δ 9.63–9.65 (dd, J = 6.8, J = 1.2 Hz, 1H, 7-H), 9.03–9.04 (dd, 1H, J = 4.4, J = 2.8 Hz, 5-H), 7.63–7.66 (dd, 1H, J = 6.8, J = 4.8 Hz, 6-H), 7.51–7.55 (t, 1H, Jo = 7.6 Hz, Ph-5′-H), 7.36–7.38 (d, 1H, Jo = 7.6 Hz, Ph-6′-H), 7.29–7.31 (m, 2H, Ph-2′,4′-H), 3.83 (s, 3H, Ph-3′-OMe), 2.23 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 185.89, 159.88, 157.17, 147.31, 146.60, 140.15, 138.12, 130.67, 121.53, 119.66, 119.54, 113.83, 113.74, 55.99, 15.25.
HRMS (ESI) m/z: 268.1003 [M + H]+.
Elemental analysis: calcd. for C15H13N3O2: C, 67.40; H, 4.90; N, 15.72% found: C, 67.52; H, 4.66; N, 15.97%.
4.1.1.7 1-(2′-Methoxyphenyl)-1-(2-methylimidazo[1,2-a]pyrimidin-3-yl)methanone (5g). White solid; m.p. 229 °C; yield: 0.213 g (80%); IR (KBr) νmax (cm−1): 1645 (C
O).1H NMR (400 MHz, DMSO-d6): δ 9.64–9.66 (dd, 1H, J = 6.8, J = 2.0 Hz, 7-H), 9.06–9.07 (dd, 1H, J = 4.4, J = 2.0 Hz, 5-H), 7.67–7.70 (dd, 1H, J = 6.8, J = 4.4 Hz, 6-H), 7.52–7.56 (t, 1H, J = 8.0 Hz, Ph-4′-H), 7.37–7.39 (d, 1H, J = 7.6 Hz, Ph-6′-H), 7.30–7.33 (m, 2H, Ph-3′,5′-H), 3.84 (s, 3H, Ph-2′-OMe), 2.25 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 185.89, 159.92, 157.59, 146.23, 140.03, 138.34, 130.76, 130.71, 121.68, 119.75, 119.70, 114.17, 113.81, 56.05, 15.05.
HRMS (ESI) m/z: 268.1003 [M + H]+.
Elemental analysis: calcd. for C15H13N3O2: C, 67.40; H, 4.90; N, 15.72% found: C, 67.71; H, 4.56; N, 15.46%.
4.1.1.8 1-(Thiophen-2′-yl)-1-(2-methylimidazo[1,2-a]pyridin-3-yl)methanone (5h). White solid; m.p. 244 °C; yield: 0.193 g (79%); IR (KBr) νmax (cm−1): 1654 (C
O).1H NMR (400 MHz, DMSO-d6): δ 9.43–9.45 (dd, 1H, J = 6.8, J = 1.2 Hz, 7-H), 9.02–9.03 (dd, 1H, J = 4.0, J = 1.2 Hz, 5-H), 8.24–8.25 (d, 1H, J = 4.4 Hz, thienyl-5′-H), 7.93–7.94 (d, 1H, J = 3.2 Hz, thienyl-3′-H), 7.61–7.64 (dd, 1H, J = 6.8, J = 4.4 Hz, 6-H), 7.33–7.35 (t, 1H, J = 4.4 Hz, thienyl-4′-H), 2.51 (s, 3H, 2-CH3).
13C NMR (100 MHz, DMSO-d6): δ 177.02, 157.36, 145.97, 144.53, 142.68, 138.04, 137.19, 136.61, 129.26, 119.49, 113.74, 14.97.
HRMS (EI) m/z: 244.0467 [M + H]+.
Elemental analysis: calcd. for C12H9N3OS: C, 59.24; H, 3.73; N, 17.27% found: C, 59.12; H, 3.62; N, 17.49%.
4.2 Binding studies
Data availability
Data for this article, including experimental data, (1H, 13C, [1H–13C] HMBC, and [1H–13C] HSQC) for final compounds have been included as part of the ESI.†Author contributions
R. A. and P. K.; conceptualization, supervision, reviewing and editing. M. S. and G. S.; methodology, investigation, computational calculations, writing and original draft preparation.Conflicts of interest
The author(s) confirm that this article's content has no conflict of interest.Acknowledgements
The authors are thankful to their respective institutions for providing the required facility to complete this work. We thank the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for providing financial assistance to Manisha Sharma (Grant 09/105(0274)/2018-EMR-I) as SRF.References
- R. Aggarwal and G. Sumran, An insight on medicinal attributes of 1,2,4-triazoles, Eur. J. Med. Chem., 2020, 205, 112652, DOI:10.1016/j.ejmech.2020.112652
.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25(8), 1909, DOI:10.3390/molecules25081909
- F. Doganc, A. S. Aydin, E. Şahin and H. Göker, Regioselective N-alkylation of some 2 or 6-chlorinated purine analogues, J. Mol. Struct., 2023, 1272, 134200, DOI:10.1016/j.molstruc.2022.134200
- H. T. Abdel-Mohsen, A. Abood, K. J. Flanagan, A. Meindl, M. O. Senge and H. I. El Diwani, Synthesis, crystal structure, and ADME prediction studies of novel imidazopyrimidines as antibacterial and cytotoxic agents, Arch. Pharm., 2020, 353(3), 1900271, DOI:10.1002/ardp.201900271
- Á. Ramírez-Trinidad, K. Carrillo-Jaimes, J. A. Rivera-Chávez and E. Hernández-Vázquez, Synthesis and cytotoxic/antimicrobial screening of 2-alkenylimidazo[1,2-a]pyrimidines, Med. Chem. Res., 2023, 32(1), 144–157, DOI:10.1007/s00044-022-02997-6
- M. Bayanati, M. Khoramjouy, M. Faizi, M. A. Movahed, M. Mahboubi-Rabbani and A. Zarghi, Novel Benzo[4,5]imidazo[1,2-a]pyrimidine derivatives as selective Cyclooxygenase-2 Inhibitors: Design, synthesis, docking studies, and biological evaluation, Med. Chem. Res., 2023, 32(3), 495–505, DOI:10.1007/s00044-023-03022-0
- S. Alqarni, L. Cooper and J. Galvan Achi, et al., Synthesis, Optimization, and Structure–Activity Relationships of Imidazo[1,2-a]pyrimidines as Inhibitors of Group 2 Influenza A Viruses, J. Med. Chem., 2022, 65(20), 14104–14120, DOI:10.1021/acs.jmedchem.2c01329
- N. M. Panchani and H. S. Joshi, Green and Catalyst-Free Synthesis of Some New Benzo[4,5]imidazo[1,2-a]pyrimidine Derivatives as Antimicrobial and Antitubercular Agents, Russ. J. Org. Chem., 2022, 58(4), 604–611, DOI:10.1134/S1070428022040200
- R. Kumar, R. Singh and A. das Chagas Almeida, et al., Imidazo[1,2-a]pyrimidine as a New Antileishmanial Pharmacophore against Leishmania amazonensis Promastigotes and Amastigotes, ACS Omega, 2023, 8(43), 40613–40621, DOI:10.1021/acsomega.3c05441
- P. Prasad, A. G. Kalola and M. P. Patel, Microwave assisted one-pot synthetic route to imidazo[1,2-a]pyrimidine derivatives of imidazo/triazole clubbed pyrazole and their pharmacological screening, New J. Chem., 2018, 42(15), 12666–12676, 10.1039/C8NJ00670A
- E. Koti Reddy, C. Remya, A. M. Sajith, K. V. Dileep, C. Sadasivan and S. Anwar, Functionalised dihydroazo pyrimidine derivatives from Morita–Baylis–Hillman acetates: synthesis and studies against acetylcholinesterase as its inhibitors, RSC Adv., 2016, 6(81), 77431–77439, 10.1039/C6RA12507G
- F. Peytam, G. Takalloobanafshi and T. Saadattalab, et al., Design, synthesis, molecular docking, and in vitro α-glucosidase inhibitory activities of novel 3-amino-2,4-diarylbenzo[4,5]imidazo[1,2-a]pyrimidines against yeast and rat α-glucosidase, Sci. Rep., 2021, 11(1), 11911, DOI:10.1038/s41598-021-91473-z
- M. Azzouzi, Z. E. Ouafi and O. Azougagh, et al., Design, synthesis, and computational studies of novel imidazo[1,2-a]pyrimidine derivatives as potential dual inhibitors of hACE2 and spike protein for blocking SARS-CoV-2 cell entry, J. Mol. Struct., 2023, 1285, 135525, DOI:10.1016/j.molstruc.2023.135525
- S. C. Goodacre, L. J. Street and D. J. Hallett, et al., Imidazo[1,2-a]pyrimidines as Functionally Selective and Orally Bioavailable GABA A α2/α3 Binding Site Agonists for the Treatment of Anxiety Disorders, J. Med. Chem., 2006, 49(1), 35–38, DOI:10.1021/jm051065l
- K. C. Rupert, J. R. Henry and J. H. Dodd, et al., Imidazopyrimidines, potent inhibitors of p38 MAP kinase, Bioorg. Med. Chem. Lett., 2003, 13(3), 347–350, DOI:10.1016/S0960-894X(02)01020-X
- M. L. S. O. Lima, C. B. Braga and T. B. Becher, et al., Fluorescent Imidazo[1,2-a]pyrimidine Compounds as Biocompatible Organic Photosensitizers that Generate Singlet Oxygen: A Potential Tool
for Phototheranostics, Chem.–Eur. J., 2021, 27(20), 6213–6222, DOI:10.1002/chem.202004957
- M. Rawat and D. S. Rawat, Copper oxide nanoparticle catalysed synthesis of imidazo[1,2-a]pyrimidine derivatives, their optical properties and selective fluorescent sensor towards zinc ion, Tetrahedron Lett., 2018, 59(24), 2341–2346, DOI:10.1016/j.tetlet.2018.05.005
- S. Pareek, D. Jain and S. Hussain, et al., A new insight into corrosion inhibition mechanism of copper in aerated 3.5 wt.% NaCl solution by eco-friendly imidazopyrimidine dye: experimental and theoretical approach, Chem. Eng. J., 2019, 358, 725–742, DOI:10.1016/j.cej.2018.08.079
- K. Karami, N. Jamshidian and M. Zakariazadeh, Synthesis, characterization and molecular docking of new C,N -palladacycles containing pyridinium-derived ligands: DNA and BSA interaction studies and evaluation as anti-tumor agents, Appl. Organomet. Chem., 2019, 33(3), 4728, DOI:10.1002/aoc.4728
- S. Siddiqui, F. Ameen, I. Jahan, S. M. Nayeem and M. Tabish, A comprehensive spectroscopic and computational investigation on the binding of the anti-asthmatic drug triamcinolone with serum albumin, New J. Chem., 2019, 43(10), 4137–4151, 10.1039/C8NJ05486J
- T. A. Wani, A. H. Bakheit and S. Zargar, et al., Toxicity Study and Binding Analysis of Newly Synthesized Antifungal N-(4-aryl/cyclohexyl)-2-(pyridine-4-yl carbonyl) hydrazinecarbothioamide Derivative with Bovine Serum Albumin, Int. J. Mol. Sci., 2023, 24(5), 4942, DOI:10.3390/ijms24054942
- H. Dezhampanah and A. M. Moghaddam Pour, Multi technique investigation on interaction between 5-(2-thiazolylazo)-2,4,6-triaminopyrimidine and HSA and BSA, J. Biomol. Struct. Dyn., 2022, 40(18), 8143–8154, DOI:10.1080/07391102.2021.1906751
- V. S, A. K. Das, Y. Bylappa, A. Nag and M. Dolai, A dual-functional rhodamine B and azo-salicylaldehyde derivative for the simultaneous detection of copper and hypochlorite: synthesis, biological applications and theoretical insights, Anal. Methods, 2024, 16(47), 8164–8178, 10.1039/D4AY01758G
- P. Singla, V. Luxami, R. Singh, V. Tandon and K. Paul, Novel pyrazolo[3,4-d]pyrimidine with 4-(1H-benzimidazol-2-yl)-phenylamine as broad spectrum anticancer agents: Synthesis, cell based assay, topoisomerase inhibition, DNA intercalation and bovine serum albumin studies, Eur. J. Med. Chem., 2017, 126, 24–35, DOI:10.1016/j.ejmech.2016.09.093
- J. Liu, Y. He and D. Liu, et al., Characterizing the binding interaction of astilbin with bovine serum albumin: a spectroscopic study in combination with molecular docking technology, RSC Adv., 2018, 8(13), 7280–7286, 10.1039/C7RA13272G
- Y. Lv, Q. Liang, Y. Li, X. Liu, D. Zhang and X. Li, Study of the binding mechanism between hydroxytyrosol and bovine serum albumin using multispectral and molecular docking, Food Hydrocoll., 2022, 122, 107072, DOI:10.1016/j.foodhyd.2021.107072
- S. Khatun and Q. F. A. Riyazuddeen, In-vitro binding analysis of bovine serum albumin with sulindac/chlorpromazine: Spectroscopic, calorimetric and computational approaches, J. Mol. Liq., 2020, 299, 112124, DOI:10.1016/j.molliq.2019.112124
- R. Goel, V. Luxami and K. Paul, Synthetic approaches and functionalizations of imidazo[1,2-a]pyrimidines: an overview of the decade, RSC Adv., 2015, 5(99), 81608–81637, 10.1039/C5RA14795F
- Y. Vara, E. Aldaba, A. Arrieta, J. L. Pizarro, M. I. Arriortua and F. P. Cossío, Regiochemistry of the microwave-assisted reaction between aromatic amines and α-bromoketones to yield substituted 1H-indoles, Org. Biomol. Chem., 2008, 6(10), 1763, 10.1039/b719641e
- K. M. Bonger, R. J. van den Berg and L. H. Heitman, et al., Synthesis and evaluation of homo-bivalent GnRHR ligands, Bioorg. Med. Chem., 2007, 15(14), 4841–4856, DOI:10.1016/j.bmc.2007.04.065
- A. Herath, R. Dahl and N. D. P. Cosford, Fully Automated Continuous Flow Synthesis of Highly Functionalized Imidazo[1,2-a] Heterocycles, Org. Lett., 2010, 12(3), 412–415, DOI:10.1021/ol902433a
- M. Mantipally, M. R. Gangireddy, R. Gundla, V. N. Badavath, S. R. Mandha and V. C. Maddipati, Rational design, molecular docking and synthesis of novel homopiperazine linked imidazo[1,2-a]pyrimidine derivatives as potent cytotoxic and antimicrobial agents, Bioorg. Med. Chem. Lett., 2019, 29(16), 2248–2253, DOI:10.1016/j.bmcl.2019.06.031
- I. Pravst, M. Zupan and S. Stavber, Solvent-free bromination of 1,3-diketones and β-keto esters with NBS, Green Chem., 2006, 8(11), 1001–1005, 10.1039/B608446J
- R. Aggarwal and G. Sumran, A facile [hydroxy(tosyloxy)iodo]benzene mediated synthesis of 2-arylimidazo[1,2-a]pyrimidines and their conversion into 3-bromo-2- arylimidazo[1,2-a]pyrimidines, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45(12), 2690–2695, DOI:10.1002/chin.200714170
- R. N. Rao, B. MM, B. Maiti, R. Thakuria and K. Chanda, Efficient Access to Imidazo[1,2-a]pyridines/pyrazines/pyrimidines via Catalyst-Free Annulation Reaction under Microwave Irradiation in Green Solvent, ACS Comb. Sci., 2018, 20(3), 164–171, DOI:10.1021/acscombsci.7b00173
- B. Das, N. Bhunia and M. Lingaiah, A Simple and Efficient Metal-Free Synthesis of Tetrasubstituted Pyrroles by Iodine-Catalyzed Four-Component Coupling Reaction of Aldehydes, Amines, Dialkyl Acetylenedicarboxylates, and Nitromethane1, Synthesis, 2011, 2011(21), 3471–3474, DOI:10.1055/s-0030-1260228
- R. Aggarwal, M. Hooda and P. Kumar, et al., Visible-Light-Prompted Synthesis and Binding Studies of 5,6-Dihydroimidazo[2,1-b]thiazoles with BSA and DNA Using Biophysical and Computational Methods, J. Org. Chem., 2022, 87(6), 3952–3966, DOI:10.1021/acs.joc.1c02471
- G. Sumran and R. Aggarwal, A convenient [hydroxy(tosyloxy)iodo]benzene-mediated one-pot synthesis of 2-arylimidazo[2,1-b[benzothiazoles]], J. Sulfur Chem., 2015, 36(2), 170–177, DOI:10.1080/17415993.2014.996221
- R. Aggarwal, M. Hooda, P. Kumar and M. C. Torralba, Visible-light-mediated regioselective synthesis of novel thiazolo[3,2-b] [1,2,4]triazoles: Advantageous synthetic application of aqueous conditions, Org. Biomol. Chem., 2022, 20(3), 584–595, 10.1039/d1ob02194j
- S. Siddiqui, F. Ameen, S. ur Rehman, T. Sarwar and M. Tabish, Studying the interaction of drug/ligand with serum albumin, J. Mol. Liq., 2021, 336, 116200, DOI:10.1016/j.molliq.2021.116200
- R. Patel, N. Maurya and M. U. D. Parray, et al., Esterase activity and conformational changes of bovine serum albumin toward interaction with mephedrone: Spectroscopic and computational studies, J. Mol. Recognit., 2018, 31(11), 2734, DOI:10.1002/jmr.2734
- A. Anish Babu, K. Karthick, R. Subramanian and K. Swarnalatha, Exploring the structural interaction of BSA with amine functionalized ruthenium(II) metal complex, J. Biomol. Struct. Dyn., 2020, 38(13), 4032–4039, DOI:10.1080/07391102.2019.1683071
- B. Pastrello, G. C. dos Santos, L. C. da Silva-Filho, A. R. de Souza, N. H. Morgon and V. F. Ximenes, Novel aminoquinoline-based solvatochromic fluorescence probe: Interaction with albumin, lysozyme and characterization of amyloid fibrils, Dyes Pigments, 2020, 173, 107874, DOI:10.1016/j.dyepig.2019.107874
- J. Lazniewska, M. Agostino and S. M. Hickey, et al., Spectroscopic and Molecular Docking Study of the Interaction between Neutral Re(I) Tetrazolate Complexes and Bovine Serum Albumin, Chem.–Eur. J., 2021, 27(44), 11406–11417, DOI:10.1002/chem.202101307
- M. H. Gehlen, The centenary of the Stern-Volmer equation of fluorescence quenching: From the single line plot to the SV quenching map, J. Photochem. Photobiol. C Photochem. Rev., 2020, 42, 100338, DOI:10.1016/j.jphotochemrev.2019.100338
- K. Paliwal, P. Haldar, P. K. S. Antharjanam and M. Kumar, Mixed Ligand Mononuclear Copper(II) Complex as a Promising Anticancer Agent: Interaction Studies with DNA/HSA, Molecular Docking, and In Vitro Cytotoxicity Studies, ACS Omega, 2022, 7(25), 21961–21977, DOI:10.1021/acsomega.2c02354
- A. V. Ardhapure, V. Gayakhe and S. Bhilare, et al., Extended fluorescent uridine analogues: synthesis, photophysical properties and selective interaction with BSA protein, New J. Chem., 2020, 44(34), 14744–14754, 10.1039/D0NJ02803G
- R. Ma, H. Pan and T. Shen, et al., Interaction
of Flavonoids from Woodwardia unigemmata with Bovine Serum Albumin (BSA): Application of Spectroscopic Techniques and Molecular Modeling Methods, Molecules, 2017, 22(8), 1317, DOI:10.3390/molecules22081317
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01795e |
| This journal is © The Royal Society of Chemistry 2025 |