
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the enhanced structural, elastic, mechanical, and optoelectronic properties of BaWO4 via oxygen vacancies and europium doping: a DFT + U insight into tailored energy applications
Shah Hussaina,
Raj Walib,
Sikander Azam *c,
Qaiser Rafiqd,
Mehmoona Nisard,
Wilayat Khand,
Yasir Saeed*a and
Mohammed A. Amine
*c,
Qaiser Rafiqd,
Mehmoona Nisard,
Wilayat Khand,
Yasir Saeed*a and
Mohammed A. Amine
aDepartment of Physics, Abbottabad University of Science and Technology, Abbottabad, Pakistan. E-mail: yasir.saeed@kaust.edu.sa
bNational Water and Energy Center, United Arab Emirates University, Al Ain, 15551, United Arab Emirates
cDepartment of Physics, Riphah International University, Islamabad, Pakistan. E-mail: sikander.physicst@gmail.com
dDepartment of Physics, Bacha Khan University, Charsada, Pakistan
eDepartment of Chemistry, College of Science, Taif University, P.O. Box 11099, Taif, 21944, Saudi Arabia
First published on 4th June 2025
Abstract
In this study, we examine the structural, electronic, and optical properties of Eu-doped BaWO4 using the full potential linearized augmented plane wave (FPLAPW) method, within the framework of density functional theory (DFT). The calculations are performed using the Generalized Gradient Approximation with an optimized effective Hubbard parameter ‘U’ (GGA + U), implemented in WIEN2K software. The introduction of oxygen vacancies and Eu doping significantly impacts the elastic properties of BaWO4, including its elastic constants, bulk modulus, shear modulus, and Poisson's ratio. These modifications result in a predictable reduction in stiffness and rigidity but enhance the material's optoelectronic functionality. By adding the Hubbard parameter term ‘U’, with a value of U = 7 eV, a more accurate description of the system is achieved, particularly in systems with a strong correlation of d- and f-electronic states. In contrast to the wide band gap (4.885 eV) of parent BaWO4, the electronic band gap decreases to 2.80 eV for Eu-doped BaWO4. Additionally, the creation of O-deficiency in BaWO4 results in a reduction in the band gap value to 0.8 eV (spin-up) and 2.6 eV (spin-down). The partial density of states (PDOS) reveals that the Eu-f (Eu-d) state dominates the valence band maximum, while the conduction band minimum is attributed to the W-d state for spin-up and spin-down channels, respectively. Further analysis of the optical response, including the dielectric constant ε(ω), absorption coefficient I(ω), reflectivity R(ω), refractive index n(ω), and optical energy loss functions L(ω), with different incident photon energies, is presented. When Eu atoms are added to the BaWO4 sample, the gap between optical bands narrows, indicating the development of intermediate energy levels. The calculated band gaps confirm that the Eg of oxygen vacancy (V_O) < oxygen vacancy shows good agreement with optoelectronic devices.
1 Introduction
Dye-sensitized solar cells (DSSCs) have gained popularity in recent decades as a green energy source due to their low cost, high stability, ease of processing, and high conversion efficiencies (PCEs), making them a part of the next generation of photovoltaic cells.1–4 DSSCs typically use a photoelectrode, a counter electrode, and a redox electrolyte comprising iodide and tri-iodide ions.5,6 The photoelectrode plays a crucial role in the generation of photo-excited electrons in DSSC performance. O'Regan and Gratzel published the first studies on DSSCs in 1991,7 based on a visible light-sensitizing material using TiO2 nanoparticles, with a conversion efficiency of 7%. Methods such as surface modification, surface treatment, semiconductor recombination, luminous materials, and structural optimization have been explored to improve photoelectrode efficiency.8–12 The amount of light energy absorbed by the DSSC is strongly influenced by the photoelectrode's light absorption spectrum.13Recently, luminescent conversion materials have been proposed as a viable option for extending light absorption in DSSCs.14 These materials can shift or convert a broad spectrum of light into photons of a single wavelength. Up-conversion (UC), down-conversion (DC), and down-shifting (DS) luminescence techniques are used to transform infrared or ultraviolet light into visible light.15
Down-shifting is a single-photon process that involves converting one high-energy absorbed photon into a low-energy photon. The Stokes Law is demonstrated by the Stokes shift, which represents a wavelength change. One high-energy photon can be converted into one or more low-energy photons using down-converting materials. Both techniques help reduce the energy loss caused by the thermalization of hot charge carriers after high-energy photon absorption.16,17 Rare earth (RE) ions have been effectively employed in dye-sensitized solar cells to improve light scattering and absorption spectra of dye-loaded photoactive electrodes in DSSCs.18 As a result, down-converting materials have the potential to enhance the efficiency of DSSCs.14,19 Praseodymium (Pr3+)-doped inorganic host materials are among the most promising white-light-emitting nanophosphor materials because they can provide broad and intense luminescence in both visible and infrared regions, making them more suitable for a wider range of optical applications.20
Due to their applications in sensor technology, laser materials, color displays, optoelectronics, white light-emitting diodes, bio-imaging, and security applications, barium tungstate (BaWO4 or BWO) is the primary host material for rare earth ions.21–24 Rare earth ion-doped barium tungstate possesses exceptional thermal and chemical stability, along with distinctive electrical and optical properties resulting from different energy level configurations. BaWO4 has been widely used as a host material for various phosphor powders, including green and bluish phosphors. Barium tungstate (BaWO4) is the most well-known and heaviest member of the alkaline earth tungstate family. BaWO4 is one of the most reactive alkaline earth tungstates, widely investigated due to its excellent electrical conductivity and photoluminescence properties. These compounds play a significant role in various technological applications such as light-emitting diodes (LEDs), scintillator detectors, and solar cells. Eu- and Sm-doped BaWO4 is synthesized using the solid-state method and characterized by X-ray diffraction. Experimental findings, such as optical properties, luminescence intensity, photoluminescence (PL) lifetime, stimulated emission cross-section, bandwidth, and quantum efficiency of doped BaWO4, were improved with Eu and Sm doping.25 Cho26 experimentally studied the structural and optical properties of Eu-doped BaWO4, finding that Eu incorporation increased emission intensity. S. Cho27 developed low-cost BaWO4:RE3+ (RE = Eu or Sm) phosphor powders using a solid-state reaction with various doping concentrations of RE3+. They studied the surface morphology and photoluminescence spectra of the Eu3+-doped BaWO4. The photoluminescence spectra of Eu3+-doped BaWO4 occurred at 274 nm, while for Sm-doped BaWO4, it occurred at 568, 603, and 649 nm, respectively. Deng et al.28 prepared a quasi-single crystalline thin film, which is a promising material for white-light-emitting diodes (w-LED) for solid-state lighting, displays, and photoelectric detectors. They discussed the mechanism of energy transition and luminescence kinetics of the doped BaWO4 matrix. George et al.29 presented a new perspective on the highly stable rare-earth-doped BaWO4 nanofibers, emphasizing the reversibility of fluorescence quenching, which allows precise reuse during the sensitive detection of explosives, compared to other materials. Although much experimental work has been done, little theoretical work has been conducted to explore the physical properties of pure BaWO4. Carvalho et al.30 computed the phonon density of states, specific heat at constant volume, entropy, and Gibbs free energy of the three MWO4 (M = Ca, Sr, or Ba) compounds. It was found that the magnitude of the specific heat at constant volume at high temperature follows the order: BaWO4 > CaWO4 > SrWO4. Additionally, a relationship between the M2+ ionic radius in the scheelite MWO4 (M = Ba, Sr, or Ca) and their entropy, Gibbs free energy, and Helmholtz free energy was verified. Ashraf et al.31 investigated the electronic and optical properties of BaWO4 using Density Functional Theory (DFT) within the framework of CASTEP at an optimized cut-off energy value of 1200 eV. They found that the highest band gap value of BaWO4 corresponds to the highest loss function and the smallest value of the dielectric constant. High absorption in the UV range and a large refractive index make BaWO4 suitable for the formation of sensors, filters, transparent conducting films for window layers on solar cells, solar control coatings, and scintillators. Despite many experimental and theoretical works, the role of O-deficiency on the optoelectronic behavior of Eu-doped BaWO4 using the Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) approach remains unexplored. This research investigates the structural, electronic, and optical properties of the compound Ba1−xEuxWO4 (x = 2.08%) with oxygen vacancy (V_O). Here, Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) is employed to account for d-state or f-state corrections to the electronic structure. Adding the Hubbard potential term “U” further enhances the physical properties, in addition to the electronic structure, including structural and optical properties. The objective of this work is to provide a comprehensive description and correction of the structure, electronic, and optical properties of Eu-doped BaWO4 and O-defect when the Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) approximation is employed in WIEN2K software. This study will help establish a roadmap for the development of new optoelectronic devices and light-emitting diodes (LEDs). By using Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) in WIEN2K software, we compare the results of electronic properties, including the partial density of states (PDOS), the total density of states (TDOS), and optical properties of Eu-doped BaWO4 and O-defects with those of pure BaWO4.
2 Computational methodology and structural details
2.1 Structural properties
In this work, we investigate the three-dimensional tetragonal structure of BaWO4 with the space group I41/a (88) and crystallographic parameters a = 15.11, b = 14.43, c = 29.699, and α, β, γ. We also study doped BaWO4 with Eu, along with oxygen vacancy (V_O), which adopts a monoclinic layered crystal structure with the crystallographic parameters a = 14.43, b = 15.11, c = 26.72, and α, β, γ. The pure crystal structure of BaWO4 is shown in Fig. 1(a). Fig. 1(b) illustrates the doping of Eu (x = 2.08%) in the investigated compound Ba1−xEuxWO4. In this research, we also introduce oxygen vacancy (V_O & 2V_O), and as the oxygen vacancy increases, the band gap of Ba1−xEuxWO4 also increases. Fig. 1(c and d) shows the oxygen vacancy (V_O & 2V_O) in the Ba1−xEuxWO4 compound. It is well-recognized that while the GGA + U method helps in correcting the self-interaction errors associated with localized d and f states, it may still underestimate the bandgap. More recently, self-interaction-corrected (SIC) methods have been developed, which offer a significant improvement in the prediction of electronic properties at a lower computational cost compared to hybrid functionals. These methods have demonstrated better agreement with experimental bandgaps in complex systems, as reported in previous studies.32,33 However, the main focus of the present study is not on achieving the most accurate bandgap values but rather on understanding the trends in structural, electronic, and optical properties upon Eu doping in BaWO4. All theoretical calculations of Eu-doped BaWO4 (x = 2.08%) and oxygen vacancies (V_O & 2V_O) were carried out under the framework of Density Functional Theory (DFT). To study the electronic and optical properties of the Ba1−xEuxWO4 compound, we used the all-electron full potential linearized augmented plane wave (FP-LAPW) method as implemented in the WIEN2K simulation package software.34 To address the issue of underestimating the band gap of materials, including 3d, 4d, 5d, and 4f electrons within DFT, we employed the generalized gradient approximation + optimized effective Hubbard parameter (U) (GGA + U)35 to account for the d- and f-transition states. Here, we doped Eu (x = 2.08%) in BaWO4 and introduced oxygen deficiencies in Ba1−xEuxWO4, reporting a brief description of its photoluminescence and electronic properties using Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) within the framework of DFT. We also investigate the band gap of the Ba1−xEuxWO4 compound and gain deeper insight into its structural and optoelectronic properties. In this study, we focus on the three-dimensional structures of BaWO4, Ba1−xEuxWO4, and oxygen deficiency.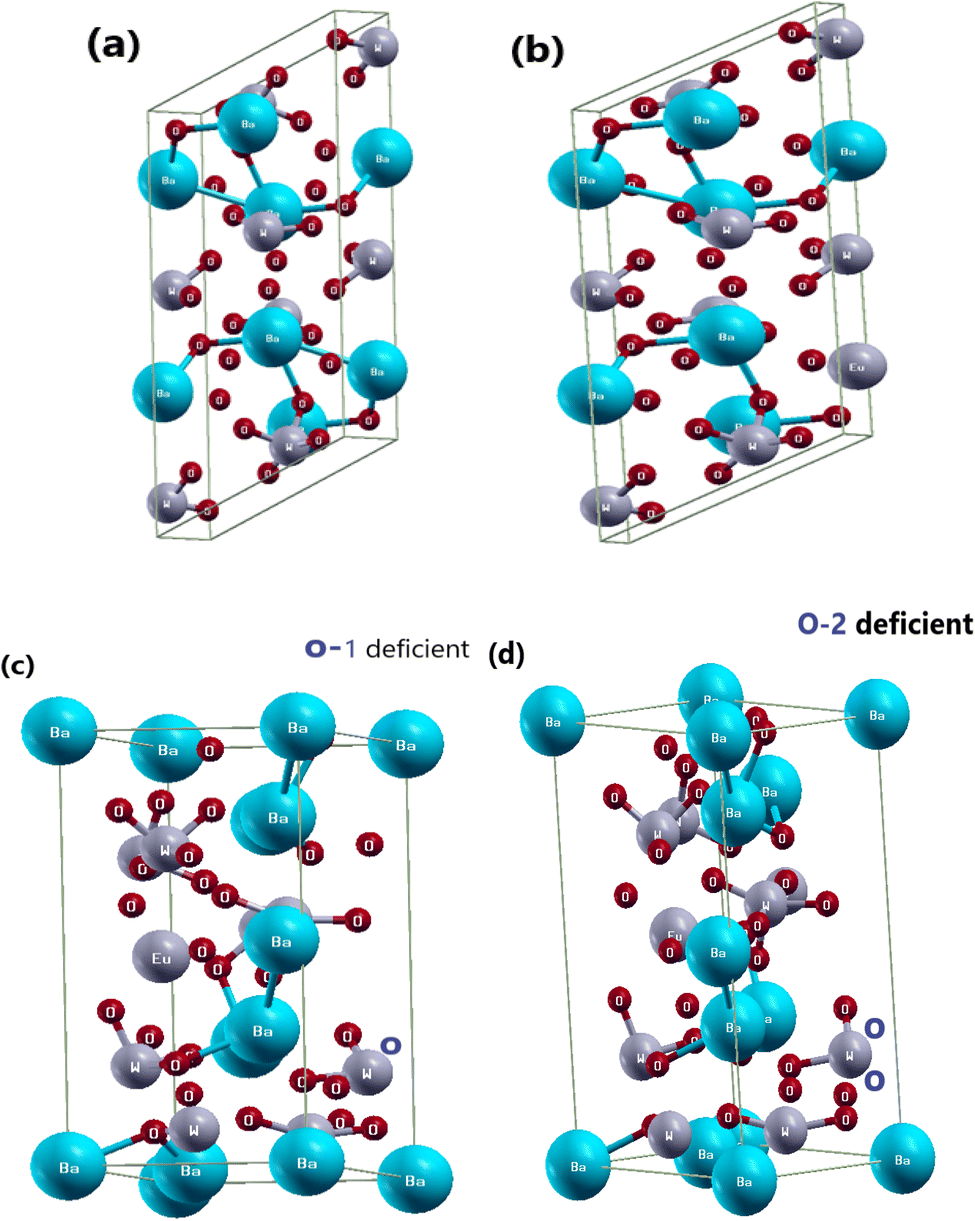 | ||
| Fig. 1 Optimized unit cell structure, (a) parental structure of BaWO4, (b) Eu-doped with Ba1−xEuxWO4 compound, (c) & (d) 1-oxygen atom deficient (V_O) & 2-oxygen atoms deficient (0–2) in Ba1−xEuxWO4 compound. | ||
2.2 Optimized energy and volume
The structural properties of BaWO4 are obtained through the volume optimization technique. The optimization procedure in WIEN2K is highly accurate, allowing the calculation of the structural properties of materials. Structural optimization, based on Murnaghan's equation, was performed to obtain a relaxed structure. In this procedure, the optimized energy is extracted from a particular optimized volume, and these data are fitted using Murnaghan's equation of state to derive structural parameters such as the minimum total energy (Eo), equilibrium volume (Vo), bulk modulus (B, GPa), and B′.The crystal structure and optimization curve of BaWO4, plotting energy (RY) versus volume (Å3), are shown in Fig. 2(b). In this figure, it can be seen that the maximum energy is −5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 348328.00 eV at a volume of 4000 Å3. This E–V curve indicates that as the volume increases, the energy decreases, and when the volume approaches 4900 Å3, a maximum decrease in energy is observed at −5348341.92 eV. At higher values of volume, a rapid increase in energy is observed. The optimized ground-state properties, such as Vo, B (GPa), B′, and the minimum energy (Eo), are shown in Table 1. Additionally, the charge neutrality in Eu-doped BaWO4 is achieved by the creation of O-vacancies. The volume optimization of Eu doping, along with oxygen vacancies (V_O & 2V_O), at different positions in BaWO4 was performed. The minimized energy of the most relaxed crystal structures of Eu doping and oxygen vacancies (V_O & 2V_O) in the lattice are shown in Fig. 1(b–d).
348328.00 eV at a volume of 4000 Å3. This E–V curve indicates that as the volume increases, the energy decreases, and when the volume approaches 4900 Å3, a maximum decrease in energy is observed at −5348341.92 eV. At higher values of volume, a rapid increase in energy is observed. The optimized ground-state properties, such as Vo, B (GPa), B′, and the minimum energy (Eo), are shown in Table 1. Additionally, the charge neutrality in Eu-doped BaWO4 is achieved by the creation of O-vacancies. The volume optimization of Eu doping, along with oxygen vacancies (V_O & 2V_O), at different positions in BaWO4 was performed. The minimized energy of the most relaxed crystal structures of Eu doping and oxygen vacancies (V_O & 2V_O) in the lattice are shown in Fig. 1(b–d).
 | ||
| Fig. 2 (a) Schematic of the single unit cell structure of BaWO4 (b) variation of total energies by volume for BaWO4 compound. | ||
| Current work | Volume (Vo) | Bulk modulus B (GPa) | BP | Minimum energy (Eo) |
|---|---|---|---|---|
| Generalized gradient approximation (GGA) + optimized effective Hubbard parameter (U) (GGA + U) | 4904.1699 | 165.8149 | 5.0000 | −5348341.72 eV |
Furthermore, the formation energy for the dynamic stability of Ba1−xEuxWO4 with Eu doping and oxygen vacancy (V_O) is calculated using the following equations.36
Where, ExEuxWO4 with Eu doping and oxygen vacancy (V_O) denotes the total energy of Ba1−xEuxWO4 with Eu doping and oxygen vacancy (V_O). ExBa, ExEu, ExW, ExO, and nxBa, nxEu, nxW, and nxO denote the energy and number of atoms of Ba, Eu, W, and O, respectively. The negative sign of formation energy confirms their stability; higher negativity indicates greater stability. Consequently, Oxygen vacancy (V_O) is more stable than Ba1−xEuxWO4.
2.3 Elastic properties of BaWO4 under the influence of oxygen vacancies and Eu doping
Elastic properties are essential in understanding the structural integrity and response of materials to external stress. These properties determine how materials behave under various loading conditions, influencing their suitability for specific applications. Barium tungstate (BaWO4), with its tetragonal scheelite structure, is a material of interest due to its promising optoelectronic and mechanical characteristics. Modifications such as oxygen vacancies and doping with europium (Eu) can significantly influence these elastic properties, affecting both mechanical stability and functional performance.The intrinsic elastic properties of a material are described by parameters like elastic constants (C11, C12, and C44), bulk modulus (B), shear modulus (G), and Young's modulus (E). These parameters are vital for applications where materials are exposed to mechanical stresses, such as in sensors, scintillators, and light-emitting devices. However, introducing oxygen vacancies and dopant atoms into BaWO4 can disrupt its lattice structure, leading to changes in bonding interactions and, consequently, in its elastic properties.
| Property | Pristine BaWO4 | BaWO4 with oxygen vacancies | Eu-doped BaWO4 |
|---|---|---|---|
| C11 (GPa) | 71 | 64 | 62 |
| C12 (GPa) | 52 | 49 | 42 |
| C44 (GPa) | 100 | 91 | 44 |
| Bulk modulus (B, GPa) | 98 | 87 | 82 |
| Young's modulus (E, GPa) | 144 | 131 | 125 |
| Poisson's ratio (ν) | 0.27 | 0.29 | 0.32 |
| Hardness (H, GPa) | 5.0 | 4.5 | 4.0 |
For future advancements, balancing mechanical and functional properties is essential. Strategies such as co-doping or advanced synthesis methods can mitigate the drawbacks associated with reduced elastic moduli while further enhancing the material's performance. Computational modeling and experimental validation can provide insights into optimal doping concentrations and defect configurations to achieve this balance. The elastic properties of BaWO4, including its elastic constants, bulk modulus, shear modulus, and Poisson's ratio, are significantly influenced by the introduction of oxygen vacancies and Eu doping. These modifications result in a predictable reduction in stiffness and rigidity but enhance the material's optoelectronic functionality. Understanding and optimizing these trade-offs are critical for tailoring BaWO4 for specific applications, particularly in the fields of photonics and energy storage.
2.4 Electronic band structures
The investigation explores Janus MnSeTe and MnSTe electronic and magnetic characteristics under biaxial strain conditions while also studying doping effects on spin polarization and magnetic anisotropy which results in spin gapless semiconductivity when strain is compressive.37 Bilayers of Janus FeClF exhibit crucial valleytronic application features that include both bipolar magnetic semiconducting properties and anomalous valley Hall effects.38 Strain and doping elements transform Janus FeClF into an optimal material for spintronic technologies because they boost the Curie temperature together with perpendicular magnetic anisotropy.39To gain a better understanding of the relationship between band structures and optical properties of the investigated compound BaWO4 with Eu doping (x = 2.08%) and oxygen deficiency, Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) has been used within the framework of DFT. The electronic band structure analysis suggests that the band gaps for indirect-band-gap semiconductors in Ba1−xEuxWO4 with Eu doping (x = 2.08%) and oxygen vacancy (V_O) are slightly smaller than those determined from the absorption spectrum.40 Recent findings emphasize the potential of these indirect-band-gap semiconductors for photocatalytic applications due to their lower radiative recombination rate.41 Furthermore, observations of flat bands within the conduction band (CB) indicate the presence of a second phase, possibly explaining the emergence of superconductivity.42–49 This insight underscores the potential of Ba1−xEuxWO4 with Eu doping (x = 2.08%) and oxygen vacancy (V_O) for advanced applications in ultra-powerful magnet technology. Despite the observable range of the bands, their broadening in the presence of oxygen vacancy (V_O) further enhances their suitability for advanced applications. Traditionally, solar cells utilize silicon (Si) and germanium (Ge) due to their abundance, self-passivation, and indirect band-gap nature. The operation of an indirect band-gap semiconductor in a solar cell involves both a photon and a phonon, inducing changes in momentum and energy during the transition from the valence band (VB) to the conduction band (CB). The inverse relationship between minority carrier diffusion length and absorber depth highlights the importance of having a sufficiently thick absorbing layer for efficient solar cells.50
Despite the lower light absorption characteristic of indirect semiconductors, their advantages, including longer recombination lifetimes and larger diffusion lengths, make them suitable for efficient solar cells. Indirect band-gap semiconductors find applications in thin-film solar cells due to their weaker light absorption, enabling a broader photon energy range, particularly accommodating lower-energy solar photons.51–53 Additionally, their defect tolerance and reduced heat generation make them crucial for durable solar cells, especially in concentrator photovoltaic systems. Tandem solar cells, incorporating materials with different band gaps, further optimize energy conversion across the solar spectrum, leveraging the benefits of indirect-band-gap materials.54–56
In the context of double perovskite halides, the term effective mass of charge carriers plays a crucial role in understanding the behavior of electrons or holes. The effective mass varies depending on the material and its composition, influenced by the band structure and the types of phosphors and elements present in LED materials. The calculation of the effective mass of charge carriers (electrons or holes) in Ba1−xEuxWO4 with Eu doping (x = 2.08%) and oxygen vacancy (V_O) at high symmetry points of VBM and CBM involves the use of the E–K dispersion curve. The formula utilized for determining the effective mass is as follows:
| m* = (ℏ2/(d2E/dK2)) |
The electronic structure of BaWO4 with Eu doping (2.08%) and oxygen vacancy along high symmetry direction points of the Brillouin Zone, such as Γ, X, and M, with energy plotted in electron volts on the y-axis for the spin-up and spin-down states, is depicted in Fig. 3 (a–d), respectively. The band gap of the parent compound BaWO4 is 4.885 eV. It is evident that in the Ba1−xEuxWO4 compound with Eu doping (x = 2.08%), the spin-up and spin-down configuration graphs show that the maxima of the valence band and the minima of the conduction band do not lie on the same symmetric line, indicating that it is a semiconductor material with an indirect band gap, as shown in Fig. 3(a & b). The band gap energy is 2.8 eV for spin-up and 3.0 eV for spin-down states. From the band structures of the Eu-doped compound, we observe that the Fermi level shifts towards the valence band, which clearly indicates that the material is a p-type semiconductor, as depicted in Fig. 3(a & b).
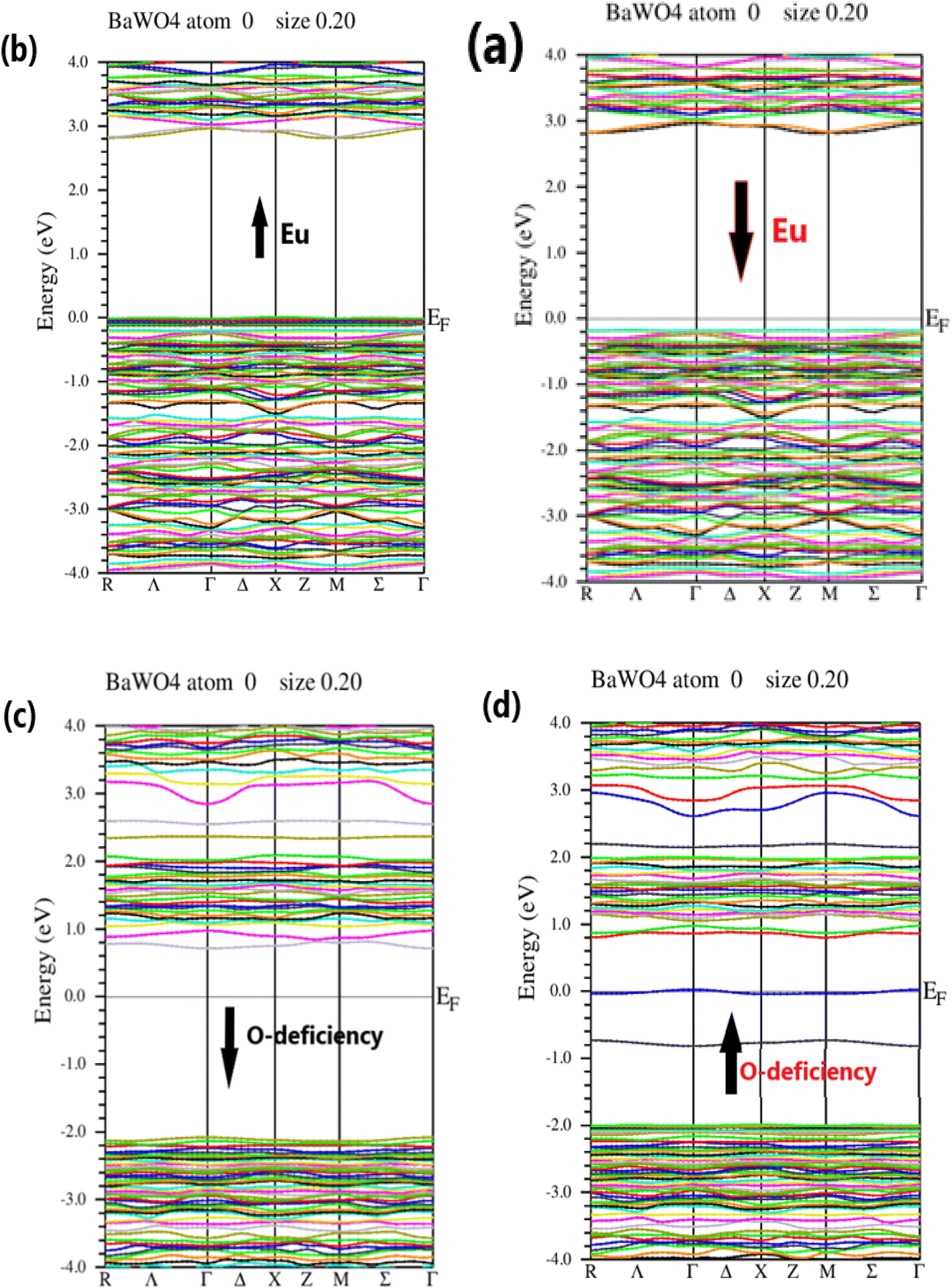 | ||
| Fig. 3 Energy Bandgap Structures, (a) & (b) represent the Ba1−xEuxWO4 with Eu doped (x = 2.08%) for spin up state and spin down state, (c) & (d) represent the oxygen vacancy (V_O) in BaWO4 for spin up and spin down state along symmetry direction in Brillouin Zone. | ||
Fig. 3(c & d) show the electronic band structure of the oxygen vacancy (V_O) in the BaWO4 compound for spin-up and spin-down states. This figure indicates that the maxima of the valence band and the minima of the conduction band lie on the same symmetric line, showing that it is a semiconductor material with a direct band gap. The band gap values for the spin-up and spin-down channels are 0.8 eV and 2.6 eV, respectively. It can also be observed that the Fermi level is slightly shifted towards the conduction band, indicating that the material is an n-type semiconductor for both spin-up and spin-down states. In the electronic band structure of oxygen deficiency, it is evident that there is a reduction in the band gap compared to the Eu-doped electronic band structure. The findings from the calculated band structures of the Eu-doped compound reveal not only a reduction in the band gap but also the formation of an intermediate band between the valence band (VB) and conduction band (CB) due to the doping of Eu into the host compound BaWO4. As the concentration of Eu atoms increases, the intermediate band also increases, and the fundamental band gap decreases. Consequently, this reduces the optical threshold, enhances the optical transition, and shifts it to the visible region, proving that Eu-doped BaWO4 is a promising material for optical devices.
2.5 Density of states
Semiconducting materials demonstrate the typical electronic band structure, determined by the contribution of different electronic states in the conduction band and valence band. To study the changes in the electronic band structure, we calculated and plotted the total density of states (TDOS), elemental density of states (ETDOS), and partial density of states (PDOS) for the parent compound BaWO4, Ba1−xEuxWO4 with Eu doping (x = 2.08%), and oxygen vacancy (V_O). The density of states (DOS) helps us understand the band energy nature and electronic parameters, such as the particular atomic and orbital origins of the material. To resolve the inconsistency between the minority and majority spin-up and spin-down states, the total density of states for the Ba1−xEuxWO4 compound with Eu doping (x = 2.08%) has been computed using the Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) approximation under the framework of Density Functional Theory (DFT). We also calculated the elemental total density of states (ETDOS) and partial density of states (PDOS) for the parent material BaWO4, Eu-doped (x = 2.08%), and oxygen-deficient materials depicted in Fig. 4 and 5. Partial density of states (PDOS) describes the contribution of different orbital states in both valence and conduction bands. The zero-energy level is known as the Fermi energy level. The region on the right side with positive energy values represents the conduction band, while the region on the left side with negative energy values represents the valence band. The graph below shows the spin-up and spin-down symmetries of the concerned material. In Fig. 4(a), the total density of states (TDOS) for the parent material BaWO4 reveals the maximum contribution near the Fermi level in the valence band, ranging from −5 eV to 0 eV. In the conduction band, the maximum contribution is observed in the region ranging from 10 eV to 12 eV. Fig. 4(b) represents the graphical representation of elemental total density of states (ETDOS) energy, ranging from −12 eV to 12 eV. It has been observed that in the valence band, the maximum contribution of oxygen near the Fermi level ranges from −4 eV to 0 eV, with a small contribution from tungstate (W) in the region from −5 eV to 0 eV. Also, a high contribution from barium (Ba) is observed in the region from −11 eV to −10 eV in the lower valence band. Fig. 4(c) is the graphical representation of orbital contributions from Ba (s-state & p-state), tungstate (s-state & p-state), and oxygen (s-state & p-states). Fig. 4(d) represents the graphical contributions of Ba (d-state) and tungstate (d-state & f-state) transition atoms in the valence and conduction bands of the compounds BaWO4 under investigation. Here, partial DOS was computed using the Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) approximation for strongly correlated d-states and f-states of Ba and W in energy ranges from −12 eV to 12 eV. In Fig. 4(c), the valence band maximum contribution of the O-p state and the very small contribution of the O-s state can be seen near the Fermi level in the energy range from −4 eV to 0 eV, while the large contribution of the Ba-p state is observed in the energy range from −11 eV to −10 eV in the lower valence bands. In the conduction band, energy ranges from 0 eV to 4 eV, with a very small contribution from the O-p state. In Fig. 4(d), in the valence band, the major contribution of the W-d state is observed near the Fermi level in the energy range from −4 eV to 0 eV. In the conduction band, energy ranges from 3 eV to 4 eV with a maximum contribution from the W-d state, and the maximum contribution from Ba-d is seen in the energy range from 3 eV to 8 eV. | ||
| Fig. 4 (a) Calculated total density of states, (b) the elemental resolved density of states, and (c & d) the orbital resolved density of states for BaWO4. | ||
 | ||
| Fig. 5 Density of states (states per eV per unit cell vs. energy (eV)) for the Ba1−xEuxWO4 material with Eu doping (x = 2.08%). The figure includes (a) and (b) showing the Elemental Density of States (EDOS), elemental TDOS for Ba, W, O, and Eu, and the Partial Density of States (PDOS) for both spin-up and spin-down states. | ||
In Fig. 5(b), the elemental density of states (EDOS) for Ba1−xEuxWO4 with Eu doping (x = 2.08%) for spin-up and spin-down states reveals that the maximum contribution near the Fermi level in the valence band occurs in the energy range from 0 eV to −1 eV. Furthermore, in the conduction band, the maximum contribution is observed in the energy range from 5 eV to 9 eV. Fig. 5(b) represents the elemental density of states (EDOS), ranging from −4 eV to 12 eV. It can be observed that in the valence band, the maximum contribution of oxygen for both spin-up and spin-down channels is near the Fermi level, while Eu contributes only to the spin-up channel and is completely absent in the spin-down channel. Tungstate (W) also contributes to the spin-up channel near the Fermi level. Here, it can be observed that the Eu total contribution is entirely absent in the spin-down state in the valence band, indicating that there is no mirror symmetry. Additionally, the elemental density of states (EDOS) in the conduction band shows a maximum contribution from W and a very small contribution from Eu and O near the Fermi level. In the energy range from 5 eV to 9 eV, Eu and W show strong hybridization for both spin-up and spin-down states.
Fig. 5(c) presents the graphical representation of the orbital contributions from Ba (d-state), tungstate (d-state & f-state), and Eu (d-state and f-states) transition atoms in the valence and conduction bands. Here, partial DOS was computed using the Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) approximation for strongly correlated d-states and f-states of Ba, W, and Eu for both spin-up and spin-down states in energy ranges from −4 eV to 12 eV. In the valence band, the maximum contribution of the Eu-f state and a very small contribution of the Eu-d state for the spin-up channel can be seen near the Fermi level in the energy region from −1 eV to 0 eV, while a large contribution from the W-d state is observed in the energy range from −4 eV to −3 eV for both spin-up and spin-down channels. In the conduction band, the major contribution from the W-d state for both spin-up and spin-down states is observed near the Fermi level. The Eu-d state is slightly present in both channels of the conduction band. In the energy range from −0.6 eV to 8.5 eV, the Ba-d state is the maximum contributor for the spin-up state, and the Eu-d orbital shows a major contribution for both channels. In this energy range, Ba-d, W-d, and Eu-d states show strong hybridization. The maximum peaks of Ba-d for the spin-up state and Eu-d for both the spin-up and spin-down states can be observed. It can be noted that there is no mirror symmetry, and some magnetic moment exists in both spin-up and spin-down states. The magnetic moment increases with the concentration of Eu doping (i.e., Eu1: 7.00005, Eu2: 14.00010). On the other hand, the creation of oxygen vacancy also increases the magnetic moment: as shown in Table 3: for V_O deficiency, the magnetic moment is 9.00029, and for 2V_O deficiency, it is 11.00057.
| Eu = 2.08% | Eu = 4.16% | Oxygen deficiency (V_O) | Oxygen deficiency (2V_O) | Eu = 2.08% | Eu = 4.16% | Oxygen deficiency (V_O) | Oxygen deficiency (2V_O) | ||
|---|---|---|---|---|---|---|---|---|---|
| μINT | 0.10175 | 0.20387 | 1.24118 | 2.37775 | μO27 | −0.00286 | −0.00287 | 0.04135 | −0.00432 |
| μBa1 | −0.00001 | 0.00814 | 0.01441 | μO28 | 0.00000 | −0.00157 | 0.00108 | −0.00125 | |
| μEu1 | 6.92971 | μO29 | −0.00016 | −0.00485 | −0.00444 | 0.02033 | |||
| μBa2 | 0.00000 | 0.00004 | 0.00636 | 0.01440 | μO30 | −0.00469 | −0.00485 | −0.00150 | 0.01667 |
| μBa3 | 0.00004 | 0.00004 | 0.00013 | 0.00017 | μO31 | −0.00156 | −0.00157 | 0.01953 | 0.00328 |
| μBa4 | −0.00000 | 0.00004 | 0.00708 | 0.00749 | μO32 | −0.00000 | −0.00017 | −0.00346 | 0.00336 |
| μBa5 | −0.00000 | 0.00015 | 0.00060 | 0.00751 | μO33 | −0.00358 | −0.00381 | 0.00301 | 0.00223 |
| μBa6 | 0.00015 | 0.00015 | 0.00389 | 0.00383 | μO34 | −0.00024 | −0.00381 | 0.00037 | 0.00198 |
| μBa7 | 0.00004 | 0.00004 | 0.00004 | 0.00370 | μO35 | −0.00016 | −0.00017 | 0.00249 | −0.00094 |
| μW8 | 0.00001 | −0.00037 | 0.68261 | 0.69100 | μO36 | 0.00000 | −0.00004 | 0.00002 | 0.00025 |
| μW9 | 0.00002 | −0.00038 | 0.00139 | 0.69155 | μO37 | −0.00013 | −0.00141 | −0.00080 | −0.01109 |
| μW10 | −0.00039 | −0.00038 | 0.01346 | 0.01265 | μO38 | −0.00127 | −0.00141 | −0.00011 | −0.01187 |
| μW11 | −0.00038 | −0.00037 | −0.00024 | 0.01210 | μO39 | −0.00004 | −0.00004 | 0.00462 | −0.00020 |
| μW12 | 0.00000 | 0.00031 | 0.04772 | 0.04751 | μO40 | −0.00006 | −0.00409 | −0.00496 | −0.00449 |
| μW13 | 0.00012 | 0.00014 | 0.00040 | 0.04720 | μO41 | −0.00015 | −0.00006 | −0.00786 | |
| μW14 | 0.00002 | 0.00014 | 0.00892 | 0.00796 | μEu41 | 6.92979 | |||
| μW15 | 0.00031 | 0.00031 | 0.00063 | 0.00832 | μO42 | 0.00009 | −0.00006 | −0.00774 | |
| μO16 | −0.00002 | −0.00258 | −0.00030 | 0.00392 | μEu42 | 9.00029 | |||
| μO17 | −0.00007 | −0.00082 | 0.00380 | 0.00509 | μO43 | −0.00402 | −0.00409 | −0.00195 | 0.00640 |
| μO18 | −0.00075 | −0.00082 | −0.00249 | 0.00489 | μO44 | −0.00012 | −0.00521 | 0.00003 | 0.00104 |
| μO19 | −0.00254 | −0.00258 | 0.00499 | 0.00005 | μO45 | −0.00018 | −0.00017 | −0.00020 | |
| μO20 | 0.00000 | −0.00351 | −0.00016 | −0.00341 | μO46 | 0.00001 | −0.00017 | −0.00400 | |
| μO21 | −0.00009 | −0.00008 | −0.00006 | 0.00209 | μO47 | −0.00510 | −0.00521 | −0.00604 | |
| μO22 | 0.00002 | −0.00008 | −0.00339 | 0.00166 | μO48 | 0.00006 | |||
| μO23 | −0.00350 | −0.00351 | 0.00217 | 0.00143 | μEu48 | 6.92984 | 6.92971 | ||
| μO24 | 0.00000 | −0.00287 | −0.00035 | −0.00134 | μTOT | 7.00005 | 14.00010 | 9.00029 | 11.00057 |
| μO25 | −0.00030 | −0.00030 | 0.00168 | 0.05032 | |||||
| μO26 | 0.00000 | −0.00030 | −0.00283 | 0.05011 |
One can observe that after Eu doping, the properties of the material are altered. In both spin-up and spin-down states, the valence and conduction regions are displaced from the Fermi level, indicating that the material behaves as a semiconductor.
3 Optical properties
The study of optical properties is essential for materials to be used in optoelectronic devices such as LEDs, solar cells, and other applications. To analyze the behavior of compounds for optical applications, a number of in-depth and comprehensive analyses have been conducted to determine their optical properties. These optical properties include dielectric function dispersion ε(ω), reflectivity R(ω), refractive index n(ω), absorption coefficient I(ω), and optical energy loss L(ω).3.1 Dielectric function dispersion
Optical properties play a crucial role in understanding the nature of the material Ba1−xEuxWO4 with Eu doping (x = 2.08%) and oxygen vacancies (V_O). The optical properties of a material represent the frequency response of its optical parameters, such as energy band structures, lattice vibrations, impurity levels, and magnetic excitations. Energy band structures and the dielectric dispersion function are interdependent parameters. Both interband and intraband transitions are important for defining the optical response of the material. Generally, intraband transitions are related to metals, while interband transitions, such as direct and indirect transitions, are associated with semiconductor materials. These transitions are highly sensitive to electromagnetic (EM) interactions. In our calculations, scattering phonons contribute to interband transitions (indirect), which are responsible for the emission of electromagnetic interactions. The dielectric constant ε(ω), reflectivity R(ω), refractive index n(ω), absorption coefficient I(ω), and optical energy loss L(ω) are commonly used to analyze the optical response. The complex optical susceptibility dispersion ε(ω) consists of two parts: the real part (Re(ω)) and the imaginary part (Im(ω)), which describe the photon interaction (E = hv) with electrons and show the material's response.The absorption coefficient I(ω) and refractive index n(ω) are associated with dispersed dielectric functions, ε1(ω) and ε2(ω) through Kramer kronig relation i.e.

| ε(ω) = ε1(ω) + iε2(ω) |
Dispersed dielectric function ε(ω) includes both real ε1(ω) and imaginary parts ε2(ω).
| ε1(ω) = n2 − k2 |
Equation describes the real part of the permittivity, where ε1(ω) represents the material's response to an electric field at frequency ω. Here, n is the refractive index, indicating the material's light bending ability, and k is the extinction coefficient, which measures how much light is absorbed by the material. This equation links the material's optical properties, such as refraction and absorption, to its overall electromagnetic response. The real part (Re(ω)) explains the dispersion and polarization when impinging light falls on these materials.
While the imaginary part (Im(ω)) represents the absorption of light, energy loss, and the sum of all transitions between occupied and unoccupied states. It also involves the calculations of the electronic wave function and energy eigenvalues.
| ε2(ω) = 2nk |
Equation represents the imaginary part of the complex permittivity, where ε2(ω) describes the material's absorption response at frequency ω. In this equation, n is the refractive index, indicating how much light is refracted in the material, and k is the extinction coefficient, which quantifies the material's absorption of light. This equation links the material's absorption characteristics to both its refractive index and extinction coefficient, crucial for understanding energy dissipation in optical materials.
3.2 Absorption coefficient I(ω)
The absorption coefficient explains the penetration of light into the material before it is absorbed (or fully immersed into the material). Electronic transport cannot occur from the valence energy band to the conduction energy band below the band gap of the incident energy. Fig. 6 depicts the relationship between the absorption coefficient and energy (eV) for the parent structure BaWO4, oxygen-deficient, and Ba1−xEuxWO4 compounds with Eu doping (x = 2.08%).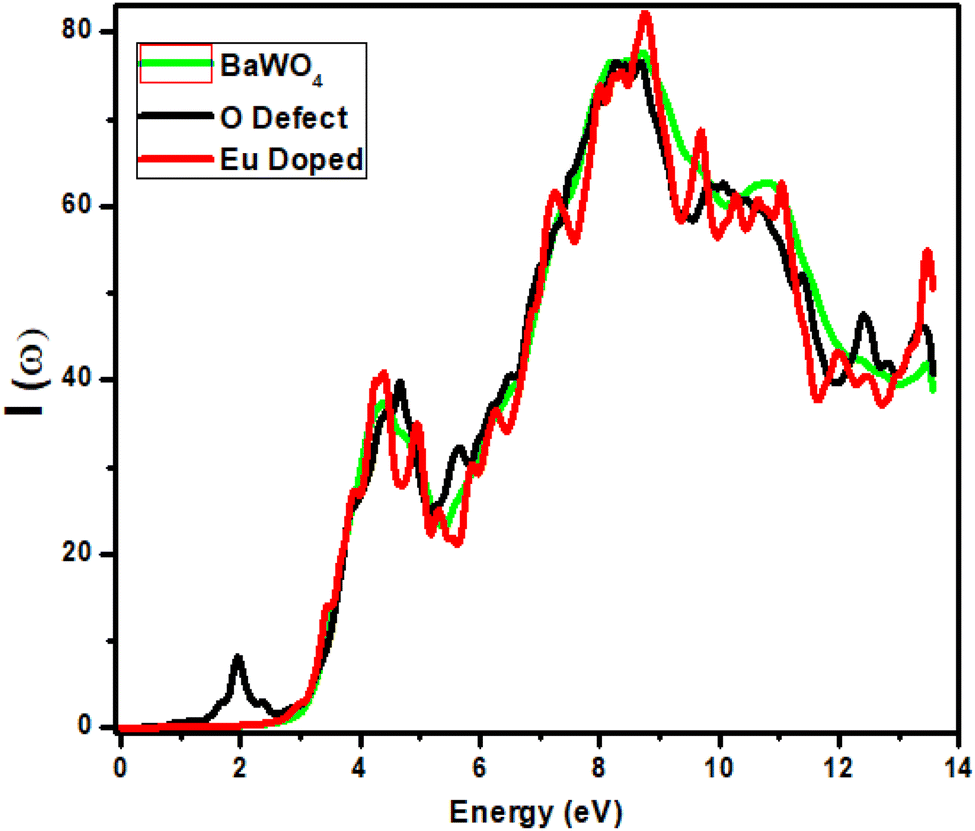 | ||
| Fig. 6 Calculated absorption coefficient I(ω) of Ba1−xEuxWO4 with Eu-doped (x = 2.08%) and oxygen vacancy (V_O). | ||
For BaWO4 and Ba1−xEuxWO4 compounds, the static graph is observed from 0–2.8 eV. We can see that no absorption occurs in the infrared (IR) region. Absorption begins in the visible region for the parent compound BaWO4 and the doped compound Ba1−xEuxWO4 after 2.8 eV, with the first peak observed at 4.5 eV, showing a small peak difference. For Eu-doped, the maximum peak value is observed at 9 eV in the ultraviolet region, where the absorption coefficient is 81%. For oxygen-deficient material, a static region (with no variation) is observed up to 1.5 eV, and the first peak appears in the visible region at 2 eV, where the absorption coefficient is 8%, which reduces the band gap and is beneficial for optoelectronic devices. For the parent material BaWO4 and the oxygen-deficient material, the maximum peak is observed at the same energy value 8.2 eV in the ultraviolet region, where the corresponding absorption coefficient is 75%, due to the forbidden band gap between the valence bands and conduction bands. All materials exhibit maximum absorption in the ultraviolet region. After the energy value of 8.2 eV, a decrease is observed in all graphs, with small fluctuations.
It can be seen that the band gap is reduced by 0.8 eV in the oxygen-deficient material, compared to the parent material BaWO4 and Eu-doped (x = 2.08%) Ba1−xEuxWO4 compound (2.8 eV). The absorption coefficient I(ω) is formulated as:
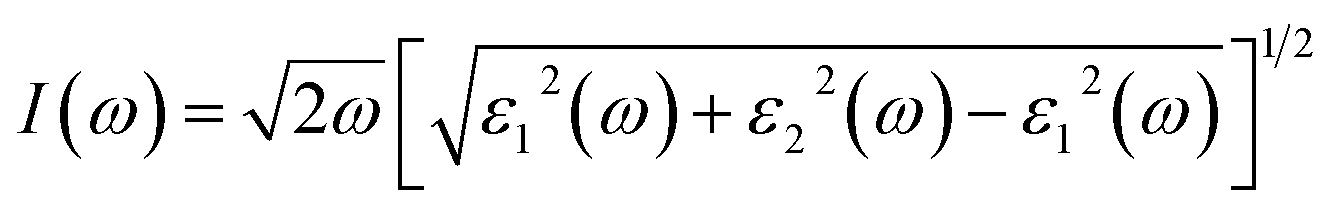
3.3 Energy loss function L(ω)
The interaction of fast-moving electrons with material/matter results in energy loss of moving particles, given by the energy loss function L(ω). Fig. 7 shows the energy loss for the oxygen-deficient material shows the first peak at 2 eV in the visible region. The same material exhibits peaks at 4.8 eV, 9 eV, and 11.6 eV, and suddenly increases at an energy value of 13 eV. The major peaks for the parent material BaWO4 have been observed at 5 eV, and 11.8 eV, with a sudden increase at the energy value 13 eV. The major peaks observed for Eu-doped material are at 2 eV, 5 eV, 9.2 eV, and 11.9 eV, with a sudden increase at 13 eV. The overall graphs show that energy loss increases at higher energy values, and it is greater for the parent and oxygen-deficient materials as compared to the Eu-doped material. In the visible region, the maximum loss can be seen in the case of oxygen, where the maximum absorption is observed, indicating that oxygen is suitable for optoelectronic applications. | ||
| Fig. 7 Calculated energy loss L(ω) of Ba1−xEuxWO4 with Eu-doped (x = 2.08%) and oxygen vacancy (V_O). | ||
The relation of energy loss function L(ω) is

3.4 Real and imaginary part dielectric function dispersion
The real part (Re(ω)) shows the refractive index of the material and describes the dispersion behavior of light. The real value (Re(ω)) of dispersed optical susceptibility is shown in Fig. 8(a). The dielectric function of the compound Ba1−xEuxWO4 with Eu doping (x = 2.08%) and oxygen vacancy (V_O) is shown in Fig. 8(a) and (b). The real part of the dielectric constant (Re(ω)) first experiences a dramatic increase until it reaches its maximum value, after which it begins to decrease. The graph shows the band gap energy between 0–14 eV. The static values for BaWO4 and Eu-doped are 2.4, and for the O-defect are 2.7. As the static value of the O-defect is greater than the other two, it indicates that the O-defect is better suited for optoelectronic devices. The higher value of the dielectric constant of the O-defect shows that it has a smaller band gap, which further favors it over other materials for optoelectronic devices. The main peak is observed in the energy region from 2 eV to 4 eV. For Eu-doped, it is 3.1, for BaWO4 it is 3.2, and for the O-defect, it is 3.3, respectively. With a further increase in energy, the value of the real part (Re(ω)) decreases and enters the negative region at 8.9 eV, where the material shows metallic behavior.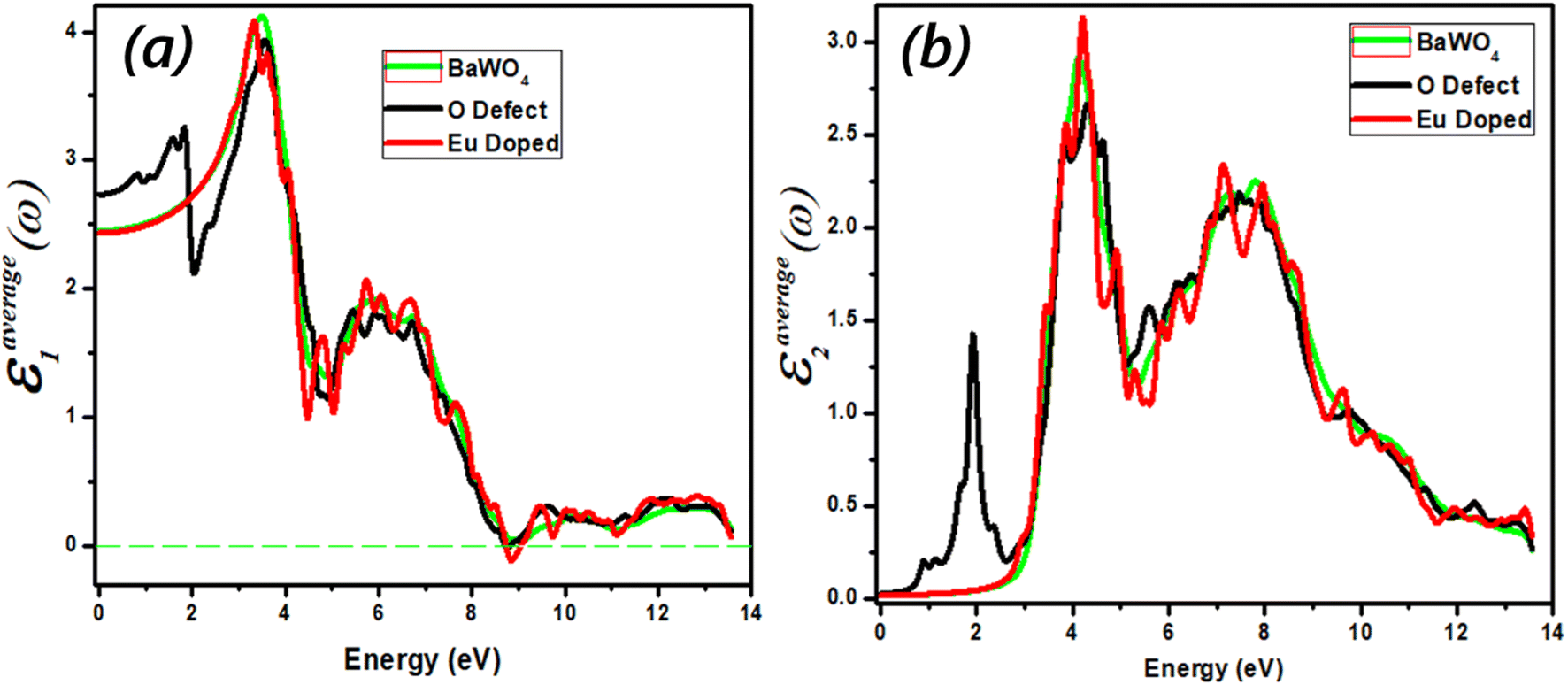 | ||
| Fig. 8 (a and b) Calculated optical spectra of Ba1−xEuxWO4 with Eu-doped (x = 2.08%) and oxygen vacancy (V_O) (a) the real part ε1(ω) of the dielectric function, (b) the imaginary part ε2(ω) of the dielectric function. | ||
The dielectric behavior of Ba1−xEuxWO4 is slightly enhanced compared to oxygen vacancy (V_O). In the analysis of the compounds, there is a minimal variation in the energy loss function (L(ω)) within the range of −1.16 to −2 for energies greater than 10 eV. The negative values of (L(ω)) in this energy range indicate the metallic character of the compounds. Notably, the substitution of Ba1−xEuxWO4 with oxygen vacancy (V_O) results in increased absorption peak intensities, shifting them to a lower energy region. This shift is attributed to the greater electronic influence of O compared to Ba1−xEuxWO4 with Eu doping (x = 2.08%) and oxygen vacancy (V_O).
The relationship between static polarization and band gap follows Penn's model:55

 quantifies the influence of the charge carrier's oscillations relative to the energy gap, affecting the material's dielectric response at low frequencies. This equation is particularly useful for modeling the optical properties of materials, especially semiconductors, under different conditions of frequency. As energy increases, the real part of the dielectric function becomes positive again, indicating the metallic nature of the material in the plasmon region. Plasmon oscillations arise from interband transitions and are associated with the dispersive part of the material in the optical spectra, causing electromagnetic waves to reflect back.
quantifies the influence of the charge carrier's oscillations relative to the energy gap, affecting the material's dielectric response at low frequencies. This equation is particularly useful for modeling the optical properties of materials, especially semiconductors, under different conditions of frequency. As energy increases, the real part of the dielectric function becomes positive again, indicating the metallic nature of the material in the plasmon region. Plasmon oscillations arise from interband transitions and are associated with the dispersive part of the material in the optical spectra, causing electromagnetic waves to reflect back.
The imaginary part (Im(ω)) provides insights into the absorption coefficient of the material. It is crucial as it determines the maximum absorption area and governs interband transitions within the materials used for device fabrication. Due to the limitations of Density Functional Theory (DFT), the transition points (from the valence band to the conduction band) exhibit a slight deviation from the band structure and are illustrated in Fig. 8(b). The maximum value of (Im(ω)), known as the first absorption peak (FAP), corresponds to electronic transitions at the Fermi level. Fig. 8(b) represents the imaginary part of dispersed optical susceptibility (Im(ω)). Absorption starts in the IR region, and we observe a distinct main peak for oxygen-deficient material at 1.8 eV, with the corresponding dispersion value of 1.4, whereas there is no change in the behavior of other dopant materials. From 0 to 2.8 eV, no transition occurs for the Eu-doped material and the parent material BaWO4, due to the incident photon having less energy than the bandgap energy; in this region, no electron moves from the valence band to the conduction band. For the parent material BaWO4 and doped material, absorption starts after 2.8 eV in the visible region, and the maximum absorption is observed at 4 eV in the UV region. Maximum absorption is observed in the visible region for oxygen-deficient material.
3.5 Reflectivity Ravg(ω) spectral
Reflectivity (R(ω)) versus photon energy gap (eV) is shown in Fig. 9. A constant value of reflectivity (R(ω)) has been observed in the energy range 0–1 eV in the infrared region for both the parent and Eu-doped materials, whereas for the oxygen-deficient material, it has been observed at 0.6 eV. The first peak for the oxygen-deficient material occurs in the visible region at 1.9 eV, known as the lossless region. For the parent material, the maximum reflectivity of 0.17 has been observed at an energy of 8.7 eV. For Eu-doped material, the maximum reflectivity of 0.20 is observed at an energy value of 8.85 eV, and for the oxygen-deficient (V_O) material, a high peak is observed at an energy value of 8.7 eV in the ultraviolet region, where the corresponding R(ω) is 0.18. A remarkable increase in reflectivity was observed for all materials after 5–9 eV and 13 eV, with small fluctuations associated with the indirect transition. A smaller value of reflectivity is observed in the visible region for the oxygen-deficient material at 2 eV, which suggests that the oxygen-deficient material is suitable for optoelectronics, due to the reduced bandgap of oxygen compared to Eu-doped materials. All materials show maximum reflectivity in the ultraviolet (UV) region; however, this reflectivity is insufficient for use in materials designed for shielding.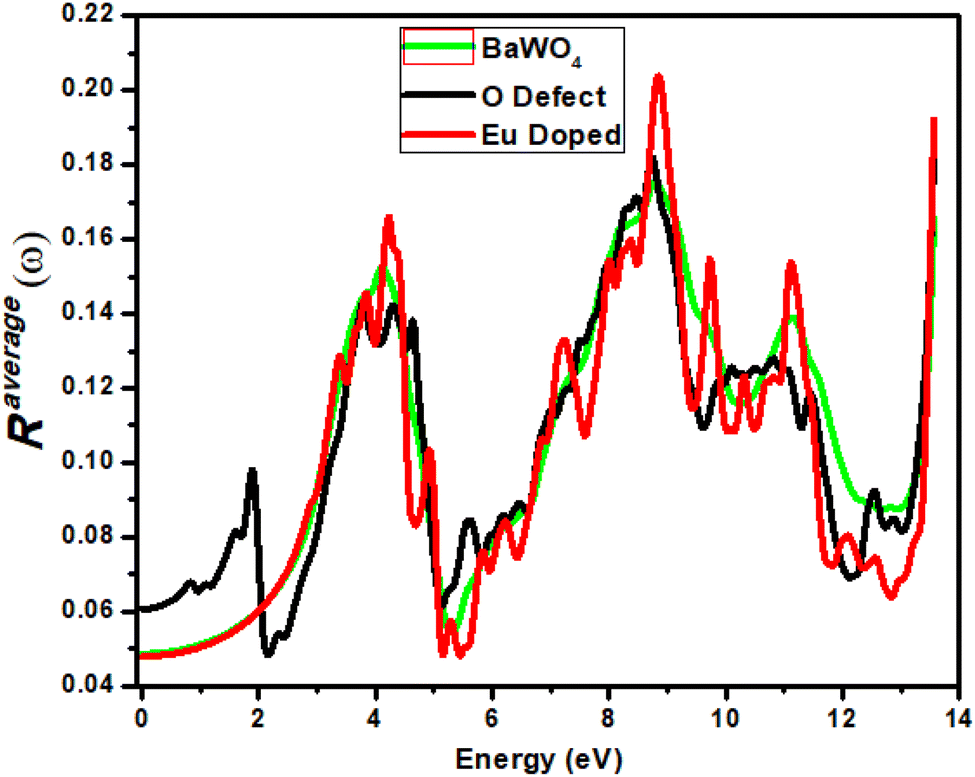 | ||
| Fig. 9 Calculated reflectivity Ravg(ω) of Ba1−xEuxWO4 with Eu-doped (x = 2.08%) and oxygen vacancy (V_O). | ||
Reflectivity Ravg(ω) is calculated by using the following eq.
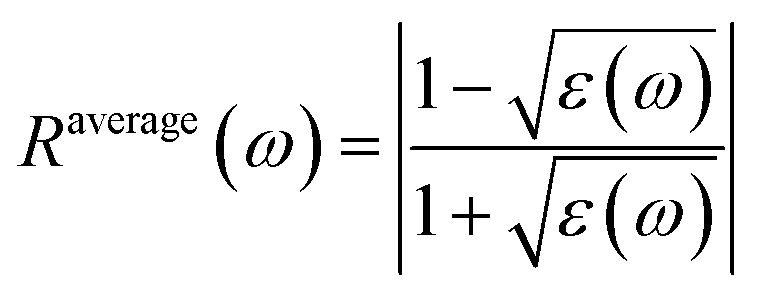
3.6 Refractive index n(ω)
The graph below shows the refractive index n(ω) in the energy range 0–14 eV. For the parent and Eu-doped materials, the static region lies between the energy ranges 0–1.7 eV in the infrared region. The static value of the refractive index n(ω) for oxygen-deficient material is 1.65, and for the parent compound BaWO4 and Eu-doped compound, it is 1.59. Fig. 10 shows the parent material graph reaches a maximum value of n = 2.1 at 3 eV. A decline in the graph for the parent and Eu-doped materials is observed at 5.2 eV. However, in the visible region, a sudden rise in the refractive index is observed from photon energies ∼2.1 eV, with an increase in the energy value. The maximum peak is observed at n = 2 at photon energy ∼3.5 eV in the ultraviolet region for both oxygen-deficient (V_O) and Eu-doped materials. A sudden decline in the refractive index is observed until it reaches 5.2 eV for both oxygen-deficient (V_O) and Eu-doped materials. From photon energies 5.2 eV to 7.2 eV, the refractive index increases with small fluctuations for the parent material, oxygen-deficient, and Eu-doped materials. The fluctuations occur due to various inner band transitions. Above 7.2 eV, the refractive index decreases for all materials. One can see that the graph of the oxygen-deficient refractive index is constant with small fluctuations, but in the visible region, a sudden decrease is observed in their values from 2 eV to 2.1 eV. | ||
| Fig. 10 Calculated the refractive index of Ba1−xEuxWO4 with Eu-doped (x = 2.08%) and oxygen vacancy (V_O). | ||
The optical activities are considered suitable for n(ω) values ranging from 2.0 to 4.0,51 and the materials under investigation fall within this range. The observation of refractive index values exceeding 1 strongly supports the semiconducting nature of these materials. Additionally, the refractive index provides insight into bond strength, with values above unity indicating stronger covalent bonding compared to ionic interactions. The presence of covalent bonds, whether static or dynamic, in these materials is attributed to photon deceleration and an increase in electron density.52
It can be expressed by means of the following relation

4 Conclusion
We have reported the theoretical investigation of the structural and electronic properties of Eu-doped BaWO4 material and the creation of oxygen vacancies in the parent compound BaWO4, using the Full potential linearized augmented plane wave (FPLAPW) method, within the framework of Density Functional Theory (DFT). The elastic properties of BaWO4, including its elastic constants, bulk modulus, shear modulus, and Poisson's ratio, are significantly influenced by the introduction of oxygen vacancies and Eu doping. These modifications result in a predictable reduction in stiffness and rigidity but enhance the material's optoelectronic functionality. The calculations are made within the Generalized Gradient Approximation (GGA) + Optimized Effective Hubbard Parameter (U) (GGA + U) approach, employed in the WIEN2K software. Adding the Hubbard parameter U with a value of 7 eV provides a better description of the system with a strong correlation of d electronic states and f-electrons. To examine the electronic structure of Eu-doped BaWO4 material and the creation of oxygen vacancy (V_O) in the parent compound BaWO4, we have studied their band structures. The direct band gap is calculated for oxygen-deficient material, and the indirect band gap for Eu-doped BaWO4, with band gap values of 0.8 eV and 2.8 eV, respectively. We have computed the total, elemental, and partial densities of states (PDOS), and band structures for the optimization of the structure of Eu-doped BaWO4, which are discussed in detail. In partial density of states (PDOS), the strong influence of W: Ba: 4d, O, and Eu were noted as the foremost contributors in the hybridization process, thus enhancing the electronic and optical properties. The band structures and density of states reveal the semiconductor nature of the material. The structural properties, including bulk modulus, lattice constant (a), and cohesive energy, have been measured. For oxygen-deficient material, the least optical loss is seen in the visible region when the first maximum absorption peak is observed at 2 eV. In this region, the reflectivity is minimum, confirming that oxygen vacancy is beneficial for optoelectronic devices. Maximum reflectivity is observed in the ultraviolet region, but it is insufficient for use as a shielding material against ultraviolet radiation. The calculated results reveal that BaWO4: V_O is more suitable for optoelectronic devices due to its direct band gap semiconductor nature and smaller band gap values. The results calculated in our study are consistent with the literature.Data availability
All the data supporting the findings of this study are included in the article.Author contributions
All authors contributed equally in the article in conceptualization, investigation, analysis, writing original draft, review and editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
Funding received from “National Water and Energy Center, United Arab Emirates University, Al Ain, 15551, United Arab Emirates”. The authors extend their appreciation to Taif University, Saudi Arabia for supporting this work through project number (TU-DSPP-2024-01)References
- P. S. M. Gharavi and M. R. Mohammadi, The improvement of light scattering of dye sensitized solar cells aided by new dandelion-like TiO2 nanostructures, Sol. Energy Mater. Sol. Cells, 2015, 137, 113–123 CrossRef.
- J. Yu, D. Wang, Y. Huang, X. Fan, X. Tang and C. Gao, et al., A cylindrical core–shell-like TiO2 nanotube array anode for flexible fiber-type dye-sensitized solar cells, Nanoscale Res. Lett., 2011, 6, 94 Search PubMed.
- A. H. G. Niaki, A. Bakhshayesh and M. Mohammadi, Double-layer dye-sensitized solar cells based on Zn-doped TiO2 transparent and light scattering layers: Improving electron injection and light scattering effect, Sol. Energy, 2014, 103, 210–222 CrossRef.
- M. Grätzel, Recent advances in sensitized mesoscopic solar cells, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- M. Freitag, J. Teuscher, Y. Saygili, X. Y. Zhang, F. Giordano and P. Liska, et al., Dye-sensitized solar cells for efficient power generation under ambient lighting, Nat. Photonics, 2017, 11, 372–379 CrossRef CAS.
- R. Katoh and A. Furube, Electron injection efficiency in dye-sensitized solar cells, J. Photochem. Photobiol., A, 2014, 20, 1–16 CrossRef CAS.
- B. O'Regan and M. Gratzel, A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films, Nature, 1991, 353, 737–740 CrossRef.
- V. Kumar, S. K. Swami, A. Kumar, O. M. Ntwaeaborwa, V. Dutta and H. C. Swar, Eu3+ doped down-shifting TiO2 layer for efficient dye-sensitized solar cells, J. Colloid Interface Sci., 2016, 484, 24–32 CrossRef CAS PubMed.
- M. Yu, H. Xu, Y. Li, Q. Dai, G. Wang and W. Qin, Morphology, luminescence, and photovoltaic performance of lanthanide-doped CaWO4 nanocrystals, J. Colloid Interface Sci., 2020, 559, 162–168 CrossRef CAS PubMed.
- M. Luoshan, L. Bai, C. Bu, X. Liu, Y. Zhu and K. Guo, et al., Surface plasmon resonance enhanced multi-shell-modified upconversion NaYF4:Yb3+, Er3+@SiO2@Au@TiO2 crystallites for dye-sensitized solar cells, J. Power Sources, 2016, 307, 468–473 CrossRef CAS.
- Y. Zhang, N. Zhou, K. Zhang and F. Yan, Plasmonic copper nanowire@TiO2 nanostructures for improving the performance of dye-sensitized solar cells, J. Power Sources, 2017, 342, 292–300 CrossRef CAS.
- H. Hafez, M. Saif and M. S. A. Abdel-Mottaleb, Down-converting lanthanide doped TiO2 photoelectrodes for efficiency enhancement of dye-sensitized solar cells, J. Power Sources, 2011, 196, 5792–5796 CrossRef CAS.
- N. G. Park, Perovskite solar cells: An emerging photovoltaic technology, Mater. Today, 2015, 18, 65–72 CrossRef.
- M. Mursal, N. Azmi and S. Fatmiyah, Synthesis of TiO2-based photoelectrode and natural dye for dye-sensitized solar cell (DSSC), J. Phys.: Conf. Ser., 2021, 1882, 012006 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal halide perovskites as visible-light sensitizers for photovoltaic cells, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef PubMed.
- Q. Y. Zhang and X. Y. Huang, Recent progress in quantum cutting phosphors, Prog. Mater. Sci., 2010, 55, 353–427 CrossRef.
- X. Y. Huang, S. Y. Han, W. Huang and X. G. Liu, Enhancing solar cell efficiency: The search for luminescent materials as spectral converters, Chem. Soc. Rev., 2013, 42, 173–201 RSC.
- X. Hou, T. Xuan, H. Sun, X. Chen, H. Li and L. Pan, High performance perovskite solar cells by incorporating a ZnGa2O4:Eu3+ nanophosphor in the mesoporous TiO2 layer, Sol. Energy Mater. Sol. Cells, 2016, 149, 121–127 Search PubMed.
- H. Lai, Y. Wang, G. Dua, W. Li and W. Han, Dual functional YVO4:Eu3+,Bi3+@SiO2 submicron-sized core–shell particles for dye-sensitized solar cells: Light scattering and down conversion, Ceram. Int., 2014, 40, 6103–6108 CrossRef.
- Z. Liu, H. Ma, L. Zhao, W. Bian, X. Xu and X. Fan, et al., Luminescence quenching properties of Sr2Ga2GeO7:Pr3+ with and without traps participation, J. Solid State Chem., 2019, 271, 23–28 Search PubMed.
- F. Xue, Y. Hun, L. Chen, G. Ju, T. Wang and S. Zhang, et al., Luminescence properties of a novel greenish-blue emission long persistent phosphor Sr3TaAl3Si2O14:Pr3+, Ceram. Int., 2016, 42, 11039–11044 CrossRef.
- W. B. Dai, Y. F. Lei, J. Zhou, M. Xu, L. L. Chu and L. Li, et al., Near-infrared quantum-cutting and long-persistent phosphor Ca3Ga2Ge3O12:Pr3+, Yb3+ for application in in vivo bioimaging and dye-sensitized solar cells, J. Alloys Compd., 2017, 726, 230–239 CrossRef.
- L. Jinsheng, Y. Hangying, Z. Shaoan, J. Jinlong, Q. Bao and H. Haiping, et al., Synthesis and luminescence properties of BaWO4:Pr3+ microcrystal, J. Rare Earths, 2011, 29, 623 CrossRef.
- P. Kumar, S. Kanika, R. Singh, R. Lahon, A. Gundimeda and G. Gupta, et al., A strategy to design lanthanide doped dual-mode phosphor mediated spectral converter for solar cell applications, J. Lumin., 2018, 196, 207–213 CrossRef.
- C. Bouzidi, et al., Spectroscopic properties of rare-earth (Eu3+, Sm3+) doped BaWO4 powders, J. Lumin., 2015, 161, 448–455 CrossRef.
- S. Cho, Annealing effect on properties of BaWO4:Eu3+ phosphor thin films grown on glass substrates by radio-frequency magnetron sputtering, Appl. Surf. Sci., 2017, 202–206 Search PubMed.
- S. Cho, Synthesis and photoluminescence properties of BaWO4:RE3+ (RE = Eu or Sm) phosphors, J. Korean Phys. Soc., 2018, 72, 959–965 CrossRef CAS.
- W. Deng, et al., Structural and optical investigations of quasi-single crystal Eu3+-doped BaWO4 thin films, Langmuir, 2018, 34, 8499–8507 CrossRef CAS PubMed.
- G. George, et al., A novel reversible fluorescent probe for the highly sensitive detection of nitro and peroxide organic explosives using electrospun BaWO4 nanofibers, J. Mater. Chem. C, 2019, 7, 14949 RSC.
- I. P. Carvalho, A. F. Lima and M. V. Lalic, Ab initio study of the phonon and thermodynamic properties of the scheelite MWO4 (M = Ba, Sr or Ca) compounds, Solid State Commun., 2021, 333, 114290 CrossRef CAS.
- R. Ashraf, et al., DFT based investigations of BaWO4: Electronic and optical properties, Phys. B, 2021, 621, 413309 CrossRef CAS.
- B. G. Janesko, Replacing hybrid density functional theory: Motivation and recent advances, Chem. Soc. Rev., 2021, 50, 8470–8495 RSC.
- R. Shinde, S. S. R. K. C. Yamijala and B. M. Wong, Improved band gaps and structural properties from Wannier-Fermi-Löwdin self-interaction corrections for periodic systems, J. Phys. Condens. Matter, 2021, 33, 115501 CrossRef CAS.
- P. Blaha, K. Schwarz, G. Madsen, D. Kvasnicka and J. Luitz, WIEN2k, an Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties, ed. K. Schwarz, Technische Universität Wien, Austria, 2001 Search PubMed.
- A. I. Li, V. I. Anisimov and J. Zaanen, Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef.
- Q. Rafiq, S. Azam, I. Boukhris and N. Tamam, Predictive analysis of Cu and Ni substitution effects on the structural, optoelectronic and thermoelectric behavior of CdS: A first-principles approach, J. Phys. Chem. Solids, 2025, 205, 112769 CrossRef.
- L. Zhang, Y. Zhao, Y. Liu and G. Gao, High spin polarization, large perpendicular magnetic anisotropy, and room-temperature ferromagnetism by biaxial strain and carrier doping in Janus MnSeTe and MnSTe, Nanoscale, 2023, 15, 18910 RSC.
- L. Zhang, Y. Liu and G. Gao, Electric-field- and stacking-tuned antiferromagnetic FeClF bilayer: The coexistence of bipolar magnetic semiconductor and anomalous valley Hall effect, Adv. Funct. Mater., 2023, 35, 2417857 CrossRef.
- L. Zhang, Y. Liu, Z. Xu and G. Gao, Electronic phase transition, perpendicular magnetic anisotropy, and high Curie temperature in Janus FeClF, 2D Mater., 2023, 10, 045005 CrossRef.
- T. Saha, M. M. H. Babu, M. Arifuzzaman and J. Podder, Thermodynamic and dynamic stability in a new potential Cs2AgAsCl6 perovskite: Insight from DFT study, Phys. Chem. Chem. Phys., 2022, 24, 26609–26621 RSC.
- Q. Rafiq, S. S. Hayat, S. Azam, R. Neffati and M. Amami, Quantum control of substitutional effects on the S-involved optoelectronic properties of BeSxSe1-x ternary alloys, Comput. Condens. Matter., 2023, 36, e00819 CrossRef.
- B. Ehrler, E. Alarcon-Lladó, S. W. Tabernig, T. Veeken, E. C. Garnett and A. Polman, Photovoltaics reaching for the Shockley-Queisser limit, ACS Energy Lett., 2020, 5, 3029–3033 CrossRef.
- B. Wu, Y. Zhou and G. Xing, et al., Long minority-carrier diffusion length and low surface-recombination velocity in inorganic lead-free CsSnI3 perovskite crystal for solar cells, Adv. Funct. Mater., 2017, 27, 1604818 CrossRef.
- W. Gao, C. Ran and J. Xi, et al., High-quality Cs2AgBiBr6 double perovskite film for lead-free inverted planar heterojunction solar cells with 2.2% efficiency, ChemPhysChem, 2018, 19, 1696–1704 CrossRef PubMed.
- M. S. Shadabroo, H. Abdizadeh and M. R. Golobostanfard, Elpasolite structures based on A2AgBiX6 (A: MA, Cs, X: I, Br): Application in double perovskite solar cells, Mater. Sci. Semicond. Process., 2021, 125, 105639 CrossRef.
- M. Wang, P. Zeng and S. Bai, et al., High-quality sequential-vapor-deposited Cs2AgBiBr6 thin films for lead-free perovskite solar cells, Sol. RRL, 2018, 2, 1800217 CrossRef.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, A bismuth-halide double perovskite with long carrier recombination lifetime for photovoltaic applications, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- S. Das, M. Debbarma and S. Chattopadhyaya, Calculations of physical properties of Ba2GdSbO6 (BGSO) double perovskite for thermoelectric and solar cell applications, Phys. B, 2023, 664, 414979 CrossRef CAS.
- R. Sa, Y. Wei, W. Zha and D. Liu, A first-principle study of the structural, mechanical, electronic, and optical properties of vacancy-ordered double perovskite Cs2TeX6 (X = Cl, Br, I), Chem. Phys. Lett., 2020, 754, 137538 CrossRef CAS.
- R. D. Grimes and E. R. Cowley, A model dielectric function for semiconductors, Can. J. Phys., 1975, 53, 2549–2554 CrossRef CAS.
- H. H. Hegazy, G. M. Mustafa, A. Nawaz, N. A. Noor, A. Dahshan and I. Boukhris, Tuning of direct bandgap of Rb2ScTlX6 (X = Cl, Br, I) double perovskites through halide ion substitution for solar cell devices, J. Mater. Res. Technol., 2022, 19, 1271–1281 CrossRef CAS.
- S. A. Dar and B. Want, DFT study of structural, mechanical, and opto-electronic properties of scandium-based halide double perovskite Cs2ScInBr6 for optoelectronic applications, Micro Nanostruct., 2022, 170, 207370 CrossRef CAS.
- Q. Rafiq, M. T. Khan, S. S. Hayat, S. Azam, A. U. Rahman and H. O. Elansary, et al., Adsorption and solar light activity of noble metal adatoms (Au and Zn) on Fe(111) surface: a first-principles study, Phys. Chem. Chem. Phys., 2024, 26, 17118–17131 RSC.
- Q. Rafiq, S. S. Hayat, S. Azam and A. Mahmood, First-principles calculations to investigate electronic, magnetic, and optical properties at (110) and (111) surfaces of Ni adsorption on CdO, Mol. Phys., 2024, e2341117–e2341135 CrossRef.
- Q. Rafiq, S. S. Hayat, S. Azam, A. U. Rahman and M. Irfan, Tailoring of optoelectronic and thermal properties of silver doped tin selenide alloy for optoelectronic applications, Phys. B, 2023, 666, 415095 CrossRef CAS.
- Q. Rafiq, S. Azam, S. S. Hayat, U. Javed, A. Mahmood and S. A. Khan, Investigating the chemical and physical aspects: optical and electrical properties of TiO2, TiO2:Fe, and Fe/TiO2 (111) through DFT analysis, Chem. Pap., 2023, 78, 283–294 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |