
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced supercapacitor performance using EG@COF: a layered porous composite†
Junaid Khan *abd,
Anique Ahmedc and
Abdullah A. Al-Kahtanie
*abd,
Anique Ahmedc and
Abdullah A. Al-Kahtanie
aDepartment of Physics, Government Postgraduate Collage No. 1, Abbottabad, Khyber Pakhtunkhwa, Pakistan. E-mail: junaidkhan.nanotech@gmail.com
bDepartment of Higher Education Achieves and Libraries, Government of Khyber Pakhtunkhwa, Pakistan
cFaculty of Engineering Sciences, GIK Institute of Engineering Sciences and Technology, Topi 23640, Khyber Pakhtunkhwa, Pakistan
dDepartment of Chemical and Bilogical Engineering, Gachon University, 1342 Seongnam-daero, Seongnam 13120, Republic of Korea
eChemistry Department, Collage of Science, King Saud University, P. O. Box 2455, Riyadh-22451, Saudi Arabia
First published on 11th April 2025
Abstract
In this work, to address the issue of poor conductivity in COFs, a layered porous composite (EG@COF) was successfully synthesized. A redox-active COF (DAAQ-TFP COF) was grown on the surface of expanded graphite (EG) through a solvent-free in situ synthesis. SEM analysis displayed that the obtained composite (EG@COF) possessed a layered porous structure. Further investigations revealed that EG not only improved electrical conductivity but also regulated the pore size of the COFs. This structure was highly conducive to enhancing the specific capacitance of the electrode material. An electrochemical study demonstrated that the specific capacitance of EG@COF-3 reached 351 C g−1 at 1 A g−1, with 94.4% capacitance retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. The excellent capacitance retention was attributed to the stable backbone of the COF. Meanwhile, an asymmetric supercapacitor (ACS) comprising activated carbon (AC) and EG@COF exhibited an energy density of 16.4 W h kg−1 at a power density of 806.0 W kg−1.
000 cycles. The excellent capacitance retention was attributed to the stable backbone of the COF. Meanwhile, an asymmetric supercapacitor (ACS) comprising activated carbon (AC) and EG@COF exhibited an energy density of 16.4 W h kg−1 at a power density of 806.0 W kg−1.
1. Introduction
Supercapacitors (SCs) have emerged as a class of highly promising energy storage devices, distinguished by their exceptional rapid charge–discharge capabilities, superior power density compared to conventional batteries (e.g., lead-acid batteries), and remarkable cycling stability, making them well-suited for diverse energy storage applications.1,2 However, a critical challenge remains: how to enhance their energy density without compromising their inherent high-power performance.3–5 At the heart of this challenge lies the selection of electrode materials, which play a pivotal role in dictating the electrochemical performance of SCs. Thus, the development of advanced electrode materials is of paramount importance.6,7Covalent organic frameworks (COFs) represent a unique class of porous crystalline polymers formed via the covalent assembly of organic building blocks.8 Since their pioneering discovery by Yaghi in 2005,9 COFs have garnered widespread attention due to their exceptional chemical stability, tunable structures, and high specific surface areas.10 These advantageous properties have enabled their extensive applications in energy storage, gas separation, catalysis, and beyond.10,11 Nevertheless, COFs face considerable challenges in electrochemical energy storage, primarily due to their inherently low electrical conductivity, which hinders their electrochemical performance. Moreover, the availability and utilization of redox-active sites in COFs significantly impact their efficacy as supercapacitor materials.
A notable example is the work by Dichtel's group in 2013,12 where they successfully synthesized a two-dimensional COF (DAAQ-TFP COF) with an ultrahigh specific surface area using 1,3,5-trimethylformylresorcinol (TFP) and redox-active 2,6-diaminoanthraquinone (DAAQ) monomers. Despite exhibiting a higher capacitance than non-redox-active COFs, only 2.5% of its redox-active sites were effectively utilized, highlighting the necessity of increasing both the density and utilization efficiency of redox-active sites to enhance electrochemical performance.
In recent years, various strategies have been explored to improve COF-based supercapacitor materials. For instance, Zeng et al.13 developed a carbon-based material (COF-T800) by carbonizing COFs at high temperatures, which resulted in a 3.3-fold increase in electrical conductivity compared to its pristine COF precursor. Similarly, Sun et al.14 synthesized COF/GO composites via in situ and ex situ methods, subsequently carbonizing them to obtain C/rGO materials. Their findings revealed that the in situ approach yielded a higher nitrogen content, thereby increasing the density of redox-active sites and significantly improving specific capacitance. Notably, the C/rGO material obtained via the in situ method exhibited a remarkable capacitance of 234 F g−1 at 0.8 A g−1, vastly outperforming its ex situ counterpart (43.5 F g−1). Additionally, the synthesis of COFs, often achieved through Schiff-base condensation reactions, is influenced by several parameters, including reaction time, temperature, and pressure.15 Consequently, there remains substantial scope for further exploration and optimization of COF-based materials for supercapacitor applications.16,17
Expanded graphite (EG) is a lightweight, high-surface-area material derived from graphite through thermal or chemical expansion, resulting in an interconnected, porous structure with excellent electrical conductivity. When combined with covalent organic frameworks (COFs), the resulting hybrid material benefits from the synergistic combination of EG's conductivity and COFs' highly ordered porosity, structural tunability, and redox-active sites. This enhances ion transport, charge storage capacity, and cycling stability, making the composite an excellent candidate for supercapacitor applications. The presence of COFs introduces additional functional groups that improve electrolyte interaction, while EG provides a robust conductive network, reducing internal resistance and boosting overall electrochemical performance.
In this study, we successfully synthesized an expanded graphite-supported COF (EG@COF) via a solvent-free in situ approach, facilitating the uniform growth of a redox-active DAAQ-TFP COF on the surface of expanded graphite. The resulting EG@COF exhibited a distinctive layered porous architecture, with scanning electron microscopy (SEM) analyses confirming that its morphology and thickness could be finely tuned by adjusting the proportion of expanded graphite. Optimal conditions enabled the uniform growth of the COF on the EG surface, yielding a well-defined laminated porous structure. The as-prepared EG@COF-3 demonstrated an impressive specific capacitance coupled with outstanding cycling stability. These findings underscore the potential of this synthesis strategy for developing high-performance electrode materials, offering valuable insights into the design of next-generation supercapacitors.
2. Experimental
2.1. Materials
Expandable graphite (EG), 2,6-diaminoanthraquinone (DAAQ), 2,4,6-triformylphloroglucinol (TFP), and p-toluenesulfonic acid (PTSA) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Methanol, acetone, and ethanol were provided by Xilong Chemical Co., Ltd. All reagents were used as received without any further purification.182.2. Pretreatment of expandable graphite
The purchased expandable graphite (1 g) was placed in a crucible and heated in a muffle furnace at 1000 °C for one minute to obtain expanded graphite.2.3. Preparation of COF and EG@COF
Pure COF was prepared based on the report19 with modifications. Briefly, DAAQ (0.107 g), TFP (0.063 g), and PTSA (0.5 g) were mixed in 1.0 mL of water. The mixture was then ground for 30 min. The obtained slurry was reacted in an oven at 100 °C for 72 h. The product was washed sequentially with acetone and methanol to obtain pure COF.The EG@COF composites were synthesized using a solvent-free in situ synthesis method. As shown in Scheme 1, DAAQ (0.107 g) and PTSA (0.5 g) were weighed and added to EG in different amounts (0.0085 g, 0.0119 g, 0.0153 g, 0.0187 g), and the mixture was thoroughly combined in an agate mortar. Subsequently, TFP (0.063 g) was added to the agate mortar, and the mixture was ground for 30 min. The slurry was then heated at 100 °C for 72 h in an oven. The solid obtained was ground and washed with methanol and deionized water to remove PTSA and unreacted reagents. Finally, the powder was vacuum-dried at 60 °C overnight. The prepared composites were labeled as EG@COF-1, EG@COF-2, EG@COF-3 and EG@COF-4, based on the varying amounts of EG used.
 | ||
| Scheme 1 Schematic of the synthesis of EG@COF. | ||
2.4. Electrochemical measurements
All electrochemical measurements were carried out using a CHI660E workstation (Chenhua, Shanghai) in a 6 M KOH electrolyte solution. The mass-specific capacitance (Cg, F g−1) was calculated based on the constant current charge/discharge test data and the following eqn (1):20
 | (1) |
For practical applications, an asymmetric supercapacitor (ACS) was assembled using a two-electrode system. The energy density (E, W kg−1) and power density (P, W h kg−1) was calculated using the following eqn (2) and (3):22,23
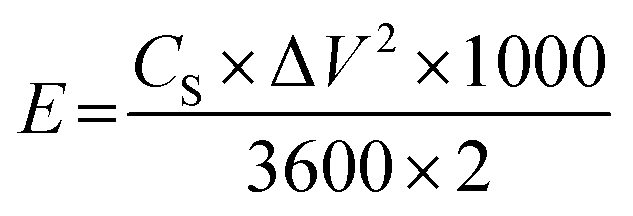 | (2) |
 | (3) |
3. Results and discussion
The crystal structures of pure COF and EG@COF were analyzed by powder X-ray diffraction (Fig. 1a). It could be seen that there was a well-defined peak at 2θ ≈ 3.6° for all samples, corresponding to the (100) reflection plane of COF, confirming the successful synthesis of COF. For pure COF, there was a broad and weak peak at 2θ ≈ 26.6°, attributed to the π–π stacking of the (001) plane of the COF material, which was also consistent with the literature.24 For EG@COFs, a distinct characteristic peak appeared at 2θ ≈ 26.6° in their XRD curves, resulting from the overlap of the reflection peaks of EG (002) and the COF (001). This characteristic peak became more pronounced as the proportion of EG increased.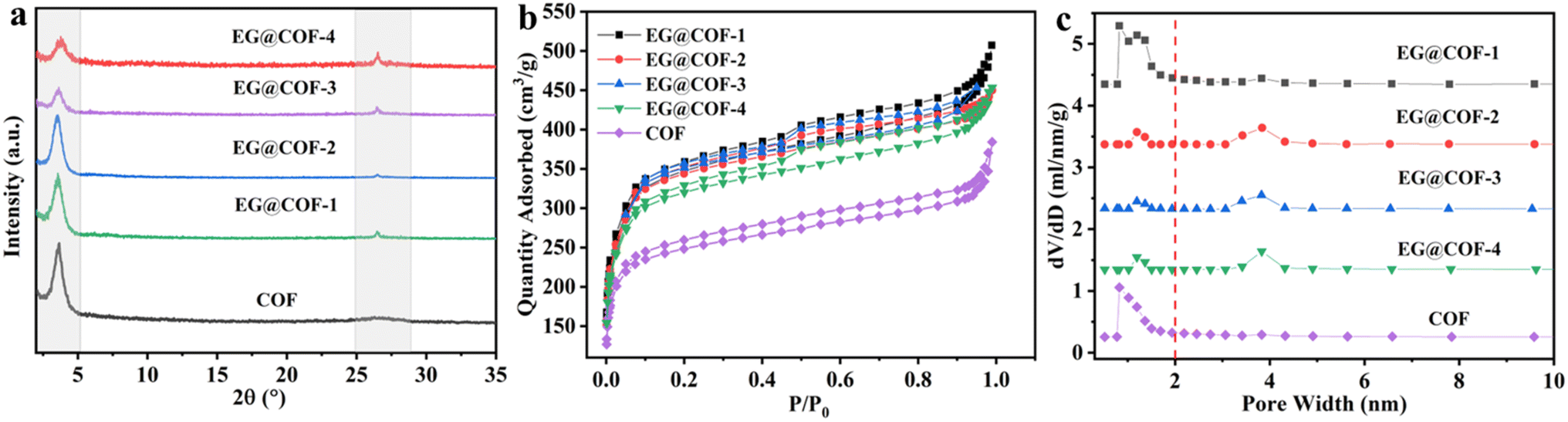 | ||
| Fig. 1 (a) Powder X-ray diffraction patterns of pure COF and EG@COFs, (b) N2 adsorption and desorption isotherms recorded at 77 K, (c) pore size distributions of pristine COF and EG@COFs calculated from the adsorption isotherms. | ||
The porosity of pure COF and EG@COF was measured by N2 adsorption–desorption at 77 K (Fig. 1b). It could be seen that all the adsorption and desorption curves exhibited steep adsorption peaks at low relative pressures (P/P0 < 0.5), characteristic of type I adsorption and desorption isotherms, indicating the presence of micropores in the material. In contrast, the adsorption and desorption curves with the addition of EG showed an obvious hysteresis loop near P/P0 = 0.5, indicating that the material contained mesopores. The pore size distribution of the adsorption–desorption isotherms (Fig. 1c) showed that only micropores were present in the pure COF material, whereas both micropores and mesopores were present in EG@COFs. Combined with the data in Table 1, it could be observed that the addition of EG changed the average pore size of the composites, and the specific surface area increased from 956 m2 g−1 for pure COF to 1397 m2 g−1 for EG@COF-3. Among the EG@COFs, the specific surface area of EG@COF-4 decreased, as too much EG was added, preventing the COF from growing uniformly on its surface. Based on the above analysis, EG@COF-3 was selected for further study.
| Material | SBET (m2 g−1) | Pore size (nm) | Coverage ratio (%) |
|---|---|---|---|
| EG@COF-1 | 1316 | 0.822 | 37.7 |
| EG@COF-2 | 1320 | 3.829 | 38.1 |
| EG@COF-3 | 1397 | 3.823 | 46.2 |
| EG@COF-4 | 1218 | 3.416 | 27.4 |
| COF | 956 | 0.757 | Reference |
The FT-IR spectra of DAAQ, TFP, COF, and EG@COF-3 are shown in Fig. 2a. In the DAAQ curve, the peaks near 3502 and 3450 cm−1 indicated symmetric and asymmetric stretching vibrations of –NH2. The peaks near to the value of 1635 cm−1 correspond to the asymmetric and symmetric stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond corresponding to the redox monomer. The CC stretching vibration of the anthraquinone ring and the C–N stretching vibration of the amino group appeared at 1566 and 1286 cm−1, respectively. Furthermore, the disappearance of the N–H absorption peaks (3502 and 3450 cm−1) and the generation of a new C–N stretching vibration peak at 1382 cm−1 for pure COF and the composite EG@COF indicated that an amidation reaction occurred. Compared to the CO absorption peaks of DAAQ and TFP (1635 and 1641 cm−1, respectively), pure COF and composite EG@COF exhibited a lower energy β-ketoenamine CO stretching peak at 1622 cm−1, indicating the successful synthesis of COF and EG@COF. In addition, the C–O peak in TFP did not appear in the FT-IR spectrum of COF, overall indicating the absence of both DAAQ and TFP monomers.
O bond corresponding to the redox monomer. The CC stretching vibration of the anthraquinone ring and the C–N stretching vibration of the amino group appeared at 1566 and 1286 cm−1, respectively. Furthermore, the disappearance of the N–H absorption peaks (3502 and 3450 cm−1) and the generation of a new C–N stretching vibration peak at 1382 cm−1 for pure COF and the composite EG@COF indicated that an amidation reaction occurred. Compared to the CO absorption peaks of DAAQ and TFP (1635 and 1641 cm−1, respectively), pure COF and composite EG@COF exhibited a lower energy β-ketoenamine CO stretching peak at 1622 cm−1, indicating the successful synthesis of COF and EG@COF. In addition, the C–O peak in TFP did not appear in the FT-IR spectrum of COF, overall indicating the absence of both DAAQ and TFP monomers.
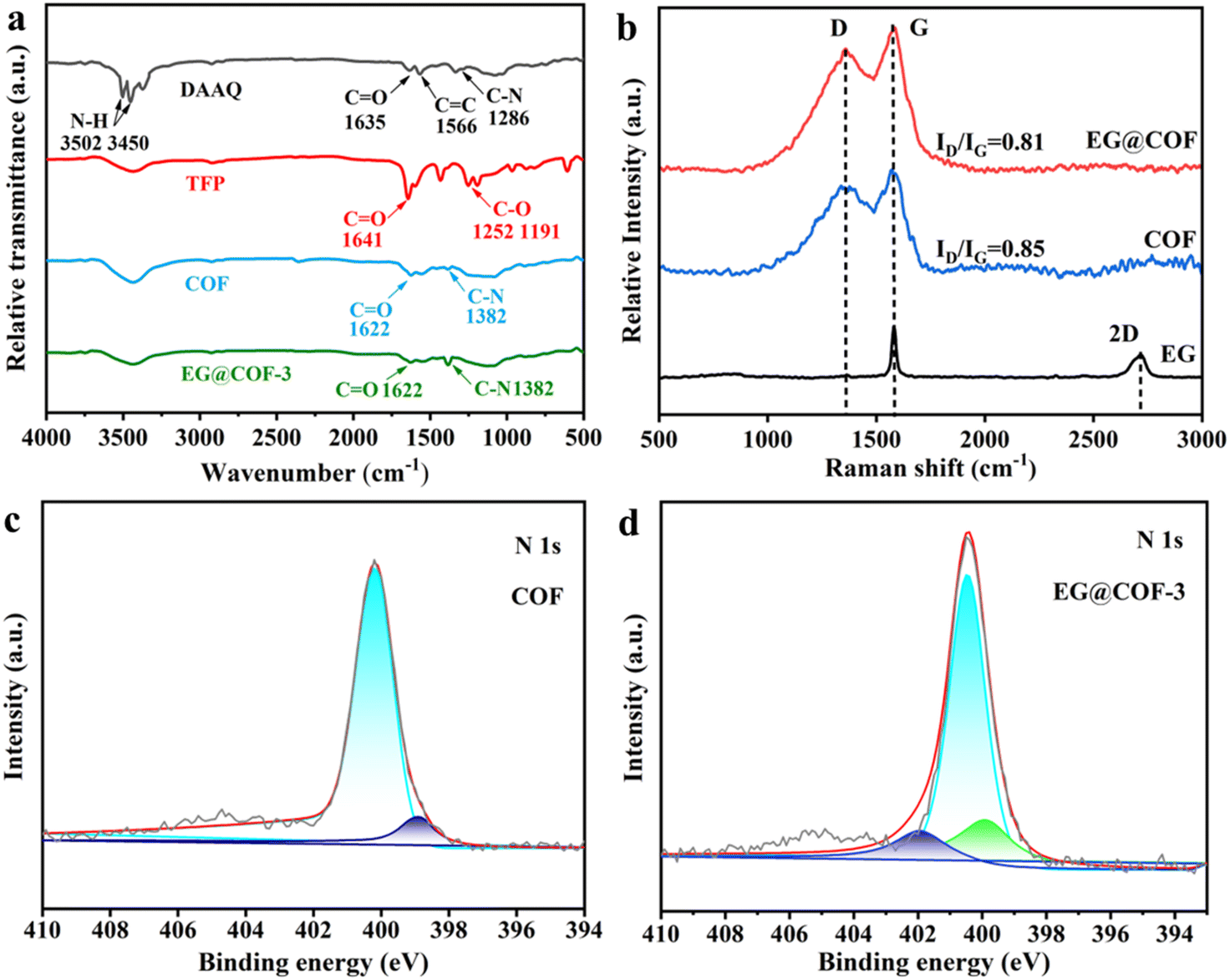 | ||
| Fig. 2 (a) FT-IR spectra of DAAQ, TFP, COF, and EG@COF-3, (b) Raman spectra of EG, pure COF, and EG@COF-3, (c) high-resolution N 1s X-ray photoelectron spectra of pure COF, (d) high-resolution N 1s X-ray photoelectron spectra of EG@COF-3. | ||
Fig. 2b shows the Raman spectra of EG, pure COF, and EG@COF-3. In the Raman spectra of carbon materials, the D peak indicates the lattice defects of carbon atoms, and the G peak corresponds to the stretching vibration of sp2 hybridized carbon atoms.25 The value of ID/IG is typically used to evaluate the degree of defects in carbon materials. In the Raman spectrum of EG, the G peak (2700 cm−1) appeared, while the D peak was less prominent, indicating a higher degree of regularity. The ID/IG ratio of EG@COF-3 was 0.81, which was slightly lower than that of COF (0.85). This was due to the large number of oxygen functional groups on the surface of EG, which underwent non-covalent interactions (such as hydrogen bonding, van der Waals forces, etc.) with COF. To further understand the bonding state of EG@COF-3, X-ray photoelectron spectroscopy (XPS) analysis was performed. For pure COF (Fig. 2c), the high-resolution XPS spectrum of N 1s could be deconvoluted into two peaks at 399.3 eV and 400.4 eV, corresponding to the N atoms in –NH2 and –NH–C, respectively. When EG bound to COF (Fig. 2d), the binding energy of N 1s shifted towards higher energies, with three distinct peaks at 399.8 eV, 400.8 eV, and 401.9 eV. The new peaks that appeared could be attributed to the reaction of the oxygen-containing carboxylic acid groups on the surface of EG with the amine group in DAAQ.26 This indicated a change in the bonding pattern of N atoms in EG@COF, proving the chemical connection between EG and COF. This also corresponded to a decrease in the degree of defects in EG@COF, as observed in the Raman spectrum.
The scanning electron microscopy (SEM) images of expanded graphite before and after milling are shown in Fig. 3a and b, respectively. The morphology of expanded graphite exhibited a worm-like structure, with large pores between the layers (Fig. 3a). After grinding, it transformed into a smooth flake structure (Fig. 3b). The COF material, on the other hand, was granular, with a morphology that showed an aggregated structure formed by the growth of 2D planes and longitudinal stacking (Fig. 3c). The SEM image of EG@COF-1 did not differ significantly from that of pure COF and still showed the stacking morphology of COF (Fig. 3d). In the SEM image of EG@COF-2, it could be seen that this stacking decreased with an increase in EG addition, and the flake structure of expanded graphite, along with the grown COF particles, was observable (Fig. 3e). In the SEM image of EG@COF-3 (Fig. 3f), more holes were observed, indicating that the stacking of COF was reduced and that the COF was able to grow uniformly on the surface of EG. Meanwhile, by observing the side of EG@COF-3 (Fig. 3g), it could be seen that COF particles also grew between the layers of EG. With further increasing the amount of EG, COF could not grow uniformly on the EG surface (Fig. 3h). Based on the SEM image of EG@COF-4, it could be observed that the smoother lamellar structures belonged to EG. The above results implied that an appropriate amount of EG was beneficial for the uniform growth of COF.
 | ||
| Fig. 3 Scanning electron microscopy images of: (a) and (b) expanded graphite, (c) pure COF, (d) EG@COF-1, (e) EG@COF-2, (f) and (g) EG@COF-3, (h) EG@COF-4. | ||
It was clear that EG@COF-3 had the optimum morphology. This structure was conducive to increasing the ion exchange rate in the material, which in turn improved the conductivity and electrochemical properties of the material. In addition, the elemental analysis of EG@COF-3 (Fig. S1†) was also carried out, where a uniform distribution of the three elements C, N, and O were observed, further supporting the above analysis.
The electrochemical properties of the samples were tested using a three-electrode system. Fig. 4a shows the CV curves of COF and EG@COFs at 10 mV s−1. It could be seen that all of them exhibited two redox peaks. According to ref. 26, it is known that the redox (charge/discharge) mechanism of COF in EG@COF involves the reversible quinone to hydroquinone transformation, as follows (4):
 | (4) |
 | ||
| Fig. 4 (a) Comparison of cyclic voltammetry (CV) curves of EG@COFs and pure COF at a scan rate of 10 mV s−1, (b) GCD curves of EG@COFs and pure COF at different current densities, (c) comparison of specific capacitance values of different materials in this work, (d) CV curves of EG@COF-3 with different scan rates, (e) GCD curves of EG@COF-3 with different current densities, (f) electrochemical impedance spectra of EG@COFs and pure COF. | ||
In addition, the curve of EG@COF-3 had the largest peak area, indicating a higher specific capacitance with the largest charge transfer in its electrochemical reaction. Fig. 4b presents their GCD curves at 1 A g−1. It was evident that EG@COF-3 had the longest discharge time, and the specific capacitances of COF, EG@COF-1, EG@COF-2, EG@COF-3, and EG@COF-4 were 407, 404, 462, 501 and 477 F g−1, respectively (Fig. 4c). The specific capacitance was recorded as 285, 283, 351 and 334 C g−1 respectively. The CV curves of EG@COF-3 at different scanning rates are summarized in Fig. 4d. These CV curves showed no obvious changes at scanning rates of 5–50 mV s−1, indicating that EG@COF-3 had good electrochemical reversibility. However, the CV of EG@COF-3 showed signs of polarization only when the scanning rate reached 100 mV s−1. According to the GCD plots of EG@COF-3 (Fig. 4e), they did not show distortion with the increase in current density (even at 8 A g−1). Moreover, the specific capacity was 375, 351, 329, 295, and 288 C g−1 at 0.5, 1, 2, 5, and 8 A g−1, respectively.
In addition, the capacitance of EG@COF-3 prepared in this study exceeded the capacitance values previously reported in the literature (Table 2). Furthermore, the specific capacitance of EG@COF-3 retained 82% of the original specific capacitance in the range of 0.5–8 A g−1. The excellent specific capacitance and retention rate were attributed to two main factors: first, the expanded graphite provided a good growth substrate, altering the pore size of the COF and improving the ionic conductivity of the composites; second, the increase in the specific surface area promoted the exposure of the active sites and facilitated the faradaic reaction. Subsequently, the kinetics of EG@COFs were further investigated using the equivalent circuit model and electrochemical impedance spectroscopy (EIS) spectra (Fig. 4f). According to Fig. 4f, EG@COF-3 exhibited the lowest charge transfer resistance (Rct, 0.15 Ω). In contrast, the Rct of EG@COF-1, EG@COF-2, EG@COF-4, and COF were slightly higher (0.20 Ω, 0.25 Ω, 0.19 Ω, and 0.33 Ω, respectively). As shown in the low-frequency region, it was clear that EG@COF-3 had a steeper slope, while COF showed the smoothest slope. This proved that EG@COF-3 had the lowest diffusion resistance. The addition of expanded graphite enhanced the conductivity of EG@COF, and the amount added also influenced the magnitude of the diffusion resistance. Additionally, a further cycling stability test of EG@COF-3 was carried out (Fig. S2†). The result showed that EG@COF-3 had a 94.4% capacitance retention after 10000 cycles.
| Material | Electrolyte | Potential window (V) | Current density (A g−1) | Specific capacitance (F g−1) | Ref. |
|---|---|---|---|---|---|
| DAAQ-TFP COF | 1 M H2SO4 | −0.3–0.3 | 0.1 | 48 ± 10 | 12 |
| N-Doped C/rGO | 6 M KOH | 0.2–0.6 | 0.8 | 234 | 14 |
| cCNT@COF | 0.5 M H2SO4 | −0.3–0.3 | 0.5 | 376 | 27 |
| DAAQ-COFs/GA | 1 M H2SO4 | −0.5–0.5 | 1 | 378 | 27 |
| PDC-MA-COF | 6 M KOH | 0–0.5 | 1 | 335 | 28 |
| COFBTA-DPPD–rGO | 2 M KOH | 0–0.5 | 0.5 | 239.1 | 29 |
| EG@COF | 6 M KOH | −1–0.3 | 1 | 501 | This work |
To further investigate the electrochemical behavior of EG@COF, an asymmetric supercapacitor (AC//EG@COF-3) was prepared following the method by Zhu30 (Fig. 5a). The GCD curve of the ACS is shown in Fig. 5b. The specific capacity of the ACS was calculated to be 72 C g−1 at 1 A g−1. Its CV curves at different scanning rates are summarized in Fig. 5c. The non-rectangular shape of the curves indicated the simultaneous presence of pseudo-capacitance and double-layer capacitance. Additionally, the relationship between the peak current (i) and scanning rate (v) can be described by eqn (5):
| i = avb | (5) |
| i(V) = k1v + k2v1/2, | (6) |
 | ||
| Fig. 5 (a) Schematic of the AC//EG@COF device, (b) GCD plots, (c) CV plots, (d) b-values, (e) capacitive and diffusion contribution, (f) CV plots for different voltage windows. | ||
Usually, the b-value of 0.5 indicates linear diffusion control, while a b-value of 1.0 indicates capacitive charge storage without diffusion control. As shown in Fig. 5d, the b-values were calculated to be 0.75 and 0.66 based on the oxidation and reduction peaks of EG@COF. This implied that the electrochemical behavior of EG@COF was both capacitive and diffusion-controlled. The ratio of capacitive and diffusive contributions was calculated by eqn (6). The proportions of capacitive and diffusive contributions for scan rates of 5–50 mV s−1 are shown in Fig. 5e. The capacitive contribution values were found 35%, 49%, 53%, 56% and 70%, respectively. The result illustrated that the capacitive contribution gradually increased and dominated as the scan rate increased. Fig. 5f summarizes the CV curves of the AC//EG@COF device with different voltages at a scan rate of 20 mV s−1. No significant polarisation was observed even at 1.6 V, verifying the feasibility of the voltage range selection.
In practical applications, energy density and power density are two indicators used to evaluate the performance of materials. As shown in Fig. 6b, the energy density of EG@COF//AC was 16.4 W h kg−1 at a power density of 806.0 W kg−1, exceeding the electrode materials reported in ref. 19 and 31. Fig. 6a shows that the capacitance retention of EG@COF//AC reached 74% after 10000 cycles at 4 A g−1, while the coulombic efficiency remained at 98%. The above results proved that EG@COF had good cycling stability. In addition, it could also be observed from the inset that the GCD curves before and after cycling remained stable. Fig. 6c demonstrated that a GUET panel consisting of 29 LEDs could be lit using two EG@COF//AC devices connected in series.
 | ||
| Fig. 6 (a) Cycling stability test, (b) Ragone plot of the ASC, (c) digital photograph of LEDs powered by ASC. | ||
4. Conclusion
Layered porous material EG@COF was successfully synthesized by a solvent-free in situ synthesis method. Here, EG was used as the growth substrate of COF, enabling effective charge transport within EG (as a conductive skeleton), thereby improving the conductivity of the COF and modulating the pore size of COF to a certain extent. Therefore, more redox active sites could be utilized. The ion exchange rate was accelerated by the synergistic effect of EG and COF. Electrochemical tests demonstrated that the specific capacitance of EG@COF-3 was as high as 351 C g−1 at 1 A g−1. The capacitance retention was 94.4% after 10000 cycles. It exhibited good performance when used as an EG@COF//AC device. The ongoing optimization of COFs focuses not only on the adjustment of pore size but also on the development of COF species and the utilization of redox sites. This enhancement of COF conductivity and regulation of the pore size of COF through a carbon growth substrate, as demonstrated in this study, also provides new ideas for the application of COF-based composites in the field of energy storage.
Data availability
The data will be made available upon request.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.Acknowledgements
The authors thank the “Researchers Supporting Project Number (RSP2025R266), King Saud university, Riyadh, Saudi Arabia” for their financial support.References
- A. K. Yedluri and H.-J. Kim, Wearable super-high specific performance supercapacitors using a honeycomb with folded silk-like composite of NiCo2O4 nanoplates decorated with NiMoO4 honeycombs on nickel foam, Dalton Trans., 2018, 47(43), 15545–15554 RSC.
- K. D. Kumar, T. Ramachandran, Y. A. Kumar, A. A. Mohammed and M. Kang, Hierarchically fabricated nano flakes-rod-like CoMoO–S supported Ni-foam for high-performance supercapacitor electrode material, J. Phys. Chem. Solids, 2024, 185, 111735 CrossRef CAS.
- Y. A. Kumar and H.-J. Kim, Preparation and electrochemical performance of NiCo2O4@NiCo2O4 composite nanoplates for high performance supercapacitor applications, New J. Chem., 2018, 42(24), 19971–19978 RSC.
- Y. A. Kumar and H.-J. Kim, Effect of time on a hierarchical corn skeleton-like composite of CoO@ZnO as capacitive electrode material for high specific performance supercapacitors, Energies, 2018, 11(12), 3285 CrossRef CAS.
- A. Dixit, J. A. Abraham, M. Manzoor, M. Altaf, Y. A. Kumar and R. Sharma, A comprehensive DFT analysis of the physical, optoelectronic and thermoelectric attributes of Ba2lnNbO6 double perovskites for eco-friendly technologies, Mater. Sci. Eng., B, 2024, 307, 117530 CrossRef CAS.
- A. K. Yedluri and H.-J. Kim, Enhanced electrochemical performance of nanoplate nickel cobaltite (NiCo2O4) supercapacitor applications, RSC Adv., 2019, 9(2), 1115–1122 RSC.
- Y. Guo, K. Wang, Y. Hong, H. Wu and Q. Zhang, Recent progress on pristine two-dimensional metal–organic frameworks as active components in supercapacitors, Dalton Trans., 2021, 50(33), 11331–11346 RSC.
- J. Khan and A. Ahmed, Benzene-1,4-dicarboxylic acid-based Ni-MOF for efficient battery-supercapacitor hybrids: electrochemical behavior and mechanistic insights, J. Energy Storage, 2024, 100, 113455 CrossRef.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Porous, crystalline, covalent organic frameworks, Science, 2005, 310(5751), 1166–1170 CrossRef CAS PubMed.
- Q. Gu, J. Zha, C. Chen, X. Wang, W. Yao and J. Liu, et al., Constructing chiral covalent-organic frameworks for circularly polarized light detection, Adv. Mater., 2024, 36(17), 2306414 CrossRef CAS PubMed.
- Q. Gu, X. Lu, C. Chen, X. Wang, F. Kang and Y. Y. Li, et al., High-performance piezoelectric two-dimensional covalent organic frameworks, Angew. Chem., Int. Ed., 2024, 63(39), e202409708 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, β-Ketoenamine-linked covalent organic frameworks capable of pseudocapacitive energy storage, J. Am. Chem. Soc., 2013, 135(45), 16821–16824 CrossRef CAS PubMed.
- R. Zhang, Y. Zeng, M. Hu, J. Zhang, Y. Yang and H. Liu, et al., Electrochemical aptasensor with signal amplification strategy of covalent organic framework-derived carbon material for ultrasensitive determination of carbendazim, Microchem. J., 2024, 200, 110450 CrossRef CAS.
- M. Ibrahim, H. N. Abdelhamid, A. M. Abuelftooh, S. G. Mohamed, Z. Wen and X. Sun, Covalent organic frameworks (COFs)-derived nitrogen-doped carbon/reduced graphene oxide nanocomposite as electrodes materials for supercapacitors, J. Energy Storage, 2022, 55, 105375 CrossRef.
- N. Shakeel and J. Khan, Morphology-driven electrochemical attributes of Cu-MOF: a high-performance anodic material for battery supercapacitor hybrids, RSC Adv., 2024, 14(46), 33941–33951 RSC.
- J. Khan and A. Ahmed, Experimental and theoretical insights into benzene-1,4-dicarboxylic acid based Co-MOFs: an anodic material for expedient battery-supercapacitor hybrids, Sustainable Energy Fuels, 2024, 8(18), 4355–4364 RSC.
- S. Xu, J. Wu, X. Wang and Q. Zhang, Recent advances in the utilization of covalent organic frameworks (COFs) as electrode materials for supercapacitors, Chem. Sci., 2023, 14(47), 13601–13628 RSC.
- J. Khan, Zincronization-induced surface modification of CoMn phosphate for improved electrochemical performance in battery-supercapacitor hybrid systems, J. Energy Storage, 2024, 81, 110432 CrossRef.
- A. M. Khattak, Z. A. Ghazi, B. Liang, N. A. Khan, A. Iqbal and L. Li, et al., A redox-active 2D covalent organic framework with pyridine moieties capable of faradaic energy storage, J. Mater. Chem. A, 2016, 4(42), 16312–16317 RSC.
- D. K. Kulurumotlakatla, A. K. Yedluri and H.-J. Kim, Hierarchical NiCo2S4 nanostructure as highly efficient electrode material for high-performance supercapacitor applications, J. Energy Storage, 2020, 31, 101619 CrossRef.
- J. Khan and A. Ahmed, From design to efficiency: cobalt-based MOFs for efficient and stable electrocatalysis in hydrogen and oxygen evolution reactions, RSC Adv., 2025, 15, 8420–8429 RSC.
- T. Anitha, A. E. Reddy, Y. A. Kumar, Y.-R. Cho and H.-J. Kim, One-step synthesis and electrochemical performance of a PbMoO4/CdMoO4 composite as an electrode material for high-performance supercapacitor applications, Dalton Trans., 2019, 48(28), 10652–10660 RSC.
- Y. A. Kumar, K. D. Kumar and H.-J. Kim, A novel electrode for supercapacitors: efficient PVP-assisted synthesis of Ni3S2 nanostructures grown on Ni foam for energy storage, Dalton Trans., 2020, 49(13), 4050–4059 RSC.
- J. Gong, W. Luo, Y. Zhao, J. Wang, S. Wang and C. Hu, et al., Surface engineering of Ni wires and rapid growth strategy of Ni-MOF synergistically contribute to high-performance fiber-shaped aqueous battery, Small, 2022, 18(42), 2204346 CrossRef CAS PubMed.
- Y. A. Kumar, K. D. Kumar and H.-J. Kim, Reagents assisted ZnCo2O4 nanomaterial for supercapacitor application, Electrochim. Acta, 2020, 330, 135261 CrossRef CAS.
- N. Graf, E. Yegen, T. Gross, A. Lippitz, W. Weigel and S. Krakert, et al., XPS and NEXAFS studies of aliphatic and aromatic amine species on functionalized surfaces, Surf. Sci., 2009, 603(18), 2849–2860 CrossRef CAS.
- X. Kong, S. Zhou, M. Strømme and C. Xu, Redox active covalent organic framework-based conductive nanofibers for flexible energy storage device, Carbon, 2021, 171, 248–256 CrossRef CAS.
- L. Li, F. Lu, R. Xue, B. Ma, Q. Li and N. Wu, et al., Ultrastable triazine-based covalent organic framework with an interlayer hydrogen bonding for supercapacitor applications, ACS Appl. Mater. Interfaces, 2019, 11(29), 26355–26363 CrossRef CAS PubMed.
- L. Xu, F. Wang, X. Ge, R. Liu, M. Xu and J. Yang, Covalent organic frameworks on reduced graphene oxide with enhanced electrochemical performance, Microporous Mesoporous Mater., 2019, 287, 65–70 CrossRef CAS.
- Y. Zhu, J. Lao, F. Xu, L. Sun, Q. Shao and Y. Luo, et al., A structurally controllable flower-shaped phosphide derived from metal-organic frameworks for high-performance supercapacitors, J. Electroanal. Chem., 2024, 966, 118376 CrossRef CAS.
- B. Sun, J. Liu, A. Cao, W. Song and D. Wang, Interfacial synthesis of ordered and stable covalent organic frameworks on amino-functionalized carbon nanotubes with enhanced electrochemical performance, Chem. Commun., 2017, 53(47), 6303–6306 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01653c |
| This journal is © The Royal Society of Chemistry 2025 |