
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic carbonaceous materials: preparation methods and applications
Ming-ming Chen
 *a,
Cheng-yang Wang
a,
Masahiro Toyoda
b and
Michio Inagaki
c
*a,
Cheng-yang Wang
a,
Masahiro Toyoda
b and
Michio Inagaki
c
aKey Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300350, P. R. China. E-mail: chmm@tju.edu.cn
bFaculty of Science and Technology, Oita University, 700 Dannoharu, Oita, 870-1192, Japan
cProfessor Emeritus of Hokkaido University, 228-7399 Nakagawa, Hosoe-cho, Hamana-ku, Hamamatsu 431-1304, Japan
First published on 21st May 2025
Abstract
Although most carbon materials are hydrophobic, their oxidation would add hydrophilic functional groups onto their surfaces, making them amphiphiles. In this review, amphiphilic carbonaceous materials are reviewed, focusing on their preparation methods, compositions, and properties associated with their structural models. In addition, a variety of applications arising from their amphiphilicity are mentioned, including organic-free carbon coatings, fabrication of nanoporous structures in carbons, carbon supports for metal nanoparticles, environmental remediation, monitoring of toxic gases, dispersing agents, and biomedical applications.
 Ming-ming Chen | Mingming Chen earned her PhD in Chemical Technology from Tianjin University in China in 2003. Now, she is a Full Professor at Tianjin University and has been honored as the Chief Expert of a National 863 Project sponsored by the Chinese Government. She serves in the committee of Supercapacitor and Energy Storage Division in the Chinese Electrotechnical Society. Her research is primarily centered on renewable energy storage, focusing on the study of carbon-based and non-carbon energy storage materials. Her key research areas encompass the development of electrode and electrolyte materials for lithium–sulfur batteries, lithium-ion batteries, sodium-ion batteries, supercapacitors, and solid-state batteries. |
 Cheng-yang Wang | Chengyang Wang is a Professor at the School of Chemical Engineering and Technology, Tianjin University. He obtained his PhD degree from Hokkaido University in 1998. He has been engaged in research on pitch-based carbons since 1982 when he worked at the Institute of Coal Chemistry in China, focusing on studies of “Preparation, structure and properties of mesophase pitches and carbon fibers thereof”, “Thermal behavior of pitch during mesophase formation”, “Rheological characteristics of pitches” and so on. |
 Masahiro Toyoda | Masahiro Toyoda is a Professor Emeritus of Oita University in Japan. His research interests have been related to carbon materials and energy storage for the past 30 years. In 2006, he received an academic award from the Carbon Society of Japan for his research on the exfoliation of carbon fibers, and in 2012, he was awarded the Carbon Excellent in Review Award from the International Journal of Carbon. He has published more than 200 research papers and many reviews in journals and ten books, focusing on the sorption of heavy oils into carbon materials and carbon materials in photocatalysis. |
 Michio Inagaki | Michio Inagaki is a Professor Emeritus of Hokkaido University in Japan and is working on carbon materials for more than 60 years now. He won the SGL Carbon Award (2005) for his contribution to carbon science and technology and the Peter A. Thrower Award (2011) for exceptional contribution to the international carbon community. He has published more than 500 research papers and many reviews in journals, as well as the following books with Elsevier: “New Carbons-Control of Structure and Functions” (2000), “Materials Science and Engineering of Carbon: Fundamentals” (2014), “Advanced Materials Science and Engineering of Carbon” (2013), “Materials Science and Engineering of Carbon: Characterization” (2017), “Graphene: Preparations, Properties, Applications, and Prospects” (2020) and “Porous Carbons: Syntheses and Applications” (2022). |
1. Introduction
Most carbon materials are hydrophobic, as represented by all types of graphites, including synthesized graphite materials and natural graphites. Hydrophobicity of these materials endows them with excellent chemical stabilities. Owing to their chemical stability, or inertness, hydrophobic carbon materials have attracted significant attention and are widely studied in some new application fields, such as in the biomedical field.1,2 However, it has been recognized that carbon materials with hydrophilicity are required for certain applications. The modification of the surface of carbon is usually performed in solutions, mostly aqueous solutions.Some carbon materials can be well dispersed in alkaline aqueous solution and even in neutral solution and some organic solutions but precipitate in acidic solution. This can be understood as the amphiphilic nature of materials. Amphiphilic carbonaceous materials (ACMs) refer to a class of 2D materials rich in sp2 C domains and are composed of hydrophilic functional groups together with hydrophobic conjugated aromatic carbon domains, which are supposed to be inherited from their precursors. ACMs were first synthesized from a carbonaceous mesophase in 1987 and called “aqua-mesophase”.3 Owing to graphite oxides (GtOs) or graphene oxide (GnO)4 derived from graphite via various oxidation processes, ACMs recalled the attention of scientists almost three decades right after Fujii et al.3 firstly announced them and brought forward the concept of ACMs. From then on, scientists from several research fields have focused on all types of ACMs, mainly because of their processability in solutions, particularly in aqueous solutions.5
Some ACMs can be found in nature, for instance, humic acids. However, most ACMs are chemically synthesized from carbon-enriched precursors, including coal,6 pitch,7 green coke,8,9 biomass10 and graphite,11 via different preparation methods. Oxidation is an indispensable process and induces reasonable elemental composition changes, as summarized in Table 1. ACMs have particular bulk chemical composition. Compared with their pristine raw materials, ACMs have decreased carbon contents and much higher oxygen contents of approximately 30–40 wt% and 50–70 wt%, respectively. Although the carbon content is lower than that of the precursors, the C/H atomic ratio is still around 2 or higher but less than 3 owing to their abundant conjugated aromatic structure, distinguishing ACMs from common C- and O-based polymers. Besides the C/H ratio, the C/O atomic ratio of ACMs is between 2 and 3 (some are around 2). For example, in the study by Wang et al.,7 oxidation caused coal tar pitch to separate into two parts. Although both parts originated from the same precursor, the part possessing 53.1 wt% of C (C/H atomic ratio of 1.77 and C/O of 2.03) was water-dispersible, belonging to the ACM category, while the other part with 80.3 wt% C (C/H ratio of 3.34 and C/O ratio of 7.82) could not be dispersed in water and was not an ACM. Besides elemental analysis, when XPS was applied for surface characterization, it seems that the composition range was wide, as shown in Table 1. For example, in the study by Wang et al.,9 the C/O atomic ratio of ACMs obtained from green needle coke or calcined coke was calculated to be higher than 4, even approaching to 5 based on the published XPS data.
| By chemical conversion | Ref. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pristine precursor | ACMs obtained from the precursor | |||||||||||||
| Material | C | H | N | O | S | C/Ha | C | H | N | O | S | C/Ha | C/Oa | |
| a Atomic ratio.b Atomic percentage of elements determined using XPS.c Containing metallic ash of 0.21%, obtained after heating at 1000 °C for 2 h.d Containing metallic ash of 4.4%.e After purification by removing impurities of bio- or natural-ash. | ||||||||||||||
| Raw coke | 92.9 | 3.7 | 1.0 | 1.7 | 0.7 | 2.09 | 51.5 | 1.5 | 6.5 | 40.1 | 0.4 | 2.86 | 1.71 | 12 |
| Raw coke | 94.1 | 3.5 | 1.3 | 1.1 | 0 | 2.24 | 53.5 | 1.6 | 7.4 | 37.5 | 0 | 2.79 | 1.90 | 13,14 |
| Calcined coke | 96.2 | 2.96 | 0.23 | 0.19 | 0.42 | 2.71 | 80.6b | — | 2.8b | 16.3b | 0.3b | — | 4.94 | 9 |
| Green coke | 94.6 | 3.83 | 0.45 | 0.26 | 0.86 | 2.06 | 78.0b | — | 2.3b | 19.3b | 0.4b | — | 4.04 | |
| Coal tar pitch | 90.5 | 4.3 | 0.9 | 3.6 | 0.7 | 1.75 | 53.1 | 2.5 | 4.8 | 34.8 | 4.8 | 1.77 | 2.03 | 7 |
| Coal tar pitch | 57.5d | 3.1d | 1.0d | 29.8d | 4.2d | 1.55d | 2.57 | 15,16 | ||||||
| Petroleum cokec | 65.6 | 2.4 | 3.4 | 26.2 | 2.4 | 2.28 | 3.34 | 17 | ||||||
| Brown coal | 69.1 | 6.0 | — | — | — | 0.96 | — | 18 | ||||||
| Bituminous coal | 62.4 | 2.8 | 4.5 | 29.3 | 0.3 | 1.86 | 2.84 | 6 | ||||||
| Bituminous coal | 62.4 | 3.21 | 4.56 | 29.28 | 0.55 | 19 | ||||||||
In water, ACMs are independent and separated from each other, and thus regarded as primary particles. Outside water, they aggregate because of electrostatic attractions. AFM demonstrated that when draining the water out of an ACM-CP (ACMs derived from coal tar pitch) aqueous mixture, ACM-CP aggregated immediately as a ball with a width of 252 nm and thickness of 10 nm,7 as shown in Fig. 1a and b. Besides, GnO with lateral dimensions smaller than 100 nm was reported to stack like coins.20
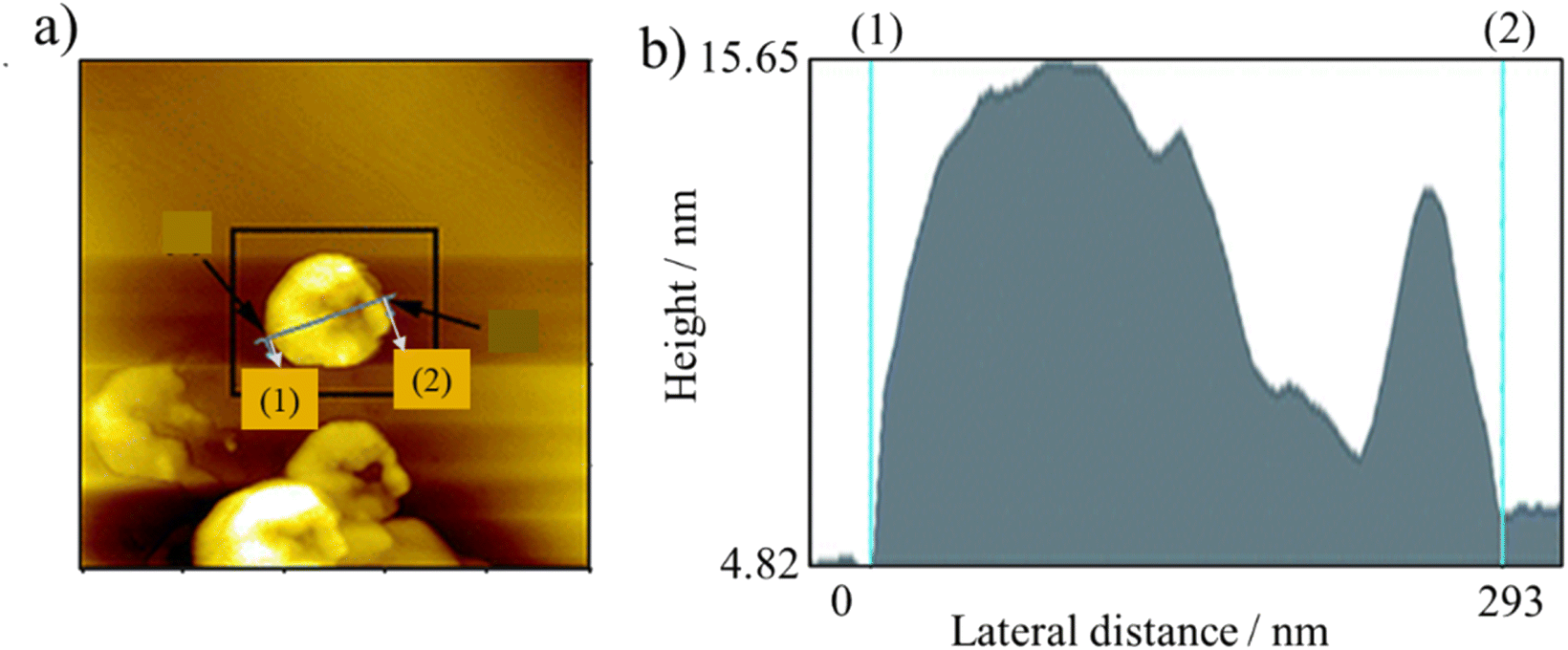 | ||
| Fig. 1 (a) AFM image in tapping mode of ACM-CP on a mica flake and (b) height distribution of the particle in (a).7 Reproduced from ref. 7 with permission from the Royal Society of Chemistry, copyright 2013. | ||
The size of the primary particles of ACMs, in terms of lateral dimensions and thickness, is the most important factor for achieving amphiphilicity. The lateral dimensions of ACMs cover a wide range from micrometers11 to less than 100 nm.20 Consequently, primary particles of ACMs have very different solution properties including surface activity11 and colloidal stability.20,21 GnO sheets with lateral dimensions of ∼8 μm are actually amphiphilic with an edge-to-center distribution of hydrophilic and hydrophobic domains. Thus, GnO can adhere to interfaces, and thus lower the interfacial energy, acting as a surfactant.11 In contrast, as reported by the same research group, aqueous GnO with lateral dimensions smaller than 100 nm was a stable nano-colloid, rather than a surfactant, owing to the high charge density originating from its high edge-to-area ratio. Furthermore, nano GnO can even act as a better dispersing agent for some insoluble materials (e.g., carbon nanotubes) in water, creating a more stable colloidal dispersion.20 Chen et al. also reported that the surface tension of ethylene glycol was only slightly reduced from 45.18 to 41.08 mN m−1 (ref. 21) when mixed with ACM-CP (small in lateral dimensions), demonstrating that small ACMs show low surface activity.
Besides size, the C/O atomic ratio definitely has some impact on amphiphilic behavior, which is a reasonable deduction. Logically, a lower C/O atomic ratio, such as the aforementioned 2–3, indicates sufficient hydrophilic O-containing functional groups per carbon surface for hydrophilic behavior, while a higher C/O atomic ratio more than 5 or 6 more-likely indicates hydrophobic behavior caused by the presence of hydrophobic carbon. Unfortunately, only a few publications clearly reported carbon materials with amphiphilic behavior together with their elemental composition and functional group categories and quantity. Thus, more detailed research is appealing, especially studies specifying each functional group and its quantity, together with the surface tension and critical micelle concentration values for the determination of amphiphilicity.
The simple preparation process in solution, and particularly the size-dependent properties of ACMs make them attractive for researchers and engineers because of their diversity and various possible applications. Here, the processes of the preparation of ACMs and their characteristics are reviewed and classified into three categories based on their raw materials. The first category is ACMs obtained from pitch including green coke. The second is ACMs derived from graphite and the third is ACMs obtained from biomass including coal. Their applications are also reviewed, focusing on the production of porous carbons, carbon-coated metal oxides, supports for metal nanoparticles, additives for crystal growth, modifiers for carbon materials, materials for environmental protection, etc.
2. Preparation and relevant characteristics
2.1 ACMs obtained from pitch and green coke
Raw coke (green coke in particular) is a suitable starting material for the synthesis of ACMs12,14,22,23 is satisfactory yields. However, coke calcined at around 1300 °C only produced a total yield of ACMs of less than 7%.9 Both isotropic pitch7,8 and anisotropic mesophase pitch24 were reported to be converted to ACMs although the resultant ACMs differed in chemical composition from that derived from raw coke, as shown in Table 1. The procedures for the preparation of ACMs are shown as a flow diagram in Fig. 2. Raw coke, for example, is oxidized in a mixed acid of concentrated H2SO4 and HNO3 (7/3 in volume) at 80 °C for 3 h under stirring. Then, the mixture can be poured into cold water to precipitate crude ACMs. The crude ACMs can be purified by firstly dissolving them in a 1 M NaOH aqueous solution, and secondly adding 1 M HCl to the decanted supernatant. Finally, purified ACMs are obtained by filtering and repeatedly washing the precipitates with de-ionized water. The yield of ACMs using this procedure is about 30 wt%.12,14 | ||
| Fig. 2 Flow diagram for the preparation of ACMs. | ||
Two ACMs, ACM-CP and ACM-GC, were prepared from a coal tar pitch (CP) and green needle-like coke (GC), respectively. They were subjected to structural investigation by FTIR, XPS, TPD, etc. It is noticeable that both nitrogen and oxygen atoms were introduced in both ACMs during oxidation, as shown in Table 1. The FTIR and N 1s XPS spectra shown in Fig. 3 demonstrate that the N introduced into these two ACMs existed in different chemical states, similar to the introduced O. The FTIR band located at 1720 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the carboxylic group, whereas C–O in the carboxylic and ether groups is identified by the peak at 1200 cm−1 in the fingerprint region. The –NO2 group chemically bonded with aromatic rings can be characterized by the peaks at 1530 and 1345 cm−1, corresponding to the asymmetric and symmetric stretching vibrations, respectively. All the data (Fig. 3a) suggest the successful introduction of N and O by oxidation. In addition, deconvolution of the N 1s XPS spectra showed that there are three types of C–N bonds existing in both ACMs, that is, cyanide functional group (C
O stretching vibration of the carboxylic group, whereas C–O in the carboxylic and ether groups is identified by the peak at 1200 cm−1 in the fingerprint region. The –NO2 group chemically bonded with aromatic rings can be characterized by the peaks at 1530 and 1345 cm−1, corresponding to the asymmetric and symmetric stretching vibrations, respectively. All the data (Fig. 3a) suggest the successful introduction of N and O by oxidation. In addition, deconvolution of the N 1s XPS spectra showed that there are three types of C–N bonds existing in both ACMs, that is, cyanide functional group (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N: the N atom gets bonded to a C atom at the edge of the layer plane), as suggested by the peak located at 398.2 ± 0.5 eV (pyrrolic N 1s), N atoms replacing C atoms in the layer plane by the peak of graphitic N 1s at 400.7 ± 0.5 eV, and chemisorbed nitrogen oxide species by peak nitrogen oxide N 1s at 405.4 ± 0.2 eV (Fig. 3b).
N: the N atom gets bonded to a C atom at the edge of the layer plane), as suggested by the peak located at 398.2 ± 0.5 eV (pyrrolic N 1s), N atoms replacing C atoms in the layer plane by the peak of graphitic N 1s at 400.7 ± 0.5 eV, and chemisorbed nitrogen oxide species by peak nitrogen oxide N 1s at 405.4 ± 0.2 eV (Fig. 3b).
 | ||
| Fig. 3 ACMs derived from coal tar pitch (CP) and green coke (GC): (a) FTIR and (b) N 1s XPS spectra.7,9 Reproduced from ref. 7 with permission from the Royal Society of Chemistry, copyright 2013. Reproduced from ref. 9 with permission from Science Press, copyright 2015. | ||
In ACM-CP (prepared from coal tar pitch), most of the N atoms are bonded to C atoms mainly in pyridinic N, together with graphitic and pyrrolic N and a relatively small amount of nitrogen oxides. Alternatively, in ACM-GC (prepared from green coke), large amounts of chemisorbed nitrogen oxides were detected, and only a small amount of graphitic and pyrrolic N were identified. These two ACMs also varied in terms of O-containing species and their quantities, as demonstrated by their temperature programmed desorption (TPD) profiles in Fig. 4.25 ACM-GC and ACM-CP contained comparable amounts of epoxy and carboxyl groups. However, the content of anhydride in ACM-GC was found to be almost twice that in ACM-CP. In addition, a considerable amount of lactone and a small amount of thermal-stable quinone or carbonyl appeared in ACM-GC. The difference in chemical composition between ACM-GC and ACM-CP may have resulted from their precursors. Green coke is reasonably considered to consist of a larger amount of aromatic conjugated structures than coal tar pitch because of the high temperature coking, which prevents N and O from directly bonding with C in green coke. The large amount of nitrogen oxides in ACM-GC (Fig. 3b) is due to the presence of a variety of surface functional groups in the pristine green coke, as evidenced by the FTIR spectra in Fig. 3a.
 | ||
| Fig. 4 TPD profiles of (a and b) ACM-CP25 and (c and d) ACM-GC. Reproduced from ref. 25 with permission from Elsevier Ltd, copyright 2016. | ||
All ACMs are dispersible in water, but they have different particle size distributions. The hydrodynamic diameter of the ACM-CP and ACM-GC particles in alkaline solutions was measured by dynamic light scattering (DLS). ACM-CP consisted of particles smaller than 200 nm (mainly around dozens of nm), whereas ACM-GC contained a broad distribution of large particles bigger than 10 μm although it formed a stable dispersion. The stable dispersion of these ACMs can be attributed to their ionization and negatively charged particles. The zeta potential at pH 6.7 was measured to be −61.3 mV for ACM-CP and −51.8 mV for ACM-GC. Thus, compared to the pristine coal tar pitch, which possessed a zeta potential of −5.2 mV, the zeta potentials of these two ACMs ensured that they formed water-stable colloids.7
From another perspective, it can be inferred from the above-mentioned size distributions combined with the inherent nature of the precursors that ACM-GC and ACM-CP must have different lateral sizes, although the authors did show any visual images of them. Besides, their edge-to-area ratios were different, which could be objectively deduced from their zeta potentials. ACM-CP exhibited a higher edge-to-area ratio than ACM-GC although the synthesis conditions were same for these two ACMs. Despite these differences, both ACM-CP and ACM-GC were reported to be stable in aqueous colloids. However, this does not mean that oxidation will make all the resulting materials stable colloids. For example, Wang et al.7 reported the preparation of an oxidation-synthesized material possessing 80.3 wt% C (C/H ratio of 3.34 and C/O ratio of 7.82), which could not be dispersed in water (not amphiphilic). From our viewpoint, to form a colloidal stable, both the lateral size and edge-to-area ratio should be within a certain range. The lateral size affects the hydrophobicity given that the lateral moieties are composed of π–π* conjugated carbon rings. Alternatively, the edge-to-area ratio affects the hydrophilicity, which is induced by the ionization of the functional groups located at the edge. Thus, these two aspects should be balanced to achieve amphiphilicity. However, the threshold cannot be derived from the existing publication at present.
Based on these measurements, a structural model for ACM-CP was proposed, as shown in Fig. 5.21 ACM-CP is composed of small flaky particles, in which polyaromatics constitute the main hydrophobic moieties, while surface functional groups containing heteroatoms such as O, S and N act as hydrophilic moieties, which guarantee its stable dispersion in aqueous solution. However, when removed from solvents, they have a strong tendency to aggregate, probably resulting from the π–π* conjugation of the hydrophobic moieties.7
 | ||
| Fig. 5 Structural model for ACM-CP.21 Reproduced from ref. 21 with permission from the American Chemical Society, copyright 2014. | ||
ACMs could be changed to a gel in NH3·H2O solution at 25 °C, where the gelation time strongly depended on the concentration and pH of the ACM solution, decreasing markedly with an increase in the concentration at the pH of 11.5 than at 13.0, as shown in Fig. 6.12 The carbonaceous gel of ACM became a sol above 70 °C, but it reversed to a gel at room temperature. By adding urea to the ACM solution, the gelation time became longer, being prolonged to 4 and 9 days by the addition of 0.3 and 0.6 mol per dm3 urea, respectively. Alternatively, the addition of 0.1 mol per dm3 glyoxal reduced the gelation time to a quarter of that of the solution without any additives. This result suggests the formation of a hydrogen-bond network in the ACM gels, which was probably accelerated by the carboxyl and hydroxyl groups present in the ACM.14 Additionally, the gelation of ACMs is possible in polar organic solvents, such as N,N-dimethylacetamide (DMAc), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and ethylene glycol.14,26
 | ||
| Fig. 6 Effects of concentration of ACM and pH on the gelation time of a mesophase-pitch-derived ACM in an NH3·H2O aqueous solution at 25 °C.12 Reproduced from ref. 12 with permission from Elsevier Ltd, copyright 1991. | ||
The ACM prepared from raw coke and its dry gel (prepared in NH3·H2O) were studied for their high-temperature behaviors up to 2800 °C by thermal analyses (DTA and TG) up to 600 °C.14 Their TG and DTA curves are shown in Fig. 7a and b, respectively. The ACM and its dry gel exhibited similar curves, showing a broad DTA peak at around 330 °C, which probably due to the elimination of nitro, carboxylic and anhydride functional groups. However, their structural changes with high temperature treatment were different in terms of the d002 interlayer spacing and crystallite size along the Lc(002) c-axis, as determined by XRD and shown in Fig. 7c.14 Specifically, the structural improvement, in other words, graphitization, was depressed in the ACM, and even more markedly in its dry gel. The severe oxidation and quite random aggregation of nano-sized ACM particles in the dry gel seemed to strongly interrupt the essential structural change to graphite. The results showed that the resulting dry gel had a non-graphitizing nature, although the pristine raw coke has graphitizing nature. The dry gel prepared in organic media, such as DMF (organo-dry gel), exhibited slightly better graphitization compared to the dry gel prepared in NH3·H2O, but still far from that of the pristine raw coke.27
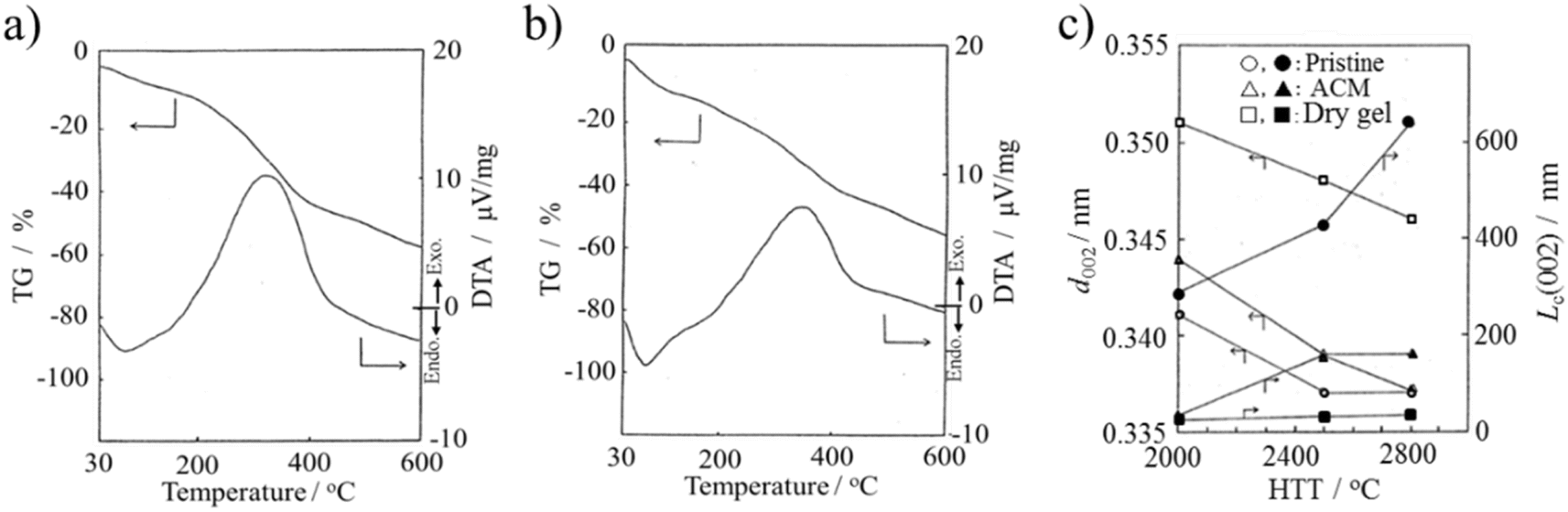 | ||
| Fig. 7 ACMs prepared from raw coke: TG and DTA curves (10 °C min−1) for (a) ACM and (b) its dry gel prepared in NH3·H2O, and (c) changes in d002 interlayer spacing and Lc(002) crystallite size with heat treatment temperature for the pristine raw coke, ACM and its dry gel.14 Reproduced from ref. 14 with permission from the Carbon Society of Japan, copyright 1993. | ||
2.2 From graphite
Graphite is oxidized by strong oxidizing agents such as KMnO4 in concentrated H2SO4. This process is usually called Hummers' method.28 By applying this procedure carefully, particularly under strong stirring or sonication, complete exclusion of the residual MnO4− and MnO2, and using supernatant, other types of ACMs can be generated, namely, few-layered graphene oxide (GnO) or even single graphene layers.Few-layered GnO contains carboxylic groups at the edges of its flakes, while phenol, hydroxyl and epoxide groups are mainly located at its basal plane. One of the models proposed for GnO is shown in Fig. 8a. The apparent thickness of GnO flakes is about several nm, while their lateral sizes are on the scale of common colloidal particles, ranging from nanometers to micrometers.11 Thus far, similar processes have been used in industry to produce exfoliated graphite and flexible graphite sheets.29
 | ||
| Fig. 8 Chemical structure models of (a) graphene oxide (GnO) and (b) reduced GnO (rGnO).11 Reproduced from ref. 11 with permission from De Gruyter as part of a partnership with IUPAC, copyright 2011. | ||
Reduced graphene oxide (rGnO) was prepared via the further reduction of GnO. A model of rGnO is shown in Fig. 8b. The lateral sizes of both GnO and rGnO flakes could be tuned by selecting their starting precursors and oxidation time.20,30 It must be noted that the oxidation–exfoliation–reduction process turns the super-hydrophobic graphite powder into hydrophilic analogues including GnO and some rGnO. Thus, this process has attracted attention as a route to prepare a large quantity of graphene flakes.4,31–35 Although the resultant graphene layers were defective, as illustrated in Fig. 8b, most of the π-conjugated carbon networks of the precursors were restored in the basal plane, thus resulting in partially recovered e-conductivity.
GnO flakes prepared through Hummers' method contain only a negligible amount of N atoms, which may be a characteristic of rGnOs in different ACMs. However, it is possible to dope N into GnO flakes using mixed acids of H2SO4 and HNO3, which is known as Brodie's method,36 although it is rarely applied for the synthesis of GO.
GnO has long been considered hydrophilic due to its excellent water dispersibility. However, it is actually amphiphilic with an edge-to-center distribution of hydrophilic and hydrophobic functional groups.11,37 Its amphiphilicity varies with pH due to the change in the ionization degree at its edge groups such as –COOH. As shown in Fig. 9, the zeta potential of GnO decreased with an increase in pH value. Specifically, its charge density increased with a decrease in its size from micrometers to less than 100 nm, and also with a decrease in the degree of its reduction (from few-layer rGnO to GnO). This suggests that the amphiphilicity of GnO is tunable not only by the solution pH value, but also by size of the dispersed particles and their reduction degree.
 | ||
| Fig. 9 Changes in the zeta potential with pH for GnOs.11 Reproduced from ref. 11 with permission from De Gruyter, as part of a partnership with IUPAC, copyright 2011. | ||
GnO flakes could be formed into thin film by drop-casting an aqueous dispersion GnO onto an Si3N4/SiO2 substrate38 or ITO-coated glass substrate.39 Once GnO shifted from separated flakes to a film, the measurement of its electrical property became feasible.39 The AC conductivity (σAC) of the GnO film at different temperatures in the range of 25–100 °C was found to strongly depend on both temperature and frequency ω, as shown in Fig. 10a and b, respectively. Pressure-assisted self-assembly is another method employed for the fabrication of amphiphilic GnO membranes.40 A porous PAN membrane was prepared by casting an N-methyl-2-pyrrolidone (NMP) solution with 15 wt% polyacrylonitrile (PAN) onto a nonwoven polyester fabric, the surface of which was hydrolyzed using 2 M NaOH aqueous solution at 50 °C to enhance its hydrophobicity. A 400 ppm GnO aqueous suspension was placed on the porous PAN membrane in a Buechner funnel, and then pressurized under 5 kg cm−2 to get self-assembled GnO and porous PAN composite membranes. Vacuum filtration is also helpful for composing almost GnO paper from colloidal suspensions.41 The resulting GnO sheets possessed the mean lateral dimensions of approximately 1 μm and thicknesses ranging from 1 to 25 μm. The stress–strain curve for the film under tension, as shown in Fig. 11a, is divided into three regions, where region I is due to straightening of the GnO flakes, region II is due to elastic deformation and region III is due to plastic deformation of the film. In region I, certain residual strain was observed after unloading, as shown in Fig. 11b. The modulus of the GnO film was determined to be 32 GPa as the average and 4262 GPa as the highest value.
 | ||
| Fig. 10 AC conductivity (σAC) of a GnO film: (a) temperature dependence at 100 Hz and (b) frequency, ω, dependence at different temperatures.39 Reproduced from ref. 39 with permission from Elsevier Ltd, copyright 2015. | ||
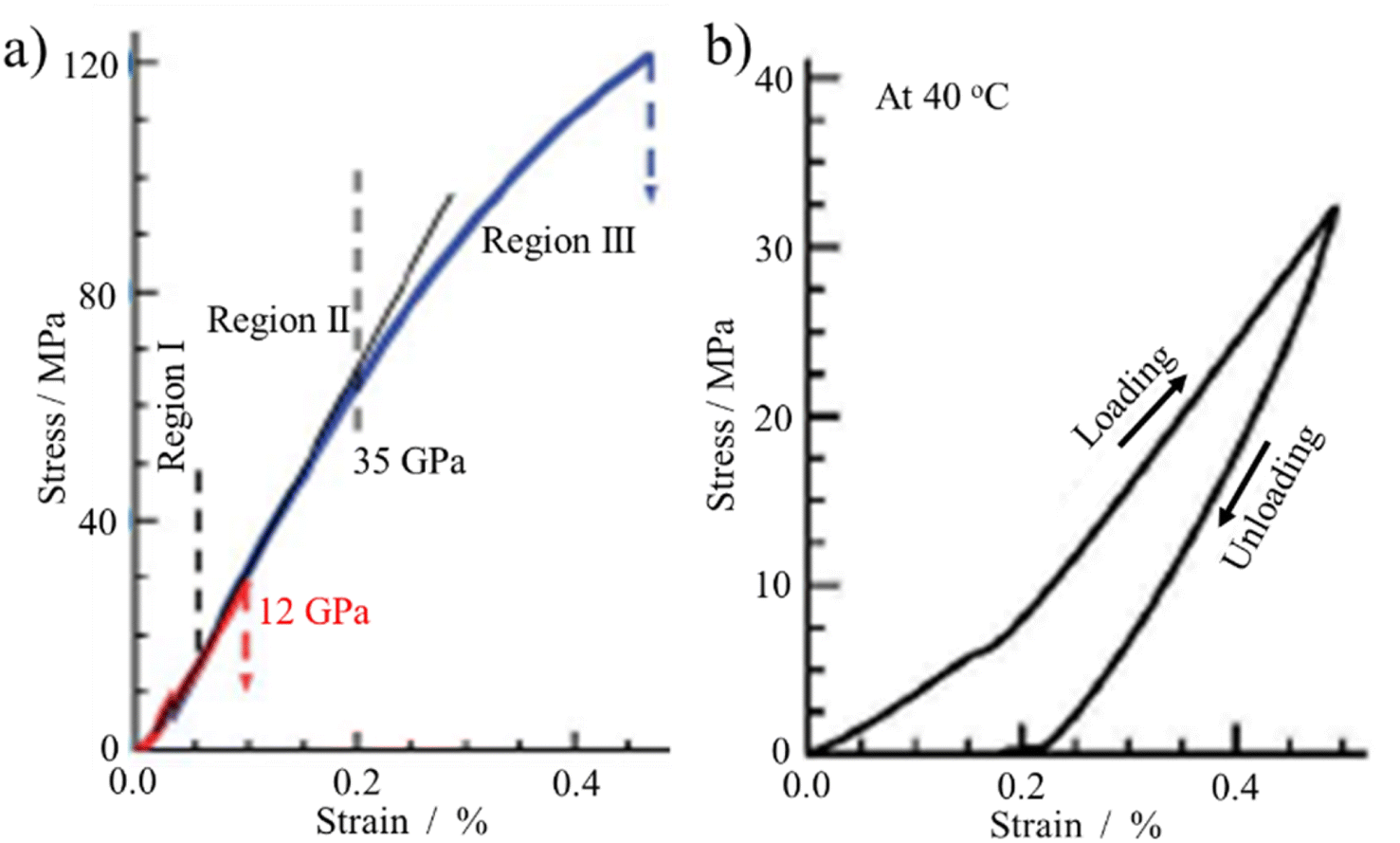 | ||
| Fig. 11 Stress–strain behavior of GnO films under tension: (a) stress–strain curve and (b) loading-unloading in region I in (a).41 Reproduced from ref. 41 with permission from Springer Nature Publishing Group, copyright 2007. | ||
The functional groups in GnO can be further modified by either thermal treatment or microwave irradiation, resulting in the modification of its amphiphilicity.42 The XPS spectra of the thin GnO film on an Si3N4/SiO2 substrate and after annealing at 500 °C are compared in Fig. 12.38 On the as-deposited film, the peaks assigned to C–OH, CO and OC–OH bonds are clearly observed in both the C 1s and O 1s spectra, in addition to that for the CC and C–C bonds in the C 1s spectra. Upon annealing at 500 °C in an Ar/H2 atmosphere, aiming to reduce the GO film, the peaks corresponding to C–OH and CO bonds were still clearly observed. Solvothermal treatment is another type of thermal annealing.43 The hydrophilicity of GnO flakes could be enhanced by mixing their aqueous suspension with alkylamine in ethanol, followed by treatment at 90 °C in a Teflon-lined autoclave. GnO flakes in a diluted ammonia aqueous solution with pH of about 10 were discretely irradiated by microwaves at 450 W for 1–20 min at intervals of 30 s to exclude thermal effects and keep the solution at 30 °C.42 OC–OH group is the most sensitive group to microwave. By irradiation, the OC–OH groups decreased drastically, while the other groups decrease gradually, as shown in Fig. 13a and b. As a result of the reduction of the OC–OH groups, the hydrophilicity decreased, and then the water uptake decreased to about a half of that before microwave irradiation. The contact angle of water droplets also increased from −29.7° to −69.9°.
 | ||
| Fig. 12 XPS spectra for GnO films prepared using Hummers' method after deposition on Si3N4/SiO2 substrate and after heat-treatment at 500 °C: (a) C 1s and (b) O 1s spectra.38 Reproduced from ref. 38 with permission from Elsevier Ltd, copyright 2010. | ||
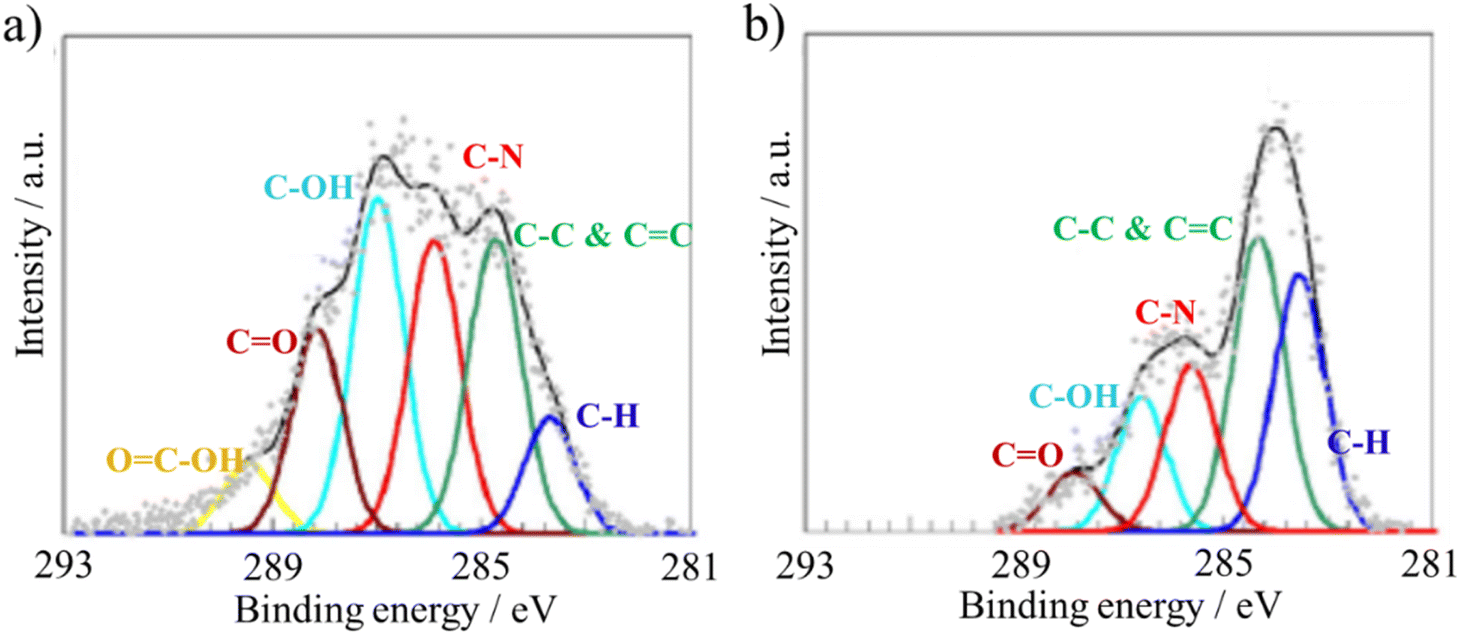 | ||
| Fig. 13 XPS C 1s spectra of (a) as-prepared GnO film and (b) after 450 W microwave irradiation for 20 min 42. Reproduced from ref. 42 with permission from Elsevier Ltd, copyright 2015. | ||
Graphene-based amphiphilic Janus nanosheets (AJNs) were prepared through temporary immobilization on the surface of starch microspheres.44 In an aqueous dispersion of starch powder (derived from white tapioca), an aqueous dispersion (1 mg mL−1) of GnO prepared using H2SO4, KMnO4 and H2O2 was slowly added and stirred for 8 h at room temperature to get starch microspheres coated by GnO (GnO-immobilization) by hydrogen bonds. After washing with water and ethanol, the GnO-coated starch microspheres were mixed with an absolute ethanol solution of alkylamine, and then mildly stirred for 12 h at room temperature to complete the amination of the GnO surfaces on the starch microspheres. After washing with ethanol, the resultant microspheres were again dispersed in absolute ethanol and alternately subjected to sonication and heating. The supernatant of the fluid system was separated from the starch microspheres (precipitated) by filtration, in which AJNs were dispersed. The procedure for the synthesis of the graphene-based AJNs is illustrated in Fig. 14. The wetting nature of the surface of the resultant AJNs is demonstrated in Fig. 15a and b. The surface facing starch was hydrophilic because it was basically the unmodified GnO surface, while the surface modified by alkylamine was hydrophobic. Janus sulfonated graphene oxide nanosheets (JSGO) were synthesized in a GnO aqueous dispersion with a kerosene solution of octadecylamine (ODA) and aminopropyl sulfonic acid (ASP) aqueous solution of pH 7–8 under stirring for 18 h at room temperature.45 The modified GnO sheets (JSGO nanosheets) were obtained after washing with ethanol and deionized water, followed by vacuum freeze-drying. The contact angle of a water droplet on the hydrophobic surface of JSGO was 102°, which is much larger than that of 37° on the hydrophilic surface of JSGO, as shown in Fig. 15c and d. These Janus and amphiphilic characteristics could further enable the formation of some special assemblies with unique capabilities. For example, a spongy polyvinyl alcohol/Janus graphene oxide (J-GnO) hybrid hydrogel (SPJH) was reported to be assembled by the aid of the strong interfacial interactions between hydrogen bonds and hydrophobic interactions.46 The strong interfacial interactions strengthened the hierarchical microstructure of the SPJH, permitting its use even in harsh environments. The SPJH possessed excellent photothermal conversion ability, exhibiting a solar absorption rate of about 99.7%.
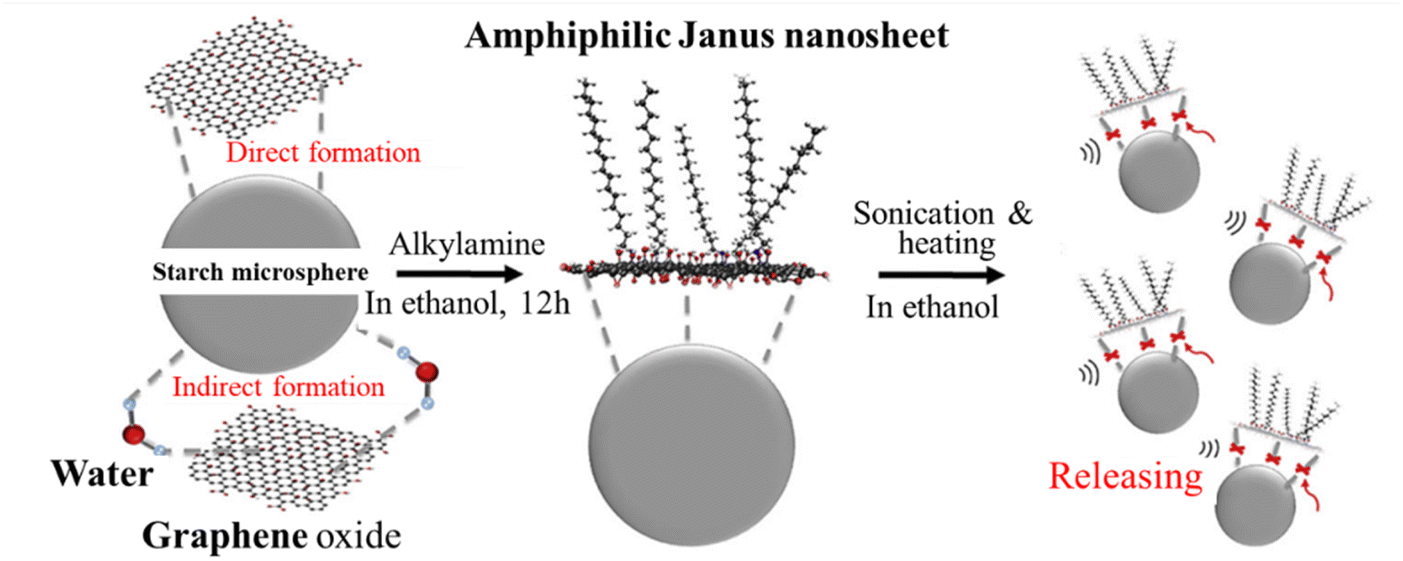 | ||
| Fig. 14 Illustration of the synthesis of graphene-based AJNs using temporary substrates of starch microspheres.44 Reproduced from ref. 44 with permission from Elsevier Ltd, copyright 2018. | ||
 | ||
| Fig. 15 Contact angles of water droplets on surfaces of AJN (a and b)44 and JSGO (c and d).45 (a and c) are on hydrophilic surfaces while (b and d) are on hydrophobic surfaces. Reproduced from ref. 44 with permission from Elsevier Ltd, copyright 2018; reproduced from ref. 45 with permission from Elsevier Ltd, copyright 2022. | ||
2.3 From biomass and coal
The acidic components in bio-degraded biomass are called humic acids.47,48 In this case, supercritical fluid CO2 technology helps to efficiently remove non-humic substances from biomass and only leave humic acid with hours. In comparison, the traditional extraction requires several days and sequentially utilizes ether, acetone, ethanol and dioxane as extraction solvents.49 Humic acids are mainly composed of carbon (52–56%), hydrogen, nitrogen and oxygen. They are complex mixtures of many different acidic compounds containing carboxyl and phenol groups, not a single acid.10,50 Given that they are soluble in alkaline aqueous solution and precipitated by acidic solution, humic acids are essentially amphiphilic.The structure model shown in Fig. 16 was proposed for a humic acid having the composition of C308H328O90N5 with the molecular weight of 5540 Da based on various structural analyses including spectroscopic, thermal and colloid chemical, together with electron microscopy.51 This model represents the structural characteristics of humic acid, i.e., assembly of covalently linked aromatic and aliphatic moieties, carrying carboxyl, phenolic and alkoxy groups. From a structural point of view, naturally occurring humic acids have less aromatic moieties than the ACMs derived from pitch and raw coke, although they all are amphiphilic. The atomic C/H ratio of ACMs derived from coal and biomass is lower than 1.9, mostly in the range of 0.77 to 1.86, whereas the C/H ratio of ACMs from coke is above 2.28 up to 2.86 (Table 1). This obviously suggests that the polyaromatic moieties in ACMs derived from coke and pitch are more predominant than that in humic acids.
 | ||
| Fig. 16 Structural model for humic acid with the chemical composition of C308H328O90N5.51 Reproduced from ref. 51 with permission from Springer Berlin Heidelberg, copyright 1993. | ||
In fact, humic acids vary in terms of ratio of aromatic (rigid) segments to linear (flexible) aliphatic segments. Synchronous scan fluorescence was used to characterize the differences in aromaticity among six humic acids.52 Besides synchronous scan fluorescence, FTIR can also be a guide, through which Qiao et al. found that leonardite humic acid (LHA) differs from biotechnology humic acid (BHA).17 LHA has more aromatic and cyclic hydrocarbons than BHA, which was identified by the broad band at 1605 cm−1 (aromatic CC stretching) in its FTIR spectrum, as shown in Fig. 17, while BHA has a more lignocellulose-type structure, presenting infrared absorption bands at 1050–1150 cm−1 and 1456 cm−1. These micro-segmental differences account for their different TG behaviors.17 The pyrolysis of LHA was gradual, finally leaving 60.1 wt% residual mass at 1000 °C. However, BHA was sequentially pyrolyzed, firstly in a low temperature (<400 °C) range, and then in a high temperature range (600–800 °C), resulting in 38.2 wt% residual mass.
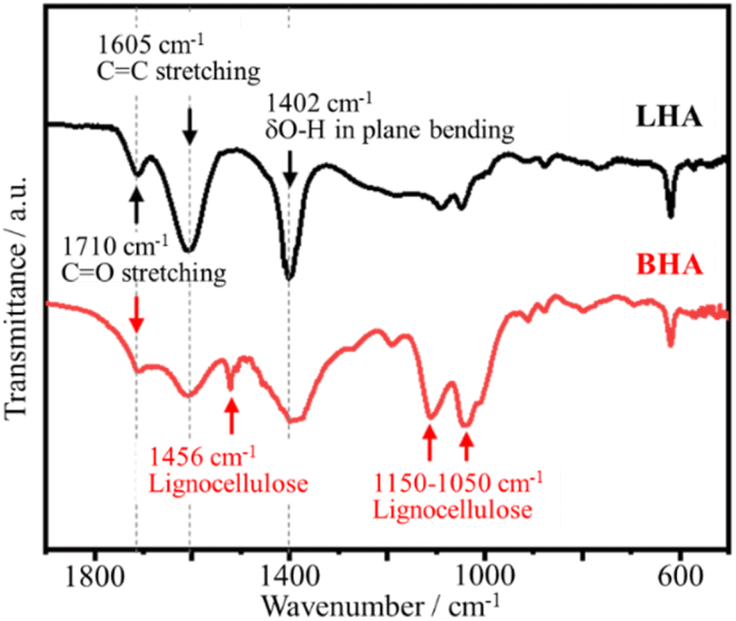 | ||
| Fig. 17 FTIR spectra of biotechnology humic acid (BHA) and leonardite humic acid (LHA).17 Reproduced from ref. 17 with permission from Elsevier Ltd, copyright 2014. | ||
The pyrolysis residual mass of humic acids cannot be denoted as carbon residual mass given that most humic acids come from nature, which inevitably results in the introduction of non-carbon impurities. For example, metallic ash of 2.13 and 3.17 wt% was reported in LHA and BHA, respectively, while a humic acid isolated from lignite was confirmed to possess 5.22 wt% ash.53 Thus, additional purification becomes necessary before a certain application (mentioned later).
The polycondensation of low-molecular-weight compounds is a feasible way to prepare humic acids. In this case, amino acids, sugars and phenols are the preferred precursors and catalysts of clay minerals such as nontronite,54 kaolinite and natural zeolites55 are necessary. When the precursor changes to high-molecular-weight species such as coal of the high volatile bituminous rank,19,56,57 brown coal,18,58 and even lignite,59,60 analogous synthesis procedures are performed, including partial oxidation followed by extraction with alkali solution and precipitation with dilute acid. The type of coal precursor accounts for the aromatic carbon content in humic acids, which is as high as 65–75% and higher than that from low-molecular-weight precursors. Besides, the elemental composition varies not only because of the precursor but also due to the use of oxidation agents. Sulfuric acid left sulfur in the resulting humic acid, increasing its S content from originally 14.5 to 32.5 mg g−1.57 Fungal-transformation is responsible for the increase in N content of 47.36% compared to the pristine lignite.60 Regardless of the method used, the yield of the humic acid is reported to be much higher than that of ACMs derived from pitch and coke, even reaching 89 wt%.19
Oxidation, especially long-term atmospheric oxidation, has a vital effect on the chemical composition of humic acids. For example, in pyrite-free bituminous coal weathering (lasting 15 days) to humic materials,61 based on the analysis of the resulting pyrolyzed humid acid, it was found that the CO2 evolution in the range of 150–450 °C was up to 13.9 wt%, while the total CO2 evolution up to 950 °C was 20.1 wt%; the CO evolution was less than 0.2 wt%, while the total CO was 2.1 wt%.6 Neither fusion nor evolution of volatile aromatic fragments was observed. In total, 13 wt% carbon in the humic acid was converted to volatile species.
The zeta potential of humic acids represents their degree of ionization due to the presence of hydrophilic surface groups in aqueous solution. Therefore, zeta potential has been used as a measure for the hydrophilicity of humic acids, depending on the humic acid concentration in aqueous solutions.53 As shown in Fig. 18a, the zeta potentials of LFA, BHA and LHA at pH = 11 decreased rapidly with an increase in their concentration up to 0.2 g L−1, for example, −28.4 mV for BHA at 0.05 g L−1 and −50.3 mV at 0.2 g L−1. When the concentration increased above 0.2 g L−1, the zeta potentials of leonardite fulvic acid (LFA), LHA and BHA at pH = 11 all became lower than −45 mV, which is a common criterion for stable colloids. In addition, after 0.2 g L−1, their changing tendencies plateaued. In comparison with petroleum coke-based ACMs (denoted as ACM in Fig. 18) and ACM-CP,7 humic acids have higher charge density, and thus better hydrophilicity. Below 0.2 g L−1, the pH significantly influences the ionization of humic acid, illustrated by LFA in Fig. 18.
 | ||
| Fig. 18 Changes in zeta potentials with the concentration of aqueous solutions of leonardite humic acid (LHA), biotechnology humic acid (BHA), and leonardite fulvic acid (LFA) in comparison to that of a coke-derived ACM.53 Reproduced from ref. 53 with permission from Elsevier Ltd, copyright 2014. | ||
2.4 From organic precursors
Some organic chemicals, those in the form of ring-type structure in particular, were reported as perfect precursors for amphiphiles. However, when these chemicals are employed, a surfactant seems to be indispensable for the preparation of ACMs.Sulfonated carbon with amphiphilic properties was synthesized via the hydrothermal carbonization of a mixture of 8 g of furfural, 8 g of sodium dodecylbenzene sulfonate (SDBS) and a small amount of sulfuric acid at 185 °C for 24 h in an autoclave. The obtained sulfonated carbon (coded as F–S) possessed both hydrophobic segments (long flexible carbon chains) and hydrophilic parts (O-containing functional groups).62 However, in the absence of the SDBS surfactant, only hydrophilic carbon (coded as F–T) was obtained due to the presence of O-containing functional groups. The possible structure models for F–S and F–T are illustrated in Fig. 19.
 | ||
| Fig. 19 Possible structural models for (a) F–S and (b) F–T.62 Reproduced from ref. 62 with permission from the Royal Society of Chemistry, copyright 2014. | ||
Amphiphilic carbon dots (ACDs) were synthesized from nonionic surfactants, commercially available alkyl glycosides with different alkyl chains, APG06, APG10, and APG1214, by heating at 180 °C for 9 h, followed by dissolving in xylene, and then centrifuging at 8000 rpm for 10 min to remove any traces of undissolved material (mainly APG1214). After drying at 60 °C for 24 h, ACDs in shape of powder (coded as C6-ACD, C10-ACD and C1214-ACD) were obtained.63 The TEM image of ACDs and their particle size distribution are shown in Fig. 20a and b, respectively, exhibiting a uniform dispersion without apparent aggregation. C1214-ACD demonstrated a distinct lattice structure with a periodicity of 0.22 nm, as shown in the inset of Fig. 20a. The resultant ACDs had similar surface tension curves as conventional surfactants, as shown in Fig. 20c. Their surface activity depended on the alkyl chain length of the precursor APGs. With an increase in the ACD concentration, the surface tension of the aqueous ACD solutions gradually decreased and became constant through a breakpoint. The critical micelle concentration (CMC) of C6-, C10-, and C1214-ACDs calculated from the breakpoint was 4000.00, 1500.00, and 40.00 mg L−1 with the surface tension values of 30.58, 30.14, and 27.18 mN m−1, respectively. In another report, ACDs were synthesized using the cationic surfactant cetylpyridinium chloride, anionic surfactant SDBS, and nonionic surfactant APG12 with MWCNTs.65 2.0 g of citric acid and 2.0 g of dodecylamine were dissolved in 50 mL of absolute ethanol and heated at 180 °C for 3 h, followed by centrifuging at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min to remove the undissolved material. The product was dissolved in ultrapure water after removing ethanol by reducing the distillation pressure and dialyzing with a 1 kDa dialysis membrane for 12 h. Finally, C12-ACDs were obtained. C6-ACD, C8-ACD and hydroxyl carbon dot H-ACD were synthesized by replacing dodecylamine with hexylamine, n-octylamine and ethylenediamine, respectively. To enrich the surface functional groups of ACDs, C12-ACDs were further sulfonated by a sulfamic acid-methanol binary mixture, followed by reflux at 70 °C for 5 h (C12-SACDs). The particle sizes of C12-SACDs were distributed in the range of 1.0–4.0 nm, and the average sizes of C12-ACD and C12-SCD were calculated to be 2.6 and 2.0 nm, respectively. The typical thickness of C12-ACD and C12-SACD were measured to be about 1.80 and 1.73 nm, respectively. Sulfonation resulted in a slight change in the particle size and morphology of the ACD particles. The surface tension curve for C12-ACD is similar to that of the C6-, C10- and C1214-ACDs (Fig. 20c). A comparison with the pristine surfactants, nonionic APG12, anionic-SDBD and H-ACD, is shown in Fig. 20d. The CMCs of C6-, C8-, and C12-ACDs were determined to be 5,000, 2,500, and 100 mg L−1, respectively. Their surface tension was 37.82, 34.62, and 27.45 mN m−1, respectively.
000 rpm for 15 min to remove the undissolved material. The product was dissolved in ultrapure water after removing ethanol by reducing the distillation pressure and dialyzing with a 1 kDa dialysis membrane for 12 h. Finally, C12-ACDs were obtained. C6-ACD, C8-ACD and hydroxyl carbon dot H-ACD were synthesized by replacing dodecylamine with hexylamine, n-octylamine and ethylenediamine, respectively. To enrich the surface functional groups of ACDs, C12-ACDs were further sulfonated by a sulfamic acid-methanol binary mixture, followed by reflux at 70 °C for 5 h (C12-SACDs). The particle sizes of C12-SACDs were distributed in the range of 1.0–4.0 nm, and the average sizes of C12-ACD and C12-SCD were calculated to be 2.6 and 2.0 nm, respectively. The typical thickness of C12-ACD and C12-SACD were measured to be about 1.80 and 1.73 nm, respectively. Sulfonation resulted in a slight change in the particle size and morphology of the ACD particles. The surface tension curve for C12-ACD is similar to that of the C6-, C10- and C1214-ACDs (Fig. 20c). A comparison with the pristine surfactants, nonionic APG12, anionic-SDBD and H-ACD, is shown in Fig. 20d. The CMCs of C6-, C8-, and C12-ACDs were determined to be 5,000, 2,500, and 100 mg L−1, respectively. Their surface tension was 37.82, 34.62, and 27.45 mN m−1, respectively.
 | ||
| Fig. 20 Amphiphilic carbon dots (ACDs) synthesized from nonionic surfactants, alkyl glycosides with different alkyl chains including APG06, APG10, and APG1214 (coded as C6-ACD, C10-ACD and C1214-ACD, respectively): (a) TEM image with a crystalline lattice image (inset) and (b) particle size distribution of C1214-ACD;63 (c) surface tension curves (25 °C) of C6-ACD, C10-ACD and C1214-ACD comparing with nonionic APG1214;63 (d) surface tension curves of C1214-ACD in comparison with the pristine surfactants including H-ACD,63 nonionic APG121463 and anionic SDBD.64 Reproduced from ref. 63 with permission from Elsevier Ltd, copyright 2024; reproduced from ref. 64 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Graphitic carbon nitride (g-C3N4), as a visible-light-active photocatalyst, has received increasing attention because of its metal-free nature, low cost and high stability. Its modification to a amphiphilic material as well as delamination into ultrathin two-dimensional (2D) nanosheets was performed to enhance its photocatalytic performances. Amphiphilic g-C3N4 nanosheets were reported to be synthesized via covalent grafting of hydrophobic 4-(trifluoromethyl)benzyl (TFMB) groups on the surface of chemically exfoliated carbon nitride nanosheets (CNNSs).64 Specifically, CNNSs were synthesized using g-C3N4 powder prepared from melamine via thermal polycondensation at 520 °C for 4 h through an alkali treatment (chemical exfoliation) under reflux conditions (in 0.2 mol per L NaOH aqueous solution at 100 °C for 12 h). Then, TFMB-groups were grafted onto their surface by mixing 1 g of CNNSs and 2.5 g of K2CO3 with 4-(trifluoromethyl)benzyl bromide (TFMBB) in tetrahydrofuran under reflux at 80 °C for 12 h. Finally, the obtained TFMB-CNNS was an amphiphilic material derived from g-C3N4. It is promising for the effective utilization of visible light because of its possibility to be dispersed in both water and organic solvents.
3. Applications
3.1 Precursors for nanoporous carbons
Nanoporous carbons have been synthesized from various ACMs. Compared with commercial activated carbons derived from plant fruit shells or mineral raw materials including coal and petroleum coke, the principal advantage of employing ACMs as carbon precursors is that they are finely dispersed in KOH aqueous solution, which is an indispensable activation agent, resulting in the highly efficient usage of KOH. If the relevant technology is commercialized, a reduced amount of KOH will be needed, thus creating a more environmentally friendly process. In addition, when ACMs are used for the preparation of nanoporous carbon, activation at high temperature is coupled with carbonization for the simplification of the process.7,8,15,16,66 This is beneficial to achieve a lower cost and easy scaling up.Nanoporous carbons were synthesized by mixing ACM-GC with KOH in water at different KOH/ACM mass ratios, followed by heat-treatment at 700–900 °C (simultaneous carbonization and activation).8 The N2 adsorption/desorption isotherms at 77 K are shown in Fig. 21. These isotherms exhibit that the pore development in the resultant carbons was markedly enhanced with an increase in the mixing ratio, temperature and time. Notably, the development of mesopores was significant. For example, abundant mesopores were created under mild KOH activation conditions (KOH/ACM ratio of 4/1, 800 °C and 1 h activation). The Smeso reached up to 900 m2 g−1, accounting for 27% of the total SBET. Due to their large Smeso, a symmetric electric double-layer capacitor (EDLC) based on the ACM-GC derived nanoporous carbons could perform at very high charge–discharge rate.
 | ||
| Fig. 21 N2 adsorption/desorption isotherms at 77 K for mesoporous carbons prepared from ACM-GC: effects of (a) KOH/ACM ratio, (b) heat treatment temperature and (c) heating time.8 Reproduced from ref. 8 with permission from Elsevier Ltd, copyright 2011. | ||
The pore structure parameters of the carbons synthesized from ACM-CP by KOH activation at 800 °C for 1 h are summarized in Table 2 and compared with the pristine pitch and the precipitates during oxidation in HNO3/H2SO4 (water-insoluble part).7 ACM-CP, which is the water-soluble part, can deliver a highly porous carbon having almost the same amounts of mesopores and micropores. In the case of a symmetric EDLC with 6 M KOH aqueous electrolyte composed of ACM-CP that was prepared with a KOH/ACM ratio of 1.5 at 800 °C for 1 h, no marked evidence of pseudo-capacitance was detected during charge/discharging (Fig. 22a) and a capacitance of 160 F g−1 was retained even at current density as high as 100 A g−1 (Fig. 22b). Nanoporous carbons derived from a commercially available humic acid by carbonization and KOH-activation were also reported to be mesoporous, and consequently delivered a high-rate performance in aqueous electrolytes, including H2SO4, Na2SO4 and KOH.67 They demonstrated high cycling stability, with 92% retention even after 9000 cycles at a rate of 1 A g−1.
| Precursor | KOH/precursor ratio (by mass) | SBET/(m2 g−1) | Smeso/(m2 g−1) | Vtotal/(cm3 g−1) | Vmicro/(cm3 g−1) | Vmeso/(cm3 g−1) |
|---|---|---|---|---|---|---|
| Pristine pitch (CP) | 3/1 | 1574 | 201 | 0.65 | 0.54 | 0.11 |
| ACM-CP (water soluble part) | 3/1 | 3413 | 1821 | 1.76 | 0.90 | 0.86 |
| 1.5/1 | 2575 | 1128 | 1.43 | 0.71 | 0.65 | |
| 1/1 | 2115 | 621 | 1.27 | 0.61 | 0.50 | |
| Water insoluble part | 3/1 | 1721 | 432 | 0.76 | 0.50 | 0.26 |
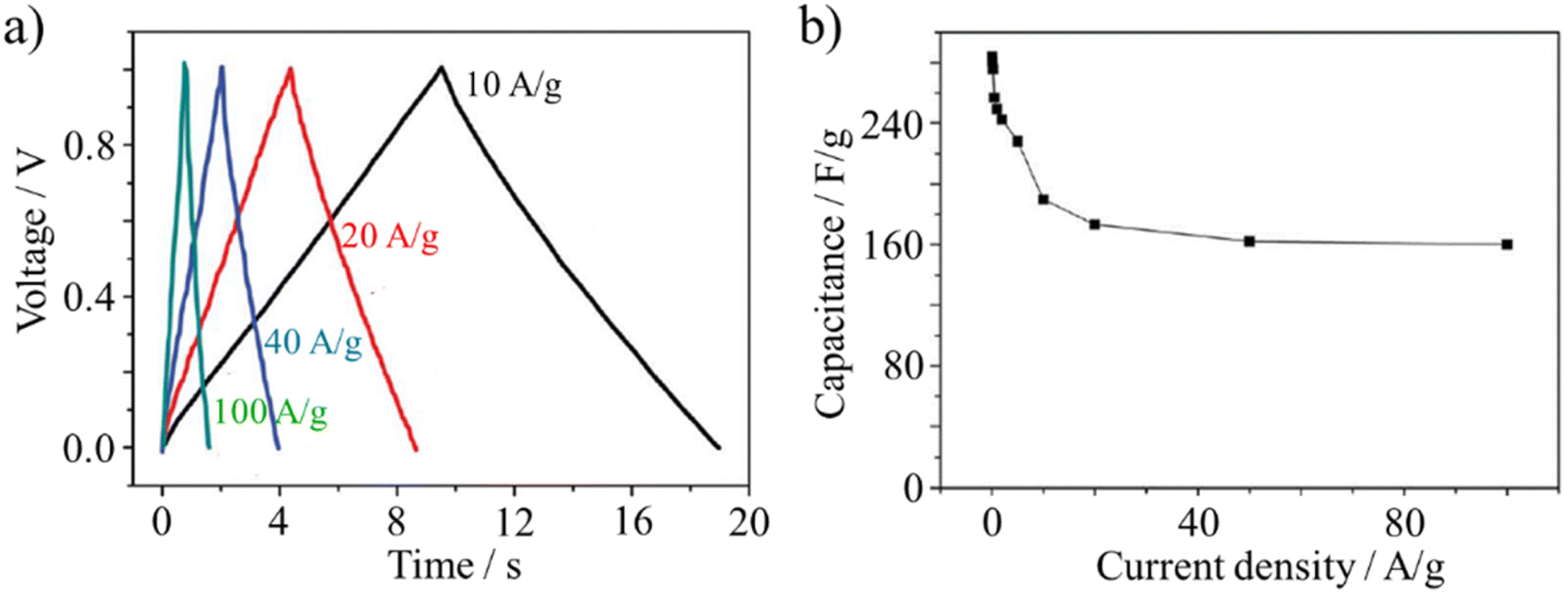 | ||
| Fig. 22 Symmetric EDLC of nanoporous carbons derived from ACM-CP with KOH/ACM ratio of 1.5/1 with 6 M KOH aqueous electrolyte: (a) charge/discharge curves at different current densities and (b) rate performance up to 100 A g−1.7 Reproduced from ref. 7 with permission from the Royal Society of Chemistry, copyright 2013. | ||
The high specific capacity of the ACM-derived nanoporous carbons makes them competent capacitive electrodes that could couple with a faradaic MnO2 electrode to construct a hybrid capacitor (an asymmetric cell consisting of MnO2|1 M Na2SO4 aqueous electrolyte|ACM-derived carbon). Even in aqueous electrolyte, the asymmetric cell had a significantly high operation voltage over 1.0 V, resulting in a high energy density.7,68,69 No obvious overcharge appeared in the charge/discharge curve (Fig. 23a) and a high-rate performance was maintained up to 40 A g−1 (Fig. 23b) for the cell with a 1/1 mass ratio of electrochemically active materials in two electrodes.7 The energy density for this asymmetric cell was 26 W h−1 kg−1 at a current density of 0.5 A g−1, and 22 W h−1 kg−1 at 2 A g−1. Thus, the optimization of the mass ratio of the active materials in these two electrodes is expected improve the performance of the cell.
 | ||
| Fig. 23 Asymmetric capacitor of MnO2|1 M Na2SO4 aqueous electrolyte|ACM-CP-derived carbon: (a) charge/discharge curves at different current densities and (b) rate performance.7 Reproduced from ref. 7 with permission from the Royal Society of Chemistry, copyright 2013. | ||
Employing commercially available sulfonated pitch mixed with KOH in water, nanoporous carbon powders were synthesized by carbonization and activation at 800 °C.15 The SBET of the resultant carbons increased with an increase in the KOH/pitch ratio from 1600 m2 g−1 at the ratio of 1 to 3500 m2 g−1 at 3, which was associated with the marked increase in Smeso from 317 to 2100 m2 g−1. Consequently, they exhibited a high EDLC performance in aqueous electrolyte, including 6 M KOH and 1 M Li2SO4, as well as in 1 M TEABF4/PC non-aqueous electrolyte. By employing the spray drying method, nanoporous carbon spheres were fabricated using the same mixtures.16 The EDLC composed of the resultant carbon spheres with 1 M TEABF4/PC electrolyte exhibited an energy density of 24 W h−1 kg−1 at the power density of 25 kW kg−1.
Commercially available humic acids, LHA and BHA, were carbonized and activated at 800 °C for 1 h after mixing with KOH in their aqueous solutions at a mass ratio of 1.5/1 to obtain nanoporous carbons.17 The pore structure parameters of the resultant carbons are shown in Table 3. LHA-derived carbon was obtained with a much higher carbon yield than the BHA-derived carbon. These carbons possessed a very similar pore structure, both having almost the same SBET as 2000 m2 g−1, but the former was rich in mesopores, whereas the latter was rich in micropores. However, the former had much higher porosity, as measured by mercury porosimetry (Smacro and Vmacro), than the latter. The electrochemical performances of these carbons in symmetric EDLCs with 6 M KOH aqueous electrolyte are shown in Fig. 24. The BHA-derived carbon exhibited a slightly higher capacitance at low current density than LHA-derived one, but a little lower capacitance at high current density as 100 A g−1. Both carbons have not only stable rate performances at high current densities, as high as 100 A g−1, but also high cycle performances up to 10000 cycles at a current density of 100 A g−1. Commercially available humic acid was converted to nanoporous carbon having a Vmicro of 0.238 cm3 g−1 in Vtotal of 0.757 cm3 g−1 by mixing with KOH in water and carbonization at 800 °C, which was used to load MnO2 nanoparticles for the fabrication of an EDLC electrode.70
| Carbon | Yield/(wt%) | SBET/(m2 g−1) | Smicro/(m2 g−1) | Vtotal/(cm3 g−1) | Vmeso/(cm3 g−1) | Vmicro/(cm3 g−1) | Smacro/(m2 g−1) | Vmacro/(cm3 g−1) | Porosity/(%) |
|---|---|---|---|---|---|---|---|---|---|
| LHA-derived | 51 | 2040 | 1164 | 1.35 | 0.62 | 0.62 | 41.7 | 12.7 | 78.1 |
| BHA-derived | 17 | 2060 | 1503 | 1.05 | 0.27 | 0.74 | 13.4 | 6.4 | 37.6 |
 | ||
| Fig. 24 Rate and cycle performances (inset) of LHA- and BHA-derived nanoporous carbons.17 Reproduced from ref. 17 with permission from Elsevier Ltd, copyright 2014. | ||
ACM-derived nanoporous carbons were demonstrated to be particularly suitable for the fabrication of EDLCs. Several reports7,17 confirmed that they have outstanding advantages in terms of double layer energy storage, independent of the ACM precursor used. Whether the precursor is biomass17 or coal tar pitch,7 the resulting porous carbons exhibit extremely excellent energy storage capabilities. The EDLC capacitance could reach over 160 F g−1 even at 100 A g−1, with over 60% capacity retention vs. capacitance at 0.05 A g−1 (Fig. 22b and 24). This performance is attributed to the properties of the ACMs. Their amphiphilicity ensured that even at a very low KOH consumption, the mesopore-to-micropore ratio in the porous carbon was 1:1 or when Smicro exceeds1000 m2 g−1 (Tables 2 and 3), which not only facilitates ion transfer because of the abundant mesopores, but also guarantees sufficient energy storage sites in the micropores. To date, no commercial activated carbons have been found to be comparable.
Besides KOH activation/carbonization, other methods have been used to prepare ACM-derived nanoporous carbons. Brown coal-derived humic acids were carbonized at 900 °C, and then activated at 800 °C in steam up to 25–75 wt% burn-off to obtain nanoporous carbons.18 Their pore structure parameters are shown in Table 4. The resultant carbons were microporous, and the highest Smicro of 1500 m2 g−1 was obtained with a high burn-off of 75 wt%.
| Burn-off/(wt%) | SBET/(m2 g−1) | Smicro/(m2 g−1) | Smeso/(m2 g−1) | Vmicro/(cm3 g−1) | Vmeso/(cm3 g−1) | Smacro/(m2 g−1) | Vmacro/(cm3 g−1) |
|---|---|---|---|---|---|---|---|
| 25 | 802 | 867 | 52 | 0.308 | 0.082 | 4 | 0.216 |
| 50 | 1154 | 1184 | 130 | 0.421 | 0.224 | 6 | 0.320 |
| 75 | 1497 | 1519 | 233 | 0.540 | 0.575 | 10 | 0.512 |
Pre-oxidation is frequently used for the preparation of porous carbons from various carbon precursors. However, the literature clearly indicates that the transformation of carbon precursors into ACMs via pre-oxidation is limited.71,72 In fact, usually after pre-oxidation, O-containing groups are definitely introduced on the surface of the precursors, making them ACMs.
Pre-oxidation functions well in the preparation of nanoporous carbon. Coal (semi-anthracite) pre-oxidized in air, followed by steam activation at 850 °C to 50 wt% burn-off exhibited high microporosity (SBET of 847 m2 g−1 and Vtotal of 0.384 cm3 g−1), which was higher than that without pre-oxidation (168 m2 g−1 and 0.088 cm3 g−1).73 Anthracite was oxidized in 70% HClO4 aqueous solution either by quick heating to 165 °C or step-by-step heating to 160 °C, followed by carbonization at 850 °C in a CO2 flow, resulting in an SBET as high as 1600 m2 g−1 and Vtotal of ∼1 cm3 g−1.74,75 The effects of pre-oxidation on anthracite were studied in detail by measuring the volumes of ultra-micropores (<0.7 nm), super-micropores (0.7–2 nm) and mesopores (2–50 nm) (Vultra, Vsuper and Vmeso, respectively) via CO2 and N2 adsorption isotherms and that of macropores (>50 nm) (Vmacro) via mercury porosimetry.76 In the case of pre-oxidation, concentrated HNO3 was found to be more effective than air, as shown in Fig. 25. The effects of the severity of the oxidation were studied.71 As shown in Fig. 26a, a high concentration of HNO3 was required to get humic acid, at least 0.5 N. To achieve an SBET as high as 1500 m2 g−1, the KOH/humic acid ratio must be more than 1, as shown in Fig. 26b, where the SBET is shown as a function of HNO3 concentration and KOH/coal ratio. It was also pointed out that activated carbon having an SBET close to 2000 m2 g−1 can be obtained by mixing only 1.5 times KOH when the coal was pre-oxidized in 0.5 N HNO3. This pre-oxidation resulted in principally humin (NaOH insoluble part), together with only a small amount of humic acid (NaOH soluble). In fact, the so-called pre-oxidation is a route to prepare ACMs.
 | ||
| Fig. 25 Histogram of the different types of pores in carbons prepared from anthracite under various pre-oxidation conditions. The number shown for each column is total oxygen content (μmol g−1) after pre-oxidation.76 Reproduced from ref. 76 with permission from Elsevier Ltd, copyright 1997. | ||
 | ||
| Fig. 26 Oxidation and activation of a bituminous coal: (a) yield of humin and humic acid as a function of HNO3 normality and (b) SBET of the resultant activated carbons as a function of KOH/coal ratio and HNO3 normality used for oxidation.71 Reproduced from ref. 71 with permission from Elsevier Ltd, copyright 1995. | ||
Green petroleum coke was oxidized in 20% H2O2 aqueous solution, and then carbonized and activated by KOH at 730 °C.77 Compared with carbons from the non-pre-oxidation process, pre-oxidation caused the SBET and Vtotal of the resultant carbons to markedly increase, as shown in Table 5. Jiang et al. also demonstrated the contribution of pre-oxidation to the pore characteristics of green-petroleum-coke-derived carbons. Specifically, 15% H2O2 or 20% HClO4 aqueous solutions were the selected oxidation reagents and compared to non-pre-oxidation.78 Pre-oxidation was very effective to develop pore structures in the resultant activated carbon, particularly by H2O2, as shown in Fig. 27. HNO3 is another oxidation reagent, which was employed to convert peach stone to carbon molecular sieves with a selectivity for CO2/CH4 separation, which was much higher than that of the activated carbon prepared from the same precursor without pre-oxidation.72,79
| Green coke | Oxidation | KOH activation yield at 730 °C/(wt%) | SBET/(m2 g−1) | Vtotal/(cm3 g−1) |
|---|---|---|---|---|
| As-received | Without | 61.4 | 1763 | 1.113 |
| In 20% H2O2 | 40.5 | 2744 | 1.649 | |
| After calcination | Without | 76.5 | 586 | 0.407 |
| In 20% H2O2 | 68.7 | 1282 | 0.863 |
 | ||
| Fig. 27 N2 adsorption isotherms of the activated carbons derived from petroleum coke with or without pre-oxidation.78 Reproduced from ref. 78 with permission from Elsevier Ltd, copyright 2008. | ||
Besides the hydrophilic surface functional groups on ACMs, the hydrophobic conjugated aromatic carbon domains in ACM particles are very useful when self-assembly is induced by sp2 C–C bonds. Hollow carbon microbeads were prepared using urea and an ACM prepared from raw coke in a water-in-oil emulsion at 80 °C, followed by carbonization at 500–1000 °C.80 The diameter and wall thickness of the microbeads could be tuned based on the ACM dosage. Their diameter ranged from 11.9 to 13.5 μm, while their wall thickness was 3.0–3.5 μm when the ACM concentration increased. Even graphene flakes were reported to be formed by annealing LHA droplets on a copper substrate at 1100 °C in Ar/H2.81 The authors demonstrated the formation of single crystalline graphene. However, regrettably, no experimental evidence for the crystallinity of the formed graphene flake was presented.
3.2 Precursors for carbon coatings
Carbon coating is one of effective ways to improve the performances of materials. Therefore, it is widely applied to various functional materials including metals and metal oxides.82 The water dispersibility of ACMs makes the relevant carbon coating environment-friendly and easier to apply, especially in comparison to that using organic solvents. Furthermore, this process does not sacrifice the carbon yield.ACM coating is usually achieved via a wet process. For example, artificial graphite particles were carbon coated by dispersing them in an aqueous solution containing ACM-GC of 5 g L−1, and then heat-treated at 2800 °C after water evaporation.83 The resulting carbon-coated material was used as an anode material in lithium-ion rechargeable batteries (LIBs). The charge/discharge behaviors and cyclic performances of the LIBs using the pristine graphite and the carbon-coated anodes are compared in Fig. 28. Employing the carbon coating using ACM-GC, the discharge capacity in the first cycle was significantly enhanced to 366.4 mA h g−1 in comparison to 309.0 mA h g−1 without the coating. This was the same for the coulombic efficiency (Fig. 28a). The retention of the discharge capacity after 30 cycles at 50 mA g−1 was as high as 95.5% for the carbon-coated graphite compared to 74.6% for the pristine graphite (Fig. 28b). Compared to some dry coating methods,84–86 coating in an ACM-containing slurry enables the coating layer to be very uniform and tunable via the ACM concentration.
 | ||
| Fig. 28 LIBs using graphite anode with or without carbon-coating by ACM-GC: (a) charge/discharge curves and (b) cyclic performances at 50 mA g−1.83 Reproduced from ref. 83 with permission from Elsevier Ltd, copyright 2010. | ||
Another LIB anode material, Li4Ti5O12 (LTO), was successfully coated by ACM-GC dispersed in an aqueous solution, followed by carbonization at 800 °C.87 The dosage of 10 mg ACM-GC per gram LTO provided a homogeneous coating layer with a thickness of 3 nm, as shown in Fig. 29. This accounts for the markedly improved rate performance as well as cyclic performances, as shown in Fig. 30.
 | ||
| Fig. 29 (a) SEM image and (b)TEM image of carbon-coated Li4Ti5O12 using ACM-GC.87 Reproduced from ref. 87 with permission from Elsevier Ltd, copyright 2012. | ||
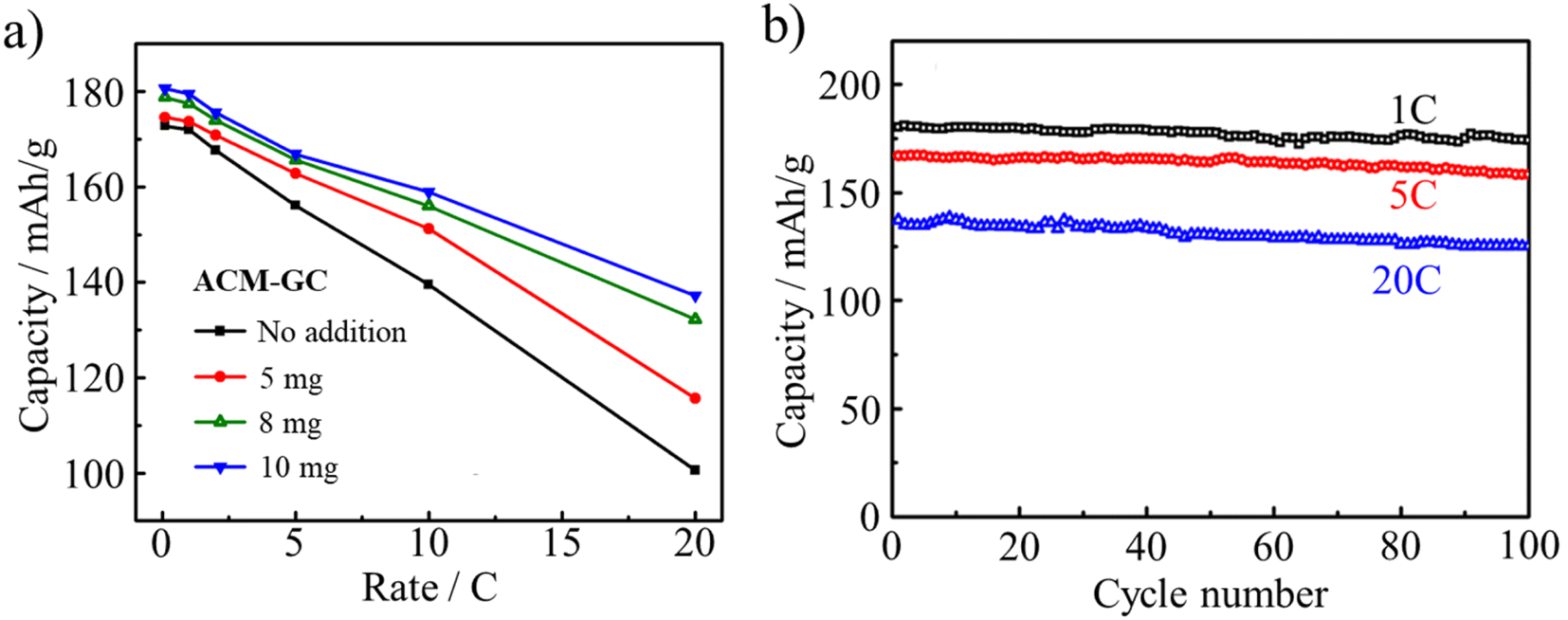 | ||
| Fig. 30 LIB performances of Li4Ti5O12 (LTO) anode coated with ACM-GC: (a) rate performances as a function of ACM-GC dosage and (b) cycle performances of LTO coated with ACM-GC at 10 mgACM/gLTO dosage at different rates.87 Reproduced from ref. 87 with permission from Elsevier Ltd, copyright 2012. | ||
Besides LIB anode materials, cathode materials such as LiFePO4 were also reported to be coated by coal tar pitch-derived ACMs, called ACM-CP,88 using a wet method. The LIBs were composed of the carbon-coated LiFePO4 as the working electrode (cathode) and Li metal as the counter electrode with 1 M LiPF6 electrolyte in EC/DEC (1/1 by volume). The LIB using LiFePO4 coated by 6.8% carbon delivered discharge capacities of 151 mA h g−1 at 1C rate and 132 mA h g−1 at 10C (Fig. 31a) with 80% retention even after1000 cycles at a high rate of 10C (Fig. 31b).
 | ||
| Fig. 31 Cyclic performances of LIBs with carbon-coated LiFePO4 cathode by ACM-CP: (a) discharge capacities at different current densities from 0.1C to 60C and (b) discharge capacity and coulombic efficiency vs. cycle numbers up to 1000 cycles at 10C rate.88 Reproduced from ref. 88 with permission from Elsevier Ltd, copyright 2014. | ||
Coating using ACMs is based on chemical interaction rather than simple physical covering. The carbon layer formed from humic acid on the surface of ZnO nanoparticles was found to enhance the stability of the composite via electrostatic and steric interactions.89 The interaction could be controlled based on the humic acid concentration, preventing the complete or partial aggregation of the ZnO nanoparticles. Cotton fabric, which is originally hydrophilic, exhibited enhanced hydrophobicity after being coated by amphiphilic multi-layer GnO. GnO was in an aqueous solution with different concentrations ranging from 0.05 to 0.4 mg mL−1.90–92 The GnO concentration not only tuned the thickness of the coating layers,92 but also altered the degree of hydrophobicity of the GnO-coated cotton fabric, as characterized by its water contact angle. The coating markedly increased the water contact angle from 0° for the fabric without coating to 143° for the fabric coated in the 0.4 mg per mL GnO dispersion. Obviously, this is beyond what physically covering hydrophilic GnO can provide. The coated amphiphilic multi-layered GnO exhibited chemical interactions with the cotton fabric, as confirmed by FTIR spectroscopy. Consequently, the chemical interactions led to a change in the photocatalytic and antibacterial activities of the fabric.90 Other researchers also reported the effects of chemical interactions between coating layers and cores on the overall properties of composites. For example, the electrical resistivity markedly decreased to 91.8 kΩ sq−191 when the coated GnO layer was reduced to multi-layered rGnO by ascorbic acid, principally changing the chemical interactions between the core and the shell.
Humic acid was even applied as an in situ coating agent, which increased the capacity retention of the carbon electrodes due to the change in the electrode–electrolyte interfaces.93 This positive effect was found to be the maximum when humic acid content was 5 wt%.
3.3 Precursors for carbon supports for metal nanoparticles
The metal binding abilities of ACMs were reported, as illustrated by an ACM-containing aerogel dried with supercritical CO2 after substitution of the water in the gel with acetone.94,95 ACMs are negatively charged in aqueous solution, attracting positively charged metal ions. Based on electrostatic attraction, metal nanoparticles are easily loaded onto ACM-derived carbons by mixing organo-metallic compounds with ACMs, followed by carbonization.27,96–98 Pt nanoparticles (about 22 nm in size) were loaded in carbon microbeads (10–20 μm in diameter) prepared in a water-in-oil emulsion.98 The catalytic performance of these Pt-loaded carbon spheres was studied using the hydrogenation of cyclohexene as a function of Pt content and CO2 activation time.99 Other organo-carbonaceous gels were prepared by mixing ACM with organic metal compounds, such as Pd(acetonate)2, Ru(acetonate)3 and Ni(acetonate)2 in N,N-dimethylformamide (DMF) at 80 °C. After carbonization at a temperature above 500 °C, nanosized metal particles (Pd of 20–30 nm, Ru of 40 nm and Ni of 10 nm) were uniformly dispersed on the ACM-derived carbon.97 Electrocatalytic electrodes were prepared by depositing Pt-loaded ACM in DMF on a glassy carbon plate, followed by carbonization at 500–1000 °C.99,100 The current density for proton reduction at the potential of −0.2 V (vs. Ag/AgCl) increased with an increase in the concentration of Pt and with an increase in the heat treatment temperature up to 700 °C, after which the dramatic growth of the Pt particle size took place (shown in Fig. 32). Due to the electrostatic attraction between Pt2+ and negatively charged ACMs, Pt particles are prevented from moving around, and thus their aggregation is depressed markedly, particularly at high temperature. | ||
| Fig. 32 Diameter distribution of Pt particles loaded on ACMs after heat treatment at different temperatures of (a) 500 °C, (b) 700 °C and (c) 1000 °C.100 Reproduced from ref. 100 with permission from Elsevier Ltd, copyright 1996. | ||
Humic acid-derived carbons as catalyst carriers could enable the loading of metals on them smaller than that detectable by X-ray diffraction. Carbons containing metallic Co nanoparticles in humic acid were prepared from mixtures of a humic acid with either cobalt phthalocyanine or cobalt chloride, followed by carbonization at 800 °C.101 In this Co/carbon composite, the presence of crystalline Co could not be confirmed by X-ray diffraction, suggesting a high dispersion of nano-sized Co metals. Even more interesting, after exclusion of Co, the humic acid-derived carbon alone exerted catalytic activity for the oxygen reduction reaction, which is not rare. Sulfonated carbon with amphiphilic properties was reported to exhibit catalytic activity for the esterification of various organic acids with long carbon-chains and steric compounds such as pivalic acid.62 It was synthesized via the hydrothermal carbonization of a mixture of furfural, sodium dodecylbenzene sulfonate (SDBS) and sulfuric acid at 185 °C for 24 h and coded as F–S. The catalytic activities for pivalic acid conversion were compared on various catalysts, as shown in Fig. 33a and b. The F–S catalyst delivered higher activity than not only two commercial catalysts, Amberlyst-15 and Nafion-50, but also that synthesized using p-toluenesulfonic acid instead of SDBS (named F–T).
 | ||
| Fig. 33 Conversion of pivalic acid over sulfonated carbon catalysts (F–S and F–T) in comparison with two commercially available catalysts (Amberlyst-15 and Nafion-15) at 70 °C: (a) conversion kinetics using various catalysts (catalyst: 0.5 g, pivalic acid: 1.02 g and methanol: 1.60 g) and (b) effect of catalyst amount (pivalic acid: 1.02 g and methanol:1.60 g).62 Reproduced from ref. 62 with permission from the Royal Society of Chemistry, copyright 2014. | ||
3.4 Additives for crystal growth
Besides precursors of porous carbons, carbon coating and carbon supports, ACMs were found to successfully change the morphology of MnO2 particles by intervening in their crystal growth.102 MnCO3/ACM nanospheres were obtained by adding NaHCO3 to an aqueous mixture of MnSO4 with ACM-CP. When oxidized by KMnO4, MnCO3/ACM was chemically converted to MnO2/ACM hollow spheres (Fig. 34d–f). Without ACMs, MnO2 hollow cubes rather than hollow spheres were obtained, as shown in Fig. 34a–c. Besides the change in morphology, the existence of ACM-CP up to 13 wt% apparently improved the electrochemical energy storage performances, particularly the cyclic stability, which is very appealing for MnO2-containing electrodes. The well dispersed nano-sized and negatively charged ACMs in water provide numerous electrostatic attraction sites to capture Mn2+ ions to be further oxidized to MnO2. The obtained MnO2 particles could be anchored on the ACMs, consequently relieving the dissolution of MnO2 effectively and promoting the stability of the MnO2/ACM composite. Compared to double-shelled MnO2 hollow spheres,103 the MnO2/ACM composite exhibited improved cyclic stability because of the contribution of ACMs. | ||
| Fig. 34 SEM and TEM images of MnO2 particles prepared from MnCO3/ACM mixture: (a–c) hollow cubes and (d–f) hollow spheres. (a and d) SEM images, and (b, c, e and f) TEM images.102 Reproduced from ref. 102 with permission from the American Chemical Society, copyright 2014. | ||
Negatively charged ACM particles in aqueous solution were applied for the fabrication of crystalline materials.24,88 An aqueous mixture of hexadecyl trimethyl ammonium bromide (HTAB), NaOH, and ACMs derived from mesophase pitch was heated in an autoclave at 100 °C.24 The precipitated powder obtained was stabilized by heating in air up to 300 °C, and then carbonized at 800 °C. Due to the electrostatic interactions between the negatively charged ACMs and the positively charged HTAB micelles, ordered or partially ordered assembles were obtained. Most of the resultant vesicle particles possessed a worm-like shape with a diameter of 80–200 nm and hollow cavity, as shown in Fig. 35. ACM-CP was also used in the synthesis of LiFePO4.88 By adding ACMs to an aqueous solution of LiOH, FeSO4 and H3PO4, well-crystallized LiFePO4 nanocrystals with a lateral size of 90–250 nm and thickness of 25–35 nm were successfully synthesized under hydrothermal conditions at 180 °C. ACMs suppressed the crystal growth along the b-axis by adhering to the ac facets of the LiFePO4 nanoplatelets through electrostatic interactions. Consequently, they intervened in the growth of LiFePO4 nanoplatelets. The synthesized powder contained about 1.1 wt% C, which was reasonably supposed to come from ACMs and assisted in maintaining the valence of Fe ions as 2. ACMs could even help control the crystal size of LiFePO4 to as low as 20 nm.88
 | ||
| Fig. 35 SEM and TEM images of carbon vesicles: (a and b) before carbonization and (c and d) after carbonization. Reproduced from ref. 24 with permission from Elsevier Ltd, copyright 2004. | ||
Humic acid played an important role in the synthesis of LiCoO2 in a liquid-phase reaction because of its amphiphilic nature and unique multidentate complexing ability for metal ions.104 Employing humic acid, the synthesis temperature of LiCoO2 was lower than that for solid-state synthesis and its morphology was induced to be a well-developed layered structure. Consequently, this layer-structured LiCoO2 displayed a higher capacity than that prepared by a solid-state reaction. Besides, in liquid-phase synthesis, even in solid-state reactions, humic acid was reported to work well as a bi-functional additive, a reduction agent as well as a morphology-guiding agent in the synthesis of an LIB cathode composite, Li3V2(PO4)3/C.105 Humic acid aided in the formation of an Li3V2(PO4)3/carbon composite merging with each other, while carbon black, another carbon source, guided Li3V2(PO4)3 wrapped with small carbon particles. This merging morphology guaranteed that the cathode material decayed slowly during charging–discharging cycling, still maintained 145.2 mA h g−1 after 200 cycles at 1C.
3.5 Modifiers for carbon materials
Humic acid modification makes carbon electrodes sensitive to trace impurity cations, including Fe2+, Cu2+ and Ni2+.106 Linear relationships between the current on electrodes modified by humic acid and the concentration of these divalent metallic cations were obtained, while no signals were obtained for the electrode without humic acid modification.Humic acid modification on carbon greatly changes its surface characteristics, and thus introduces new properties in composites. The adsorption of U4+ by activated carbon was found to be stimulated by the adsorption of humic acid in advance. When 41.5 mg g−1 humic acid coexisted with U4+ in water or when the activated carbon was saturated in advance by humic acid, the saturated adsorption capacity of U4+ on the carbon became 100 mg g−1 for and 21.1 mg g−1 for humic acid, respectively.107 The adsorption of humic acids is known to stabilize carbon nanotube (CNT) dispersions in water.108 Four humic acids with different acidities, 6.19, 5.40, 4.72 and 4.08 mmol g−1, were sequentially extracted from peat soil. They were adsorbed onto multi-walled CNTs (MWCNTs) with different diameters of <10, 20–40 and 60–100 nm, respectively, and stabilized in water. The adsorption of humic acid normalized by the surface area of the MWCNTs increased with an increase in the outer-diameter of the MWCNTs and decrease in the polarity of the humic acids. The stabilization of MWCNTs in water increased with the increase in the adsorption of humic acid, leveling-off at a threshold that was markedly lower than the maximum adsorption. The removal of MWCNTs stabilized in water by the adsorption of humic acid was studied because of the ecological requirement of maintaining the aquatic environment.109 The presence of humic acid and salt (MgCl2) in water was reported to increase the size of agglomeration of carbon nanoparticles (fullerene C60 and carbon nanotubes).110
Amphiphilic few-layered GnO possesses molecule-particle duality because of its two different length scales, i.e., thickness of about 1 nm and lateral sizes ranging from nanometers to hundreds of micrometers.11 Owing to its molecular nature, few-layered GnO itself can adhere to the surface of the water/air interface and form a thin GnO film.37 Alternatively, with its particle nature, few-layered GnO can be used as an agent to create colloidal dispersions of insoluble materials in solutions, especially π-conjugated aromatics, CNTs and graphite, and thus enhance their solution processability. By adhering to or even wrapping an insoluble particle, few-layered GnO lowered the interfacial energy of the solid–liquid interfaces and stabilized an emulsion system such as toluene in water and CNTs in water.37 Few-layered GnO could stably disperse MWCNTs in water by mixing them in CNT/GnO in a mass ratio of 1/3 after sonication, as shown in Fig. 36a.37 This is probably because few-layered GnO flakes wrap CNTs by π–π* interaction and the hydrophilic functional groups on GnO help their dispersion. The stability of the colloidal suspension of CNTs with GnO in water was monitored by its optical absorbance. In the presence of GnO, the CNT colloidal suspensions remained nearly constant over 24 h, as shown in Fig. 36b.
 | ||
| Fig. 36 Few-layered graphene oxide (GnO) helps dispersion of multi-walled carbon nanotubes in deionized water: (a) immediately after sonication and (b) absorbance of the two suspensions in (a) vs. standing still time.37 Reproduced from ref. 37 with permission from the American Chemical Society, copyright 2010. | ||
The surface modification provided by few-layered GnO enabled the pervaporation separation of an isopropanol/water mixture through porous PAN membranes with high efficiency. The composite membrane was fabricated by self-assembling amphiphilic GnO layers on porous PAN membranes, resulting in 99.5 wt% water permeation at 2047 g m−2 h−1 permeation flux.40 The authors illustrated the separation mechanism. Specifically, water molecules are adsorbed preferentially on the oxygen-containing functional groups on the few-layered GnO, and then penetrate the ‘‘empty’’ space between the GnO layers (Fig. 37). Then, water diffuses rapidly due to its low friction contact with the hydrophobic central region, where the water molecules form a ‘‘passage’’, facilitating their transport through the GnO film.
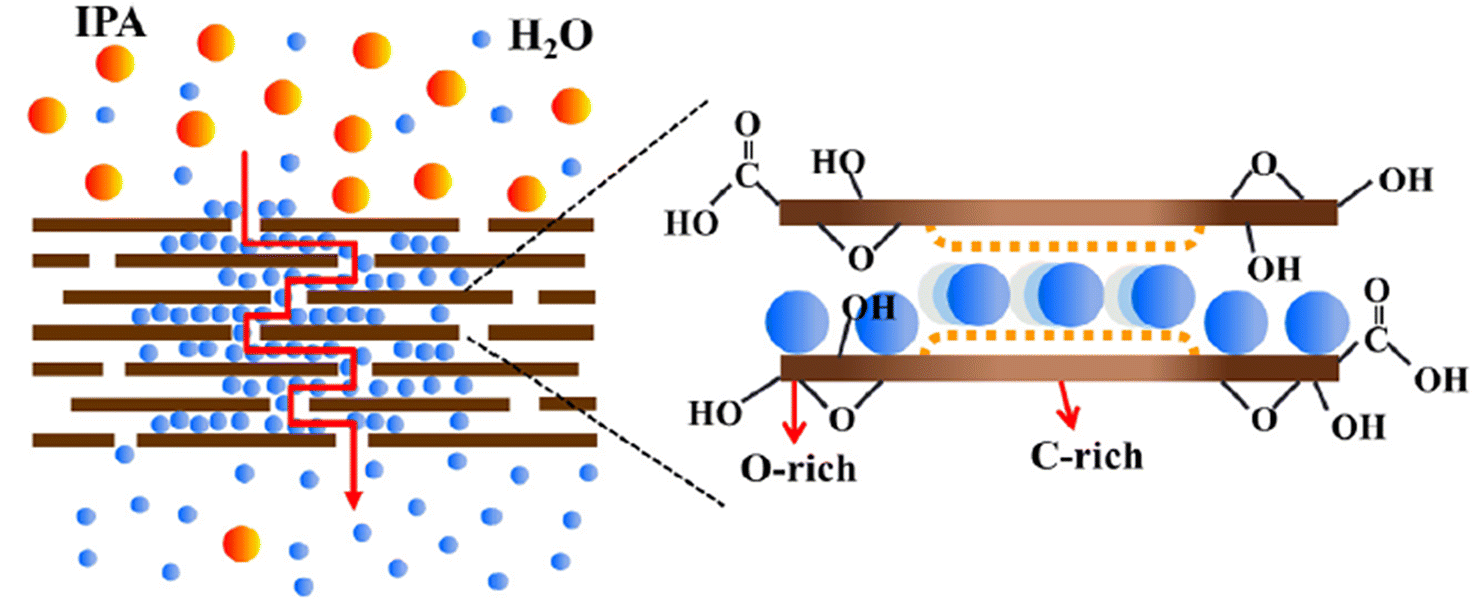 | ||
| Fig. 37 Schematic of the mechanism for separating H2O from isopropanol using a pressure-assisted self-assembled few-layered GnO film.40 Reproduced from ref. 40 with permission from Elsevier Ltd, copyright 2014. | ||
Amphiphilic g-C3N4 nanosheets (TFMB-CNNS) were synthesized by grafting 4-(trifluoromethyl)benzyl (TFMB) groups on the surface of chemically exfoliated carbon nitride nanosheets (CNNSs). TFMB-CNNS contained hydrophobic and hydrophilic functionalities.64 Its photocatalytic activity was confirmed by the reduction of nitrobenzene to aniline at oil–water biphasic interfaces. Its reduction performances for nitrobenzene at the boundary of the oil–water bi-phase are shown in Fig. 38a–c. Fig. 38a and b demonstrate how the photocatalytic activity of TFMB-CNNS is effectively enhanced by delamination (exfoliation to CNNS) and TFMB-grafting.
 | ||
| Fig. 38 Phase boundary photocatalysis of TEMB-CNNS for nitrobenzene reduction: production of aniline (a) in water phase, (b) under various conditions, and (c) for different oil phases.64 Reproduced from ref. 64 with permission from the Royal Society of Chemistry, copyright 2019. | ||
3.6 Environment remediation and toxic gas surveillance
GnOs have attracted attention as new carbon materials to effectively remove various gaseous and aqueous pollutants by adsorption. Their potential has been demonstrated in various reports, as reviewed.111 On few-layered GnO, adsorption is mainly governed by the interactions between the negatively charged acidic functional groups on its surface and cationic ions in either gaseous or aqueous pollutants, while on graphene, the π–π interaction between graphene nanosheets and the adsorbate plays a dominant role in adsorption.Many researchers proved that few-layered GnO may be a suitable adsorbent for heavy metal ions including Cd2+ and Co3+ ions,112 Pb2+,113 U4+114 and Eu3+115 in aqueous solution. The adsorption of heavy ions was experimentally demonstrated112–114 or proven by modeling techniques115 to be dependent on the pH value and independent or less dependent on ionic strength. Cu2+ in solution pushed few-layered GnO sheets to fold up and form large aggregates.116 GnO was also used as an adsorbent for the removal of tetracycline antibiotics due to (1) the π–π interactions between the ring structure in the tetracycline molecule and the aromatic moieties in GnO and (2) the cation–π interactions between the amino group on the ring C4 of tetracycline and conjugated π-electron-rich structures.117 Aminotriazines were selected as model chemicals for DFT calculations on the interactions between chemicals and graphene.118 Their strong affinity for the graphene layer due to the specific attractive interactions of NR2 (R representing an alkyl group) groups with the surface of graphene is expected to exhibit a strong driving force for adsorption.
Composites containing few-layered GnO exhibited better performances in contaminant removal from large volumes of wastewater and pollutant gases. Although these nanocomposites possessed disordered structures, improvements in contaminant retention compared to the parent GnO were observed owing to the formation of complexes.119 The sorption of Co2+ on a magnetite/multi-layered GnO composite originated from inner-sphere surface complexation at low pH values, whereas Co2+ removal was accomplished by simultaneous precipitation and inner-sphere surface complexation at high pH values.120 The removal of Cu2+ and fulvic acid (FA) from aqueous solution was reported to be performed via the formation of few-layered GnO/Fe3O4 composites.121 FA modified the surface of the GnO/Fe3O4 composites, enhancing their adsorption capacity for Cu2+ at pH 5.3 because of the additional complexation, but weakening the adsorption at higher pH because of the destroyed complexation. Au3+ and Pd2+ could be spontaneously adsorbed by chitosan/GnO composites with the maximum adsorption capacity of 1076 mg g−1 for Au3+ and 217 mg g−1 for Pd2+, respectively.122 Owing to the formation of complexes between NH3 and Keggin polyanions (also called polyoxometalates), NH3 was strongly adsorbed on the surfaces of GnO/polyoxometalate nanocomposites with less than 1 wt% of gas released by air purging.119 An optimized glucose-modified Zn-based MOF/multi-layered GnO nanosheet composite exhibited high H2S uptake of up to 130.1 mg g−1.123
In the case of water purification, a graphene-based composite membrane was synthesized by depositing GnO on a modified polyacrylonitrile substrate via the pressure-assisted self-assembly technique, showing excellent performances during the pervaporation separation of a 70 wt% isopropyl alcohol/water mixture.124 Given that GnO is amphiphilic, water molecules are adsorbed at the hydrophilic edges, and then rapidly diffuse through the hydrophobic cores, forming a water passage channel, which promotes a high permeation flux. Consequently, up to 99.5 wt% water permeated with a permeation flux of 2047 g m−2 h−1. Micrometer-sized GnO nanosheets were laminated to form a film with sub-micrometer thickness. Although it was completely impermeable to liquids, vapors and gases including He, this graphene-based carbon membrane could permeate H2O at least 1010 times faster than He, which was explained by the amphiphilic nature of few-layered GnO.125 Free-standing GnO films with a thickness of <1 μm prepared by spray- or spin-coating (Fig. 39a) were impermeable to He, whereas could permeate water vapor as an open aperture, although they were completely impermeable to ethanol and hexane vapors. The rGnO film due to its virtual hydrophobicity was used for comparison, showing impermeability to steam (Fig. 39b).
 | ||
| Fig. 39 Water-permeable GnO films: (a) digital photograph of the film and (b) weight loss of water for a container sealed with a GnO film (film thickness ≈ 1 μm; aperture area ≈ 1 cm2). No loss was detected for ethanol, and hexane, but water was evaporated from the container as freely as through an open aperture.125 Reproduced from ref. 125 with permission from the American Association for the Advancement of Science, copyright 2012. | ||
GtO, which was grafted with a bifunctional oligoester, showed enhanced dispersibility in aprotic polar solvents such as DMF. The introduction of the oligoester on GtO made it a chemical environment-responsive material.126 The interactions of graphene derivatives with natural amphiphiles including peptides, cellulose and palmitic acid were systematically simulated.127 Owing to the differences in the hydrocarbon content, conformational flexibility and molecular geometry of amphiphiles, these natural amphiphiles interact with graphene derivatives with different binding affinities. Peptides and cellulose form a soft nanosheet-amphiphilic bio-corona because of their weak binding with GnO, while palmitic acid binds strongly with GnO to form a hard bio-corona. Actually, it is the whole bio-corona rather than only the few-layered GnO substrate that has considerable effects on the biological and environmental sensitivity.127
Both thermally reduced few-layered GnO and ZnO decorated chemically reduced few-layered rGnO were reported to exhibit high room-temperature sensing properties for toxic gases in the air. Based on few-layered rGnO thermally reduced at 200 °C,128 a miniaturized device showed a sensitive and fast response for 100 ppm NO2, besides 100% recovery after 30 min exposure to dry air. In addition, the deposition of sensitive ZnO particles on chemically reduced few-layered rGnO improved the sensibility. The ZnO/rGnO composite presented a large response and quick recovery with a selectivity towards industrial toxins, CO and NH3, for concentrations as low as 1 ppm at room temperature.129
Amphiphilic carbon dots (ACDs) prepared from a nonionic surfactant (C6-ACD, C10-ACD and C1214-ACD) exhibited high oil removal efficiencies.63 The removal efficiency of an oil film was measured by immersing a glass plate covered with a crude oil film into ACD solutions. The oil area shrinkage was found to be 51.0%, 46.6%, 56.2%, and 78.1% in C6-, C10- and C1214-ACDs, respectively.
3.7 Biomedical applications
Few-layered GnO and graphene, including rGnO, have recently been considered as promising drug delivery carrier and antibacterial material candidates. Recent efforts employing GnO and graphene in biomedical applications were reviewed.1,2,130–132Water-dispersible GnO was demonstrated to work as a platform for the sensitive and selective detection of DNA and proteins through fluorescence emission in the near-infrared region.133–135 A GnO-based DNA sensor was designed for multiplex, sequence-specific DNA detection.135 The large planar surfaces of single-layer and/or few-layered GnO enabled the simultaneous detection of multiple molecular targets in the same solution.136 GnO films were prepared on stainless steel substrates via the electrophoretic deposition of a few-layered GnO suspension containing Mg(NO3)2. The resulting material was called positively charged GnO nanowalls. Another material called rGnO nanowalls was prepared via the chemical reduction (hydrazine vapor) of GnO. Both nanowalls showed high antibacterial activity toward two bacteria, Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus), acting as models for Gram-negative and Gram-positive bacteria, respectively, as shown in Fig. 40.
 | ||
| Fig. 40 Antibacterial activities of GnO and rGnO nanowalls towards: (a) E. coli and (b) S. aureus.136 Reproduced from ref. 136 with permission from the American Chemical Society, copyright 2010. | ||
The negatively charged GnO could bind the cationic polyethylenimine (PEI) by electrostatic interactions. The resultant PEI/GnO complexes were reported to be stable and highly enriched in positive charges, enabling the effective loading of plasmid DNA via layer-by-layer assembly.137 Few-layered GnO was employed as a 2-dimensional nano-vehicle for gene loading and transfection.138,139
3.8 Carbon foam
The spontaneous bubble method is a technique for the preparation of carbon foam from ACMs. In general, it involves the pyrolysis of ACMs in a limited space, accompanied by a surge in pressure caused by gas production during the process. Yang and co-workers reported that by an efficient atmospheric pressure self-foaming technique, carbon foams were prepared from three highly bonded bituminous coal as raw materials.140 Obviously, the chemical structures of these precursors affected the formation, growth, and rupture of the pores in the carbon foam.In fact, not all coal is suitable for the preparation of carbon foam. Only strongly bonded bituminous coal with more cyclic aliphatic fraction and high mobility is preferred as a carbon foam precursor. To tune the fluidity of coals, additives including biomass, agricultural products and thermoplastic polymers141 are needed given that they are chemically rich in cyclic aliphatic moieties that produce sufficient volatile chemicals for foaming at a certain temperature.
The rapid freeze-drying method is another useful way to make rGnO-derived carbon foam, which is especially effective for that derived from aqueous solution. A 7 mg per mL GnO aqueous solution was mixed with ethanol at a volume ratio of 30:1 in a mold directly attached to liquid nitrogen for 10 min, long enough to be completely freeze-dried.142 After laser reduction, rGnO foam was obtained. Sometimes, to well arrange all those 2D GnO sheets, a surfactant such as CTAB143 is needed.
In recent years, the applications of carbon foam were fully reviewed in the field of electromagnetic interference shielding144 and solar steam generation,145,146 among which some are ACM-derived carbon foam. In addition, after doping non-metallic or metallic elements, the carbon foam composites are enabled with some new capabilities, for example, sensitivity to some small molecules, which is useful for environmental protection.147 Furthermore, carbon-integrated polymer composite foams have several uses in the field of biomedical engineering.148
4. Concluding remarks
ACMs have been synthesized from various raw materials, ranging from organics as biomass extracted from brown coal or peat, pitch and green coke to crystalline graphite. Alternatively, some ACMs occur in nature as humic acids, which often coexist with some impurities, such as minerals. Depending on their raw materials, ACMs have a wide elemental composition, which is limited to 50–70 wt% C and 30–40 wt% O for most ACMs, except GtO and few-layered GnO originating from graphite. Considering that ionizable surface functional groups are responsible for the hydrophilicity of ACMs, while conjugated polyaromatic moieties make them hydrophobic, their amphiphilicity can be controllable and adjusted by excluding some surface functional groups and changing the size of their aromatic moieties. Once the amphiphilicity of ACMs becomes precisely tunable, it will promote their applications markedly. However, it must be pointed out that controlling and adjusting the amphiphilicity of ACMs have not been successfully achieved to date. The hydrothermal processing of biomass149 may enable this by changing the pH value of the aqueous environment and choosing different precursors for ACMs, focusing on their aromatic carbon percentage.It has been reviewed that carbon coating of electrode materials, including graphite and Li4Ti5O12, is effective to improve the reversible capacity, coulombic efficiency and cycle performances of LIBs.82 Besides the carbon coating precursors usually used in organic solvents, for example pitch, ACMs make carbon coating practical in aqueous solution owing to their hydrophilicity. ACM-associated coating techniques make it possible to avoid the use of organic solvents, which are associated with pollution and safety risks. They are also economic and facile pathways with high carbon yields from various raw materials.150 In particular, for these coating processes in alkali solution, ACMs may be introduced in the synthesis system immediately after the formation of the core substances, without solid–liquid separation, rinsing, drying and pulverization, which are usually necessary before carbon coating in an organic solvent. From a technology point of view, this process simplification offers engineers a promising improvement in controllability.
As an amphiphile, ACMs are theoretically and practically suitable for the preparation of carbon-supported catalysts, which has attracted scarce attention to date. Besides their potential gelation in aqueous solution, ACMs are Lewis bases with multi and scattered negatively charged active sites, and consequently can interact strongly with metal ions (regarded as Lewis acids). This capture provided by ACMs will prevent the aggregation of metal ions usually observed during catalytic processes, inspired by the research on a long-life MnO2 electrode.103 In addition, based on a conceptual analysis, porous carbon carriers may be formatted during the calcination of catalysts via desorption of their surface functional groups, rather than during pre-synthesis.
To apply ACMs in the fields of biomedicine and environment remediation, particularly few-layered GnO, high reproducibility in their fabrication is required. Various routes for the synthesis of graphene intermediated by few-layered GnO from graphite by oxidation have been reported,32,151 but processes for the preparation of few-layered GnO as an amphiphilic material has to be explored to control its amphiphilicity.
Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge Dr Qiao Zhijun for providing us with Zeta potential data of humic acids, Dr Wang Jiuzhou and MSc. Wang Liqun for sharing their FTIR results of ACM-GC and ACM-CP with us, Dr Zhang Yanan for some image modification. We also would like to appreciate Lin Weiteng, Jia Ke, Zhang Yating, Li Xuan and Lu Hongbing for their help in the layout of the paper. This work was financially supported by the National Natural Science Foundation of China (NSFC 51372168, 22078228).References
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Biomedical applications of graphene and graphene oxide, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS.
- J. Lee, J. Kim, S. Kim and D.-H. Min, Biosensors based on graphene oxide and its biomedical application, Adv. Drug Delivery Rev., 2016, 105, 275–287 CrossRef CAS PubMed.
- M. Fujii, Y. Yamada, T. Imamura and H. Honda, Preparation of aqua-mesophase by nitration or sulfonation of carbonaceous mesophase and properties of carbon material made from it, 18th Biennial Conference of Carbon. Worcester Polytechnic Institute, American Carbon Society, Massachusetts. USA, 1987, pp. 405–406 Search PubMed.
- M. Inagaki and K. Takai, Terminology for graphene to graphite and for graphene oxide to graphite oxide, Carbon Reports, 2022, 1, 59–69 CrossRef.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The chemistry of graphene oxide, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- M.-D. Ahmed and C. R. Kinney, Pyrolysis of humic acids prepared from oxidized bituminous coal, J. Am. Chem. Soc., 1950, 72, 556–559 CrossRef CAS.
- L. Wang, J. Wang, F. Jia, C. Wang and M. Chen, Nanoporous carbon synthesized with coal tar pitch and its capacitive performance, J. Mater. Chem. A, 2013, 1, 9498–9507 RSC.
- J. Wang, M. Chen, C. Wang, J. Wang and J. Zheng, Preparation of mesoporous carbons from amphiphilic carbonaceous material for high-performance electric double-layer capacitors, J. Power Sources, 2011, 196, 550–558 CrossRef CAS.
- J. Wang, L. Wang, M. Chen, C. Wang, C. Zhang and F. He, Nanoporous carbons from oxidized green needle coke for high performance supercapacitor, New Carbon Mater., 2015, 30, 141–149 CrossRef.
- M. N. Jones and N. D. Bryan, Colloidal properties of humic substances, Adv. Colloid Interface Sci., 1998, 78, 1–48 CrossRef CAS.
- L. J. Cote, J. Kim, V. C. Tung, J. Luo, F. Kim and J. X. Huang, Graphene oxide as surfactant sheets, Pure Appl. Chem., 2011, 83, 95–110 CAS.
- D. Tateishi, K. Esumi and H. Honda, Formation of carbonaceous 22 gel, Carbon, 1991, 29, 1296–1298 CrossRef CAS.
- K. Esumi, S. Takahashi, H. Sugii, D. Tateishi and H. Honda, Characterization of organo carbonaceous gel, Carbon, 1993, 31, 1303–1306 CrossRef CAS.
- D. Tateishi, K. Esumi, H. Honda and H. Oda, Characterization of carbonaceous gel and gel beads, Tanso, 1993, 157, 59–68 CrossRef CAS.
- Y. Guo, Z. Shi, M. Chen and C. Wang, Hierarchical porous carbon derived from sulfonated pitch for electrical double layer capacitors, J. Power Sources, 2014, 252, 235–243 CrossRef CAS.
- Y. Zhu, Y. Guo, C. Wang, Z. Qiao and M. Chen, Fabrication of conductive carbonaceous spherical architecture from pitch by spray drying, Chem. Eng. Sci., 2015, 135, 109–116 CrossRef CAS.
- Z. Qiao, M. Chen, C. Wang and Y. Yuan, Humic acids-based hierarchical porous carbons as high-rate performance electrodes for symmetric supercapacitors, Bioresour. Technol., 2014, 163, 386–389 CrossRef CAS PubMed.
- A. Jankowska, T. Stiemieniewska, K. Tomkov, M. Jasienko-Halat, J. Kaczmarczyk, A. Albiniak, J. J. Freeman and M. Yates, The pore structure of activated chars of brown coal humic acids obtained at increased rate of carbonization, Carbon, 1993, 31, 871–880 CrossRef CAS.
- H. B. Charmbury, J. W. Eckerd, J. S. La Torre and C. R. Kinney, The chemistry of nitrogen in humic acids from nitric acid treated coal, J. Am. Chem. Soc., 1945, 67, 625–628 CrossRef CAS.
- J. Luo, L. J. Cote, V. C. Tung, A. T. L. Tan, P. E. Goins, J. Wu and J. X. Huang, Graphene oxide nanocolloids, J. Am. Chem. Soc., 2010, 132, 17667–17669 CrossRef CAS.
- M.-M. Chen, X.-Y. Zhang, L.-Q. Wang and C.-Y. Wang, Effects of amphiphilic carbonaceous nanomaterial on the synthesis of MnO2 and its energy storage capability as an electrode material for pseudocapacitors, Ind. Eng. Chem. Res., 2014, 53, 10974–10981 CrossRef CAS.
- D. Tateishi, K. Esumi, H. Honda and H. Oda, Preparation of carbonaceous gel beads, Carbon, 1992, 30, 942–944 CrossRef CAS.
- J. Huang, W. Xing, F. Subhan, X. Gao, P. Bai, Z. Liu, Y. Wang, Q. Xue and Z. Yan, Functionalization of petroleum coke-based mesoporous carbon for synergistically enhanced capacitive performance, J. Mater. Res., 2017, 32, 1248–1257 CrossRef CAS.
- Z. Li, W. Yan and S. Dai, A novel vesicular carbon synthesized using amphiphilic carbonaceous material and micelle templating approach, Carbon, 2004, 42, 767–770 CrossRef CAS.
- P.-P. Chang, K. Matsumura, C.-Y. Wang, T. Kinumoto, T. Tsumura, M.-M. Chen and M. Toyoda, Frame-filling structural nanoporous carbon from amphiphilic, Carbon, 2016, 108, 225–233 CrossRef CAS.
- D. Tateishi, K. Esumi and H. Honda, Preparation of organo-carbonaceous gel, Carbon, 1992, 30, 944–945 CrossRef CAS.
- K. Esumi, S. Takahashi, H. Sugii, D. Tateishi and H. Honda, Characterization of organo-carbonaceous gel, Carbon, 1993, 31, 1303–1306 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, Preparation of graphite oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- M. Inagaki, F. Kang and M. Toyoda, Exfoliation of graphite via intercalation compounds, Chem. Phys. Carbon, 2005, 29, 1–69 Search PubMed.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Electrochemistry of graphene and related materials, Chem. Rev., 2014, 114, 7150–7188 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Graphene and graphene oxide: Synthesis, properties, and applications, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- M. Inagaki, Y. A. Kim and M. Endo, Graphene: Preparation and structural perfection, J. Mater. Chem., 2011, 21, 3280–3294 RSC.
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, A roadmap for graphene, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Recent advances in graphene-based biosensors, Biosens. Bioelectron., 2011, 26, 4637–4648 CrossRef CAS.
- M. Toyoda, S. Hou, Z.-H. Huang and M. Inagaki, Exfoliated graphite: room temperature exfoliation and their applications, Carbon Lett., 2022, 33, 335–362 CrossRef.
- C. Brodie, Sur ie poids atomique du graphite, Ann. Chim. Phys., 1860, 59, 466–472 Search PubMed.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, Graphene oxide sheets at interfaces, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- O. Akhavan, The effect of heat treatment on formation of graphene thin films from graphene oxide nanosheets, Carbon, 2010, 48, 509–519 CrossRef CAS.
- M. Yasin, T. Tauqeer, S. M. H. Zaidi, S. E. San, A. Mahmood, M. E. Köse, B. Canimkurbey and M. Okutan, Synthesis and electrical characterization of graphene oxide films, Thin Solid Films, 2015, 590, 118–123 CrossRef CAS.
- W. S. Hung, Q. F. An, M. De Guzman, H. Y. Lin, S. H. Huang, W. R. Liu, C. C. Hu, K. R. Lee and J. Y. Lai, Pressure-assisted self-assembly technique for fabricating composite membranes consisting of highly ordered selective laminate layers of amphiphilic graphene oxide, Carbon, 2014, 68, 670–677 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Preparation and characterization of graphene oxide paper, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- R. Rasuli, Z. Mokarian, R. Karimi, H. Shabanzadeh and Y. Abedini, Wettability modification of graphene oxide by removal of carboxyl functional groups using non-thermal effects of microwave, Thin Solid Films, 2015, 589, 364–368 CrossRef CAS.
- G. Wang, B. Wang, J. Park, J. Yang, X. Shen and J. Yao, Synthesis of enhanced hydrophilic and hydrophobic graphene oxide nanosheets by a solvothermal method, Carbon, 2009, 47, 68–72 CrossRef CAS.
- D. Luo, F. Wang, B. V. Vu, J. Chen, J. Bao, D. Cai, R. C. Willson and Z. Ren, Synthesis of graphene-based amphiphilic Janus nanosheets via manipulation of hydrogen bonding, Carbon, 2018, 126, 105–110 CrossRef CAS.
- J. Cao, Y. Chen, X. Wang, J. Zhang, Y. Li, S. Wang, X. Wang and C. Liu, Janus sulfonated graphene oxide nanosheets with excellent interfacial properties for enhanced oil recovery, Chem. Eng. J., 2022, 443, 136391 CrossRef CAS.
- C. Ma, Q. Liu, Q. Peng, G. Yang, M. Jiang, L. Zong and J. Zhang, Biomimetic hybridization of Janus-like graphene oxide into hierarchical porous hydrogels for improved mechanical properties and efficient solar desalination devices, ACS Nano, 2021, 15, 19877–19887 CrossRef CAS PubMed.
- S. Salati, G. Papa and F. Adani, Perspective on the use of humic acids from biomass as natural surfactants for industrial applications, Biotechnol. Adv., 2011, 29, 913–922 CrossRef CAS PubMed.
- W.-W. Tang, G.-M. Zeng, J.-L. Gong, J. Liang, P. Xu, C. Zhang and B.-B. Huang, Impact of humic/fulvic acid on the removal of heavy metals from aqueous solutions using nanomaterials: A review, Sci. Total Environ., 2014, 468–469, 1014–1027 CrossRef CAS PubMed.
- A. Radwan, R. J. Willey, G. Davies, A. Fataftah, E. A. Ghabbour and S. A. Jansen, Supercritical fluid CO2 extraction accelerates isolation of humic acid from live Pilayella littoralis (Phaeophyta), J. Appl. Phycol., 1996, 8, 545–551 CrossRef CAS.
- J. Schellekens, P. Buurman, K. Kalbitz, A. van Zomeren, P. Vidal-Torrado, C. Cerli and R. N. J. Comans, Molecular Features of Humic Acids and Fulvic Acids from Contrasting Environments, Environ. Sci. Technol., 2017, 51, 1330–1339 CrossRef CAS.
- H.-R. Schulten and M. Schnitzer, A state of the art structural concept for humic substances, Naturwissenschaften, 1993, 80, 29–30 CrossRef CAS.
- M. Erhayem and M. Sohn, Effect of humic acid source on humic acid adsorption onto titanium dioxide nanoparticles, Sci. Total Environ., 2014, 470–471, 92–98 CrossRef CAS.
- Y. Zhou, Y. Zhang, P. Li, G. Li and T. Jiang, Comparative study on the adsorption interactions of humic acid onto natural magnetite, hematite and quartz: Effect of initial HA concentration, Powder Technol., 2014, 251, 1–8 CrossRef CAS.
- M. C. Wang and P. M. Huang, Catalytic power of nontronite, kaolinite and quartz and their reaction sites in the formation of hydroquinone-derived polymers, Appl. Clay Sci., 1989, 4, 43–57 CrossRef CAS.
- S. Fukuchi, A. Miura, R. Okabe, M. Fukushima, M. Sasaki and T. Sato, Spectroscopic investigations of humic-like acids formed via polycondensation reactions between glycine, catechol and glucose in the presence of natural zeolites, J. Mol. Struct., 2010, 982, 181–186 CrossRef CAS.
- Z. Wang, T. Wu, Z. Wu, Z.-K. Li, J. Yan, H. Yan, C. Pan, S. Kang, Z. Lei, S. Ren and H. Shui, A low carbon footprint method for converting low-rank coals to oxygen-containing chemicals, Fuel, 2022, 315, 123277 CrossRef CAS.
- A. K. Mittal and C. Venkobachar, Uptake of cationic dyes by sulfonated coal: Sorption mechanism, Ind. Eng. Chem. Res., 1996, 35, 1472–1474 CrossRef CAS.
- R. Alvarez-Puebla, R. Aroca, C. Valenzuela-Calahorro and J. Garrido, Retention of cobalt on a humin derived from brown coal, J. Hazard. Mater., 2006, 135, 122–128 CrossRef CAS.
- V. Calemma, P. Iwanski, R. Rausa and E. Girardi, Changes in coal structure accompanying the formation of regenerated humic acids during air oxidation, Fuel, 1994, 73, 700–707 CrossRef CAS.
- L. Dong, Q. Yuan and H. Yuan, Changes of chemical properties of humic acids from crude and fungal transformed lignite, Fuel, 2006, 85, 2402–2407 CrossRef CAS.
- S. Chang and R. A. Berner, Humic substance formation via the oxidative weathering of coal, Environ. Sci. Technol., 1998, 32, 2883–2886 CrossRef CAS.
- R. Jia, J. Ren, X. Liu, G. Lu and Y. Wang, Design and synthesis of sulfonated carbons with amphiphilic properties, J. Mater. Chem. A, 2014, 2, 11195–11201 RSC.
- R. Lu, S. He, T. Wang, L. Lai and M. Zhao, Low-cost preparation of temperature-resistant and salt-tolerant amphiphilic carbon dots from a nonionic surfactant and its application in enhanced oil recovery, Carbon, 2024, 225, 119104 CrossRef CAS.
- C. Yao, A. Yuan, Z. Wang, H. Lei, L. Zhang, L. Guo and X. Dong, Amphiphilic two-dimensional graphitic carbon nitride nanosheets for visible-light-driven phase boundary photocatalysis, J. Mater. Chem. A, 2019, 7, 13071–13079 RSC.
- Y. Yang, M. Zhao and L. Lai, Surface activity, micellization, and application of nano-surfactants-amphiphilic carbon dots, Carbon, 2023, 202, 398–413 CrossRef CAS.
- J. Wang, M. Chen, C. Wang, J. Wang and J. Zheng, A facile method to prepare carbon aerogels from amphiphilic carbon material, Mater. Lett., 2012, 68, 446–449 CrossRef CAS.
- J. Yin, D. Zhang, J. Zhao, X. Wang, H. Zhu and C. Wang, Meso- and micro-porous composite carbons derived from humic acid for supercapacitors, Electrochim. Acta, 2014, 136, 504–512 CrossRef CAS.
- T. Cottineau, M. Toupin, T. Delahaye, T. Brousse and D. Bélanger, Nanostructured transition metal oxides for aqueous hybrid electrochemical supercapacitors, Appl. Phys. A, 2006, 82, 599–606 CrossRef CAS.
- P. Perret, Z. Khani, T. Brousse, D. Bélanger and D. Guay, Carbon/PbO2 asymmetric electrochemical capacitor based on methanesulfonic acid electrolyte, Electrochim. Acta, 2011, 56, 8122–8128 CrossRef CAS.
- Y. Zhang, C. Zhang, G. Huang, B. Xing and Y. Duan, Synthesis and capacitive properties of manganese oxide nanoparticles dispersed on hierarchical porous carbons, Electrochim. Acta, 2015, 166, 107–116 CrossRef CAS.
- V. Verheyen, R. Rathbone, M. Jagtoyen and F. Derbyshire, Activated extrudates by oxidation and KOH activation of bituminous coal, Carbon, 1995, 33, 763–772 CrossRef CAS.
- C. Gómez-de-Salazar, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, Preparation of carbon molecular sieves by controlled oxidation treatments, Carbon, 2000, 38, 1889–1892 CrossRef.
- J. B. Parra, J. J. Pis, J. C. De Sousa, J. A. Pajares and R. C. Bansal, Effect of coal preoxidation on the development of microporosity in activated carbons, Carbon, 1996, 34, 783–787 CrossRef CAS.
- C. Daulan, S. B. Lyubchik, J.-N. Rouzaud and F. Béguin, Influence of anthracite pretreatment in the preparation of activated carbons, Fuel, 1998, 77, 495–502 CrossRef CAS.
- S. B. Lyubchik, R. Benoit and F. Béguin, Influence of chemical modification of anthracite on the porosity of the resulting activated carbons, Carbon, 2002, 40, 1287–1294 CrossRef CAS.
- B. Serrano-Talavera, M. J. Muñoz-Guillena, A. Linares-Solano and C. Salinas-Martínez de Lecea, Activated carbons from spanish coals. 3. preoxidation effect on anthracite activation, Energy Fuels, 1997, 11, 785–791 CrossRef CAS.
- C. Lu, S. Xu, M. Wang, L. Wei, S. Liu and C. Liu, Effect of pre-oxidation on the development of porosity in activated carbons from petroleum coke, Carbon, 2007, 45, 206–209 CrossRef CAS.
- B. Jiang, Y. Zhang, J. Zhou, K. Zhang and S. Chen, Effects of chemical modification of petroleum cokes on the properties of the resulting activated carbon, Fuel, 2008, 87, 1844–1848 CrossRef CAS.
- A. S. Wiryoatmojo, H. A. Mannan, R. Nasir, H. Mukhtar, D. F. Mohshim, A. Abdulrahman and Z. Man, Surface modification effect of carbon molecular sieve (CMS) on the morphology and separation performance of mixed matrix membranes, Polym. Test., 2019, 80, 106152 CrossRef CAS.
- K. Esumi, S. Eshima, Y. Murakami, H. Honda and H. Oda, Preparation of hollow carbon-microbeads from water-in-oil emulsion using amphiphilic carbonaceous material, Colloids Surf., A, 1996, 108, 113–116 CrossRef CAS.
- G. W. Beall, E.-S. M. Duraia, Q. Yu and Z. Liu, Single crystalline graphene synthesized by thermal annealing of humic acid over copper foils, Phys. E, 2014, 56, 331–336 CrossRef CAS.
- M. Inagaki, Carbon coating for enhancing the functionalities of materials, Carbon, 2012, 50, 3247–3266 CrossRef CAS.
- J. Wang, M. Chen, C. Wang, B. Hu and J. Zheng, Amphiphilic carbonaceous material modified graphite as anode material for lithium-ion batteries, Mater. Lett., 2010, 64, 2281–2283 CrossRef CAS.
- H. Nozaki, K. Nagaoka, K. Hoshi, N. Ohta and M. Inagaki, Carbon-coated graphite for anode of lithium ion rechargeable batteries: Carbon coating conditions and precursors, J. Power Sources, 2009, 194, 486–493 CrossRef CAS.
- N. Ohta, K. Nagaoka, K. Hoshi, S. Bitoh and M. Inagaki, Carbon-coated graphite for anode of lithium ion rechargeable batteries: Graphite substrates for carbon coating, J. Power Sources, 2009, 194, 985–990 CrossRef CAS.
- K. Hoshi, N. Ohta, K. Nagaoka, S. Bitoh, A. Yamanaka, H. Nozaki, T. Okuni and M. Inagaki, Production and advantages of carbon-coated graphite for the anode of lithium ion rechargeable batteries, Carbon, 2010, 48, 1322 CrossRef CAS.
- X. Guo, C. Wang, M. Chen, J. Wang and J. Zheng, Carbon coating of Li4Ti5O12 using amphiphilic carbonaceous material for improvement of lithium-ion battery performance, J. Power Sources, 2012, 214, 107–112 CrossRef CAS.
- M.-M. Chen, Q.-Q. Ma, C.-Y. Wang, X. Sun, L.-Q. Wang and C. Zhang, Amphiphilic carbonaceous material-intervened solvothermal synthesis of LiFePO4, J. Power Sources, 2014, 263, 268–275 CrossRef CAS.
- F. Mohd Omar, H. Abdul Aziz and S. Stoll, Aggregation and disaggregation of ZnO nanoparticles: Influence of pH and adsorption of Suwannee River humic acid, Sci. Total Environ., 2014, 468–469, 195–201 CrossRef CAS.
- K. Krishnamoorthy, U. Navaneethaiyer, R. Mohan, J. Lee and S.-J. Kim, Graphene oxide nanostructures modified multifunctional cotton fabrics, Appl. Nanosci., 2012, 2, 119–126 CrossRef CAS.
- S.-K. Mohammad and E. Y. Mohammad, Preparation of superhydrophobic electroconductive graphene-coated cotton cellulose, Cellulose, 2013, 20, 963–972 CrossRef.
- N. D. Tissera, R. N. Wijesena, J. R. Perera, K. M. N. de Silva and G. A. J. Amaratunge, Hydrophobic cotton textile surfaces using an amphiphilic graphene oxide (GO) coating, Appl. Surf. Sci., 2015, 324, 455–463 CrossRef CAS.
- K. Wasiński, M. Walkowiak and G. Lota, Humic acids as pseudocapacitive electrolyte additive for electrochemical double layer capacitors, J. Power Sources, 2014, 255, 230–234 CrossRef.
- A. Radwan, R. J. Willey and G. Davies, Effects of gel processing on the properties of humic acid isolated from the alga pilayella littoralis: A humic acid aerogel, J. Appl. Phycol., 1997, 9, 481–488 CAS.
- R. J. Willey, A. Radwan, M. E. Vozzella, A. Fataftah, G. Davies and E. A. Ghabbour, Humic acid gel drying with supercritical carbon dioxide, J. Non-Cryst. Solids, 1998, 225, 30–35 CrossRef CAS.
- R. N. Nickolov and D. R. Mehandjiev, On the possibility of surface modification of an alumina support with amphiphilic carbonaceous material, Adsorpt. Sci. Technol., 1998, 16, 453–463 CrossRef CAS.
- K. Esumi, H. Sugii, D. Tateishi and H. Honda, Preparation of carbon microbeads containing fine platinum particles from ‘aqua-mesophase’, Carbon, 1992, 30, 121–122 CrossRef CAS.
- K. Esumi, R. Ono, H. Sugii, H. Honda and M. Kodama, Catalytic property of platinum-dispersed carbon prepared using amphiphilic carbonaceous material, Colloids Surf., A, 1995, 94, 93–96 CrossRef CAS.
- Y. Murakami, K. Esumi and H. Honda, Preparation of the electrocatalytic electrode coated using amphiphilic carbonaceous material containing platinum compound, Carbon, 1994, 32, 1188–1189 CrossRef CAS.
- Y. Murakami, K. Esumi and H. Honda, Preparation and characterization of an electrocatalytic electrode using amphiphilic carbonaceous material containing metal compound, Carbon, 1996, 34, 463–470 CrossRef CAS.
- M. Takigami, S.-Y. Baba and J.-I. Ozaki, Preparation of carbon alloy catalysts from humic acid and their activities for the oxygen reduction reaction, Carbon, 2015, 93, 1077 CrossRef.
- M.-M. Chen, X.-Y. Zhang, L.-Q. Wang and C.-Y. Wang, Effects of amphiphilic carbonaceous nanomaterial on the synthesis of MnO2 and its energy storage capability as an electrode material for pseudocapacitors, Ind. Eng. Chem. Res., 2014, 53, 10974–10981 CrossRef CAS.
- X.-Y. Zhang, L.-Q. Han, C.-Y. Wang and M.-M. Chen, Double-shelled MnO2 hollow spheres for supercapacitors, Mater. Lett., 2014, 136, 78–80 CrossRef CAS.
- E.-D. Jeong, M.-S. Won and Y.-B. Shim, Cathodic properties of a lithium-ion secondary battery using LiCoO2 prepared by a complex formation reaction, J. Power Sources, 1998, 70, 70–77 CrossRef CAS.
- X. Zhou, Y. Liu and Y. Guo, Effect of reduction agent on the performance of Li3V2(PO4)3/C positive material by one-step solid-state reaction, Electrochim. Acta, 2009, 54, 2253–2258 CrossRef CAS.
- C. D. García and P. I. Ortiz, Characterization and application of humic acid modified carbon electrodes, Talanta, 2003, 61, 547–556 CrossRef PubMed.
- S. M. Yakout, S. S. Metwally and T. El-Zakla, Uranium sorption onto activated carbon prepared from rice straw: Competition with humic acids, Appl. Surf. Sci., 2013, 280, 745–750 CrossRef CAS.
- D. Lin, T. Li, K. Yang and F. Wu, The relationship between humic acid (HA) adsorption on and stabilizing multiwalled carbon nanotubes (MWNTs) in water: Effects of HA, MWNT and solution properties, J. Hazard. Mater., 2012, 241–242, 404–410 CrossRef CAS PubMed.
- S. Ma, C. Liu, K. Yang and D. Lin, Coagulation removal of humic acid-stabilized carbon nanotubes from water by PACl: Influences of hydraulic condition and water chemistry, Sci. Total Environ., 2012, 439, 123–128 CrossRef CAS.
- S.-R. Chae, Y. Xiao, S. Lin, T. Noeiaghaei, J.-O. Kim and M. R. Wiesner, Effects of humic acid and electrolytes on photocatalytic reactivity and transport of carbon nanoparticle aggregates in water, Water Res., 2012, 46, 4053–4062 CrossRef CAS PubMed.
- S. Wang, H. Sun, H. M. Ang and M. O. Tadé, Adsorptive remediation of environmental pollutants using novel graphene-based nanomaterials, Chem. Eng. J., 2013, 226, 336–347 CrossRef CAS.
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Few-layered graphene oxide nanosheets as superior sorbents for heavy metal ion pollution management, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- G. Zhao, X. Ren, X. Gao, X. Tan, J. Li, C. Chen, Y. Huang and X. Wang, Removal of Pb(II) ions from aqueous solutions on few-layered graphene oxide nanosheets, Dalton Trans., 2011, 40, 10945–10952 RSC.
- G. Zhao, T. Wen, X. Yang, S. Yang, J. Liao, J. Hu, D. Shao and X. Wang, Preconcentration of U(VI) ions on few-layered graphene oxide nanosheets from aqueous solutions, Dalton Trans., 2012, 41, 6182–6188 RSC.
- Y. Sun, Q. Wang, C. Chen, X. Tan and X. Wang, Interaction between Eu(III) and graphene oxide nanosheets investigated by batch and extended X-ray absorption fine structure spectroscopy and by modeling techniques, Environ. Sci. Technol., 2012, 46, 6020–6027 CrossRef CAS PubMed.
- S.-T. Yang, Y. Chang, H. Wang, G. Liu, S. Chen, Y. Wang, Y. Liu and A. Cao, Folding/aggregation of graphene oxide and its application in Cu2+ removal, J. Colloid Interface Sci., 2010, 351, 122–127 CrossRef CAS.
- Y. Gao, Y. Li, L. Zhang, H. Huang, J. Hu, S. M. Shah and X. Su, Adsorption and removal of tetracycline antibiotics from aqueous solution by graphene oxide, J. Colloid Interface Sci., 2012, 368, 540–546 CrossRef CAS.
- J. D. Wuest and A. Rochefort, Strong adsorption of aminotriazines on graphene, Chem. Commun., 2010, 46, 2923–2925 RSC.
- C. Petit and T. J. Bandosz, Graphite oxide/polyoxometalate nanocomposites as adsorbents of ammonia, J. Phys. Chem. C, 2009, 113, 3800–3809 CrossRef CAS.
- M. Liu, C. Chen, J. Hu, X. Wu and X. Wang, Synthesis of magnetite/graphene oxide composite and application for cobalt(ii) removal, J. Phys. Chem. C, 2011, 115, 25234–25240 CrossRef CAS.
- J. Li, S. Zhang, C. Chen, G. Zhao, X. Yang, J. Li and X. Wang, Removal of Cu(II) and fulvic acid by graphene oxide nanosheets decorated with Fe3O4 nanoparticles, ACS Appl. Mater. Interfaces, 2012, 4, 4991–5000 CrossRef CAS.
- L. Liu, C. Li, C. Bao, Q. Jia, P. Xiao, X. Liu and Q. Zhang, Preparation and characterization of chitosan/graphene oxide composites for the adsorption of Au(III) and Pd(II), Talanta, 2012, 93, 350–357 CrossRef CAS PubMed.
- Z.-H. Huang, G. Liu and F. Kang, Glucose-promoted Zn-based metal-organic framework/graphene oxide composites for hydrogen sulfide removal, ACS Appl. Mater. Interfaces, 2012, 4, 4942–4947 CrossRef CAS PubMed.
- W.-S. Hung, Q.-F. An, M. D. Guzman, H.-Y. Lin, S.-H. Huang, W.-R. Liu, C.-C. Hu, K.-R. Lee and J.-Y. Lai, Pressure-assisted self-assembly technique for fabricating composite membranes consisting of highly ordered selective laminate layers of amphiphilic graphene oxide, Carbon, 2014, 68, 670–677 CrossRef CAS.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Unimpeded permeation of water through helium-leak-tight graphene-based membranes, Science, 2012, 335, 442–444 CrossRef CAS.
- C. Xu, X. Wu, J. Zhu and X. Wang, Synthesis of amphiphilic graphite oxide, Carbon, 2008, 46, 386–389 CrossRef CAS.
- S. Radic, N. K. Geitner, R. Podila, A. Käkinen, P. Chen, P. C. Ke and F. Ding, Competitive binding of natural amphiphiles with graphene derivatives, Sci. Rep., 2013, 3, 2273–2280 CrossRef.
- G. Lu, L. E. Ocola and J. Chen, Gas detection using low temperature reduced graphene oxide sheets, Appl. Phys. Lett., 2009, 94, 083111 CrossRef.
- G. Singh, A. Choudhary, D. Haranath, A. G. Joshi, N. Singh, S. Singh and R. Pasricha, ZnO decorated luminescent graphene as a potential gas sensor at room temperature, Carbon, 2012, 50, 385–394 CrossRef CAS.
- C. Zhu, D. Du and Y. Lin, Graphene-like 2D nanomaterial-based biointerfaces for biosensing applications, Biosens. Bioelectron., 2017, 89, 43–55 CrossRef CAS PubMed.
- S. Syama and P. V. Mohanan, Safety and biocompatibility of graphene: A new generation nanomaterial for biomedical application, Int. J. Biol. Macromol., 2016, 86, 546–555 CrossRef CAS.
- B. Zhang, Y. Wang and G. Zhai, Biomedical applications of the graphene-based materials, Mater. Sci. Eng., C, 2016, 61, 953–964 CrossRef CAS.
- N. Mohanty and V. Berry, Graphene-based single-bacterium resolution biodevice and DNA transistor: Interfacing graphene derivatives with nanoscale and microscale biocomponents, Nano Lett., 2008, 8, 4469–4476 CrossRef CAS PubMed.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, A graphene platform for sensing biomolecules, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS.
- S. He, B. Song, D. Li, C. Zhu, W. Qi, Y. Wen, L. Wang, S. Song, H. Fang and C. Fan, A graphene nanoprobe for rapid, sensitive, and multicolor fluorescent DNA analysis, Adv. Funct. Mater., 2010, 20, 453–459 CrossRef CAS.
- O. Akhavan and E. Ghaderi, Toxicity of graphene and graphene oxide nanowalls against bacteria, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
- L. Feng, S. Zhang and Z. Liu, Graphene based gene transfection, Nanoscale, 2011, 3, 1252–1257 RSC.
- B. Chen, M. Liu, L. Zhang, J. Huang, J. Yao and Z. Zhang, Polyethylenimine-functionalized graphene oxide as an efficient gene delivery vector, J. Mater. Chem., 2011, 21, 7736–7741 RSC.
- H. Kim, R. Namgung, K. Singha, I.-K. Oh and W.-J. Kim, Graphene oxide-polyethylenimine nanoconstruct as a gene delivery vector and bioimaging tool, Bioconjugate Chem., 2011, 22, 2558–2567 CrossRef CAS PubMed.
- N. Yang, X. Gao, Y. Shen, M. Wang, L. Chang and Y. Lv, Effects of coal characteristics on the structure and performance of coal-based carbon foam prepared by self-foaming technique under atmospheric pressure, J. Anal. Appl. Pyrolysis, 2022, 164, 105516 CrossRef CAS.
- E. Rodríguez, M. A. Diez, C. Antuña-Nieto, M. A. López-Antón, R. García and M. R. Martínez-Tarazona, An insight into the role of biomass, biocompounds and synthetic polymers as additives to coal for the synthesis of carbon foams, J. Anal. Appl. Pyrolysis, 2021, 160, 105359 CrossRef.
- H. Liang, Q. Liao, N. Chen, Y. Liang, G. Lv, P. Zhang, B. Lu and L. Qu, Thermal efficiency of solar steam generation approaching 100% through capillary water transport, Angew. Chem., 2019, 58, 19041–19046 CrossRef CAS.
- X. Hu, W. Xu, L. Zhou, Y. Tan, Y. Wang, S. Zhu and J. Zhu, Tailoring graphene oxide-based aerogels for efficient solar steam generation under one sun, Adv. Mater., 2017, 29, 1604031 CrossRef.
- H. Liu, Y. Yang, N. Tian, C. You and Y. Yang, Foam-structured carbon materials and composites for electromagnetic interference shielding: Design principles and structural evolution, Carbon, 2024, 217, 118608 CrossRef CAS.
- M. Toyoda and M. Inagaki, Carbon materials for solar steam-generation, Carbon, 2023, 214, 118373 CrossRef CAS.
- H. Ghasemi, G. Ni, A. M. Marconnet, J. Loomis, S. Yerci, N. Miljkovic and G. Chen, Solar steam generation by heat localization, Nat. Commun., 2014, 5, 4449 CrossRef CAS.
- H. Zhao, X. Zhang, J. Wang and J. Wang, N, S co-doped porous carbon nanosheet foam as MALDI matrix for efficient and direct profiling of biomolecules and environmental contaminants, Appl. Surf. Sci., 2023, 623, 157052 CrossRef CAS.
- A. M. Díez-FPascual, State of the art in the antibacterial and antiviral applications of carbon-based polymeric nanocomposites, Int. J. Mol. Sci., 2021, 22, 10511 CrossRef PubMed.
- M.-M. Titirici and M. Antonietti, Chemistry and materials options of sustainable carbon materials made by hydrothermal carbonization, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- X.-F. Wang, M.-M. Chen, K. Matsumura, M. Toyoda and C.-Y. Wang, MgO-templated mesoporous carbons using a pitch-based thermosetting carbon precursor, RSC Adv., 2016, 6, 100546–100553 RSC.
- C. Botas, A. M. Pérez-Mas, P. Álvarez, R. Santamaría, M. Granda, C. Blanco and R. Menéndez, Optimization of the size and yield of graphene oxide sheets in the exfoliation step, Carbon, 2013, 63, 576–578 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |