
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation–anion co-redox induced high-capacity cathode for high energy density sodium-ion batteries†
Bing Zhangab,
Haoqing Maa,
Feng Taoa,
Chengsifan Leia,
Lei Zhangbcd,
Chaohui He*b and
Meng Huang *a
*a
aSanya Science and Education Innovation Park of Wuhan University of Technology, Sanya 572000, P.R. China. E-mail: 211808@whut.edu.cn
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P.R. China. E-mail: chaohuihe@whut.edu.cn
cHubei Longzhong Laboratory, Xiangyang, Hubei 441000, China
dZhongyu Feima New Material Technology Innovation Center (Zhengzhou) Co., Ltd, High Technology Industrial Development Zone, No. 60 Xuelan Road, Zhengzhou, 450001, P. R. China
First published on 22nd April 2025
Abstract
Iron/manganese-based layered transition metal oxides have emerged as competitive cathode candidates for sodium-ion batteries (SIBs) due to their high theoretical capacity and naturally abundant constituent elements. Nevertheless, implementation challenges persist due to irreversible phase transformations and substantial capacity fading during cycling. Herein, we present a novel P2-type Na0.73Fe0.2Mn0.52Co0.2Mg0.05Li0.03O2 (designated as P2-NFMO-CoMgLi) cathode material characterized by coupled cationic and anionic redox. The rational doping of Li+ into transition metal (TM) sites activates the reversible oxygen redox chemistry and suppresses the Jahn–Teller distortions. In addition, the doping of Mg2+ effectively inhibits Na+ vacancy ordering in low-voltage regimes (<2.5 V), and Co2+ incorporation concurrently improves specific capacity and stabilizes the TM–O bonding networks. Consequently, the P2-NFMO-CoMgLi cathode exhibits a high capacity of 178 mA h g−1 at 20 mA g−1 and a 68% capacity retention over 150 cycles at 200 mA g−1. Ex situ XPS characterization reveals that oxygen redox processes occur and provide extra capacity at high voltage. These findings provide a fundamental understanding of voltage-induced cation–anion redox in layered oxide cathodes, advancing the rational design of high-energy-density SIB systems.
1. Introduction
Sodium-ion batteries (SIBs) have emerged as promising candidates for large-scale energy storage systems and low-speed electric vehicle applications, primarily due to the natural abundance and economic viability of sodium resources coupled with their operational mechanisms analogous to lithium-ion batteries (LIBs).1,2 However, the practical implementation of SIBs faces significant challenges associated with the fundamental characteristics of Na+.3,4 Compared to their Li+ counterparts (ionic radius: 1.02 Å vs. 0.76 Å; atomic mass: 22.99 vs. 6.94 g mol−1), the larger ionic radius and heavier atomic mass of Na+ induce sluggish Na+ diffusion kinetics and irreversible structural deformation in conventional electrode materials, leading to compromised rate capability and cycling durability.5–7 Furthermore, the relatively higher standard redox potential of sodium (−2.71 V vs. SHE compared to lithium's −3.02 V) imposes intrinsic limitations on the theoretical energy density of SIB systems.8–10 The electrochemical performance of SIBs is fundamentally governed by four primary components: cathode, anode, electrolyte, and separator.11,12 Among these, cathode materials represent a critical determinant of overall battery performance, driving extensive research efforts to develop high-capacity, cost-effective cathodes with robust Na+ storage capabilities and favorable ionic transport properties.13,14 Layered transition metal oxides (TMOs) have garnered particular attention among various cathode candidates due to their intrinsic two-dimensional diffusion pathways and exceptional theoretical specific capacities (typically 200–250 mA h g−1).15,16 These TMO materials are conventionally classified into P2-type (prismatic Na+ coordination) and O3-type (octahedral coordination) structures, where the numerical designation indicates the periodic stacking sequence of transition metal layers within the unit cell.17–19 P2-type layered oxides demonstrate superior electrochemical performance compared to O3-type analogs, due to their expanded interlayer spacing (typically >5.6 Å) that enables more facile Na+ intercalation/deintercalation kinetics while maintaining structural integrity during prolonged cycling.20,21Iron–manganese-based P2-type layered oxide cathodes have drawn substantial interests for their cost-efficiency and eco-friendly merits, yet face inherent challenges, including low-voltage Na+/vacancy ordering, high-voltage P2–O2 phase transformations, and Mn3+-triggered Jahn–Teller distortions, all collectively accelerating structural degradation and capacity fade.22 While transition metal site doping (e.g., Ni2+, Cu2+, Zn2+) has been extensively employed to mitigate phase instability and electronic distortions through crystal field regulation, persistent issues remain—notably TM migration-induced P2–Z transitions, metastable P2–OP4 phase coexistence, and Na+/vacancy superlattice formation—which synergistically compromise electrochemical reversibility.23–26 Concurrent strategies involving surface engineering and morphological optimization demonstrate partial efficacy in enhancing cyclability.27–29 The selection of voltage window significantly impacts the electrochemical performance of layered cathode materials. Pang et al. demonstrated that restricting the charging voltage below 4.0 V effectively suppresses phase transitions in P2-type layered oxides under high voltages improving capacity retention. However, this approach concurrently leads to a reduction in initial capacity.30 The reversible capacity relying solely on cationic redox has reached its theoretical ceiling, which significantly impedes further energy density improvements in sodium-ion batteries (SIBs). In contrast, anionic redox chemistry—extensively studied in lithium-ion batteries (LIBs) to unlock additional capacity through oxygen or sulfur participation—provides a novel pathway for designing high-energy-density SIBs.31,32 This underscores the critical needs for integrated solutions that concurrently address phase transition, Jahn–Teller effect, and Na+/vacancy disordering—a multifaceted challenge demanding coordinated material design and fundamental investigation.
This study develops a novel P2-type Na0.73Fe0.2Mn0.52Co0.2Mg0.05Li0.03O2 (P2-NFMO-CoMgLi) cathode demonstrating coupled cationic and anionic redox. The material is able to sustain a complete solid-solution reaction even at elevated voltages up to 4.3 V. The strategic Li+ doping in transition metal (TM) layers enables capacity contributions from lattice oxygen redox while suppressing Jahn–Teller distortions. Meanwhile, the lower valence state of Li+ promotes the incorporation of a higher Na+ content (0.73) to maintain charge neutrality, which exceeds that of most reported P2-type materials and is advantageous for practical applications in full-cell configurations.33–35 In addition, Mg2+ incorporation disrupts low-voltage Na+/vacancy ordering, and Co2+ introduction enhances capacity retention through TM dissolution suppression. This synergistic multi-doping approach yields exceptional electrochemical performance, achieving a high capacity of 178 mA h g−1 at 20 mA g−1 and a high capacity retention of 68% after 150 cycles at 200 mA g−1 within 1.5–4.3 V. Systematic investigation of the Na+ storage mechanisms at various voltage windows reveals voltage-dependent performance variations (enhancing capacity via oxygen redox), providing fundamental insights for designing high-energy layered oxide SIBs through balancing anionic/cationic co-redox.
2. Experimental section
2.1. Materials
Analytical grade LiNO3 and Mn(CH3COO)2·4H2O were obtained from Macklin Reagent Company, whereas NaNO3, Fe(NO3)3·9H2O, Mg(CH3COO)2·4H2O, and Co(CH3COO)2·4H2O were sourced from Aladdin Reagent Company. Polyvinylidene fluoride (PVDF) was supplied by Solvay. N-Methyl-2-pyrrolidone (NMP, 99% purity) was sourced from Aladdin Reagent Company, while acetylene black was obtained from SCM Chem, Shanghai, China. Hard carbon, provided by XINWEI Technology Co., Ltd, was used in combination with a carbonate electrolyte solution containing 5 vol% fluoroethylene carbonate (FEC) from Duoduo Reagents Technology Co., Ltd.2.2. Synthesis of the cathode lithium replenishment material and electrodes
Stoichiometric molar equivalents of NaNO3 (Aladdin, AR), Fe(NO3)3·9H2O (Aladdin, AR), LiNO3 (Macklin, AR), Mn(CH3COO)2·4H2O (Macklin, AR), Mg(CH3COO)2·4H2O (Aladdin, AR), and Co(CH3COO)2·4H2O (Aladdin, AR) were dissolved in a PVP aqueous solution, with 5% excess Na and Li precursors added to compensate for high-temperature volatilization losses. After 12 h of stirring, the homogeneous solution was dried at 110 °C for 12 h. The resultant precursor was calcined in ambient atmosphere at 450 °C for 5 h, followed by sintering at 900 °C for 12 h to yield the final product.2.3. Electrode preparation
P2-NFMO-CoMgLi powder, acetylene black, and PVDF were mixed in NMP at a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 mass ratio to form a uniform slurry. The slurry was then cast onto aluminum foil and dried at 80 °C for 12 hours. The electrode was pressed into circular discs (10 mm diameter) and stamped, with a mass loading density of approximately 1.2–1.8 mg cm−2 relative to P2-NFMO-CoMgLi. Before button cell assembly, the cathode sheet was dried at 100 °C for 2 hours.
:2:1 mass ratio to form a uniform slurry. The slurry was then cast onto aluminum foil and dried at 80 °C for 12 hours. The electrode was pressed into circular discs (10 mm diameter) and stamped, with a mass loading density of approximately 1.2–1.8 mg cm−2 relative to P2-NFMO-CoMgLi. Before button cell assembly, the cathode sheet was dried at 100 °C for 2 hours.
The full-cell anode was made using hard carbon as the active material, Super P as the electronic conductor, and PVDF as the binder in an 8:1:1 mass ratio. A slurry of hard carbon, Super P, and PVDF was prepared and cast onto copper foil to form the electrode. The electrode was dried at 100 °C for 12 hours, then stamped into circular discs (10 mm diameter). The mass loading density of the electrode was maintained at approximately 1 mg cm−2 relative to the hard carbon.
2.4. Materials characterization
The morphology and microstructure of P2-NFMO-CoMgLi materials and electrodes were analyzed using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Elemental analysis was performed using scanning electron microscopy (SEM) with energy dispersive X-ray spectroscopy (EDS), transmission electron microscopy (TEM) with EDS, and inductively coupled plasma (ICP). The crystal structure of the samples was analyzed using X-ray diffraction (XRD, D8 Bruker). The surface elemental composition of the cycled cathode was characterized using X-ray photoelectron spectroscopy (XPS, Kratos AXIS Supra, Shimadzu). The working electrode was cleaned with dimethyl carbonate (DMC) to remove residual electrolyte from the surface and dried overnight in a glove box before measurements.2.5. Electrochemical characterization
The battery assembly process was conducted in a glove box filled with argon. The oxygen and moisture concentrations inside the glove box were maintained below 0.01 and 0.01 ppm, respectively. A half-cell was fabricated using a sodium metal as the counter electrode, GF/D as the separator, and P2-NFMO-CoMgLi as the positive electrode. 80 μL of electrolyte was injected into the half-cell, which consisted of a propylene carbonate (PC) solution with 5 vol% fluoroethylene carbonate (FEC). A full cell with an N/P ratio of approximately 1.1 was constructed using a P2-NFMO-CoMgLi cathode and hard carbon (denoted as HC) anode. Electrochemical performance was tested at 28 °C using a Land testing system. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were performed on an electrochemical workstation (Solartron Analytical).3. Results and discussion
3.1. Structural and morphological characterizations of P2-NFMO-CoMgLi
Fig. 1a outlines the synthesis protocol for the P2-Na0.73Fe0.2Mn0.52Co0.2Mg0.05Li0.03O2 compound, prepared via a polyvinylpyrrolidone (PVP)-assisted sol–gel method. X-ray diffraction (XRD) analysis confirms the typical P2-type layered structure with high purity. Rietveld refinement (Rwp = 1.69%) of the XRD pattern (Fig. 1b) validates the hexagonal P2 structure (space group: P63/mmc). The ionic radii compatibility of Co3+ (0.65 Å), Mg2+ (0.72 Å), and Li+ (0.76 Å) with Fe3+ (0.645 Å) and Mn4+ (0.53 Å) facilitates their preferential occupancy at transition metal (TM) sites.25 Refined lattice parameters (a = b = 2.8734 Å, c = 11.1989 Å, V = 80.074 Å3) and detailed crystallographic data (thermal factors, atomic coordinates, site occupancies) are tabulated in Table S1.† Morphological analysis reveals a platelet-like morphology with uniform particle sizes of 2–3 μm (Fig. 1c). High-resolution transmission electron microscope (HRTEM) images (Fig. 1d and e) demonstrate distinct lattice fringes corresponding to the (002) (d = 0.56 nm) and (102) (d = 0.23 nm) crystallographic planes, while the selected-area electron diffraction (SAED) pattern (Fig. 1f) along the [001] zone axis confirms monocrystalline characteristics of the hexagonal lattice. Energy-dispersive spectroscopy (EDS) elemental maps (Fig. 1g–m) demonstrate homogeneous distribution of Na, Mn, Fe, Co, Mg, Li, and O throughout the material. Inductively coupled plasma (ICP) analysis (Table S2†) confirms the stoichiometric consistency of the synthesized compound with theoretical values. X-ray photoelectron spectroscopy (XPS) further corroborates chemical purity, with survey spectra (Fig. S1†) exclusively containing peaks attributed to Na, Co, Mg, Li, Fe, Mn, and O.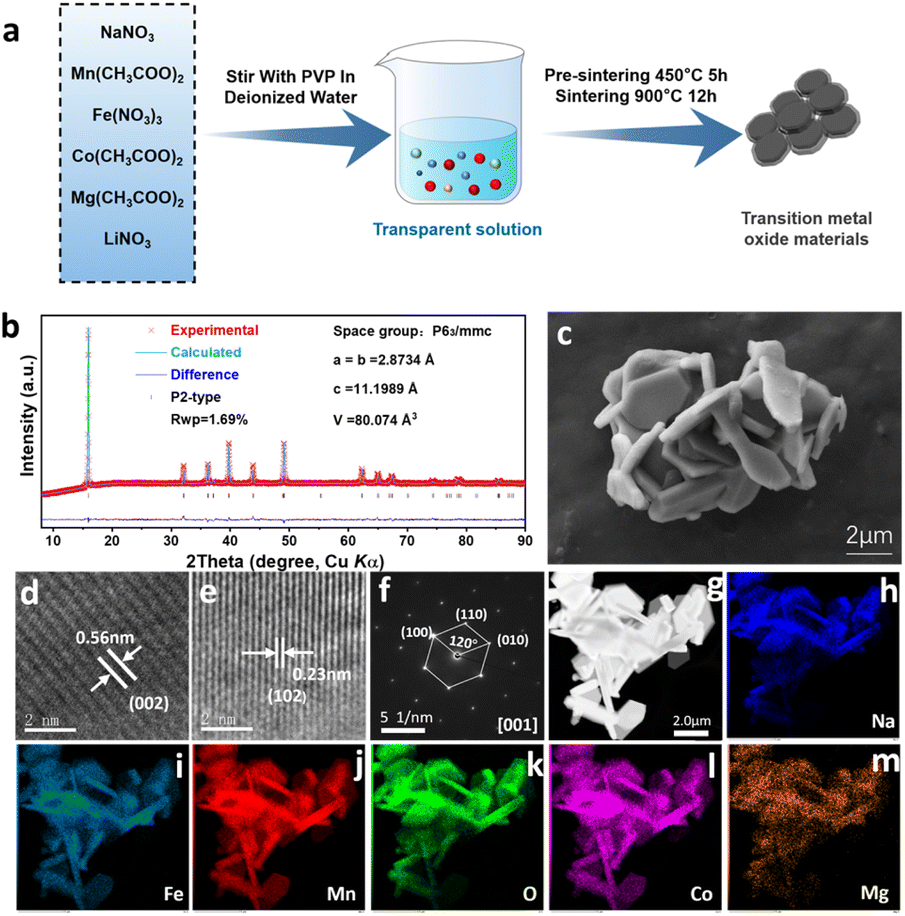 | ||
| Fig. 1 Structural and morphological characterizations: (a) schematic illustration of the fabrication process for P2-NFMO-CoMgLi. (b) Rietveld refined XRD pattern with corresponding cell parameters of the as-synthesized P2-NFMO-CoMgLi material. (c) SEM image. (d and e) HRTEM images. (f) SAED pattern of P2-NFMO-CoMgLi. (g–m) EDS mapping images of P2-NFMO-CoMgLi. | ||
3.2. Electrochemical properties of P2-NFMO-CoMgLi
The electrochemical performance of P2-NFMO-CoMgLi was systematically evaluated at various voltage windows. Cyclic voltammetry (CV) analysis (0.1 mV s−1, Fig. 2a–c) in the ranges of 1.5–4.0 V, 1.5–4.2 V, and 1.5–4.3 V vs. Na+/Na reveals reversible redox activity: peaks at 2.0/1.8 V and 2.2/2.3 V correspond to Mn3+/Mn4+ and Co2+/Co3+ transitions, respectively, while peaks at 3.5–3.8 V originate from Fe3+/Fe4+ redox. The weak intensity of Fe- and Co-related peaks reflects their low stoichiometric concentrations. Peak intensity of the CV curves shows gradual decrease, especially at 1.5–4.3 V. At elevated cutoff voltages (>4.0 V), emerging redox pairs at 3.9/4.0 V, 4.1/4.2 V, and 4.2/4.3 V are attributed to oxygen redox (O2−/(O2)n–) and show progressive intensification, which corresponds well with the O 1s XPS spectra presented later in the manuscript, as well as with the evolution of the high-voltage plateau observed in the charge/discharge profiles, indicating enhanced participation of the oxygen redox at higher potentials. Galvanostatic charge/discharge profiles (20 mA g−1, Fig. 2d) demonstrate voltage-dependent capacity, and the initial charge capacities are 60.1 mA h g−1 (1.5–4.0 V), 92.8 mA h g−1 (1.5–4.2 V), and 109.6 mA h g−1 (1.5–4.3 V). Corresponding discharge capacities are 130.8 mA h g−1, 163.1 mA h g−1, and 178.2 mA h g−1, respectively. Second-cycle profiles (Fig. 2e) exhibit rising average voltages and intensified high-voltage plateaus with expanded windows, further confirming the engagement of oxygen redox. To elucidate the superior rate capability of the P2-NFMO-CoMgLi cathode, kinetic analyses were conducted via galvanostatic intermittent titration technique (GITT) and cyclic voltammetry (CV). GITT-derived sodium diffusion coefficients (DNa), predominantly reside within 10−10–10−12 cm2 s−1 (Fig. S7†). Multi-sweep CV profiles (0.1–1.0 mV s−1, Fig. S8†) demonstrate low polarization and preserve well the curve shape with increasing scan rates, indicative of fast reaction kinetics. These findings collectively confirm the material's exceptional Na+ diffusivity, and thus high-rate performance is expected.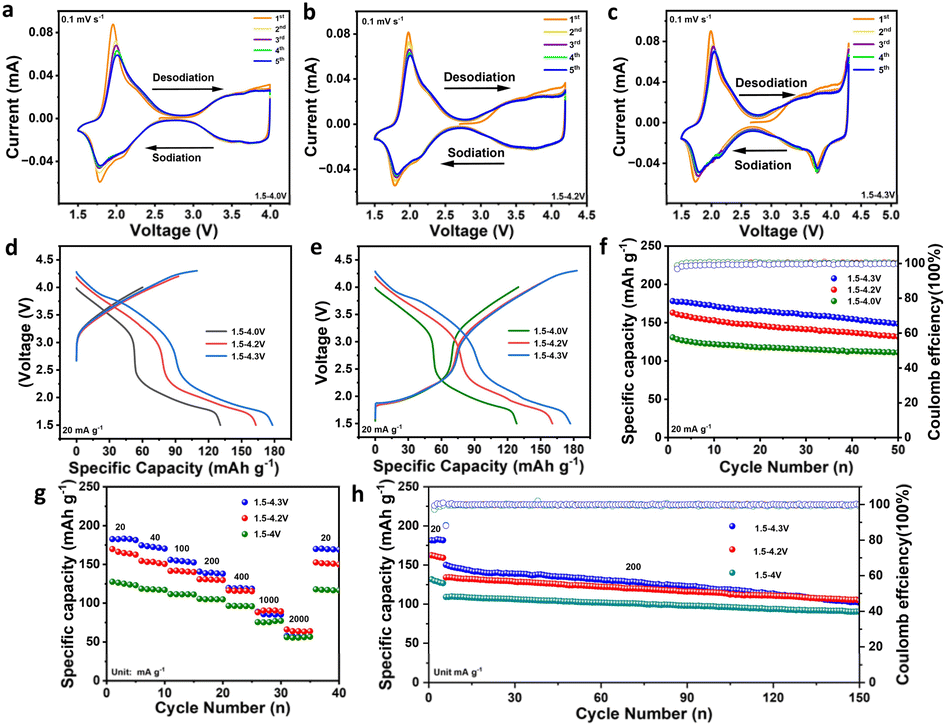 | ||
| Fig. 2 Electrochemical properties: (a–c) CV profiles at a scan rate of 0.1 mV s−1 over the voltage window of 1.5–4.0 V, 1.5–4.2 V and 1.5–4.3 V (vs. Na+/Na) of the P2-NFMO-CoMgLi cathode. (d and e) Galvanostatic charge/discharge curves for the first and second cycles at 20 mA g−1 over the voltage window of 1.5–4.2 V (vs. Na+/Na) of the P2-NFMO-CoMgLi cathode in comparison with 1.5–4.0 V and 1.5–4.3 V. (f) Cycling performance at 20 mA g−1 and (g) rate capability and (h) long-term cycling life of the P2-NFMO-CoMgLi cathode at 200 mA g−1 over the voltage window of 1.5–4.2 V (vs. Na+/Na) of the P2-NFMO-CoMgLi cathode in comparison with 1.5–4.0 V and 1.5–4.3 V. | ||
The cycling test at 20 mA g−1 (Fig. 2f) shows that P2-NFMO-CoMgLi can achieve the highest specific capacity (178.2 mA h g−1) within the voltage window of 1.5–4.3 V, compared to 1.5–4.0 V (130.8 mA h g−1) and 1.5–4.2 V (163.1 mA h g−1). After 50 cycles, the capacity retentions are 85% (1.5–4.0 V), 81% (1.5–4.2 V), and 83% (1.5–4.3 V), with the coulombic efficiency exceeding 99.5%. Fig. 2g displays the rate capability of the P2-NFMO-CoMgLi cathode at different voltage windows. Operating within the 1.5–4.3 V, the cathode delivers reversible capacities of 182.7, 174.7, 156.0, 140.7, 119.8, 88.9, and 57.8 mA h g−1 at various current densities (from 20 to 2000 mA g−1), retaining 49% of its initial capacity (20 mA g−1) at 1000 mA g−1. Narrowing the operational window to 1.5–4.2 V reduces the initial capacity to 169.8 mA h g−1 (20 mA g−1), and 88.3 mA h g−1 (52% retention) is delivered at 1000 mA g−1. At the operational window of 1.5–4.0 V, a much lower initial capacity of 127.6 mA h g−1 at 20 mA g−1 is obtained, of which 59% is maintained at 1000 mA g−1. When upper cutoff voltage augments, more oxygen redox participates. It concomitantly compromises rate capability under high-current-density operation through accelerated structural degradation. Electrochemical impedance spectra of P2-NFMO-CoMgLi (Fig. S2†) corroborate this trend, showing progressive conductivity loss at higher cutoff voltages (from 4.0 V to 4.3 V). Long-term cycling at 200 mA g−1 (Fig. 2h) yields 150 mA h g−1 initial capacity with 68% retention (1.5–4.3 V), versus 83% (1.5–4.0 V) and 78% (1.5–4.2 V) after 150 cycles, confirming that oxygen redox-enhanced capacity inversely impacts structural stability.
3.3. Na-storage mechanisms of P2-NFMO-CoMgLi
In situ XRD characterization (1.5–4.3 V vs. Na+/Na, Fig. 3a and S3†) reveals the structural evolution of P2-NFMO-CoMgLi during sodiation/desodiation. Fig. 3a illustrates continuous migration of (002) and (004) reflections toward lower angles during charging, accompanied by (100) and (102) peak shifting to higher angles, indicative of c-axis expansion (Δc = +0.18 Å, Fig. 3b) and a-axis contraction (Δa = −0.04 Å, Fig. 3b). This anisotropic lattice deformation stems from enhanced interlayer oxygen repulsion following Na+ extraction. Upon discharge processes, (002) and (004) reflections shift back to higher angles while (100) and (102) peaks shift back to lower angles, and all of them surpass their initial positions because of the quantities of Na+ reinsertion exceeding initial extraction. Meanwhile, the a = b axis expands from 2.84 Å to 2.94 Å, and c-axis shrinks from 11.32 Å to 10.90 Å, corresponding to a small volume expansion of only 3% (Fig. 3b). Additionally, the absence of phase transition and Na+/vacancy ordering throughout cycling confirms the solid-solution ion storage mechanism. Solid-solution mechanisms are also observed in reduced voltage windows (1.5–4.0 V and 1.5–4.2 V; Fig. S4†). Ex situ TEM analysis elucidates voltage-dependent structural distortions, demonstrating progressive contraction of the (102) interplanar spacing from 0.23 nm to 0.19 and 0.16 nm as the charge voltage increases from 4.0 V to 4.2 V and 4.3 V, respectively (Fig. 3c–e). Simultaneously, SAED patterns (Fig. 3f–h) illustrate more severe distortion of the intensified [001]-axis spot matrix, correlating with accumulated lattice strain that compromises cycling durability. TEM-EDS quantification (Fig. S5 and Table S3†) and ICP verification (Tables S4–S6†) confirm the voltage-proportional Na+ depletion. Elemental homogeneity persists across all charge states (EDS mapping: Fig. S6†), excluding compositional heterogeneity as a degradation factor. | ||
| Fig. 3 Na-storage mechanisms: (a) contour plots of in situ XRD patterns for the P2-NFMO-CoMgLi electrode during the first two charge/discharge cycles. (b) The evolution of lattice parameters and unit-cell volume upon charge/discharge during the first two cycles for P2-NFMO-CoMgLi. (c–e) HRTEM images, and (f–h) SAED pattern at different charged states of the P2-NFMO-CoMgLi cathode. (i) Mn 2p, (j) Co 2p, (k) Fe 2p, and (l) O 1s spectra at different charged and discharged states of P2-NFMO-CoMgLi. | ||
Ex situ XPS analysis was conducted to unravel the charge compensation mechanism in P2-NFMO-CoMgLi. Fig. 3i presents Mn 2p spectra at various charged states: the pristine electrode exhibits characteristic Mn 2p1/2 and Mn 2p3/2 peaks at 654.0 eV and 642.5 eV, respectively, indicating the initial state of Mn is at the valence of +4.33 The signatures of Co 2p3/2 at 780.6 eV and Co 2p1/2 at 795.6 eV confirms the presence of Co3+ in the initial electrode (Fig. 3j).36 Fe 2p spectra (Fig. 3k) reveal a pristine Fe 2p3/2 peak at 711.3 eV (Fe3+). O 1s spectra (Fig. 3l) exhibit lattice (529.5 eV) and surface oxygen species (533.0 eV, carbonates/C–O) at the initial state.26 When charged to 4.0 V, the Mn and Co 2p spectra remain almost unchanged. While, the peak at 711.3 eV shifts to 712.1 eV in Fe 2p spectra, revealing the oxidation of Fe3+ to Fe4+, and a new featured peak at 530.5 eV is generated, which is attributed to oxidized oxygen species (O2n−) in the O 1s spectra.37,38 This oxygen activity, facilitated by Li-induced Na–O–Li configurations and vacancy-stabilized Na–O–□ motifs, generates lone-pair oxygen states that drive reversible O2−/(O2)n– transitions, rationalizing the capacity enhancement at elevated voltages. Upon charging to 4.2 and 4.3 V, Mn, Co, and Fe 2p spectra show negligible changes, while the O2n− peak intensifies, indicating that the oxidation of O2− dominates the charge compensation. After discharging to 3.0 V, partial reduction of Mn4+ and Co3+ to Mn3+ and Co2+, respectively happens, and the reduction of Fe4+ and O2n− to Fe3+ and O2− is nearly complete. When further discharged to 1.5 V, most of the Mn4+ and Co3+ undergo the reduction process. The aforementioned outcomes reveal that Mn3+/Mn4+, Fe3+/Fe4+, and Co2+/Co3+ couples are mainly involved below 4.0 V while O2n−/O2− dominates above 4.0 V during the electrochemical processes.
3.4. Sodium-ion full batteries of P2-NFMO-CoMgLi
A proof-of-concept SIB full cell was constructed by integrating the P2-NFMO-CoMgLi with a hard carbon anode (P2-NFMO-CoMgLi//HC, Fig. 4a), employing a 1:1 cathode-to-anode mass ratio based on the cathode's discharge capacity of 178 mA h g−1 (20 mA g−1) and the anode's reversible capacity of 185 mA h g−1 (50 mA g−1, Fig. 4b and S9†). A comparative analysis of the charge–discharge profiles (Fig. 4b) and CV curves (Fig. 4c) for both electrodes are presented. The discharge plateau of P2-NFMO-CoMgLi is at ∼2.89 V, and the charge plateau of hard carbon is at ∼0.06 V. Thereafter, the operating voltage of the full cell is estimated at ∼2.83 V, which is consistent with the constructed full cell (at ∼2.75 V, Fig. 4d). The full cell delivers an initial discharge capacity of 117 mA h g−1 at 20 mA g−1, corresponding to a high energy density of 161 W h kg−1, based on both the mass of cathode and anode active materials. After, cycling for 50 times, 70% of the initial capacity is maintained (Fig. 4e). When tested at different current densities (Fig. 4f), P2-NFMO-CoMgLi//HC can deliver capacities of 112, 111, 107, 102, 96, and 82 mA h g−1 at 20, 40, 100, 200, 400, and 1000 mA g−1, respectively, with a rate retention of 73%. And the capacity recovers to 109 mA h g−1 as the current reverts to 20 mA g−1, confirming exceptional electrochemical reversibility and reta capability of the constructed full cell.
 | ||
| Fig. 4 Sodium-ion full batteries: (a) schematic configuration of the P2-NFMO-CoMgLi cathode//hard carbon anode full battery. (b) Charge/discharge pro-files and (c) CV curves of the P2-NFMO-CoMgLi cathode and hard carbon anode. (d) Galvanostatic charge/discharge profiles and (e) cycling performance at 0.1C in the voltage window of 1.4–4.1 V of the full cell (based on the cathode active mass). (f) Rate performance of the full battery. | ||
4. Conclusion
In summary, this work demonstrates a multi-ion doping strategy to engineer the novel P2-Na0.73Fe0.2Mn0.52Co0.2Mg0.05Li0.03O2 cathode, achieving high charge compensation through coupled cationic and anionic redox. Consequently, it delivers a high capacity of 150.2 mA h g−1 at 200 mA g−1 with a capacity retention of 68% over 150 cycles, and achieves exceptional rate capability (182.7 mA h g−1 at 20 mA g−1; 88.9 mA h g−1 at 1000 mA g−1). XPS analysis validates that the extra capacity derives from O2−/(O2)n– redox not the Mn/Fe/Co redox at high voltages. In situ XRD confirms the voltage-dependent single-phase solid-solution dynamics, while ex situ TEM unveils progressive structural distortions at elevated voltages (>4.0 V). A proof-of-concept full cell with hard carbon anode achieves a high capacity of 117 mA h g−1 (20 mA g−1) and a capacity retention of 70% after 50 cycles, underscoring practical viability. This study elucidates the critical trade-off between voltage window expansion (enhancing capacity via oxygen redox) and structural degradation, providing foundational insights for designing high-energy layered oxide cathodes through balanced anionic/cationic redox optimization.Data availability
Data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Project of Sanya Yazhou Bay Science and Technology City (Grant No. SKJC-JYRC-2024-64), the Hainan Provincial Joint Project of Sanya Yazhou Bay Science and Technology City (2021JJLH0069), the National Natural Science Foundation of China (22409154, and 52474333), and the Education Department of Hainan Province, project number: Hnky2024ZD-26.References
- Y. Chen, Z. Tian, J. Li and T. Zhou, Chem. Eng. J., 2023, 472, 145041 CrossRef CAS.
- M. M. Khan, Y. Zhao, Q. Liu, W. He, D. Mu, L. Li, R. Chen and F. Wu, J. Energy Storage, 2025, 116, 116038 CrossRef.
- Y.-F. Liu, K. Han, D.-N. Peng, L.-Y. Kong, Y. Su, H.-W. Li, H.-Y. Hu, J.-Y. Li, H.-R. Wang, Z.-Q. Fu, Q. Ma, Y.-F. Zhu, R.-R. Tang, S.-L. Chou, Y. Xiao and X.-W. Wu, InfoMat, 2023, 5, e12422 CrossRef CAS.
- H. Lu, S. Chu, J. Tian, Q. Wang, C. Sheng, C. Cheng, R. Liu, A. M. D'Angelo, W. K. Pang, L. Zhang, H. Zhou and S. Guo, Adv. Funct. Mater., 2023, 34, 2305470 CrossRef.
- E. Gabriel, D. Hou, E. Lee and H. Xiong, Energy Sci. Eng., 2022, 10, 1672–1705 CrossRef CAS.
- R.-M. Gao, Z.-J. Zheng, P.-F. Wang, C.-Y. Wang, H. Ye and F.-F. Cao, Energy Storage Mater., 2020, 30, 9–26 CrossRef.
- S. Gao, Z. Zhu, H. Fang, K. Feng, J. Zhong, M. Hou, Y. Guo, F. Li, W. Zhang, Z. Ma and F. Li, Adv. Mater., 2024, 36, 2311523 CrossRef CAS PubMed.
- S. Guo, Y. Sun, P. Liu, J. Yi, P. He, X. Zhang, Y. Zhu, R. Senga, K. Suenaga, M. Chen and H. Zhou, Sci. Bull., 2018, 63, 376–384 CrossRef CAS PubMed.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, J. Phys. Chem. C, 2018, 122, 13500–13507 CrossRef CAS.
- C. Jiang, Y. Wang, Y. Xin, X. Ding, S. Liu, Y. Pang, B. Chen, Y. Wang, L. Liu, F. Wu and H. Gao, Carbon Neutralization, 2024, 3, 233–244 CrossRef CAS.
- J. Li, Y. Chen, S. He, Y. Yang, Y. Wang and L. Guo, Chem. Eng. J., 2023, 452, 139311 CrossRef CAS.
- S. Sun, Y. Chen, Q. Bai, Z. Tian, Q. Huang, C. Liu, S. He, Y. Yang, Y. Wang and L. Guo, Chem. Eng. J., 2023, 451, 138780 CrossRef CAS.
- J. Li, Y. Chen, Q. Bai, S. He, Y. Yang, C. Zheng, Y. Wang and L. Guo, ACS Sustainable Chem. Eng., 2023, 11, 12631–12645 CrossRef CAS.
- S. Sun, S. Liu, Y. Chen, L. Li, Q. Bai, Z. Tian, Q. Huang, Y. Wang, X. Wang and L. Guo, Adv. Funct. Mater., 2023, 33, 2213711 CrossRef CAS.
- S. Li, Y. Sun, Y. Pang, S. Xia, T. Chen, H. Sun, S. Zheng and T. Yuan, Asia-Pac. J. Chem. Eng., 2022, 17, e2762 CrossRef CAS.
- L. Gan, X.-G. Yuan, J.-J. Han, J. Li, L. Zheng and H.-R. Yao, Carbon Neutralization, 2023, 2, 235–244 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen, C. Zou, H. Jin, S. Wang, S.-L. Chou, Y. Liu and S.-X. Dou, Adv. Funct. Mater., 2020, 30, 1909530 CrossRef CAS.
- Q. Liu, Z. Hu, W. Li, C. Zou, H. Jin, S. Wang, S. Chou and S.-X. Dou, Energy Environ. Sci., 2021, 14, 158–179 RSC.
- Y. Liu, Y.-H. Zhang, J. Ma, J. Zhao, X. Li and G. Cui, Chem. Mater., 2023, 36, 54–73 CrossRef.
- Y. Bai, L. Zhao, C. Wu, H. Li, Y. Li and F. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 2857–2865 CrossRef CAS.
- B. M. de Boisse, D. Carlier, M. Guignard, L. Bourgeois and C. Delmas, Inorg. Chem., 2014, 53, 11197–11205 CrossRef PubMed.
- J. Xu, S.-L. Chou, J.-L. Wang, H.-K. Liu and S.-X. Dou, ChemElectroChem, 2014, 1, 371–374 CrossRef.
- X. Cao, X. Li, Y. Qiao, M. Jia, F. Qiu, Y. He, P. He and H. Zhou, ACS Energy Lett., 2019, 4, 2409 CrossRef CAS.
- X. Hou, C. Li, M. Li, Y. Liu, W. Zhu, Z. Li and Y. Xu, Chin. J. Chem., 2023, 41, 2597–2603 CrossRef CAS.
- H. Jiang, G. Qian, R. Liu, W.-D. Liu, Y. Chen and W. Hu, Sci. China Mater., 2023, 66, 4542–4549 CrossRef CAS.
- L. Yang, X. Li, J. Liu, S. Xiong, X. Ma, P. Liu, J. Bai, W. Xu, Y. Tang, Y.-Y. Hu, M. Liu and H. Chen, J. Am. Chem. Soc., 2019, 141, 6680–6689 CrossRef CAS.
- M. Bianchini, E. Gonzalo, N. E. Drewett, N. Ortiz-Vitoriano, J. M. Lopez del Amo, F. J. Bonilla, B. Acebedo and T. Rojo, J. Mater. Chem. A, 2018, 6, 3552–3559 RSC.
- C. Chen, W. Huang, Y. Li, M. Zhang, K. Nie, J. Wang, W. Zhao, R. Qi, C. Zuo, Z. Li, H. Yi and F. Pan, Nano Energy, 2021, 90, 106504 CrossRef CAS.
- H. Fu, J. Li, L. Wang, X. Yang, X. Li and W. Lu, J. Phys. Chem. C, 2022, 126, 20196–20203 CrossRef CAS.
- W. K. Pang, S. Kalluri, V. K. Peterson, N. Sharma, J. Kimpton, B. Johannessen, H. K. Liu, S. X. Dou and Z. Guo, Chem. Mater., 2015, 27, 3150–3158 CrossRef CAS.
- J. Jin, Y. Liu, X. Pang, Y. Wang, X. Xing and J. Chen, Sci. China:Chem., 2021, 64, 385–402 CrossRef CAS.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- D. Yuan, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Electrochim. Acta, 2014, 116, 300–305 CrossRef CAS.
- J. T. Xu, S. L. Chou, J. L. Wang, H. K. Liu and S. X. Dou, ChemElectroChem, 2014, 1, 371–374 CrossRef.
- X. Wu, J. Guo, D. Wang, G. Zhong, M. J. McDonald and Y. Yang, J. Power Sources, 2015, 281, 18–26 CrossRef CAS.
- J.-L. Yue, W.-W. Yin, M.-H. Cao, S. Zulipiya, Y.-N. Zhou and Z.-W. Fu, Chem. Commun., 2015, 51, 15712–15715 RSC.
- Q. Shen, Y. Liu, L. Jiao, X. Qu and J. Chen, Energy Storage Mater., 2021, 35, 400–430 CrossRef.
- Y. Wang, L. Wang, H. Zhu, J. Chu, Y. Fang, L. Wu, L. Huang, Y. Ren, C.-J. Sun, Q. Liu, X. Ai, H. Yang and Y. Cao, Adv. Funct. Mater., 2020, 30, 1910327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01409c |
| This journal is © The Royal Society of Chemistry 2025 |