
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of fluorescence in novel A–D–A type phenothiazine derivatives via oxidation†
Mengdi
Lin
 *ab,
Shilong
Zhang
c,
Xuefeng
Liang
b,
Zhenhua
Shan
b,
Wenqian
Yang
b and
Haijiao
Xie
d
*ab,
Shilong
Zhang
c,
Xuefeng
Liang
b,
Zhenhua
Shan
b,
Wenqian
Yang
b and
Haijiao
Xie
d
aCollege of Pharmacy, Jinan University, Guangzhou, 511400, China. E-mail: linmd6@mail3.sysu.edu.cn
bDepartment of CMC, ApicHope Pharmaceutical Group Co., Ltd, Guangzhou, 510760, China
cNational Center for International Research on Green Optoelectronics, South China Normal University, Guangzhou, 510006, China
dHangzhou Yanqu Information Technology Co., Ltd, Hangzhou, Zhejiang Province 310003, China
First published on 21st March 2025
Abstract
1,9-Dimethyl-2,8-funtionalized phenothiazines were successfully developed through a bottom-up synthesis strategy. A high photoluminescence quantum yield of 52% was observed, upon oxidation, and a reduced PLQY of 6% was achieved which opens new avenues for designing PTZ-based push–pull systems with tailored properties.
Organic photoluminescent small molecules have gained great attention in organic photovoltaics,1–3 organic light-emitting diodes (OLEDs),4–6 and bioimaging,7–10 due to their convenience of functionalization and purification. Conventionally, a post-synthesis strategy was widely utilized in the synthesis of photoluminescent small molecules.11–17 For instance, a copper-catalyzed Ullmann-type C–N coupling reaction was implemented to afford carbazole-substituted thianthrene, exhibiting dual room temperature fluorescence–phosphorescence in air.17 Despite these significant advancements, achieving high-yield bottom-up synthesis continues to pose a considerable challenge. Consequently, the development of innovative strategies for designing tunable photoluminescent small molecules is imperative.
Phenothiazine (PTZ), a rigid yet dynamically adaptable heterocycle, exhibits a unique ability to fold along its N⋯S axis. Coupled with its exceptional electron-donating properties, PTZ was extensively utilized as a key component in the design of organic photoluminescent materials.18–20 To date, the most prevalent strategy for structural modification of the PTZ core has centered on the introduction of substituents at both the nitrogen atom and the 3,7-positions, typically achieved through post-synthetic modification approaches (A and B).11–14 For instance, a series of phenothiazine-based push–pull systems with varying sulfur oxidation states were synthesized (B and D).21 The higher oxidation states of sulfur substantially enhanced the capabilities of electron injection and transport (D). Consequently, modulating the oxidation states of the sulfur atom in the thiazine ring became a widely adopted strategy to alter properties of PTZ derivatives (Fig. 1).22–24
 | ||
| Fig. 1 Conventional functionalization and modulation strategies of PTZ systems (top) and molecular design of JNU-S with the modulation of oxidization state. | ||
However, 2,8-substituted PTZs (2,8-PTZs) were rarely reported due to synthetic difficulties and low yields, limiting their application in photoluminescence (C). An effective approach to 2,8-PTZs involved cross-coupling via the Buchwald–Hartwig reaction followed by sulfur oxidation.25 Alternatively, 2,8-PTZs could also be constructed by means of smiles rearrangement of diphenyl sulfide.26 Therefore, we envisioned that regulation of the oxidation states of sulfur in 2,8-PTZ derivatives could provide deeper insights into PTZ-based push–pull systems.
Herein, we report the facile synthesis of 2,8-functionalized-1,9-dimethyl-phenothiazines, which were further promoted to JNU-S, a nonplanar butterfly structure with electron-donating and electron-withdrawing moieties to enhance intramolecular charge transfer (ICT). Upon oxidation to JNU-SO2, an unprecedented fluorescence quench emerged, contrasting with the behavior observed in 3,7-functionalized-phenothiazine photoluminescence materials. Furthermore, the structures of both compounds were fully characterized, and their photophysical and optical properties were thoroughly investigated.
The target symmetric A–D–A type PTZ derivatives were synthesized as outlined in Scheme 1. The Buchwald–Hartwig cross-coupling of 1-bromo-3-methoxy-2-methylbenzene 1 and 3-methoxy-2-methylaniline gave compound 2 in 95% yield. 3 was synthesized with sulfur and iodine in 99% yield indicating that regioselectivity issue was effectively resolved by introducing methyl groups at 1,9-positions of PTZ. Finkelstein reaction and SN2 reaction were simultaneously conducted in one pot to afford 4 in 91% yield. Subsequent demethylation of 4 yielded 5 in 100% yield which was esterified directly with moderate yield of 85% to obtain 6. The Suzuki–Miyaura cross-coupling between 6 and (4-(methoxycarbonyl)phenyl)boronic acid was subsequently implemented, affording JNU-S in 90% yield. JNU-SO2 was quantitatively isolated through oxidation in the presence of H2O2. The structures of these compounds were unambiguously confirmed using high-resolution mass spectroscopy and NMR spectroscopy.
 | ||
| Scheme 1 Synthetic route of JNU-S and JNU-SO2. Conditions: (i) 3-methoxy-2-methylaniline, Pd(OAc)2, (±)BINAP, Cs2CO3, toluene, 90 °C, 12 h; (ii) 1,2-dichlorobenzene, S, I2, 160 °C, 3 h; (iii) bromoethane, KI, DMSO, NaOH, 70 °C, 5 h; (iv) BBr3, CH2Cl2, 25 °C, 12 h; (v) NaH, PhNTf2, THF, 25 °C, 12 h; (vi) (4-(methoxycarbonyl)phenyl)boronic acid, CsF, Pd(PPh3)4, 1,4-dioxane, 90 °C, 12 h; (vii) H2O2, AcOH, CHCl3, 25 °C, 12 h. | ||
The chemical compositions of JNU-S and JNU-SO2 were elucidated through high-resolution mass spectrometry (HRMS), which revealed prominent signal peaks at m/z 524.1898 and 556.1798, respectively. These values aligned precisely with the anticipated molecular formulas, as corroborated by the data presented in Fig. S1 and S2.† Furthermore, the structural characterization of these compounds was advanced by 1H NMR spectroscopy, with the spectra of JNU-S and JNU-SO2 depicted in Fig. 2. In the case of JNU-S, the aromatic hydrogen atoms manifested as four doublets in an intensity ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:1. These signals were meticulously assigned through Nuclear Overhauser Effect (NOE) experiments, as illustrated in Fig. S3.† A notable downfield shift of the aromatic protons in the PTZ moiety was observed, transitioning from 6.98 and 7.17 ppm to 7.28 and 7.97 ppm, a phenomenon attributed to the robust electron-withdrawing conjugation effect. Concurrently, a significant downfield shift was also noted for the methylene protons, which moved from 2.54 ppm to 3.95 ppm, further underscoring the substantial electronic influence exerted by the conjugated system.
:2:1:1. These signals were meticulously assigned through Nuclear Overhauser Effect (NOE) experiments, as illustrated in Fig. S3.† A notable downfield shift of the aromatic protons in the PTZ moiety was observed, transitioning from 6.98 and 7.17 ppm to 7.28 and 7.97 ppm, a phenomenon attributed to the robust electron-withdrawing conjugation effect. Concurrently, a significant downfield shift was also noted for the methylene protons, which moved from 2.54 ppm to 3.95 ppm, further underscoring the substantial electronic influence exerted by the conjugated system.
 | ||
| Fig. 2 Partial 1H NMR spectra of JNU-S and JNU-SO2 in CDCl3. | ||
The structures of JNU-S and JNU-SO2, prepared by recrystallization from MeCN were further explicitly confirmed by single crystal X-ray diffraction (SCXRD) analysis (see Fig. 3). Surprisingly, the dihedral angle of JNU-S was 146°, significantly smaller than 153° observed in previous studies, suggesting the realization of the regulation of the dihedral angle of PTZ. However, JNU-SO2 exhibited a dihedral angle of 160°, consistent with PTZ-S,S-dioxide, attributing to the steric effect through sulphone installation and reduced steric hindrance from methyl groups. Additionally, the orthogonal array of the PTZ core (2.915 Å) indicated significant CH–π interactions in JNU-S, suggesting a stronger electron-donating effect compared to JNU-SO2.
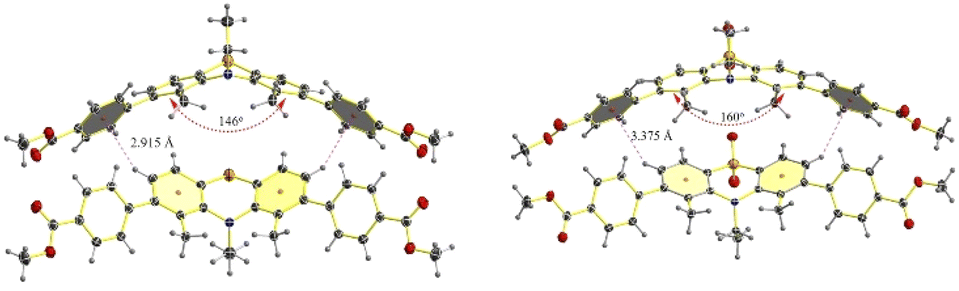 | ||
| Fig. 3 Single-crystal structures of JNU-S (left) and JNU-SO2 (right). | ||
As depicted in Fig. 4, JNU-S exhibited a single irreversible oxidation wave at 0.87 V, attributed to the strong electron-donating nature of the phenothiazine unit. In contrast, JNU-SO2 exhibited a single irreversible oxidation wave at 1.68 V, corresponding to the phenothiazine 5,5-dioxide units. This comparison highlights a notable increase in oxidation potential, reflecting the distinct electronic properties of the two compounds. This rise in oxidation potential aligned with the elevated oxidation state of sulfur and the stabilization of the highest occupied molecular orbital (HOMO) due to the presence of electron-withdrawing groups. The HOMO and LUMO energy levels of JNU-S and JNU-SO2 were determined using the onset potentials of their oxidation and reduction waves, yielding values of −5.11 eV and −5.92 eV, respectively (see Table S3†). These results revealed that the increase in the sulfur oxidation state exerted a more pronounced influence on the HOMO energy level compared to the LUMO, underscoring the significant role of sulfur oxidation in modulating the electronic structure of these compounds.
 | ||
| Fig. 4 Cyclic voltammograms of JNU (green) and JNU-SO2 (blue) in 0.1 M solution of Bu4NPF6 in dichloromethane at 50 mV s−1 scan rate versus vs. Ag/AgCl at 25 °C. | ||
Density functional theory (DFT) calculations were systematically performed to elucidate the electronic and structural characteristics of JNU-S and JNU-SO2. The inherent butterfly conformation of PTZ conferred a nonplanar structural configuration to both compounds. As illustrated in Fig. 5, the remarkable electron-donating capability of the nitrogen atom localized the highest occupied molecular orbitals (HOMOs) of JNU within the heterocyclic ring of PTZ, whereas the benzoyl groups predominantly accommodated the lowest unoccupied molecular orbitals (LUMOs). This electronic distribution engendered a significant stokes shift of 135 nm. In contrast, for JNU-SO2, the robust electron-withdrawing nature of the sulfone moiety delocalized the HOMOs across the entire PTZ core. This electronic redistribution resulted in an enlarged HOMO–LUMO energy gap, increasing from 3.93 eV to 4.16 eV, and induced a remarkable blue shift of 105 nm.
 | ||
| Fig. 5 Optimized structures of JNU-S (left) and JNU-SO2 (right), their HOMO–LUMO and orbital energies were calculated using the B3LYP/6-31G(d,p) basis set. | ||
As depicted in Fig. 6, the absorption and emission spectra of JNU-S and JNU-SO2 were investigated in chloroform. JNU-S showed an absorption band maximized at 370 nm, attributed to ICT, while JNU-SO2 exhibited a peak at 355 nm. Typically, bathochromic shifts occur due to enhanced ICT effects upon introducing electron-withdrawing groups. However, an unexpected blue shift was observed, likely due to the reduced electron-donating capability of the PTZ core in the A–D–A system. Accordingly, a homologous tendency was emerged when considering the emission maxima system.
 | ||
| Fig. 6 Normalized absorption (solid) and emission (dash) spectra of JNU-S (λex = 370 nm) and JNU-SO2 (λex = 355 nm) in CHCl3 (c = 2.0 × 10 −3 mol L−1). | ||
The fluorescence quantum yield of JNU-S was remarkable (52%) with a lifetime of 2.62 ns in chloroform, comparable to those of PTZ derivatives with high PLQY in A–D–A systems (Table 1 and Fig. S5†). Notably, while oxidation typically increased PLQY, JNU-SO2 exhibited a dramatic decrease in PLQY (6%), resulting in a fluorescence quench state. This phenomenon, attributed to the stronger electron-withdrawing conjugation in 2,8-PTZ sulfone derivatives, was observed for the first time in A–D–A and A-π-D-π-A PTZ systems, significantly enriching the fluorescence regulation family of PTZ systems. To further clarify unique property uncovered via oxidation of JNU-S, 3,7-functionalized phenothiazines JNU-SS and JNU-SSO2 were also synthesized, though PLQY decreased from 98% to 37%, fluorescence quench was not observed (Fig. S6, Scheme S1† and Table 1).
To clarify the effect of solvent, the absorption and emission spectra were systematically investigated for JNU-S and JNU-SO2 (Fig. S4†). For JNU-SO2, negligible solvent dependence was observed in both absorption and emission spectra, which can be attributed to its small dipole moment and localized excitation (LE) character. In contrast, JNU-S displayed a significant bathochromic shift and band broadening in its emission spectrum when transitioning from low-polarity toluene to high-polarity acetonitrile, a phenomenon consistent with ICT behavior.
In conclusion, 2,8-functionalized-phenothiazines, JNU-S and JNU-SO2, were synthesized through an efficient synthetic strategy. SCXRD analysis revealed that the dihedral angles of JNU-S and JNU-SO2 were significantly influenced by the oxidation state of sulfur. The oxidation potential of JNU-SO2 was notably higher than that of JNU-S, reflecting the stabilization of the highest occupied molecular orbital (HOMO) by the electron-withdrawing sulfone moiety. JNU-S exhibited a high fluorescence quantum yield (52%) in chloroform, while JNU-SO2 displayed an unprecedented fluorescence quench state (6%) due to the strong electron-withdrawing conjugation effect of the sulfone group. This phenomenon contrasted with the typical behavior of 3,7-functionalized PTZ derivatives and enriched the fluorescence regulation mechanisms in PTZ-based systems. DFT calculations confirmed the significant role of sulfur oxidation in modulating the HOMO–LUMO energy gap and electronic distribution. This work demonstrated the potential of 2,8-functionalized PTZ derivatives as versatile materials for optoelectronic applications. Future studies could explore the application of these materials in organic light-emitting diodes (OLEDs), sensors, and other photonic devices.
Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank ApicHope Pharmaceutical Group Co., Ltd for financial support.Notes and references
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- J.-L. Wang, Q.-R. Yin, J.-S. Miao, Z. Wu, Z.-F. Chang, Y. Cao, R.-B. Zhang, J.-Y. Wang, H.-B. Wu and Y. Cao, Adv. Funct. Mater., 2015, 25, 3514–3523 CrossRef CAS.
- M. J. Sung, J. Hong, H. Cha, Y. Jiang, C. E. Park, J. R. Durrant, T. K. An, S.-K. Kwon and Y.-H. Kim, Chem.–Eur. J., 2019, 25, 12316–12324 CrossRef CAS PubMed.
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- L. Duan, L. Hou, T.-W. Lee, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, J. Mater. Chem., 2010, 20, 6392–6407 RSC.
- Z. D. Popovic and H. Aziz, IEEE J. Sel. Top. Quantum Electron., 2002, 8, 362–371 CrossRef CAS.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CAS.
- H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef CAS PubMed.
- T. Terai and T. Nagano, Pflug. Arch. Eur. J. Physiol., 2013, 465, 347–359 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- J. Liu, X. Zhao, Q. Al-Galiby, X. Huang, J. Zheng, R. Li, C. Huang, Y. Yang, J. Shi, D. Z. Manrique, C. J. Lambert, M. R. Bryce and W. Hong, Angew. Chem., Int. Ed., 2017, 56, 13061–13065 CrossRef CAS PubMed.
- K. D. Thériault and T. C. Sutherland, Phys. Chem. Chem. Phys., 2014, 16, 12266–12274 RSC.
- Z. Liu, E. Shi, Y. Wan, N. Li, D. Chen, Q. Xu, H. Li, J. Lu, K. Zhang and L. Wang, J. Mater. Chem. C, 2015, 3, 2033–2039 RSC.
- K. Shanmugasundaram, R. K. Chitumalla, J. Jang and Y. Choe, New J. Chem., 2017, 41, 9668–9673 RSC.
- A. Tomkeviciene, A. Dabulienė, T. Matulaitis, M. Guzauskas, V. Andruleviciene, J. V. Grazulevicius, Y. Yamanaka, Y. Yano and T. Ono, Dyes Pigm., 2019, 170, 107605 CrossRef CAS.
- Y. Wen, H. Liu, S.-T. Zhang, G. Pan, Z. Yang, T. Lu, B. Li, J. Cao and B. Yang, CCS Chem., 2020, 3, 1940–1948 CrossRef.
- P. Pander, A. Swist, R. Turczyn, S. Pouget, D. Djurado, A. Lazauskas, R. Pashazadeh, J. V. Grazulevicius, R. Motyka, A. Klimash, P. J. Skabara, P. Data, J. Soloducho and F. B. Dias, J. Phys. Chem. C, 2018, 122, 24958–24966 CrossRef CAS.
- I. J. Al-Busaidi, A. Haque, N. K. Al Rasbi and M. S. Khan, Synth. Met., 2019, 257, 116189 CrossRef CAS.
- F. Khan and R. Misra, J. Mater. Chem. C, 2023, 11, 2786–2825 RSC.
- S. Kumar, M. Singh, J.-H. Jou and S. Ghosh, J. Mater. Chem. C, 2016, 4, 6769–6777 RSC.
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933–9943 CrossRef CAS PubMed.
- Y. Rout, A. Ekbote and R. Misra, J. Mater. Chem. C, 2021, 9, 7508–7531 RSC.
- S. Revoju, A. Matuhina, L. Canil, H. Salonen, A. Hiltunen, A. Abate and P. Vivo, J. Mater. Chem. C, 2020, 8, 15486–15506 RSC.
- I. A. Wright, M. K. Etherington, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Chem.–Eur. J., 2023, 29, e202300428 CrossRef CAS PubMed.
- W. Luo, Y. Li, L. Wang, Y. Qin, Q. Cheng, G. Hu, C. Yao and X. Song, Org. Biomol. Chem., 2024, 22, 3725–3731 RSC.
- R. L. Mital and S. K. Jain, J. Chem. Soc. C, 1969, 2148–2150, 10.1039/J39690002148.
- L. Yao, S. Sun, S. Xue, S. Zhang, X. Wu, H. Zhang, Y. Pan, C. Gu, F. Li and Y. Ma, J. Phys. Chem. C, 2013, 117, 14189–14196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2412799 (for JNU-S) and 2412800 (for JNU-SO2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra01383f |
| This journal is © The Royal Society of Chemistry 2025 |