
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical performance of porous TiO2 microspheres coated with nitrogen-doped carbon as an anode material for lithium-ion batteries†
Shimei Guo *a,
Yue Wangb,
Shubiao Xiab,
Hanwei Lia,
Siyuan Zuoa and
Wangqiong Xu*a
*a,
Yue Wangb,
Shubiao Xiab,
Hanwei Lia,
Siyuan Zuoa and
Wangqiong Xu*a
aCollege of Physics and Electronic Engineering, Qujing Normal University, Qujing, Yunnan 655011, China
bYunnan Key Laboratory of Crystalline Porous Organic Functional Materials, College of Chemistry and Environmental Science, Qujing Normal University, Qujing 655011, China
First published on 17th April 2025
Abstract
TiO2 has a robust structure and low cost and is non-toxic. However, its low electronic conductivity and lithium-ion diffusivity impede its practical application in LIBs. To improve the conductivity and lithium-ion dynamics of titanium dioxide (TiO2), we synthesized porous TiO2 microspheres coated with nitrogen-doped carbon (TiO2@C–N) through a solvothermal method combined with pyrolysis and carbonization technology. The nitrogen-doped carbon coating was prepared using a one-pot sealed carbonization method with pyrrole as the source of carbon and nitrogen. The porous TiO2 matrix in the TiO2@C–N composites provided numerous open transport pathways and storage sites for Li ions, while the nitrogen-doped carbon coating promoted the movement of electrons, leading to enhanced electrical conductivity. Undergoing 5000 cycles at 2 A g−1, the TiO2@C–N electrode delivered a cycling capacity of 71.8 mA h g−1, while the capacity of commercial graphite decayed rapidly after 3300 cycles. Rate tests of both samples under the same conditions demonstrated that the TiO2@C–N electrode was more suitable for fast charging/discharging than the graphite anode. Therefore, the TiO2@C–N composites are expected to be an alternative to commercial graphite anodes based on their electrochemical performance.
1. Introduction
Accompanied by efforts toward the exploitation and application of intermittent renewable resources, including solar, wind, and water energy, the exploration of new alternate energy storage and conversion systems has ushered in accelerated development.1–3 Among such systems, lithium-ion batteries (LIBs) have perhaps attained the most interest, particularly because of their high energy density and excellent cycling stability.4,5 Graphitic carbon, with both electrochemical stability and low cost, has remained the most popular commercial anode material for LIBs since the 1990s.6,7 However, the graphitic carbon anode has some intrinsic defects, including poor rate capability caused by large-volume changes (ca. 10%) upon battery cycling, potential risks of metallic lithium plating and dendrite formation on graphite surfaces, and rapid capacity degradation during fast charging, owing to the thermally unstable solid–electrolyte interphase (SEI) film.8–13 Therefore, with the increasing demands for fast charging/discharging, reliable safety, and long lifespan, researchers have been devoted to exploiting suitable materials to replace graphite anodes for LIBs.Titanium dioxide is considered a promising material to compete with LIBs' graphite anode because TiO2 has a robust structure, low cost, and safety, including non-toxicity.14–16 However, so far, its low electronic conductivity (∼10−12 to 10−7 S cm−1) and lithium-ion diffusivity (∼10−15 to 10−9 cm2 s−1) have impeded its actual application in LIBs,10,11 as they can result in low cycling capacities and poor rate performance.17 To address these issues, many strategies have been proposed, such as doping,17–19 surface coating,12,20,21 morphological and structure control,22–25 and constructing composites and hybrids.26–28 For example, N. Kim et al. synthesized nitrogen-doped TiO2 nanobelts (N-TNB) using a hydrothermal method with urea as the nitrogen source with TiO2 (P25) nanoparticles and reported that nitrogen doping improved the electronic conductivity and Li+-ion storage efficiency of the as-prepared N-TNB anode in LIBs, resulting in a remarkable rate capability of ∼37 mA h g−1 at 40C (1C = 335 mA g−1).29 Cheng et al. fabricated a mesoporous carbon-coated anatase (TiO2/C) nanostructure via a modular synthesis approach and found that when the TiO2 nanomaterials were used as the anodes of LIBs, they enhanced the cycling stability, facilitated high overall capacities and minimized the capacity loss compared to both their non-carbon and commercial anatase analogues.30 Ko and coworkers prepared porous anatase TiO2 1D nanotube bundles as an anode for LIBs through a hydrothermal process and post-calcination treatment. The resulting anode exhibited improved cycling and rate performance (117 mA h g−1 after the 100th cycle at 1C, 84 mA h g−1 at 10C after the 1000th cycle), benefiting from the efficient control and design of the 1D nanostructure and porous framework with a large surface area.14
Since the electrochemical performance of TiO2 can be enhanced though certain composite methods, we synthesized porous TiO2 coated with nitrogen-doped carbon (TiO2@C–N) nanocomposite microspheres using lab-made TiO2 microspheres as a precursor and pyrrole as the source of carbon and nitrogen. Pyrrole is a five-membered heterocyclic compound that contains one nitrogen heteratom, which can produce carbon and nitrogen species under sealed pyrolysis conditions. Therefore, nitrogen-doped carbon coatings were obtained by a one-pot sealed carbonization in this work, providing a new approach for the co-doping and co-coating of multiple elements in materials. As expected, the cycling and rate tests showed that the prepared TiO2@C–N porous composites displayed a longer cycle life and higher reversible capacity compared to the commercial graphite anode.
2. Experimental section
2.1 Materials
Tetrabutyl titanate (TBT, Ti (OC4H9)4, 99.0%), N-methyl-2-pyrrolidinone (NMP), polyvinylidene fluoride (PVDF), N,N-dimethylacetamide (DMA, C4H9NO, 99.0%), isopropanol (IPA, C3H8O, 99.0%) and pyrrole (C4H5N, 98.0%) were purchased from Macklin. The PVDF powder with a molecular weight of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–700000 was dissolved in N-methyl-2-pyrrolidinone (NMP) solvent at a weight ratio of 9:91. The NMP had a purity of 99.0%. Commercial graphite was purchased from Guangdong Canrd New Energy Technology Co., Ltd. The chemicals utilized were of analytical grade and were applied as-received without further purification.
000–700000 was dissolved in N-methyl-2-pyrrolidinone (NMP) solvent at a weight ratio of 9:91. The NMP had a purity of 99.0%. Commercial graphite was purchased from Guangdong Canrd New Energy Technology Co., Ltd. The chemicals utilized were of analytical grade and were applied as-received without further purification.
2.2 Synthesis of the porous TiO2 matrix and TiO2@C–N microspheres
The synthesis process for the porous TiO2 matrix and TiO2@C–N composite followed our previous work,31 and is depicted in Fig. 1. First, DMA (10 ml) and IPA (20 ml) were mixed without stirring in a Teflon container, and then 1.0 ml of TBT was rapidly dropped into the above solution and then the container was sealed in a stainless steel autoclave. The stainless steel autoclave was subsequently transferred to an oven and kept at 200 °C for 24 h. Afterwards, the oven temperature was allowed to drop naturally to below 30 °C, and then the precipitate was collected by centrifugation, cleaned six times using anhydrous ethanol, and then dried at 60 °C overnight. Finally, the TiO2 precursor was acquired through pyrolysis of the dry precipitate at 400 °C for 3 h in an oxygen-rich environment. | ||
| Fig. 1 Schematic of the synthesis process for the TiO2 precursor and TiO2@C–N. | ||
To obtain the resulting product TiO2@C–N, 1 ml pyrrole was added dropwise into 2.0 g of the TiO2 precursor, which was then sealed in a stainless steel high-pressure vessel, and then held at 550 °C in a muffle furnace for 5 h.
2.3 Characterization
The crystal structures of the precursor and the resulting products were analyzed by X-ray powder diffraction (XRD, Cu Kα radiation, λ = 0.1542 nm). The scanning rate was 4° min−1 and the scanning range was from 10° to 90°. Also, field emission scanning electron microscopy (FE-SEM, JSM-6700F) and high-resolution transmission electron microscopy (HR-TEM, JEOL JEM-2100) were used to examine the morphology and microstructure of all the samples. Alongside the N2 adsorption–desorption test, the Brunauer–Emmett–Teller (BET) model was utilized to calculate the specific surface area, while the Barrett–Joyner–Halenda (BJH) method was employed to determine the pore-size distribution. The composition and chemical states were investigated by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA). An SDT thermal-microbalance instrument (SDT Q600, TA Instruments) was used for thermogravimetric analysis (TGA) in an air atmosphere, with the samples heated from 26 °C to 800 °C at a ramp rate of 10 °C min−1. The carbon composition of the products was investigated by Raman spectrometry (532 nm laser wavelength, Thermo Fisher).2.4 Electrochemical measurements
To obtain the working electrodes, the active materials, carbon black and PVDF (7:2:1 by weight) were homogeneously dispersed in NMP solvent, and then coated onto a copper foil substrate and dried at 80 °C for more than 12 h. Finally, the thoroughly dried substrate was processed into 14 mm diameter flakes. The mass loading of active material obtained in these electrodes was about 1.5 mg cm−2. In an argon-filled environment, CR2025-type coin half cells were assembled in a glove-box using Li foil as the counter and reference electrode, and Celgard 2500 as the separator. The electrolyte was 1.0 M LiPF6 dissolved in ethylene carbonate, dimethyl carbonate, and ethylene methyl carbonate with a volume ratio of 1:1:1. The electrochemical performance of the cells was tested on a LAND CT2001A battery test system in the voltage range from 0.02 to 3.0 V at various current densities. A PARSTAT 2273 electrochemistry workstation was utilized for the cyclic voltammogram (CV) and electrochemical impedance spectroscopy (EIS) analyses. The CV was scanned at a rate of 0.1 mV s−1 in the potential range (vs. Li/Li+) from 0.01 to 3.0 V. EIS was performed in the frequency range of 100 kHz to 1.0 Hz with an AC signal amplitude of 10 mV. The test results are presented as Nyquist plots, in which the X-axis and Y-axis represent the real part Z′ and imaginary part Z′′, respectively.
3. Results and discussion
XRD was used to characterize the crystal structure of the precursor and the resulting products. As shown in Fig. 2a, all the diffraction peaks of the precursor and TiO2@C–N were in good agreement with the tetragon anatase TiO2 (JCPDS no. 21-1272), with no other diffraction peaks observed, demonstrating the high phase purity of the anatase in both samples, as well as the low content or graphitization degree of nitrogen-doped carbon in TiO2@C–N.3,32 Fig. 2b illustrates the Raman spectra of TiO2@C–N and the TiO2 precursor. Four bands could be observed at 144, 400, 510, and 640 cm−1 for both samples, which were assigned to the typical Eg, B 1g, A 1g, and Eg vibrational modes of the Ti–O bonds in anatase TiO2, respectively. The carbon characteristic peaks of the D band and G band were detected at 1328 cm−1 and 1577 cm−1, respectively.10,33 Generally, the D band indicates the disordered and imperfect structures of carbon materials, while the G band corresponds to the vibrations of carbon atoms with an sp2 electronic configuration in the graphene sheet structure.34,35 The value of IG/ID (intensity ratio of the G and D bands) represents the graphitization degree of nitrogen-doped carbon materials.12,30 In this study, IG/ID was calculated to be ca. 0.88, indicating that the nitrogen-doped carbon in the resultant product was amorphous, which was in agreement with the XRD results. | ||
| Fig. 2 (a) XRD patterns of the TiO2 precursor and TiO2@C–N; (b) Raman spectra of the TiO2 precursor and TiO2@C–N. | ||
The morphology and microstructure of the TiO2 precursor and TiO2@C–N products were observed using FE-SEM and HR-TEM. Fig. 3a shows that the TiO2 precursor comprised monodisperse microspheres with a size distribution of 1–2 μm. Upon closer examination, it became apparent that the TiO2 microspheres were actually composed of nanoflakes arranged in a two-dimensional manner to create a relaxed spherical structure and minimize the surface free energy (inset of Fig. 3a). Fig. 3b shows the HR-TEM image of the TiO2 precursor, in which the lattice stripe spacing of 0.348 nm corresponded to the (101) crystal plane of anatase TiO2 (Fig. 3b inset).36,37 After coating with nitrogen-doped carbon, a TEM image was taken to provide detailed structural information of the obtained TiO2@C–N. As displayed in Fig. 3c, the TiO2@C–N material still maintained a spherical morphology. Meanwhile, the TiO2@C–N microspheres were observed to be composed of numerous irregular TiO2 nanoparticles and open mesopores, confirming the existence of a porous structure that ensured effective contact between the internal active material and the electrolyte, as well as the rapid transport of Li+ ions. In the HR-TEM image (Fig. 3d), an amorphous phase was observed not only on the surface of the TiO2 nanocrystals but also on the inner walls of the pores, which could be assigned to the nitrogen-doped carbon species according to the Raman spectra and XRD results discussed above.35 Moreover, it was noted that the lattice fringe of anatase TiO2 disappeared at the same magnification (Fig. 3d), demonstrating that the carbon coating had a certain thickness.
 | ||
| Fig. 3 (a) SEM images of the TiO2 precursor; inset shows enlarged image. (b) TEM image of the TiO2 precursor; inset shows close-up of the marked area. (c and d) TEM images of TiO2@C–N. | ||
N2 absorption–desorption tests were also used to further confirm the porous features of the TiO2@C–N sample. The sample displayed a type IV isotherm with an H3 hysteresis loop, as shown in Fig. 4a, demonstrating that the TiO2@C–N was mesoporous in nature,14,31 consistent with the TEM observation. In addition, it was observed that the pores had a non-uniform distribution, ranging from several nanometers to 150 nm in size, as displayed in the inset of Fig. 4a. The dominant pore size was approximately 30 nm, which aligned with the findings from the HR-TEM observation. The specific surface area according to BET analysis was evaluated as 48.5 m2 g−1, with a calculated pore volume of 0.24 cm3 g−1. Fig. 4b presents the results of the thermogravimetric analysis carried out under air to determine the carbon and nitrogen contents in the composition. Below 150 °C, there was a slight weight loss of about 1.23%, which could be attributed to the evaporation of water absorbed on the surface of the porous microspheres. As the temperature increased, a main weight loss of about 16.44% occurred at 300–500 °C, which was related to the oxidation of carbon and nitrogen to form volatile species, including CO and CO2.33,34
 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms of TiO2@C–N; inset shows the distribution of pore sizes. (b) TGA plot of TiO2@C–N. | ||
Next, X-ray photoelectron spectroscopy (XPS) was utilized to analyze the composition and chemical bonding present in the TiO2@C–N composites. Fig. 5a presents the survey scan spectrum of TiO2@C–N, in which four peaks corresponding to C 1s, N 1s, Ti 2p, and O 1s could be observed, indicating the existence of Ti, O, C, and N elements with atomic contents of 10.86%, 24.03%, 58.17%, and 6.94%, respectively. As shown in Fig. 5b, the high-resolution C 1s peak could be separated into three peaks centered at ca. 284.8, 286.2, and 289.6 eV, corresponding to C–C, C–N/C–O, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.33,35,38,39 Fig. 5c illustrates two well-fitted intense peaks centered at 458.9 (Ti 2p3/2) and 464.7 eV (Ti 2p1/2), consistent with the value of Ti4+ in the TiO2 lattice.8,22 The deconvoluted peaks observed at 530.2 and 531.9 eV in Fig. 5d were assigned to O–Ti and O–C bonds, respectively.26 Fig. 5e shows three distinct peaks at approximately 398.6, 400.1, and 401.6 eV, corresponding to pyridinic N, pyrrolic N, and graphitic N, respectively, originating from the decomposition of pyrrole.40,41 The pyridinic and pyrrolic N provide numerous extrinsic defects and active sites, which promote the rapid transfer of lithium ions and generate surface-induced capacitance, while the graphitic N can improve electronic conductivity, thereby enhancing the electrochemical performance of TiO2.8,38
O bonds, respectively.33,35,38,39 Fig. 5c illustrates two well-fitted intense peaks centered at 458.9 (Ti 2p3/2) and 464.7 eV (Ti 2p1/2), consistent with the value of Ti4+ in the TiO2 lattice.8,22 The deconvoluted peaks observed at 530.2 and 531.9 eV in Fig. 5d were assigned to O–Ti and O–C bonds, respectively.26 Fig. 5e shows three distinct peaks at approximately 398.6, 400.1, and 401.6 eV, corresponding to pyridinic N, pyrrolic N, and graphitic N, respectively, originating from the decomposition of pyrrole.40,41 The pyridinic and pyrrolic N provide numerous extrinsic defects and active sites, which promote the rapid transfer of lithium ions and generate surface-induced capacitance, while the graphitic N can improve electronic conductivity, thereby enhancing the electrochemical performance of TiO2.8,38
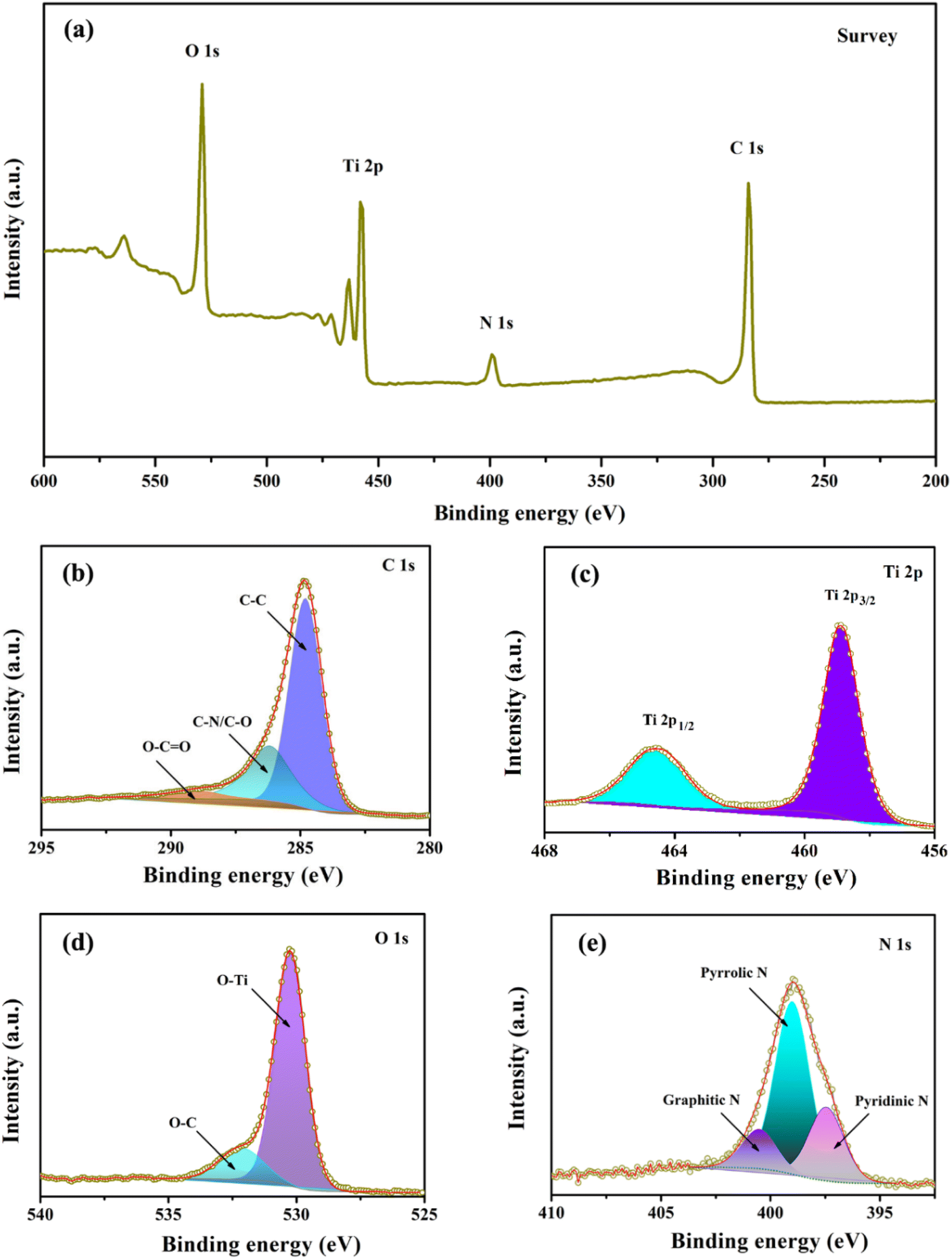 | ||
| Fig. 5 XPS spectra of TiO2@C–N. (a) Survey scan, (b) C 1s, (c) Ti 2p, (d) O 1s, and (e) N 1s. | ||
Fig. 6a displays the first three CV graphs of TiO2/C–N and its precursor. The CV was performed at a scan rate of 0.1 mV s−1 with the potential (vs. Li/Li+) in the range of 0.01 to 3.0 V. Both samples exhibited similar curves, including three reduction peaks and one oxidation peak in the initial scan. The redox peak pair around 1.6/2.1 V (ca. 1.3/2.2 V of pure TiO2) was associated with the intercalation/deintercalation of lithium ions into anatase TiO2, which was related to the biphasic transition between tetragonal anatase and orthorhombic LixTiO2 (TiO2 +xLi+ +xe− ↔ LixTiO2).8 While the other two negative peaks at 0.5 and 1.1 V (0.4 and 0.9 V of pure TiO2) disappeared in the second cycle, indicating that the intercalation and deintercalation of lithium ions were irreversible during the initial charging/discharging process, which matched well with the formation of a solid–electrolyte interphase (SEI) layer and amorphous Li2O.10 Compared to the pure TiO2 sample, the up-shift of the reduction peaks and down-shift of the oxidation peaks for TiO2/C–N indicated that the introduction of the carbon–nitrogen coating brought about a higher lithium removal/insertion activity, which is beneficial for enhancing the specific capacity and rate performance of electrodes. From the second round onwards, the CV curves showed a tendency to coincide, indicating that the stability of lithium ions intercalation and deintercalation in both anode materials was gradually enhanced. Fig. 6b shows the initial three charging and discharging curves of TiO2@C–N and pure TiO2 precursor, at a current density of 0.1 A g−1. Comparing the performance of these two samples in the first three cycles, the specific capacity of TiO2@C–N was much higher than that of its pure TiO2 precursor, demonstrating that the nitrogen-doped carbon coating is beneficial for increasing the specific cycle capacity of TiO2@C–N, which may be due to the nitrogen-doped carbon coating serving as an electron-transfer pathway, which is favorable for ensuring excellent electron contact and electrical conductivity. In addition, the appearance of two downslopes at approximately 1.6 and 0.6 V in both samples, respectively, during the first discharging process, and a voltage plateau at ca. 2.1 V in the subsequent charging were also noteworthy and consistent with the previous CV curves. At the end of the first cycle, the capacities of TiO2@C–N for discharging and charging were 546.1 and 372.2 mA h g−1, respectively, in line with an irreversible capacity loss of 31.8%, which could be attributed to the irreversible Li+ consumption caused by the formation of an SEI film. In addition, the charging/discharging curves of the second and third cycles showed very little difference, suggesting the increased cycling stability of TiO2@C–N after the initial cycle.
 | ||
| Fig. 6 (a) CV curves of TiO2@C–N and the pure TiO2 precursor; (b) galvanostatic discharge/charge curves of the initial three cycles at 0.1 A g−1 of TiO2@C–N and the pure TiO2 precursor. | ||
To evaluate its commercial application prospects, the cycling and rate performance of the TiO2@C–N composites were examined from 0.01 to 3.0 V. For comparison, the commercial graphite anode and TiO2 precursor were also tested under the same conditions. Fig. S1† and 7a show the cycling performances of all the samples at the current densities of 0.1 A g−1 and 2 A g−1, respectively. As shown in Fig. S1,† both TiO2@C–N and its precursor had a lower specific capacity at 0.1 A g−1 than commercial graphite. However, the long-term cycling performance of these three samples, as shown in Fig. 7a, at a high current density of 2 A g−1 revealed that a high reversible specific capacity of 71.8 mA h g−1 could be obtained for TiO2@C–N without any noticeable capacity degradation even after cycling 5000 times. Under the same conditions, the reversible specific capacity of the commercial graphite was only half of the value of the TiO2@C–N electrode and showed rapid decay after about 3300 cycles. The ultra-long cycle life of TiO2@C–N could be attributed to the robust structure of its TiO2 matrix. As shown in Fig S2,† the microsphere-like morphology of TiO2@C–N was well maintained, as revealed by comparing the SEM images before and after 1000 cycles at 2 A g−1. In addition, the cycling performance of the TiO2@C–N prepared in this study surpassed that of previously reported TiO2/C composites, such as TiO2@C-PAN electrode (125.5 mA h g−1 at 1C after 100 cycles),42 TiO2@NC (110 mA h g−1 after 2000 cycles at 0.5 A g−1),26 nitrogen-doped TiO2 nanobelts (67.2 mA h g−1 after 500 cycles at 1.0 A g−1).29 The TiO2 precursor, however, could only maintain a low specific capacity (ca. 48 mA h g−1) at the current density of 2 A g−1, which cannot compete with the commercial graphite, so only 1000 cycles were measured. Fig. 7b compares the rate capabilities of these three electrodes at different current densities. The reversible capacities of TiO2@C-N are 290, 240.8, 191.9, 145.7, and 95.6 mA h g−1 at current densities of 0.1, 0.2, 0.4, 0.4, and 1.6 A g−1, respectively. As expected, the rate performances of the other two samples were not satisfactory, and even the TiO2 precursor showed worse rate performance than commercial graphite. In addition to these, the reversible capacities of TiO2@C–N were lower than those of commercial graphite at low rates (e.g., 0.1 and 0.2 A g−1 current densities) and higher than that of commercial graphite at high rates (above 0.4 A g−1), suggesting the TiO2@C–N electrode is more suitable for fast charging/discharging than the commercial graphite anode. This anomaly may be due to be the synergistic effect of the increased electrical conductivity of the N-doped carbon coating and the superior structure of the porous TiO2 matrix. In the case of high-rate charge and discharge, the numerous channels in the porous TiO2 matrix not only ensured rapid ion transport but also alleviated structural stresses caused by the rapid insertion/extraction of lithium ions. Meanwhile, the N-doped carbon coating formed conductive connections inside TiO2 nanoparticles, thereby promoting the rapid movement of electrons.
 | ||
| Fig. 7 (a) Long-term cycling performance and coulombic efficiency at 2 A g−1 of the TiO2 precursor, TiO2@C–N and commercial graphite; (b) rate capabilities of the TiO2 precursor, TiO2@C–N and commercial graphite; (c) experimental and fitted Nyquist plots of commercial graphite and TiO2@C–N; inset shows the equivalent circuit model. | ||
To further reveal the underlying reasons for the unusual rate performance of these two anode materials, electrochemical impedance spectroscopy (EIS) measurements were performed after 10 cycles at 0.1 A g−1 and 1.6 A g−1. The obtained Nyquist plots and fitted equivalent electrical circuit are shown in Fig. 7c. In the figure, it is observed that a semicircular arc extending from the high-frequency to mid-frequency region was present, while a slanted line appeared in the low-frequency region. The diameter of the arc corresponded to the SEI film impedance (Rsei) and charge-transfer resistance (Rct) stemming from the impedance between the electrolyte and electrode surface,35,39 while the slope of the line was associated with the diffusion of lithium ions, corresponding to the Warburg impedance (Zw),12 which was fitted to calculate the lithium-ion diffusivity coefficient (DLi+) in each sample (Fig. S3†). Meanwhile, the electrolyte solution resistance is denoted by Re. Table 1 lists the values of Re, Rsei, Rct and DLi+ calculated from the equivalent circuit model in the inset of Fig. 7c. There was little difference in the Re values noted for all samples, but the sum of Rsei and Rct, corresponding to each semicircle diameter, decreased substantially at a higher current density. Furthermore, with the same number of cycles, the total impedance value of commercial graphite (ca. 101.15 Ω) was less than that of TiO2@C–N (about 126.42 Ω) at a current density of 0.1 A g−1, but the results were reversed at the higher current density of 1.6 A g−1 (51.61 and 27.99 Ω for commercial graphite and the TiO2@C–N electrode, respectively). The reason for this could be attributed to the nitrogen-doped carbon improving the electrical conductivity among the TiO2@C–N anode materials.43,44 Comparison of the DLi+ of each sample revealed that the lithium-ion diffusivity in TiO2@C–N at high current density was slightly faster than that at low current density, whereas the opposite was true for graphite, indicating that high-rate charging and discharging only promoted the Li+-transport dynamics in TiO2@C–N but not in commercial graphite, consistent with the long cycling and rate performance described above. This could be due to the open porous structure that promoted effective penetration of the electrolyte into the TiO2 matrix and resulted in the rapid transport of Li+ ions.
| Samples | Re (Ω) | Rsei (Ω) | Rct (Ω) | DLi+ (cm2 s−1) |
|---|---|---|---|---|
| Commercial graphite (at 0.1 A g−1) | 3.33 | 9.45 | 88.37 | 5.88 × 10−13 |
| TiO2@C–N (at 0.1 A g−1) | 3.96 | 15.56 | 106.9 | 1.18 × 10−12 |
| Commercial graphite (at 1.6 A g−1) | 2.78 | 8.59 | 40.24 | 4.49 × 10−13 |
| TiO2@C–N (at 1.6 A g−1) | 3.23 | 7.18 | 17.58 | 1.24 × 10−12 |
4. Conclusions
In summary, the TiO2@C–N porous composites were synthesized through a combination of solvothermal, pyrolysis, and carbonization technology. By performing the sealed pyrolysis of pyrrole, the nitrogen-doped carbon coating was obtained not only on the surface of TiO2 nanocrystals but also on the inner walls of the pores. As an anode material for LIBs, the porous TiO2 matrix in the composites provided numerous transport channels and active sites for Li+ diffusion and storage. Meanwhile, the nitrogen-doped carbon coating served as an electron-transfer pathway, which was favorable for ensuring excellent electron contact and electrical conductivity. As a result, the TiO2@C–N composites delivered a higher reversible specific capacity and superior rate capability than the commercial graphite anode by combining the merits of the porous TiO2 matrix and nitrogen-doped carbon coating. Thus, the TiO2@C–N porous composites are expected to be an alternative to commercial graphite anodes.Data availability
The data are available from the corresponding author upon request.Author contributions
Shimei Guo: writing – original draft, formal analysis. Yue Wang: investigation and validation. Shubiao Xia: funding acquisition. Wangqiong Xu: review and editing. Hanwei Li and Siyuan Zuo: data curation. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the financial supports from the Basic Research Special-Key Projects of Yunnan Provincial Department of Science and Technology (202401AS070001), the Fundamental Research Project of Yunnan Province (202301AT070239), and the Special Basic Cooperative Research Innovation Programs of Qujing Science and Technology Bureau & Qujing Normal University (KJLH2024YB10).References
- W. Yan, J. Wang, Q. Hu, J. Fu, M. K. Albolkany, T. Zhang, X. Lu, F. Ye and B. Liu, Nano Res., 2024, 17, 2655–2662 CrossRef CAS
.
- T. S. Munonde and M. C. Raphulu, J. Energy Storage, 2024, 78, 110064 CrossRef
- D. Zhang and H. Xu, Battery Energy, 2024, 3, 20230032 CrossRef CAS
- T. Kim, W. Song, D. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC
- C. D. Quilty, D. Wu, W. Li, D. C. Bock, L. Wang, L. M. Housel, A. Abraham, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Chem. Rev., 2023, 123, 1327–1363 CrossRef CAS PubMed
- C. Du, Z. Zhao, H. Liu, F. Song, L. Chen, Y. Cheng and Z. Guo, Chem. Rec., 2023, 23, e202300004 CrossRef CAS PubMed
- Y. Liu, A. Kouhpour, D. Hwang and H. Zarrin, Electrochim. Acta, 2024, 500, 144750 CrossRef CAS
- J. Lee, H. Ka, R. Saroha, Y. Kang, D. Kang and J. Cho, J. Energy Storage, 2023, 66, 107396 CrossRef
- X. Yan, Z. Wang, M. He, Z. Hou, T. Xia, G. Liu and X. Chen, Energy Technol., 2015, 3, 801–814 CrossRef CAS
- Z. Jia, S. Sun, A. Li, H. Wu, G. Qin, T. Liu, P. Lv and J. Chen, ACS Appl. Nano Mater., 2024, 7, 777–786 CrossRef CAS
- J. Yin, Z. Wen, J. Yu, X. Shi, G. Wang, Y.-E. Yang, J. Man, J. Sun and L. Cui, J. Alloys Compd., 2019, 808, 151728 CrossRef CAS
- Q. Ge, Z. Ma, M. Yao, H. Dong, X. Chen, S. Chen, T. Yao, X. Ji, L. Li and H. Wang, J. Colloid Interface Sci., 2024, 661, 888–896 CrossRef CAS PubMed
- S. Weng, G. Yang, S. Zhang, X. Liu., X. Zhang, Z. Liu, M. Cao, M. N. Ateş, Y. Li, L. Chen, Z. Wang and X. Wang, Nano-Micro Lett., 2023, 15, 215 CrossRef CAS PubMed
- W. Ko, Y. Wang, T. Wu, T. Wu and K. Lin, J. Chin. Chem. Soc., 2023, 70, 1168–1175 CrossRef CAS
- H. Usui, Y. Domi, Y. Yamamoto, T. Hoshi, T. Tanaka, N. Oishi, N. Nitta and H. Sakaguchi, ACS Appl. Electron. Mater., 2023, 5, 6292–6304 CrossRef CAS
- K. Pimta, T. Autthawong, W. Yodying, C. Phromma, M. Haruta, H. Kurata, T. Sarakonsri and Y. Chimupala, ACS Omega, 2023, 8, 15360–15370 CrossRef CAS PubMed
- W. Ko, T. Wu, S. He and K. Lin, Nanotechnology, 2024, 35, 175403 CrossRef CAS PubMed
- Z. H. Mahmoud, Y. Ajaj, G. K. Ghadir, H. M. Al-Tmimi, H. H. Jasim, M. Al-Salih, M. H. S. Alubiady, A. M. Al-Ani, S. S. Jumaa, S. Azat, G. F. Smaisim and E. Kianfar, Results Chem., 2024, 7, 101422 CrossRef CAS
- H. Usui, Y. Domi, T. H. Nguyen, S. Izaki, K. Nishikawa, T. Tanaka and H. Sakaguchi, Electrochemistry, 2022, 90, 037002 CrossRef CAS
- J. Li, S. Fan, H. Xiu, H. Wu, S. Huang, S. Wang, D. Yin, Z. Deng and C. Xiong, Nanomaterials, 2023, 13, 1144 CrossRef CAS PubMed
- K. Zhu, W. Feng, Z. Xue, M. Sun, S. Wu, Z. Jing and Y. Yu, ACS Sustainable Chem. Eng., 2022, 10, 10955–10965 CrossRef
- L. Luo, K. Liang, Z. Khanam, X. Yao, M. Mushtaq, T. Ouyang, M.-S. Balogun and Y. Tong, Small, 2024, 20, 2307103 CrossRef CAS PubMed
- C. W. Kim, B. Kim, C. Hyeon, L. Li, C.-Y. Chung and S.-E. Chun, Composites, Part B, 2024, 286, 111760 CrossRef
- J. Hou, F. Wang, E. Zhang, Y. Wang, P. Dong, Y. Wang, Y. Zhang, X. Li and Y. Zhang, Particuology, 2024, 93, 22–29 CrossRef CAS
- Z. Hao, J. Lyu, M. Tian, X. Zhang, K. Wang, S.-W. Yang, Y. Zhang and G. Q. Xu, Small Struct., 2024, 5, 2300442 CrossRef CAS
- J. Song, C. Su, C. Zhang, K. Wu, Z. Wu, X. Xia, W. Lei, D. Mandler, P. Du and Q. Hao, J. Alloys Compd., 2023, 967, 171615 CrossRef CAS
- U. Alli, K. McCarthy, I.-A. Baragau, N. P. Power, D. J. Morgan, S. Dunn, S. Killian, T. Kennedy and S. Kellici, Chem. Eng. J., 2022, 430, 132976 CrossRef CAS
- C. Yue, D. He, X. Zeng, Z. Ma, L. He, Z. Meng, J. Chen and W. Zhao, J. Alloys Compd., 2023, 961, 171026 CrossRef CAS
- N. Kim, M. R. Ra and G. Lee, Nanotechnology, 2020, 31, 415401 CrossRef CAS PubMed
- Y. Cheng, Q. Xia, H. Liu, M. B. Solomon, E. R. L. Brisson, L. D. Blackman, C. D. Ling and M. Müllne, ACS Appl. Mater. Interfaces, 2023, 15, 12261–12272 CrossRef CAS PubMed
- S. Guo, J. Liu, S. Qiu, W. Liu, Y. Wang, N. Wu, J. Guo and Z. Guo, J. Mater. Chem. A, 2015, 3, 23895–23904 RSC
- G. Wang, Y. Qi, D. Zhang, J. Bao, L. Xu, Y. Zhao, J. Qiu, S. Yuan and H. Li, Energy Technol., 2019, 7, 1800911 CrossRef
- S. Pan, Y. Xin, C. Miao, S. Nie and W. Xiao, Electrochim. Acta, 2024, 503, 144916 CrossRef CAS
- Y. Xing, S. Wang, B. Fang, G. Song, D. P. Wilkinson and S. Zhan, J. Power Sources, 2018, 385, 10–17 CrossRef CAS
- J. Cai, L. Chen and X. Chen, J. Nanosci. Nanotechnol., 2020, 20, 2452–2459 CrossRef CAS PubMed
- M. Zhang, A. Lu, H. Li, M. Li, J. Wang and C. Wang, ACS Appl. Energy Mater., 2023, 6, 1781–1798 CrossRef CAS
- M. Han, Y. Mu, F. Yuan, X. Bai and J. Yu, J. Power Sources, 2020, 465, 228206 CrossRef CAS
- S. Xie, Q. Ji, Y. Xia, K. Fang, X. Wang, X. Zuo and Y. Cheng, ChemistrySelect, 2021, 6, 141–153 CrossRef CAS
- Y. Zhou, P. Zhang, Y. Sun, Y. Shen and A. Xie, Energy Technol., 2018, 6, 1660–1666 CrossRef CAS
- C. Liu, Q. Lu, J. Qu, W. Feng, A. Thomas, Y. Li, I. G. G. Martinez, C. Pan and D. Mikhailov, Small, 2024, 20, 2311253 CrossRef CAS PubMed
- C. Liu, Q. Lu, M. V. Gorbunov, A. Omar, I. G. G. Martinez, P. Zhao, M. Hantusch, A. D. C. Permana, H. He, N. Gaponik and D. Mikhailova, J. Energy Chem., 2023, 79, 373–381 CrossRef CAS
- S. Hao, K. Zhang, G. Tan and J. Wang, J. Phys.: Conf. Ser., 2023, 2563, 012012 CrossRef CAS
- Md. M. Rahman, J. Wang, M. F. Hassan, D. Wexler and H. Liu, Adv. Energy Mater., 2011, 1, 212–220 CrossRef CAS
- M. Fan, Z. Yang, Z. Lin and X. Xiong, Nanoscale, 2021, 13, 2368–2372 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01379h |
| This journal is © The Royal Society of Chemistry 2025 |