
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral PCN pincer Ni(II) complex-catalyzed asymmetric hydrophosphination of 2-alkenoylpyridines with diphenylphosphine†
Jin-Ge Li,
Bing-Bo Qiu,
Hui Jiang *,
Mao-Ping Song and
Jun-Fang Gong*
*,
Mao-Ping Song and
Jun-Fang Gong*
College of Chemistry, Pingyuan Laboratory, Zhengzhou University, Zhengzhou 450001, China. E-mail: jiangh@zzu.edu.cn; gongjf@zzu.edu.cn
First published on 14th May 2025
Abstract
Herein, nine chiral PCN pincer Ni(II) complexes 2 with (phosphine)-(imidazoline) ligands and two complexes 5a and 5b bearing (phosphinite)-(imidazoline) ligands were successfully synthesized via a “one-pot” phosphination(phosphorylation)/nickelation reaction. All the new complexes were characterized using elemental analysis and NMR spectroscopy. Additionally, the molecular structures of complexes 2a, 2e and 5a were elucidated using X-ray single-crystal diffraction analysis. Their efficacy as enantioselective catalysts for the asymmetric hydrophosphination of 2-alkenoylpyridines was investigated. Using 5 mol% of complex 2a as the catalyst in the presence of Et3N, various 2-alkenoylpyridines reacted smoothly with diphenylphosphine to afford structurally diverse chiral pyridine-containing phosphine derivatives in yields up to 99% with an enantioselectivity up to 98% ee. Further transformations of the catalysis products were also studied.
Introduction
Chiral P-containing organic molecules have wide applications in the fields of asymmetric catalysis,1 materials2 and pharmaceuticals.3 A large number of methods have been developed for the construction of these compounds.4 Among them, organo-5 or transition-metal-catalyzed6,7 asymmetric hydrophosphination of electron-deficient alkenes with trivalent secondary or primary phosphines provides a direct and atom-economic approach for the synthesis of chiral phosphines, which are valuable ligands or organocatalysts for enantioselective transformations. In this regard, the cyclopalladated catalysts, particularly pincer Pd catalysts, stand out, and they have shown great potential since their first successful application in the field of asymmetric hydrophosphination in 2010.6,8,9 For example, the bis(phosphine) PCP Pd pincer complex A bearing stereogenic benzylic methylene centers (Chart 1) developed by Duan and co-workers is an extremely powerful and versatile catalyst for hydrophosphination of enones,9a,i enals,9b α,β-unsaturated N-acylpyrroles9c and carboxylic esters,9d nitroalkenes,9e α,β-unsaturated and α,β,γ,δ-unsaturated sulfonic esters9f,g with diarylphosphines. Using this catalyst, structurally diverse chiral phosphine derivatives from the 1,4- or 1,6-conjugate addition were generated in high yields with excellent enantioselectivity (≥90% ee). The PCP pincer catalyst B, an interesting complex which was prepared via catalytic asymmetric hydrophosphination followed by palladation10a performed well in hydrophosphination of α,β,γ,δ-unsaturated ketones (dienones) to afford exclusively the 1,4-addition products with excellent enantioselectivity (up to >99% ee).10b While the PCP complex C containing P-stereogenic centers instead of C-stereogenic centers exhibited good stereocontrol in the hydrophosphination of nitroalkenes11a and β,γ-unsaturated α-keto esters11b (up to 83% and 93% ee, respectively). Besides the PCP Pd pincers, bis(imidazoline) NCN D,12a the hybrid (aminophosphine)-(imidazoline) PCN E (ref. 12b) and (phosphinite)-(imidazoline) PCN F (ref. 12c) Pd pincers have been proved by us to be effective and stereoselective catalysts for the hydrophosphination of enones (up to 94% ee, 94% ee and 98% ee, respectively). Recently, we have disclosed the synthesis of a series of (phosphine)-(imidazoline) PCN Pd pincers and found that complex G (ref. 12d) showed promising enantioselectivity in the hydrophosphination of 2-alkenoylpyridines with Ph2PH (24 examples, 41–87% ee). | ||
| Chart 1 Pincer Pd(II) complexes used in asymmetric hydrophosphination. | ||
As described above, pincer Pd complexes, particularly those with P-donors, have become privileged chiral catalysts for the related asymmetric hydrophosphination. Moreover, pincer Pd-catalyzed hydrophosphination of specially designed substrates followed by the metalation reaction is a relatively simple and efficient route for the construction of new chiral metal (Pd and Ni) pincer complexes.9a,b,12c,13,14 The obtained Pd and Ni pincers either have intriguing structural characteristics or can be further employed as catalysts with high efficiency for asymmetric hydrophosphination. Notably, the unique and stable terdentate coordination mode of the pincer complexes, which effectively prevents the deactivation of catalysts caused by the coordination of the P-nucleophiles and the P-adducts, is certainly the key factor responsible for the great success of Pd pincers. Despite all these advantages, the precious nature of palladium leads to some limitations for their use in mass production. Therefore, the development of efficient but inexpensive metal catalysts with the pincer backbone for enantioselective hydrophosphination is highly desirable.
In comparison with the well-studied Pd pincer catalysts,6,9–13 chiral pincer Ni catalysts remain underdeveloped, although Ni is relatively inexpensive and abundant. In this respect, Zhang, Imamoto and co-workers reported that the PCP Ni complex H possessing P-stereogenic centers (Chart 2) could catalyze the asymmetric aza-Michael addition of α,β-unsaturated nitriles with moderate enantioselectivity (up to 46% ee).15 While Kumagai, Shibasaki and co-workers found that CCC Ni complex I afforded high levels of stereocontrol in the asymmetric addition of alkylnitriles to aldimines or aldehydes (up to 98%, 95% ee and 96% ee, respectively).16 In particular, Wang, Duan and co-workers developed unsymmetric PCP′ Ni complex J as the catalyst to realize the first highly enantioselective synthesis of P-stereogenic secondary phosphine-boranes by the asymmetric addition of primary phosphine to electron-deficient alkenes such as enones (up to 99% ee and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr).14 Recently, they have demonstrated that the PCN Ni complex K exhibited excellent enantioselectivity in the asymmetric hydrophosphination of enones with Ph2PH (up to 98% ee).17 Overall, the use of chiral pincer nickel complexes in enantioselective transformations is still limited. To develop more chiral Ni pincers as cost-effective and stereoselective catalysts for asymmetric synthesis and also in continuation of our research interest in pincer chemistry,6c,12,18 we synthesized a series of (phosphine)-(imidazoline) PCN Ni pincers 2 (Scheme 1) and investigated their potential in the catalytic asymmetric hydrophosphination of 2-alkenoylpyridines with Ph2PH. A variety of pyridine-containing phosphine derivatives were thus generated from the catalytic reactions in good yields and enantioselectivity. It is known that chiral phosphines functionalized by pyridine can serve as bidentate N,P ligands for transition metal-catalyzed asymmetric transformations.19 Consequently, the obtained catalytic products have potential use in asymmetric catalysis. The results are as follows.
:1 dr).14 Recently, they have demonstrated that the PCN Ni complex K exhibited excellent enantioselectivity in the asymmetric hydrophosphination of enones with Ph2PH (up to 98% ee).17 Overall, the use of chiral pincer nickel complexes in enantioselective transformations is still limited. To develop more chiral Ni pincers as cost-effective and stereoselective catalysts for asymmetric synthesis and also in continuation of our research interest in pincer chemistry,6c,12,18 we synthesized a series of (phosphine)-(imidazoline) PCN Ni pincers 2 (Scheme 1) and investigated their potential in the catalytic asymmetric hydrophosphination of 2-alkenoylpyridines with Ph2PH. A variety of pyridine-containing phosphine derivatives were thus generated from the catalytic reactions in good yields and enantioselectivity. It is known that chiral phosphines functionalized by pyridine can serve as bidentate N,P ligands for transition metal-catalyzed asymmetric transformations.19 Consequently, the obtained catalytic products have potential use in asymmetric catalysis. The results are as follows.
 | ||
| Chart 2 Pincer Ni(II) complexes used in asymmetric reactions. | ||
 | ||
| Scheme 1 Synthesis of the chiral (phosphine)-(imidazoline) PCN pincer Ni(II) complexes 2 and pincer Pd(II) complex 3. | ||
Results and discussion
Synthesis of chiral (phosphine)-(imidazoline) PCN pincer Ni(II) complexes
The pincer Ni(II) complexes 2a–f including 2a′ and 2e′ were successfully prepared via a “one-pot” phosphination/nickelation reaction (Scheme 1).20 Thus, chiral imidazoline ligands 1a–f with different chiral substituents and NR3 groups, which were synthesized according to the procedure reported previously,12d were first phosphinated by treatment with KPPh2 in the presence of Et3N in refluxing toluene for 12 h. Then, NiCl2 was added and refluxed for another 12 h for C–H nickelation. The corresponding PCN pincer Ni(II) chloride complexes 2a–f were isolated in 21–50% yields after purification. When NiBr2 was used for nickelation, the bromide complexes 2a′ and 2e′ were obtained in 50% and 40% yields, respectively. In addition, the Ni(II) chloride complex 2g with a t-Bu2P group instead of a Ph2P group was prepared by a similar “one-pot” phosphination/nickelation reaction. In this case, the ligand 1a first reacted with excess t-Bu2PH in MeOH at 45 °C for 24 h, followed by the addition of Et3N at room temperature and stirring for 0.5 h to complete the phosphination.21 After evaporation of the solvent under vacuum, the residue was extracted with ether and filtered through a Cellite pad. Then ether and the remaining t-Bu2PH were removed under vacuum. Next, toluene, Et3N and NiCl2 were added and refluxed for 12 h to achieve C–H nickelation. The expected pincer Ni(II) complex 2g was successfully obtained albeit in a rather low yield of 17%. Following the same procedure, but using PdCl2 instead of NiCl2, gave the pincer Pd(II) chloride complex 3 in 27% yield. This complex was prepared for comparison with the Ni complexes.All of the complexes are air- and moisture-stable in both the solid state and solution. The ligand 1c and the pincer complexes 2 and 3 are new compounds. They were well characterized by elemental analysis (HRMS for the ligand 1c), 1H NMR, 13C{1H} NMR and 31P{1H} NMR (for the complexes) spectroscopy. For example, in the 1H NMR spectra of the ligands, a singlet or a triplet with a small coupling constant of 1.8 Hz (for 1c and 1f) appears at δ in the range of 7.52–7.87 ppm for the central aryl proton located adjacent to both CH2Cl and the imidazoline ring, whereas the associated signal disappears in the 1H NMR spectra of the complexes, suggesting the formation of a Ni–C or Pd–C bond in the pincer complexes. In addition, the appearance of signals corresponding to the protons of Ph2P or t-Bu2P in the 1H NMR spectra, the extensive P–C coupling in the 13C{1H} NMR spectra and the single resonance at around 43 ppm or 84 ppm in the 31P{1H} NMR spectra of the complexes also indicate the formation of the pincer complexes.
Molecular structures of the pincer Ni complexes
The PCN pincer coordination mode in these complexes was clearly confirmed by X-ray single-crystal diffraction analysis of the crystal structures of Ni(II) complexes 2a and 2e. The molecules of 2a and 2e·CH2Cl2 are shown in Fig. 1 and 2, respectively. The selected bond lengths and angles are collected in Table 1. The P and N atoms as well as the C atom of the central aryl in the complexes 2a and 2e coordinate with the Ni(II) center to form two fused five-membered nickelacycles, where N–Ni–P has a bond angle of around 161° and C–Ni–Cl has a bond angle of around 173°. These two five-membered nickelacycles are almost coplanar with the central aromatic ring and the imidazoline ring. Both the C–Ni–P angles and the C–Ni–N angles are around 83° in the complexes, which are typical for pincers consisting of two fused five-membered metallacycles and reflect the relative steric strain of the system.12,17,20 The metal–ligand distances and bond angles around the Ni(II) center in complexes 2a and 2e are similar to the bond lengths of Ni–C, Ni–N and Ni–P being around 1.89 Å, 1.91 Å and 2.12 Å, respectively, which are comparable to those in the related chiral PCN pincer Ni complexes with (aminophosphine)-(imidazoline) ligand12b or (phosphinite)-(imidazoline) ligands.17 In addition, the metal-tridentate ligand bond lengths in the Ni complex 2a are all shorter than the corresponding Pd complex,12d following the expected pattern of Ni < Pd. | ||
| Fig. 1 Molecular structure of complex 2a with thermal ellipsoids drawn at the 50% probability level (hydrogen atoms are omitted for clarity). | ||
 | ||
| Fig. 2 Molecular structure of complex 2e·CH2Cl2 with thermal ellipsoids drawn at the 50% probability level (hydrogen atoms and the solvent molecule are omitted for clarity). | ||
| 2a | 2e·CH2Cl2 | |
|---|---|---|
| Ni–C | 1.885(4) | 1.891(4) |
| Ni–P | 2.1182(11) | 2.1408(12) |
| Ni–N | 1.913(3) | 1.918(3) |
| Ni–Cl | 2.2193(11) | 2.2130(12) |
| C–Ni–P | 82.57(12) | 83.07(13) |
| C–Ni–N | 83.17(15) | 82.29(16) |
| C–Ni–Cl | 173.41(12) | 172.81(13) |
| N–Ni–P | 160.98(11) | 160.65(11) |
| P–Ni–Cl | 96.37(5) | 100.99(5) |
| N–Ni–Cl | 99.19(10) | 94.96(11) |
Catalytic studies
To evaluate the potential of the newly obtained PCN pincer Ni(II) complexes in asymmetric hydrophosphination, the addition of Ph2PH to (E)-2-(3-phenylacryloyl)pyridine with the Ni pincers as the catalysts was first investigated (Table 2; for the convenience of operation, the P-adduct of this reaction and all the other adducts from hydrophosphination were oxidized to the corresponding phosphine oxides for analysis). Under the same conditions as the PCN Pd pincer G-catalyzed hydrophosphination of enones,12d the Ni pincer 2a, which has the same ligands as the G, afforded the product 4a only in 55% yield with 10% ee (entry 1). By contrast, 4a was obtained in >99% yield with 85% ee with the Pd pincer G as the catalyst.12d A survey of several solvents other than acetone (entries 2–7) indicated that the catalyst 2a behaved much better in CH3CN, giving the product 4a in 88% yield with 67% ee (entry 7). Lowering the temperature led to higher ee values (entries 8–10) and a good enantioselectivity of 86% ee could be achieved at 0 °C (85% yield, entry 9). With CH3CN as the solvent and at 0 °C, the performance of the other eight Ni pincers was tested (entries 11–18). The bromide counterpart of 2a (complex 2a′) gave a comparable yield (88%) and ee value (82%, entry 11). While changing the chiral substituent from (4S,5S)-diphenyl to (4S)-phenyl or benzyl resulted in rather poor enantioselectivity (entries 12 and 13). The enantioselectivity also varied with the groups on the imidazoline-N and the P donor (entries 14–18). Unfortunately, only complex 2f with N-benzoyl could provide a good enantioselectivity of 84% ee (52% yield, entry 17). Complexes with N-acetyl, N-Ts or t-Bu2P gave a racemic product or a product with low enantioselectivity (entries 14–16 and 18). Subsequently, with complex 2a as the best catalyst, various bases including Cs2CO3, NaHCO3, NaOH, KOH, and Et3N were screened (entries 19–23). Pleasingly, using Et3N as the base, both the yield and enantioselectivity were improved and excellent results were obtained (85% yield with 86% ee vs. 93% yield with 92% ee, entry 9 vs. 23). Increasing the amount of Et3N led to a further improvement in the enantioselectivity or the yield (entries 24 and 25) and the best enantioselectivity (95% ee, entry 24) was achieved with 20 mol% of Et3N. Finally, with a reduction in catalyst loading of 2a from 5 mol% to 3 mol%, the product 4a was generated in obviously decreased yields and enantioselectivity (entry 26). Consequently, on the basis of the above-mentioned results, the optimized conditions for the model reaction include using 5 mol% of complex 2a as the catalyst in the presence of 20 mol% of Et3N as the base in CH3CN at 0 °C (entry 24).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Base | Solvent | Temp. (°C) | Yieldb (%) | eec,d (%) |
| a Hydrophosphination reactions were performed with Ph2PH (0.2 mmol) and (E)-2-(3-phenylacryloyl)pyridine (0.3 mmol) in the presence of the pincer Ni complex 2 (5 mol%) and base (10 mol%) in 2 mL of solvent for 12 h.b Isolated yield.c Determined using chiral HPLC analysis.d The major enantiomer of the product 4a was determined to have the (R)-configuration by comparing its optical rotation with that of the same compound in ref. 12d.e The major enantiomer of the product 4a has the (S)-configuration.f Using 20 mol% of Et3N.g Using 30 mol% of Et3N.h Using 3 mol% of catalyst. | ||||||
| 1 | 2a | KOAc | Acetone | rt | 55 | 10 |
| 2 | 2a | KOAc | DCM | rt | 87 | 0 |
| 3 | 2a | KOAc | Toluene | rt | 93 | 20 |
| 4 | 2a | KOAc | THF | rt | 76 | 40 |
| 5 | 2a | KOAc | MeOH | rt | 99 | 0 |
| 6 | 2a | KOAc | n-Hexane | rt | 85 | 54 |
| 7 | 2a | KOAc | CH3CN | rt | 88 | 67 |
| 8 | 2a | KOAc | CH3CN | 10 | 88 | 70 |
| 9 | 2a | KOAc | CH3CN | 0 | 85 | 86 |
| 10 | 2a | KOAc | CH3CN | −10 | 78 | 84 |
| 11 | 2a′ | KOAc | CH3CN | 0 | 88 | 82 |
| 12 | 2b | KOAc | CH3CN | 0 | 75 | 9 |
| 13 | 2c | KOAc | CH3CN | 0 | 85 | 6e |
| 14 | 2d | KOAc | CH3CN | 0 | 73 | 0 |
| 15 | 2e | KOAc | CH3CN | 0 | 62 | 22 |
| 16 | 2e′ | KOAc | CH3CN | 0 | 75 | 28 |
| 17 | 2f | KOAc | CH3CN | 0 | 52 | 84 |
| 18 | 2g | KOAc | CH3CN | 0 | 91 | 2 |
| 19 | 2a | Cs2CO3 | CH3CN | 0 | 59 | 32 |
| 20 | 2a | NaHCO3 | CH3CN | 0 | 53 | 24e |
| 21 | 2a | NaOH | CH3CN | 0 | 72 | 84 |
| 22 | 2a | KOH | CH3CN | 0 | 95 | 58 |
| 23 | 2a | Et3N | CH3CN | 0 | 93 | 92 |
| 24 | 2a | Et3Nf | CH3CN | 0 | 91 | 95 |
| 25 | 2a | Et3Ng | CH3CN | 0 | 95 | 92 |
| 26 | 2ah | Et3N | CH3CN | 0 | 78 | 84 |

In our previous studies, it was found that the (phosphine)-(imidazoline) PCN Pd pincer G (Chart 1) exhibited inferior stereocontrol in most cases when compared with the related (phosphinite)-(imidazoline) PCN Pd pincer F (Chart 1) in the asymmetric hydrophosphination of 2-alkenoylpyridines.12c,d Therefore, to further evaluate the performance of the current Ni catalysts (the Ni analogues of Pd pincer G) in asymmetric hydrophosphination, the related chiral (phosphinite)-(imidazoline) pincer Ni(II) complexes 5a and 5b (the Ni analogues of Pd pincer F) were prepared according to the method reported previously (Scheme 2).12c The two new complexes were also well characterized by means of elemental analysis and NMR spectroscopy. The molecular structure of complex 5a was determined by X-ray single-crystal diffraction analysis (Fig. 3). The catalytic properties of the four complexes, namely the Pd pincer 3 and the three Ni pincers 2a, 5a and 5b, in the reaction of (E)-2-(3-phenylacryloyl)pyridine with Ph2PH under several different conditions were compared (Table 3). The choice of the conditions was based on the current (vide supra) and previous results.12c When CH3CN and KOAc were used as the solvent and base, respectively, both the Pd complex 3 and the Ni complex 2a worked well to afford the product 4a in high yields and enantioselectivity with the Pd complex 3 being slightly better (entries 1 and 2). The Ni complexes 5a and 5b showed higher activity but lower stereoselectivity, especially 5a (entries 3 and 4). Interestingly, complex 5a mainly afforded (S)-enantiomer of the product 4a, which was opposite to that of the other three complexes. With Et3N as the base, the Ni complexes 2a and 5b containing the same imidazoline ring provided good results (entries 5 and 8). However, the Pd complex 3 and the Ni complex 5a behaved poorly, either giving low yield or low enantioselectivity (entries 6 and 7). With toluene and KOAc as the solvent and base, respectively, where the Pd pincer F performed very well,12c no Ni catalysts could give satisfactory results (entries 9–14). Among them, complex 2a furnished moderate enantio-selectivity, while the other two Ni pincers even gave (almost) racemic products. Roughly, the Ni pincers 5a and 5b were more active but the Ni pincer 2a was the most stereoselective under the selected conditions.
 | ||
| Scheme 2 Synthesis of the chiral (phosphinite)-(imidazoline) PCN pincer Ni(II) complexes 5a and 5b. | ||
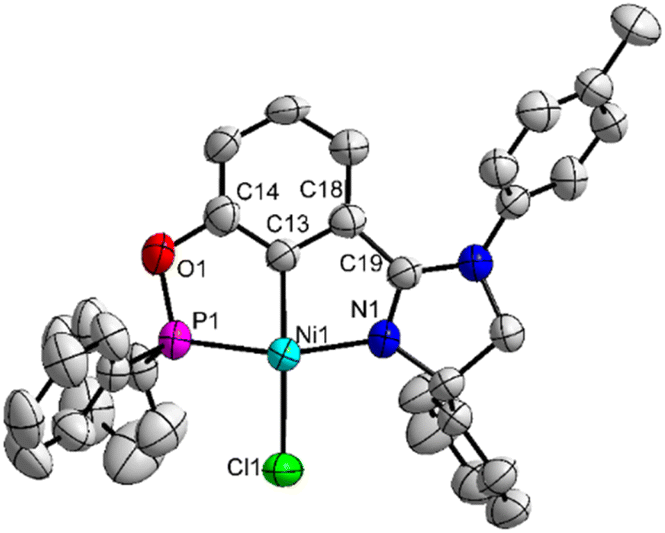 | ||
| Fig. 3 Molecular structure of complex 5a with thermal ellipsoids drawn at the 50% probability level (hydrogen atoms are omitted for clarity; one of the two independent molecules is shown). | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Base | Temp. (°C) | Yieldb (%) | eec,d (%) |
| a Hydrophosphination reactions were performed with Ph2PH (0.2 mmol) and (E)-2-(3-phenylacryloyl)pyridine (0.3 mmol) in the presence of the complex 2a, 3, or 5 (5 mol%) and Et3N (20 mol%) or KOAc (10 mol%) in 2 mL of solvent for 12 h.b Isolated yield.c Determined using chiral HPLC analysis.d The major enantiomer of the product 4a was determined to have the (R)-configuration by comparing its optical rotation with that of the same compound in ref. 12d.e The major enantiomer of the product 4a has the (S)-configuration. | ||||||
| 1 | 2a | CH3CN | KOAc | 0 | 85 | 86 |
| 2 | 3 | CH3CN | KOAc | 0 | 87 | 90 |
| 3 | 5a | CH3CN | KOAc | 0 | 91 | 30e |
| 4 | 5b | CH3CN | KOAc | 0 | 92 | 79 |
| 5 | 2a | CH3CN | Et3N | 0 | 91 | 95 |
| 6 | 3 | CH3CN | Et3N | 0 | 40 | 64 |
| 7 | 5a | CH3CN | Et3N | 0 | 99 | 27e |
| 8 | 5b | CH3CN | Et3N | 0 | 99 | 88 |
| 9 | 2a | Toluene | KOAc | 0 | 44 | 66 |
| 10 | 5a | Toluene | KOAc | 0 | 79 | 0 |
| 11 | 5b | Toluene | KOAc | 0 | 61 | 0 |
| 12 | 2a | Toluene | KOAc | −10 | 43 | 42 |
| 13 | 5a | Toluene | KOAc | −10 | 58 | 0 |
| 14 | 5b | Toluene | KOAc | −10 | 53 | 8 |

Among the twelve PCN pincers, namely one Pd and eleven Ni pincers, the Ni pincer 2a gave the best stereocontrol, therefore, with 2a as the catalyst, the asymmetric hydrophosphination of a wide range of 2-alkenoylpyridines with Ph2PH was then examined under the optimized reaction conditions (Table 2, entry 24). As shown in Table 4, substituents either electron-donating or electron-withdrawing on β-phenyl and on 6-position of pyridine in the pyridine-containing α,β-unsaturated ketone substrates were well tolerated with the current system. The substituents include Me, Et, OMe, F, Cl, Br, I and NO2 and the desired oxidized P-adducts 4b–v were produced in generally high yields (83–99%, 16 of 21 examples, entries 2–22). The ortho-substituent (Me) on β-phenyl of the 2-alkenoylpyridine led to reduced yield (79%), especially reduced enantioselectivity (50% ee), which might be likely due to steric hindrance of the ortho-position (entries 2–4). In the case of p-F substituted 2-alkenoylpyridine, both the yield and enantioselectivity of the product 4s were moderate (60%, entry 19). Gratifyingly, good to excellent enantioselectivity (78–98% ee) was achieved in all other cases, and in most cases, the enantioselectivity was higher than 90% (entries 3–18 and 20–22). It was found that the stereoselectivity varied with the position and electronic property of the substituent in the substrates. For example, 2-alkenoylpyridines bearing a meta-substituent on β-phenyl yielded the corresponding phosphine oxides with lower enantioselectivities than those possessing a para-substituent (entry 3 vs. 4, 7 vs. 8 and 9 vs. 10). However, a clear trend for the effect of electronic property of the substituent on the enantioselectivity could not be obtained. In addition to 2-alkenoylpyridines with β-phenyl and β-substituted phenyl, 2-alkenoylpyridines with β-1-naphthyl and even β-heteroaryl such as β-2-thienyl were also suitable substrates for this transformation, furnishing the oxidized P-adducts 4w and 4x in excellent yields with moderate to high enantioselectivity (90% yield with 53% ee and 98% yield with 81% ee, respectively, entries 23 and 24). Finally, 2-alkenoylpyridine containing a β-tert-butyl (alkyl) group reacted smoothly with Ph2PH, delivering the product 4y in 87% yield albeit with a rather modest ee value (20% ee, entry 25).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yieldb (%) | eec,d (%) |
| a Hydrophosphination reactions were performed with Ph2PH (0.2 mmol) and 2-alkenoylpyridines (0.3 mmol) in the presence of complex 2a (5 mol%) and Et3N (20 mol%) in 2 mL of CH3CN at 0 °C for 12 h.b Isolated yields.c Determined using chiral HPLC analysis.d The absolute configurations of the known products were assigned to be R by comparing their optical rotations with those in ref. 12d, and the configurations of the new products were assigned by analogy.e Results from ref. 12d.f Results from ref. 12c. | |||||
| 1 | H | Ph | 4a | 91 | 95(85)e(89)f |
| 2 | H | o-MeC6H4 | 4b | 79 | 50(69)e |
| 3 | H | m-MeC6H4 | 4c | 88 | 81(80)e |
| 4 | H | p-MeC6H4 | 4d | 89 | 91(77)e(73)f |
| 5 | H | p-EtC6H4 | 4e | 92 | 92(87)e |
| 6 | H | p-OMeC6H4 | 4f | 85 | 92(86)e(95)f |
| 7 | H | m-ClC6H4 | 4g | 92 | 84(82)e |
| 8 | H | p-ClC6H4 | 4h | 96 | 86 |
| 9 | H | m-BrC6H4 | 4i | 99 | 81(66)e(85)f |
| 10 | H | p-BrC6H4 | 4j | 98 | 93(67)e(87)f |
| 11 | H | p-IC6H4 | 4k | 96 | 90 |
| 12 | H | p-NO2C6H4 | 4l | 97 | 85(88)f |
| 13 | Br | Ph | 4m | 88 | 92(58)e |
| 14 | Br | p-MeC6H4 | 4n | 91 | 98(62)e |
| 15 | Br | p-EtC6H4 | 4o | 90 | 82(60)e |
| 16 | Br | p-OMeC6H4 | 4p | 71 | 78(75)e |
| 17 | Br | p-ClC6H4 | 4q | 83 | 91(41)e |
| 18 | Me | Ph | 4r | 88 | 91(85)e |
| 19 | Me | p-FC6H4 | 4s | 60 | 60 |
| 20 | Me | p-ClC6H4 | 4t | 93 | 95(80)e |
| 21 | Me | p-BrC6H4 | 4u | 79 | 91 |
| 22 | Me | p-IC6H4 | 4v | 76 | 88 |
| 23 | H |  |
4w | 90 | 53(64)e(46)f |
| 24 | H |  |
4x | 98 | 81(66)e(82)f |
| 25 | H | t-Bu | 4y | 87 | 20(n.d.)e |

It should be noted that the PCN Ni pincer 2a exhibited comparable or better stereocontrol than its Pd analogue, the Pd pincer G (Chart 1)12d in most cases (Table 4). For example, Ni catalyst 2a furnished the products 4a, 4d, 4i, 4j, 4m, 4n, 4o, 4q, 4t and 4x bearing phenyl, substituted phenyl, 6-substituted 2-pyridyl or 2-thienyl in much higher ee values than the Pd catalyst G with the same catalyst loading (95% vs. 85%, 91% vs. 77%, 81% vs. 66%, 93% vs. 67%, 92% vs. 58%, 98% vs. 62%, 82% vs. 60%, 91% vs. 41%, 95% vs. 80% and 81% vs. 66%, respectively, entries 1, 4, 9, 10, 13–15, 17, 20 and 24). Only in the cases of the products 4b and 4w with o-MeC6H4 or 1-naphthyl, the Ni complex 2a gave lower enantioselectivity than the Pd complex G (50% vs. 69% and 53% vs. 64%, respectively, entries 2 and 23). However, in comparison with the related PCN Pd pincer F (Chart 1),12c the Ni pincer 2a afforded much better enantioselectivity when the β-p-MeC6H4 enone substrate was subjected to the hydrophosphination (91% vs. 73%, entry 4). In other cases, Ni pincer 2a showed comparable stereocontrol (entries 1,6, 9, 10, 12, 23 and 24). Interestingly, both the Ni pincer 2a and the Pd pincer G yielded (R)-4, while the Pd pincer F provided (S)-4 as the major products in the catalytic reactions. In terms of the catalytic activity, the Ni pincer 2a was generally lower than the Pd pincer G. There is one exception. With 2a as the catalyst, the reaction of β-tert-butyl 2-alkenoylpyridine with Ph2PH proceeded well and the desired product 4y was obtained in 87% yield (entry 25). However, in the presence of the catalyst G the same reaction gave only trace amounts of the product 4y. Overall, the current investigations provide a more stereoselective but less expensive catalyst for the asymmetric hydrophosphination of 2-alkenoylpyridines.
To further explore the practical potential of the above-described method, transformation reactions of the catalysis products were carried out (Scheme 3). The Br-substituent on the pyridine of the oxidized P-adduct 4m could be smoothly converted to phenyl by the Pd-catalyzed Suzuki coupling reaction, furnishing the product 6a in excellent yield without any obvious loss in enantioselectivity (86% ee, Scheme 3a). The Br-substituent of 4j could also be transformed to alkynyl by the Pd/Cu-catalyzed Sonogashira coupling reaction and the desired product 6b was obtained in 83% yield without any loss of enantioselectivity (95% ee, Scheme 3b). Similar to the catalysis products, these transformed products are potentially useful chiral N,O-12c or N,P-ligands.
 | ||
| Scheme 3 (a) Suzuki and (b) Sonogashira cross-coupling reactions of 4m and 4j, respectively, to afford 6a and 6b. | ||
Based on the related studies9a,12c,14,17 as well as our experimental results, a plausible catalytic cycle for the current asymmetric hydrophosphination is proposed and shown in Scheme 4. The reaction would be initiated by replacement of the chloride in the PCN pincer Ni catalyst 2a by Et3N to generate the chiral cationic Ni–NEt3 complex. Then, transphosphination between the Ni–NEt3 complex with diphenylphosphine affords the nickel phosphido intermediate (Ni-PPh2). The subsequent intermolecular, nucleophilic addition of the diphenylphosphido group on nickel to 2-alkenoylpyridine occurs to produce the oxa-π-allylnickel intermediate, which undergoes protonolysis with Et3N+H to form the P-adduct along with the regeneration of the active pincer Ni–NEt3 catalyst.
 | ||
| Scheme 4 Proposed mechanism of asymmetric hydrophosphination. | ||
According to the stereochemical outcomes of the current hydrophosphination reactions as well as literature reports on the pincer Pd-catalyzed asymmetric hydrophosphination of enones,12c,d a possible pathway for the formation of (R)-4 was proposed (Scheme 5). To minimize the unfavorable steric repulsions between the R2 substituent at the β-position of the enone and the (4S)-phenyl group on the imidazoline ring of the catalyst, the enone substrate approaches the Ni-PPh2 intermediate with its Re-face preferentially. This facial selectivity leads to the formation of the products 4 with (R)-configuration as the major enantiomers.
 | ||
| Scheme 5 Possible stereochemical pathway for the formation of (R)-4. | ||
Conclusions
In summary, we have developed unsymmetrical chiral PCN pincer Ni(II) complexes as less expensive and stereoselective catalysts for the asymmetric hydrophosphination of 2-alkenoylpyridines with Ph2PH. During the investigations, twelve chiral pincer complexes, namely one Pd(II) and eleven Ni(II) pincers were successfully synthesized and well characterized. All the complexes were able to catalyze the stated hydrophosphination, among which the Ni complex 2a afforded the best stereocontrol. Using complex 2a as the catalyst, various pyridine-functionalized chiral phosphine oxides with structural diversity were produced in generally high yields with good to excellent enantioselectivity. Furthermore, the obtained chiral phosphine oxides could be readily transformed by metal-catalyzed coupling reactions to provide new and potentially useful chiral ligands for asymmetric catalysis.Experimental section
General information
Solvents were dried using standard methods and freshly distilled prior to use if needed. Chiral ligand 1 (ref. 12d and 20b) and enone substrates22 were prepared according to the methods reported in the literature. All other chemicals were commercially available and used without further purification. NMR spectra were recorded using a Bruker DPX-600, DPX-400, or DPX-300 spectrometer with CDCl3 as the solvent, TMS as the internal standard for 1H, 13C{1H}, and 19F{1H} NMR and 85% H3PO4 as the external standard for 31P{1H} NMR. HRMS was determined using a Waters/Agilent Q-Tof Micro MS/MS System ESI spectrometer. The enantiomeric excesses of (R)- and (S)-enantiomers were determined by HPLC analysis over a chiral column using a UV detector. The melting points were measured using a WC-1 instrument and uncorrected. Elemental analyses were performed using a Thermo Flash EA 1112 elemental analyzer. Optical rotations were recorded using an SGW®-2 polarimeter.The analytical data of the new ligand 1c are as follows.
Synthesis of the chiral (phosphine)-(imidazoline) PCN pincer nickel(II) complexes 2a–f, 2a′ and 2e′
Ligand 1 (0.5 mmol) was added to a 25 mL dry Schlenk flask in an argon atmosphere. Et3N (139 μL, 1.0 mmol), KPPh2 (1.2 mL, 0.5 M in THF) and anhydrous toluene (10 mL) were then added. The resulting mixture was refluxed for 12 h. Then, NiCl2 (194 mg, 1.5 mmol) or NiBr2 (328 mg, 1.5 mmol) was added and the reaction mixture was refluxed for another 12 h. After cooling, filtration, and evaporation, the residue was purified by column chromatography on silica gel using CH2Cl2/EtOAc (50/1) as an eluent to produce the corresponding PCN pincer complex 2a–f, 2a′ and 2e′.Synthesis of the PCN pincer metal complexes 2g and 3
Under an argon atmosphere, di-tert-butylphosphine (0.2 mL, 1.1 mmol) was added to a solution of 1a (218.1 mg, 0.5 mmol) in MeOH (5 mL, deoxygenated prior to use) in a 25 mL Schlenk flask. The solution was heated and stirred at 45 °C for 24 h and then cooled to room temperature. Degassed Et3N (0.5 mL) was added and the mixture was stirred for 0.5 h. After evaporation of the solvent under vacuum, the residue was extracted with ether and filtered through a Cellite pad. After removal of diethyl ether under vacuum, the flask was heated at 60 °C for 1 h under vacuum to remove the remaining di-tert-butyl phosphine. The residue was transferred to a 25 mL dry Schlenk tube, and then anhydrous toluene (10 mL), Et3N (83 μL, 0.6 mmol) and PdCl2 (106 mg, 0.6 mmol) or Et3N (139 μL, 1 mmol) and NiCl2 (194 mg, 1.5 mmol) were added under an argon atmosphere, and the reaction mixture was refluxed for 12 h. After cooling, filtration, and evaporation, the residue was purified by column chromatography on silica gel with CH2Cl2/EtOAc (50/1) as the eluent to produce the corresponding PCN pincer complexes 2g or 3.Synthesis of the PCN pincer nickel(II) complexes 5a and 5b
Under an argon atmosphere, to a solution of 1g or 1h (0.5 mmol) and Et3N (69.5 μL, 0.5 mmol) in toluene (5 mL) was added Ph2PCl (0.5 mmol). The resulting mixture was refluxed for 3 h. NiCl2 (194.4 mg, 1.5 mmol) was then added and the reaction mixture was refluxed for 12 h. After cooling, filtration and evaporation, the residue was purified by column chromatography on silica gel with CH2Cl2 as the eluent to produce the corresponding PCN pincer Ni(II) complexes 5a or 5b.General procedure for the enantioselective hydrophosphination of enones with diphenylphosphine
A mixture of the pincer Ni catalyst 2a (5 mol%) and Et3N (5.5 μL, 20 mol%) in anhydrous CH3CN (2 mL) was stirred at room temperature for 0.5 h under an argon atmosphere. Ph2PH (37.2 mg, 0.2 mmol) was then added and stirring was continued for 0.5 h. After addition of the enone (0.3 mmol), the resulting mixture was stirred at 0 °C for an additional 12 h and then directly oxidized with a H2O2 aqueous solution (30%, 60 μL). The mixture was stirred at room temperature for 2 h and then saturated Na2S2O3 aqueous solution was added. The organic layer was separated and the aqueous phase was extracted with CH2Cl2. The combined organic phases were dried with anhydrous Na2SO4 and the volatiles were removed under reduced pressure. Purification by column chromatography on silica gel (CH2Cl2/EtOAc: 5/1) provided the desired chiral phosphine oxides 4.Synthesis of 6a
First, 4m (98 mg, 0.2 mmol), phenylboronic acid (44 mg, 0.36 mmol), K2CO3 (60.8 mg, 0.44 mmol), Ad2BnP (1.9 mg, 2.4 mol%) and Pd(OAc)2 (0.9 mg, 2.0 mol%) were added to a 10 mL dry Schlenk tube under an argon atmosphere. Anhydrous toluene (0.1 M, 2 mL) was then added and the reaction mixture was stirred at 25 °C for 2 h. The completion of the reaction was monitored by TLC and the reaction mixture was extracted with CH2Cl2 (3 × 10 mL). The organic phase was collected, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2/EtOAc: 5/1) to give 6a as a white powder.Synthesis of 6b
Pd(dppf)Cl2 (0.01 mmol) was added to a mixture of 4j (98 mg, 0.2 mmol), CuI (0.02 mmol), phenylacetylene (0.24 mmol) and THF/NEt3 (4 mL/1 mL) under an argon atmosphere. The mixture was stirred at 60 °C overnight. The solution was then diluted with CH2Cl2, washed three times with water, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/CH2Cl2: 1/2) to give 6b as a white powder.Data availability
The data are available within the article or its ESI.† The crystallographic data for 2a, 2e and 5a have been deposited at the CCDC under numbers 2284819, 2287268 and 2424928 and can be obtained from https://www.ccdc.cam.ac.uk/structures/ (free of charge).Author contributions
Jin-Ge Li: investigation; data curation; writing (original draft). Bing-Bo Qiu: investigation. Hui Jiang: funding acquisition; supervision. Mao-Ping Song: funding acquisition; supervision. Jun-Fang Gong: conceptualization; methodology; writing (review & editing); funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (No. 22471247, 21472176 and U1904212).Notes and references
- For selected reviews, see: (a) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef PubMed; (b) W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657–1677 RSC; (c) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed; (d) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS; (e) A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS.
- For selected reviews, see: (a) M. Guinó and K. K. Hii, Chem. Soc. Rev., 2007, 36, 608–617 RSC; (b) K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS; (c) M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296–5310 RSC.
- For selected reviews, see: (a) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed; (b) L. Clarion, C. Jacquard, O. Sainte-Catherine, M. Decoux, S. Loiseau, M. Rolland, M. Lecouvey, J.-P. Hugnot, J.-N. Volle, D. Virieux, J.-L. Pirat and N. Bakalara, J. Med. Chem., 2014, 57, 8293–8306 CrossRef CAS PubMed; (c) N. Iwamoto, D. C. D. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Y. Sah, M. Meena, S. M. Standley, G. Lu, L. H. Apponi, M. Frank-Kamenetsky, J. J. Zhang, C. Vargeese and G. L. Verdine, Nat. Biotechnol., 2017, 35, 845–851 CrossRef CAS; (d) X.-L. Qin, A. Li and F.-S. Han, J. Am. Chem. Soc., 2021, 143, 2994–3002 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. S. Glueck, Chem.–Eur. J., 2008, 14, 7108–7117 CrossRef CAS; (b) D. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095–2108 RSC; (c) O. I. Kolodiazhnyi, V. P. Kukhar and A. O. Kolodiazhna, Tetrahedron: Asymmetry, 2014, 25, 865–922 CrossRef CAS; (d) V. Koshti, S. Gaikwad and S. H. Chikkali, Coord. Chem. Rev., 2014, 265, 52–73 CrossRef CAS; (e) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC; (f) B. T. Novas and R. Waterman, ChemCatChem, 2022, 14, e202200988 CrossRef CAS , for recent examples, see: ; (g) X.-T. Liu, X.-Y. Han, Y. Wu, Y.-Y. Sun, L. Gao, Z. Huang and Q.-W. Zhang, J. Am. Chem. Soc., 2021, 143, 11309–11316 CrossRef CAS; (h) Z. Yang, X. Gu, L.-B. Han and J. Wang, Chem. Sci., 2020, 11, 7451–7455 RSC; (i) Z.-H. Wu, A.-Q. Cheng, M. Yuan, Y.-X. Zhao, H.-L. Yang, L.-H. Wei, H.-Y. Wang, T. Wang, Z. Zhang and W.-L. Duan, Angew. Chem., Int. Ed., 2021, 60, 27241–27246 CrossRef CAS; (j) B. Wang, Y. Liu, C. Jiang, Z. Cao, S. Cao, X. Zhao, X. Ban, Y. Yin and Z. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202216605 CrossRef CAS; (k) C. Wang, Q. Yang, Y.-H. Dai, J. Xiong, Y. Zheng and W.-L. Duan, Angew. Chem., Int. Ed., 2023, 62, e202313112 CrossRef CAS; (l) S.-N. Yang, S.-H. Sun, C.-H. Liu, X.-T. Min, B. Wan, D.-W. Ji and Q.-A. Chen, Chin. Chem. Lett., 2023, 34, 107914 CrossRef.
- For selected examples, see: (a) G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, A. Mazzanti, L. Sambri and P. Melchiorre, Chem. Commun., 2007, 7, 722–724 RSC; (b) A. Carlone, G. Bartoli, M. Bosco, L. Sambri and P. Melchiorre, Angew. Chem., Int. Ed., 2007, 46, 4504–4506 CrossRef CAS PubMed; (c) I. Ibrahem, R. Rios, J. Vesely, P. Hammar, L. Eriksson, F. Himo and A. Córdova, Angew. Chem., Int. Ed., 2007, 46, 4507–4510 CrossRef CAS PubMed; (d) R. Maiti, J.-L. Yan, X. Yang, B. Mondal, J. Xu, H. Chai, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2021, 60, 26616–26621 CrossRef CAS.
- For reviews, see: (a) S. A. Pullarkat, Synthesis, 2016, 48, 493–503 CrossRef CAS; (b) Z. Li and W. Duan, Chin. J. Org. Chem., 2016, 36, 1805–1813 CrossRef CAS; (c) J.-K. Liu, J.-F. Gong and M.-P. Song, Org. Biomol. Chem., 2019, 17, 6069–6098 RSC; (d) J. W. K. Seah, R. H. X. Teo and P.-H. Leung, Dalton Trans., 2021, 50, 16909–16915 RSC.
- For selected examples, see: (a) I. Kovacik, D. K. Wicht, N. S. Grewal, D. S. Glueck, C. D. Incarvito, I. A. Guzei and A. L. Rheingold, Organometallics, 2000, 19, 950–953 CrossRef CAS; (b) A. D. Sadow, I. Haller, L. Fadini and A. Togni, J. Am. Chem. Soc., 2004, 126, 14704–14705 CrossRef CAS; (c) A. D. Sadow and A. Togni, J. Am. Chem. Soc., 2005, 127, 17012–17024 CrossRef CAS PubMed; (d) Y.-R. Chen, J.-J. Feng and W.-L. Duan, Tetrahedron Lett., 2014, 55, 595–597 CrossRef CAS; (e) Z. Lu, H. Zhang, Z. Yang, N. Ding, L. Meng and J. Wang, ACS Catal., 2019, 9, 1457–1463 CrossRef CAS; (f) W.-J. Yue, J.-Z. Xiao, S. Zhang and L. Yin, Angew. Chem., Int. Ed., 2020, 59, 7057–7062 CrossRef CAS; (g) Y.-B. Li, H. Tian and L. Yin, J. Am. Chem. Soc., 2020, 142, 20098–20106 CrossRef CAS; (h) Y. Zhang, Y. Jiang, M. Li, Z. Huang and J. Wang, Chem Catal., 2022, 2, 3163–3173 CrossRef CAS; (i) Q. Yang, J. Zhou and J. Wang, Chem. Sci., 2023, 14, 4413–4417 RSC; (j) D. Ji, Z. Qi and X. Li, Org. Lett., 2023, 25, 5957–5962 CrossRef CAS PubMed; (k) S. Zhang, N. Jiang, J.-Z. Xiao, G.-Q. Lin and L. Yin, Angew. Chem., Int. Ed., 2023, 62, e202218798 CrossRef CAS PubMed; (l) B. S. Daniels, X. Hou, S. A. Corio, L. M. Weissman, V. M. Dong, J. S. Hirschi and S. Nie, Angew. Chem., Int. Ed., 2023, 62, e202306511 CrossRef CAS; (m) E. G. Sinnema, T.-F. Ramspoth, R. H. Bouma, L. Ge and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2024, 63, e202316785 CrossRef CAS.
- (a) Y. Huang, S. A. Pullarkat, Y. Li and P.-H. Leung, Chem. Commun., 2010, 46, 6950–6952 RSC; (b) Y. Huang, R. J. Chew, Y. Li, S. A. Pullarkat and P.-H. Leung, Org. Lett., 2011, 13, 5862–5865 CrossRef CAS; (c) Y. Huang, S. A. Pullarkat, Y. Li and P.-H. Leung, Inorg. Chem., 2012, 51, 2533–2540 CrossRef CAS PubMed; (d) Y. Huang, S. A. Pullarkat, S. Teong, R. J. Chew, Y. Li and P.-H. Leung, Organometallics, 2012, 31, 4871–4875 CrossRef CAS; (e) R. J. Chew, K. Y. Teo, Y. Huang, B.-B. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem. Commun., 2014, 50, 8768–8770 RSC; (f) R. J. Chew, X.-R. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem.–Eur. J., 2015, 21, 4800–4804 CrossRef CAS; (g) A. Sadeer, Y.-J. Ong, T. Kojima, C. Q. Foo, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem.–Asian J., 2018, 13, 2829–2833 CrossRef CAS; (h) C. Q. Foo, A. Sadeer, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2021, 40, 682–692 CrossRef CAS.
- (a) J.-J. Feng, X.-F. Chen, M. Shi and W.-L. Duan, J. Am. Chem. Soc., 2010, 132, 5562–5563 CrossRef CAS PubMed; (b) Y.-R. Chen and W.-L. Duan, Org. Lett., 2011, 13, 5824–5826 CrossRef CAS; (c) D. Du and W.-L. Duan, Chem. Commun., 2011, 47, 11101–11103 RSC; (d) D. Du, Z.-Q. Lin, J.-Z. Lu, C. Li and W.-L. Duan, Asian J. Org. Chem., 2013, 2, 392–394 CrossRef CAS; (e) J.-J. Feng, M. Huang, Z.-Q. Lin and W.-L. Duan, Adv. Synth. Catal., 2012, 354, 3122–3126 CrossRef CAS; (f) J. Lu, J. Ye and W.-L. Duan, Org. Lett., 2013, 15, 5016–5019 CrossRef CAS PubMed; (g) J. Lu, J. Ye and W.-L. Duan, Chem. Commun., 2014, 50, 698–700 RSC; (h) M. Huang, C. Li, J. Huang, W.-L. Duan and S. Xu, Chem. Commun., 2012, 48, 11148–11150 RSC; (i) C. Li, Q.-L. Bian, S. Xu and W.-L. Duan, Org. Chem. Front., 2014, 1, 541–545 RSC; (j) J. Huang, M.-X. Zhao and W.-L. Duan, Tetrahedron Lett., 2014, 55, 629–631 CrossRef CAS; (k) G.-F. Dai, Y.-C. Song, F. Xiao and W.-L. Duan, Synthesis, 2018, 50, 3506–3512 CrossRef CAS.
- (a) X.-Y. Yang, W. S. Tay, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2015, 34, 1582–1588 CrossRef CAS; (b) X.-Y. Yang, W. S. Tay, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2015, 34, 5196–5201 CrossRef CAS.
- (a) B. Ding, Z. Zhang, Y. Xu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Lett., 2013, 15, 5476–5479 CrossRef CAS PubMed; (b) Y. Xu, Z. Yang, B. Ding, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Tetrahedron, 2015, 71, 6832–6839 CrossRef CAS.
- (a) X.-Q. Hao, Y.-W. Zhao, J.-J. Yang, J.-L. Niu, J.-F. Gong and M.-P. Song, Organometallics, 2014, 33, 1801–1811 CrossRef CAS; (b) M.-J. Yang, Y.-J. Liu, J.-F. Gong and M.-P. Song, Organometallics, 2011, 30, 3793–3803 CrossRef CAS; (c) X.-Q. Hao, J.-J. Huang, T. Wang, J. Lv, J.-F. Gong and M.-P. Song, J. Org. Chem., 2014, 79, 9512–9530 CrossRef CAS; (d) J.-J. Huang, X.-Q. Zhang, J.-J. Yang, J.-F. Gong and M.-P. Song, Dalton Trans., 2022, 51, 8350–8367 RSC.
- X.-Y. Yang, W. S. Tay, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem. Commun., 2016, 52, 4211–4214 RSC.
- C. Wang, K. Huang, J. Ye and W.-L. Duan, J. Am. Chem. Soc., 2021, 143, 5685–5690 CrossRef CAS PubMed.
- Z. Yang, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Organometallics, 2015, 34, 1228–1237 CrossRef CAS.
- (a) A. Saito, N. Kumagai and M. Shibasaki, Org. Lett., 2019, 21, 8187–8190 CrossRef CAS PubMed; (b) A. Saito, S. Adachi, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2021, 60, 8739–8743 CrossRef CAS; (c) S. Adachi, A. Saito and M. Shibasaki, Org. Lett., 2022, 24, 3901–3906 CrossRef CAS PubMed.
- C. Wang, P. Yin, Y.-H. Dai, J. Ye and W.-L. Duan, J. Organomet. Chem., 2023, 983, 122552 CrossRef CAS.
- For recent publications, see: (a) Y.-D. Wang, J.-K. Liu, X.-J. Yang, J.-F. Gong and M.-P. Song, Organometallics, 2022, 41, 984–996 CrossRef CAS; (b) N. Li, X. Yang, Y. Zhu, F. Wang, J. Gong and M. Song, Chin. Chem. Lett., 2022, 33, 2437–2441 CrossRef CAS; (c) H. Jiang, C.-Y. Zhang, J.-K. Liu, M.-P. Song and J.-F. Gong, Adv. Synth. Catal., 2023, 365, 3967–3972 CrossRef CAS; (d) X.-Q. Zhang, H.-J. Wang, H. Jiang, M.-P. Song and J.-F. Gong, Green Synth. Catal., 2024, DOI:10.1016/j.gresc.2024.08.006.
- G. Chelucci, G. Orrù and G. A. Pinna, Tetrahedron, 2003, 59, 9471–9515 CrossRef CAS.
- For the related “one-pot” phosphorylation/metalation reaction, see: (a) J.-F. Gong, Y.-H. Zhang, M.-P. Song and C. Xu, Organometallics, 2007, 26, 6487–6492 CrossRef CAS; (b) B.-S. Zhang, W. Wang, D.-D. Shao, X.-Q. Hao, J.-F. Gong and M.-P. Song, Organometallics, 2010, 29, 2579–2587 CrossRef CAS , see also ref. 12b and 12c..
- Y. Wang, C. Qin, X. Jia, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2017, 56, 1614–1618 CrossRef CAS PubMed.
- U. Shadakshari and S. K. Nayak, Tetrahedron, 2001, 57, 8185–8188 CrossRef CAS.
- W. S. Tay, X.-Y. Yang, Y. Li, S. A. Pullarkat and P.-H. Leung, RSC Adv., 2016, 6, 75951–75959 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2284819, 2287268 and 2424928. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra01336d |
| This journal is © The Royal Society of Chemistry 2025 |