DOI:
10.1039/D5RA01298H
(Paper)
RSC Adv., 2025,
15, 11478-11490
Sensitive determination of chlorpromazine in pharmaceutical formulations and biological fluids using solid phase extraction followed by HPLC-UV and spectrophotometric analysis
Received
22nd February 2025
, Accepted 4th April 2025
First published on 11th April 2025
Abstract
An environmentally friendly analytical method was developed to detect trace amounts of chlorpromazine in pharmaceutical formulations, urine, and serum samples. The method integrates magnetic solid-phase extraction (MSPE) with UV detection (MSPE-UV), utilizing environmentally friendly materials and procedures. The MSPE system employed 3-chloropropyltriethoxysilane-coated magnetic nanoparticles (Fe3O4@CPTES) as adsorbents, offering a sustainable and efficient alternative to conventional methods. The proposed technique eliminates the need for toxic organic solvents commonly used in pre-concentration steps, aligning with the principles of green chemistry. Optimization of extraction parameters, including pH, ionic strength, and adsorbent dosage, revealed high extraction efficiencies (98% for pharmaceutical solutions, 52% for urine, and 33% for serum), with corresponding enrichment factors of 102, 52, and 41, respectively, and detection limits as low as 0.08 ng mL−1 for pharmaceutical solutions. The method demonstrates excellent linearity (R2 > 0.9988) over a wide range (0.15–400 ng mL−1) and high precision (RSD < 7%). The Fe3O4@CPTES nanoparticles enable rapid, reusable, and efficient extraction, reducing both analysis time and resource consumption. While tablet analysis validates method robustness, the primary application lies in trace CPZH detection in biological matrices. This green analytical approach offers a reliable, sensitive, and eco-friendly protocol for CPZH detection, highlighting its potential for broader applications in pharmaceutical and biomedical analysis.
1. Introduction
The introduction of chlorpromazine hydrochloride (CPZH) into medical practice in the early 1950s was probably one of the major milestones in medicine and psychiatry, leading to a complete revolution in the management of mental health disorders. Further development, even more powerful than the initial, of phenothiazine-based psychopharmacological agents brought a revolution in the field, making their use a breakthrough in the management of psychiatric illnesses.1 CPZH is one of the most extensively used phenothiazine derivatives for treating many psychiatric ailments. The main uses observed are in the treatment of psychomotor disturbances, excitement and agitation and in schizophrenia and during manic episodes related to manic-depressive states. Other uses of CPZH include states of hyperkinesis and aggression conditions, and at times its prescription is found helpful in reducing anxiety and tension states in other psychiatric ailments.2 Because of their high potency, antipsychotic drugs are prescribed in low daily doses. CPZH is also extensively metabolized in the human body. In plasma, very low concentrations of CPZH have been found. From a bioanalytical as well as from a clinical point of view, the determination of bio-therapeutically optimal therapeutic levels along with the management of side effects require highly sensitive, selective, precise bio-analytical methods for determination of the amount of CPZH in biological fluids.
Various analytical methods have been employed for the determination of CPZH in different samples such as spectrophotometric,3–5 fluorescence,3 flow injection analysis,6,7 electrochemical,8 MS,9 turbidity,10,11 LLE,12 GC-MS,13 and QuEChERS-based extraction.14 The analysis and quantification of CPZH in biological matrices has been extensively studied through HPLC coupled with various detection techniques, focusing on determining its active compound composition and concentration. Several published methods have employed HPLC with diverse detection systems to measure CPZH concentrations in biological samples.15–18 However, the direct analysis of species in real biological samples poses significant challenges due to matrix interference and the limited sensitivity of instrumental detection methods for ultra-trace levels of the analyte.19 To overcome these limitations and achieve reliable, sensitive and accurate results, a preliminary separation and preconcentration step is often required prior to analysis.20 Consequently, various separation and preconcentration techniques have been developed to extract CPZH from biological fluids, including molecularly imprinted polymers (MIP),21 liquid chromatography-UV (LC-UV),22 and solid phase microextraction.23,24
A new pre-concentration technique has been developed using magnetic solid phase extraction (MSPE), enabling efficient extraction of diverse organic and inorganic analytes from various matrices. MSPE uses mixed-hemimicelles assemblies, hemimicelles/micelles, to enable sample pre-treatment and pre-concentration of a large number of target analytes before downstream analysis.24–30 The adsorbents for the development of these surface-enhanced magnetite and modified magnetite nanoparticles were surface-coated with alumina or silica to develop Raman spectroscopy-compatible Solid Phase Extraction (SPE) methods, further functionalizing these nanoparticles with ionic surfactants like SDS or CTAB. Poor extraction efficiency and time-consuming processing of large-volume samples were the drawbacks commonly observed with previously used microparticle adsorbents and were highly improved by the use of MNPs as an adsorbent and mixed-hemimicelles. The use of mixed-hemimicelles assemblies in MSPE has some advantages, such as easy elution, regeneration of the adsorbent material, large breakthrough volumes and high extraction efficiencies.31–34 Besides, the use of MNPs with a huge surface area and strong magnetism allows one to improve the adsorption capacity and decrease the time of analysis due to the possibility of fast isolation of MNPs from large volumes of samples using an external magnet.35 The MSPE technique, developed for the first time using Fe3O4 nanoparticles, demonstrated a higher enrichment factor and superior extraction efficiency compared to conventional SPE methods. The aim of this study was to develop and validate a novel, eco-friendly MSPE-UV method for the enrichment and determination of trace levels of CPZH in pharmaceutical formulations, urine, and serum samples. This method utilizes 3-chloropropyltriethoxysilane-coated magnetic nanoparticles (Fe3O4@CPTES) as adsorbents to achieve highly efficient extraction while adhering to green chemistry principles by eliminating the use of toxic organic solvents. Parameters influencing the formation of mixed hemimicelles and CPZH extraction efficiency, such as adsorbent dosage, ionic strength, pH, and elution conditions, were systematically optimized to ensure maximum recovery and sensitivity. To the best of our knowledge, this represents a novel approach for CPZH extraction and determination, offering significant potential for application in pharmaceutical and biomedical analysis.
2. Experimental
2.1. Chemicals and materials
All reagents employed in this study were of analytical grade. CPZH, with a minimum purity of >99.8%, was obtained from Sammraa Drugs Industrial (SDI, Iraq). The chemical structure of CPZH is illustrated in Fig. 1. Sodium hydroxide, ferrous chloride (FeSO4·4H2O), ferric chloride (FeCl3·6H2O), tetrahydrofuran (THF), 3-chloropropyl triethoxysilane (CPTES), triethylamine (Et3N), hydrochloric acid, HPLC-grade acetonitrile, acetone, ethanol, methanol, and NaCl were all sourced from Sigma-Aldrich (USA). Ultrapure water from a Millipore Milli-Q system was used in all experiments. The CPZH stock solution (1000 μg mL−1) was prepared by dissolving the hydrochloride salt in methanol. The stock solution was stored in a refrigerator at 4 °C. Working solutions of varying concentrations were prepared as needed by diluting the stock solution with deionized water. Methanol was selected for stock preparation due to CPZH's high solubility, while dilution with water ensures compatibility with real-sample matrices.
 |
| | Fig. 1 The chemical structure of CPZH. | |
2.2. Instruments and software
Spectrophotometric measurements were conducted using a UV-L9, UV-visible spectrophotometer (WEST TUNE, China) equipped with 1.0 cm glass cuvettes, scanning wavelengths between 200 and 800 nm. Chromatographic separations were performed on a Waters Acquity UPLC system equipped with a Binary Solvent Manager (Milford, MA, USA), a six-port switching valve from Rheodyne (Rohnert Park, CA, USA) with a 10 μL sample loop, and a Waters 2998 Photodiode Array (PDA) detector. The chromatographic data were captured and processed using Empower software, version 3.0, for subsequent analysis. The separations were carried out on a Waters BEH C18 column (100 mm × 2.1 mm, with 1.7 μm particle size). The mobile phase consisted of ammonium formate (20 mM, pH 3.5) and methanol (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 v/v), run isocratically at 0.5 mL min−1. Samples (10 μL) were analyzed with UV detection at 280 nm. The elemental composition of the samples was characterized using Energy Dispersive X-ray spectroscopy (EDX) on a Shimadzu EDX 8100 instrument (Shimadzu, Japan). Field Emission Scanning Electron Microscopy (FE-SEM) images of Fe3O4 NPs and Fe3O4@CPTES were obtained using a Teneo FE-SEM (Thermo Fisher Scientific, USA), operating at an acceleration voltage between 500 V and 30 kV. For FTIR analysis, samples were mixed with KBr powder and compressed into pellets using a hydraulic press, ensuring consistent compression to avoid variations in absorbance intensity. FTIR spectra were recorded using a Shimadzu 8400S spectrophotometer. The particle size distribution was determined by analyzing FE-SEM images using ImageJ software. Additionally, Origin Pro and SPSS software were used for figure creation and statistical analysis, respectively.
:40 v/v), run isocratically at 0.5 mL min−1. Samples (10 μL) were analyzed with UV detection at 280 nm. The elemental composition of the samples was characterized using Energy Dispersive X-ray spectroscopy (EDX) on a Shimadzu EDX 8100 instrument (Shimadzu, Japan). Field Emission Scanning Electron Microscopy (FE-SEM) images of Fe3O4 NPs and Fe3O4@CPTES were obtained using a Teneo FE-SEM (Thermo Fisher Scientific, USA), operating at an acceleration voltage between 500 V and 30 kV. For FTIR analysis, samples were mixed with KBr powder and compressed into pellets using a hydraulic press, ensuring consistent compression to avoid variations in absorbance intensity. FTIR spectra were recorded using a Shimadzu 8400S spectrophotometer. The particle size distribution was determined by analyzing FE-SEM images using ImageJ software. Additionally, Origin Pro and SPSS software were used for figure creation and statistical analysis, respectively.
2.3. Synthesis of magnetic core (Fe3O4 NPs)
Magnetic nanoparticles (Fe3O4 NPs) were synthesized via modified co-precipitation. FeCl3·6H2O (10.4 g) and FeSO4·4H2O (4.0 g) were dissolved in 50 mL deionized water. The solution was then degassed with nitrogen gas for 20 minutes. Meanwhile, 500 mL of 1.5 M NH4OH solution was heated to 40 °C in a reactor. A slow and controlled addition of the stock solution to the reactor was achieved over a period of 30 minutes using a dropping funnel, while maintaining a nitrogen gas atmosphere and vigorous agitation with a glassware stirrer. The reaction proceeded at 75 °C under nitrogen flow to exclude oxygen. The Fe3O4 NPs were magnetically separated and washed with doubly distilled water (2 × 500 mL) to remove impurities. The purified NPs were dispersed in 500 mL of degassed deionized water, resulting in a stock suspension with a concentration of approximately 15 mg mL−1. The synthesized magnetic nanoparticles (MNPs) were then analyzed using various techniques, including IR, XRD, and FE-SEM.
2.4. Functionalization of the surface of a magnetic core with CPTES
Fe3O4 magnetic nanoparticles (MNPs, 0.4 g) were dispersed in tetrahydrofuran (THF, 8.0 mL) and subjected to sonication for 10 minutes to facilitate thorough dispersion and uniform distribution of the particles. Triethylamine (Et3N, 1.16 mL, 8.33 mmol) was added to the suspension, followed by the dropwise addition of 3-chloropropyltriethoxysilane (CPTES, 3.0 mL) at room temperature. The mixture was heated and stirred at 60 °C for 12 hours. Using a magnetic field, the synthesized nanoparticles were collected, purified through successive washing with ethanol and deionized water, and dried at 60 °C, resulting in CPTES@Fe3O4 magnetic nanoparticles (MNPs).
2.5. Preparation of real samples, tablets, serum and urine for MSPE-UV
The effectiveness of the proposed MSPE-UV method for the extraction and determination of CPZH was assessed using tablet, serum, and urine samples. Urine samples were collected from five healthy male volunteers, while serum samples were provided by Al-Yarmouk Teaching Hospital (Baghdad, Iraq). Prior to analysis, the urine and serum samples were thoroughly screened to confirm that they were obtained from healthy individuals who were not taking any medications, thereby minimizing the risk of potential interference. To reduce matrix interference, all urine and serum samples underwent a 1:10 dilution with ultra-pure water before analysis. Furthermore, to precipitate and remove the proteins from serum samples, 0.5 g trichloroacetic acid was added into 50 mL of diluted serum sample; after that, centrifugation was performed at 4000 rpm for 5 minutes. CPZH tablets (one tablet contains 50 mg, commercial brand) were crushed into fine powder in a mortar and pestle for better extraction. A known quantity of the powdered CPZH was dissolved in 100 mL of distilled water and sonicated for 15 minutes to ensure complete dissolution of the compound. The mixture was then filtered using a 0.45 μm filter to remove undissolved particles, and the filtrate was diluted with double distilled water to the desired concentration. Quantification of CPZH in all real samples employed the standard addition method. The MSPE-UV procedure was then applied to the prepared samples (tablet solution, diluted urine, and protein-free serum supernatant) for CPZH extraction and analysis.
2.6. General procedure for MSPE-UV
A general procedure for MSPE-UV was employed to extract CPZH from aqueous samples. Specifically, 50 mL of an aqueous sample containing 200 ng mL−1 of CPZH (pH = 4.0) was transferred to a 100 mL glass beaker. Next, 1.0 mL of a suspension of CPTES@Fe3O4 (20 mg mL−1) was added to the sample solution, and the mixture was thoroughly mixed. The resulting mixture was then shaken and allowed to undergo extraction for 5 minutes. Subsequently, a strong Nd–Fe–B magnet (KINGS MAGNET, Type N35, 100 mm × 50 mm × 40 mm, 1.5 T) was placed at the bottom of the beaker, causing the CPTES@Fe3O4 MNPs to be isolated from the solution. Following a brief period of approximately 1 minute, the solution clarified, and the supernatant was carefully decanted. After concentrating the CPZH sample, it was separated from the adsorbent using 500 μL of a weak sodium hydroxide solution (0.05 M NaOH). A small portion of this eluate (100 μL) was then transferred to a cuvette for analysis using UV spectrophotometry. Fig. 2 illustrates the steps of process.
 |
| | Fig. 2 Schematic diagram of the MSPE-UV procedure for preconcentration and determination of CPZH. | |
2.7. Ethical approval
This study, which involved the use of human samples, was granted approval by the Institutional Ethics Committee of University of Baghdad/College of Science. Prior to sample collection, all participants provided written informed consent. The study was conducted in accordance with relevant guidelines and regulations governing the use of human specimens in biomedical research. Throughout the study, participant privacy and safety were protected through the implementation of appropriate measures during sample analysis, handling, collection and disposal.
3. Results and discussion
3.1. Characterization of synthesized Fe3O4 and CPTES@Fe3O4 NPs using FTIR, FESEM and XRD techniques
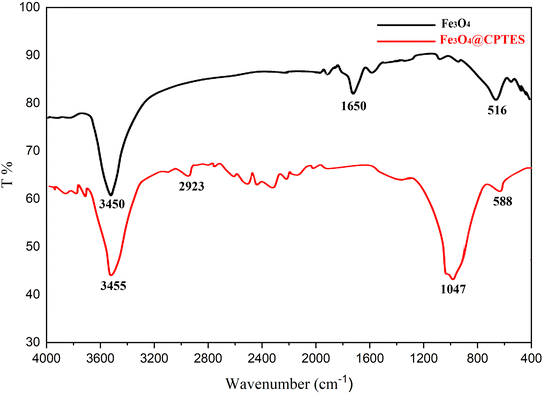
The structural and morphological properties of both synthesized Fe3O4 and Fe3O4@CPTES NPs were characterized using FTIR spectroscopy and FESEM techniques. The FTIR spectrum of the synthesized Fe3O4 NPs (as shown in Fig. 3) reveals several characteristic absorption bands, which are indicative of the specific vibrational modes present in the sample. The interpretation of these peaks can provide valuable insight into the functional groups and molecular interactions associated with Fe3O4 NPs. The FTIR spectrum presented herein eloquently illustrates the successful synthesis of Fe3O4 NPs, as evidenced by the stretching vibration of Fe–O at 516.80 cm−1 and 449.38 cm−1, which are signs of the magnetite phase. Besides that, the broadly stretched O–H stretching band at 3450.23 cm−1 and the O–H bending mode at 1650.45 cm−1 confirm that water molecules from the surrounding atmosphere or synthesis medium could, therefore, be adsorbed on the surface of the nanoparticles. Finally, after the successful synthesis of Fe3O4 NPs, their modification with (3-chloropropyl)trimethoxysilane was used to form Fe3O4@CPTES core–shell structures. This functionalization of Fe3O4 NPs by CPTES was carried out using FTIR spectroscopy. In Fig. 2, the FTIR spectrum of Fe3O4@CPTES reveals several characteristic peaks of both the magnetite core (Fe3O4) and the coating substance, CPTES. The following is the FTIR spectrum for the successful functionalization of Fe3O4 with (3-chloropropyl)trimethoxysilane. The appearance of new peaks at 2923.88 cm−1 and 2856.38 cm−1 corresponding to C–H stretching vibrations of the propyl chain, along with the characteristic Si–O–Si stretching at 1047.27 cm−1, confirms the successful formation of a CPTES layer on the surface of the Fe3O4 nanoparticles. Additionally, the Fe–O stretching vibrations below 588 cm−1 indicate that the core Fe3O4 structure is preserved after the modification, and the broad O–H stretching band at 3455.77 cm−1 suggests the presence of surface hydroxyl groups or adsorbed water. The functionalization of Fe3O4 with CPTES provides reactive surface groups (chloropropyl group), which can be further utilized for subsequent chemical modifications.
 |
| | Fig. 3 The FTIR spectra of synthesized Fe3O4 and Fe3O4@CPTES nanoparticles. | |
Fig. 4A shows the FESEM image of the synthesized Fe3O4 nanoparticles (Fe3O4 NPs). The particles display a spherical morphology with relatively smooth surfaces. The average diameter of the nanoparticles, as determined from the FESEM image, is 10.99 nm, indicating a well-controlled synthesis process that produced uniform nanoparticles. While some degree of agglomeration is observed, the individual particles remain distinguishable, suggesting that the agglomeration is primarily due to magnetic dipole interactions rather than synthesis defects.
 |
| | Fig. 4 FESEM images of (A) synthesized Fe3O4 and (B) Fe3O4@CPTES. | |
The relatively uniform size and morphology of the Fe3O4 NPs, coupled with their nanoscale dimensions, imply a high surface area-to-volume ratio, which is advantageous for various applications. These nanoparticles are particularly useful in magnetic drug delivery, catalysis, and magnetic separation processes, where particle size plays a critical role in performance. The minimal size variation further highlights the reproducibility and consistency of the synthesis method. While Fig. 4B presents the FESEM image of Fe3O4 NPs coated with (3-chloropropyl)trimethoxysilane (CPTES), forming Fe3O4@CPTES nanoparticles. After the surface functionalization with CPTES, the average diameter of the Fe3O4@CPTES nanoparticles are measured to be 11.42 nm, indicating a modest increase in particle size compared to the uncoated Fe3O4 nanoparticles (10.99 nm). This size increase is consistent with the formation of a thin CPTES coating layer around the Fe3O4 core. Morphologically, the c nanoparticles retain a largely spherical shape, similar to that of the uncoated Fe3O4 NPs, though with a slightly more aggregated appearance, which could be attributed to the coating process. The CPTES coating imparts functional groups (chloropropyl groups), which can facilitate further chemical modification or improve the particles' stability and dispersibility in various solvents or media. The successful coating is inferred from the observed increase in particle size and slight morphological changes. The well-dispersed particles, along with the observed changes in surface characteristics, suggest that the CPTES coating was successfully applied, providing a functionalized surface suitable for further covalent attachment of molecules or improved colloidal stability. These Fe3O4@CPTES NPs are promising candidates for applications in targeted drug delivery, catalysis, and surface modification due to the introduction of reactive groups on their surface.
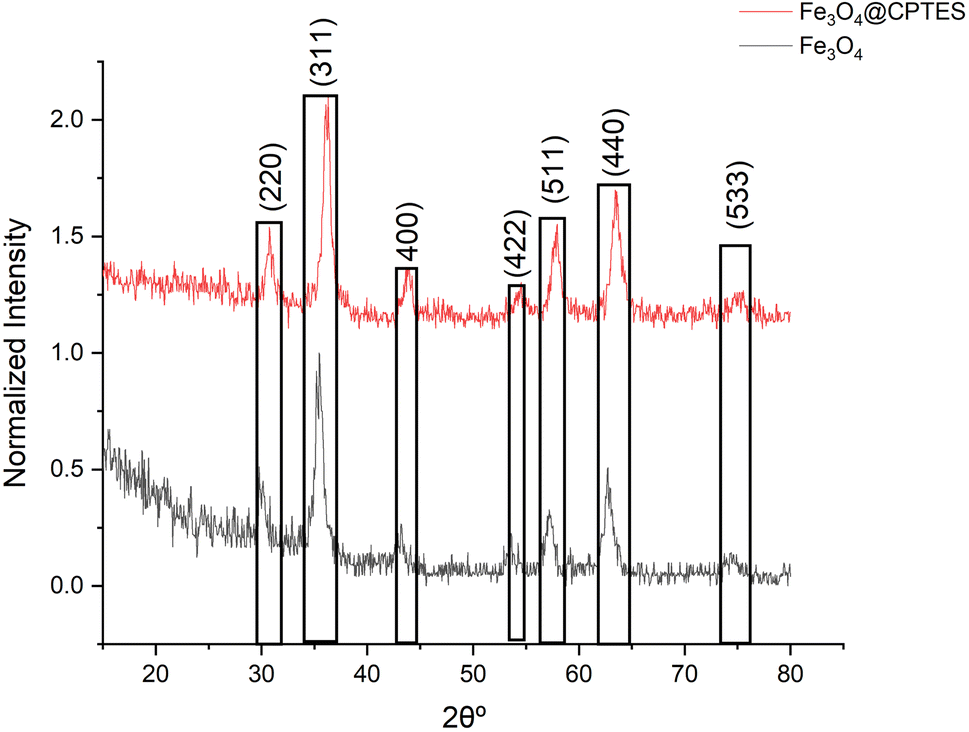
The use of X-ray diffraction (XRD) analysis provided definitive proof for the synthesis of Fe3O4 nanoparticles at both uncoated and surface-modified levels with (3-chloropropyl)triethoxysilane silane or CPTES (Fig. 5). The diffractogram shows characteristic diffraction peaks of magnetite dihedral at approximately 30.1°(220), 35.5°(311), 43.1°(400), 53.4°(422), 57.0°(511), 62.6°(440), and 74.0°(533) which according to the standards are positioned around 30.1°(220), 35.5°(311), 43.1° (400), 53.4°(422), 57.0°(511), 62.6°(440) and 74.0°(533) which correspond perfectly with JCPDS card NO 19-0629. The peak at 35.5° is particularly diagnostic of inverse spinel structure of Fe3O4. It is remarkable that there are no other peaks that would be typical for additional phases of other iron oxides or hydroxides such as gamma Fe2O3, alpha Fe2O3, Fe(OH)3 or even Fe(OH)3 was striking due to the unadulterated phases of the synthesized material. Diffraction pattern of Fe3O4@CPTES show all the sharp peaks of unmodified Fe3O4 which indicates that the crystal structure does not change after surface modification, although some broadening of peaks occurs which could relatively easily be explained by silane coating. There is little degree of doubt in the preservation of peak positions with absence of other crystalline phases in concern for the potential hydrolysis of iron(III) salts in non acid media. The synthetic approach yielded phase-pure Fe3O4 nanoparticles with the expected crystallographic structure.
 |
| | Fig. 5 The XRD pattern of synthesized Fe3O4 and Fe3O4@CPTES nanoparticles using hydrothermal method. | |
Based on the analysis of the XRD data using the Scherrer equation, the nanoparticle sizes for both samples were calculated. It was found that the Fe3O4@CPTES nanoparticles (size range 7–14.16 with an average size of 11.42 nm) are slightly larger than the Fe3O4 nanoparticles (size range 6–14.13 with an average size of 10.99 nm) by about 0.43 nm on average. This slight increase in size is consistent with what we would expect after coating Fe3O4 nanoparticles with CPTES (3-chloropropyl triethoxysilane), which adds a thin silane layer to the surface of the iron oxide nanoparticles. Both samples show relatively narrow size distributions, suggesting good control during the synthesis process.
3.2. Optimization and validation of the proposed MSPE-UV method for CPZH analysis
The development of the MSPE-UV method involved two instruments: a spectrophotometer and an HPLC-UV. Most parameters were optimized using the spectrophotometer due to its ease of use, fast analysis time, and cost-effectiveness. However, the eluent volume was optimized using the HPLC-UV due to its superior sensitivity and separation capabilities, which are essential for method validation and analyzing real samples containing complex mixtures. During optimization experiments, a solution with a concentration of 200 ng mL−1 of CPZH was used. Quantification was performed by measuring the absorption peak area using both spectrophotometry and HPLC-UV. The final elution of CPZH was carried out in 5 mL conical tubes to minimize eluent volume and enhance enrichment factors or sensitivity by improving adsorbent collection.
3.3. Impact of sample pH on CPZH adsorption
The pH of the sample solution is a crucial factor that influences the state of species (ionic or neutral forms) and the adsorption behavior of the mixed-hemimicelles system. Specifically, the charge density on the surface of the magnetic nanoparticles (MNPs) changes with pH, affecting the adsorption of CPZH.12 Fig. 6 clearly demonstrates that the maximum adsorption of CPZH occurs at a pH of 4.0. This is because, at this pH, the majority of the amino groups in CPZH are protonated, leading to a strong electrostatic attraction between the negatively charged surface of the MNPs and the positively charged CPZH molecules, resulting in high adsorption. However, when the pH is increased from 4.0 to 8.0, the amino groups become deprotonated, reducing their positive charge and weakening the electrostatic attraction. Consequently, the adsorption capacity of CPZH significantly decreases. This decrease can be explained by the electrostatic interactions between the negatively charged surface of the MNPs and the positively charged CPZH molecules, which exist in their protonated form at lower pH values, favoring adsorption, however, the insufficient number of protons at higher pH values led to a decrease in the protonated amount of CPZH, resulting in reduced adsorption. On the other hand, when the pH value was below 4.0 (in the range of 2–3), the negative charge density on the MNPs surface decreased due to the excess concentration of acid, which reduced the negative charges on the surface of the MNPs.15,20 As a result, the electrostatic attraction between the negative charges of the MNPs and the positive charges of CPZH was insufficient to facilitate hemimicelle formation, resulting in inefficient adsorption of CPZH in its cationic state (pH = 2.0–3.0).23 Therefore, a pH of 4.0 was chosen for subsequent experiments.
 |
| | Fig. 6 Effect of pH on the adsorption efficiency of CPZH using Fe3O4@CPTES. | |
3.4. The effect of ionic strength on the adsorption of CPZH
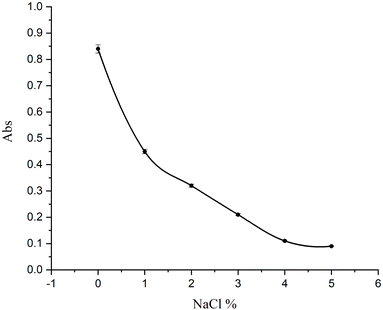
The impact of ionic strength on CPZH adsorption was examined by adding NaCl to the solution at concentrations ranging from 0 to 5% (m/v). The findings revealed that higher NaCl concentrations significantly reduced the adsorption capacity of the MNPs. This reduction can be attributed to ion–exchange reactions occurring in the solution during the adsorption process, as described in Fig. 7. High salt concentrations increase [Na+], and according to Le Chatelier's principle, this should lower CPZH adsorption. Consequently, all subsequent experiments were conducted in the absence of NaCl. Notably, in biological samples, where salts are naturally present, lower extraction efficiencies may be anticipated compared to aqueous samples.
 |
| | Fig. 7 Influence of ionic strength (NaCl concentration) on CPZH adsorption. | |
3.5. Influence of adsorbent quantity
The effect of adsorbent dosage on the extraction efficiency was evaluated by varying the amount of adsorbent used in the extraction process. Different amounts of MNPs (0–40 mg) were introduced into 50 mL of the sample solution. The results indicated that increasing the adsorbent quantity up to 20 mg caused a gradual rise in extraction recovery, after which it reached a plateau. This effect results from MNPs having a larger surface area-to-volume ratio than conventional micronized adsorbents. Consequently, satisfactory results were achieved with a relatively small amount of MNPs (20 mg), as shown in Fig. 8.
 |
| | Fig. 8 Impact of MNPs dosage on CPZH adsorption. | |
3.6. Effect of extraction time
The influence of extraction time on CPZH adsorption was also examined over a range of 1–15 minutes, as shown in Fig. 9. The results indicated that CPZH adsorption increased slightly up to 5 minutes and then remained constant. The shorter diffusion path of MNPs explains this behavior. Additionally, the CPTES-coated MNPs displayed superparamagnetic properties and high saturation magnetization, enabling quick magnetic separation (under 30 seconds) with a strong magnet. This facilitated quick extraction and elution of analytes from the MNPs, significantly shortening the analysis time. Consequently, an extraction time of 5 minutes was deemed optimal.
 |
| | Fig. 9 Effect of extraction time on CPZH adsorption efficiency. | |
3.7. Selection of a suitable eluent
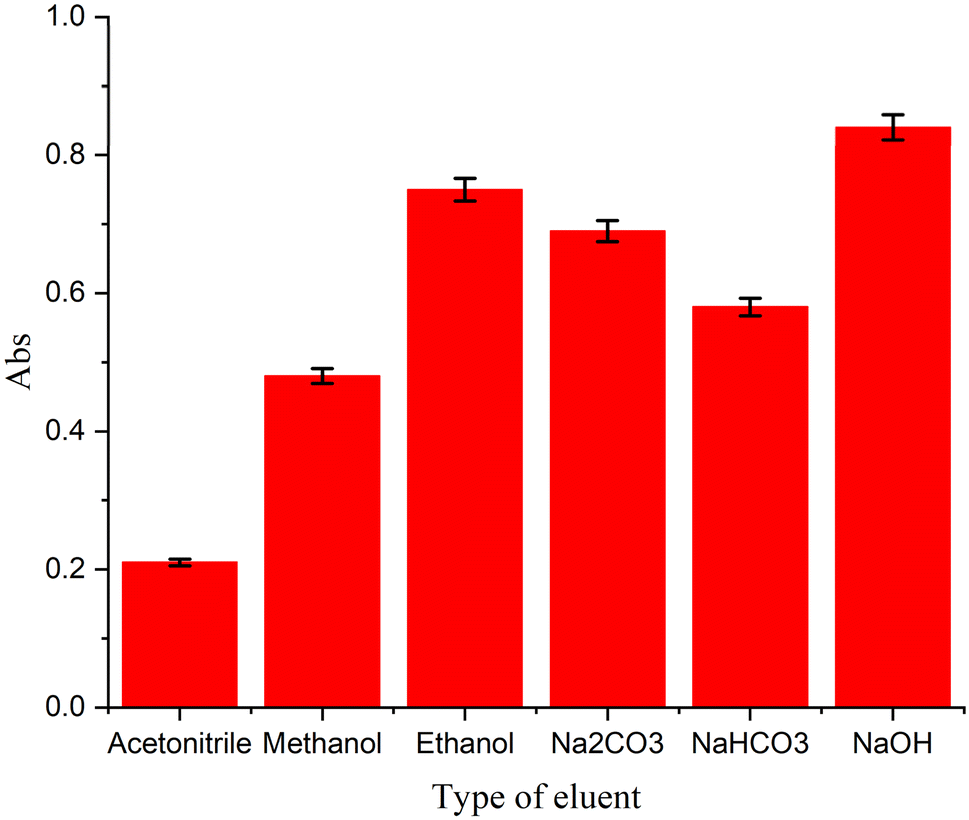
The selection of a suitable eluent for desorbing analytes from mixed-hemimicelles on coated -MNPs was investigated (Fig. 10). A series of organic solvents and aqueous solvents, known to disrupt mixed-hemimicelles were tested (acetonitrile, methanol, ethanol, Na2CO3, NaHCO3, and NaOH) for their ability to elute CPZH from CPTES-coated MNPs. The results showed that organic solvents had limited desorption efficiency compared to aqueous solvents, which exhibited excellent desorption capabilities. This can be ascribed to that at higher pH levels, particularly around the isoelectric point, the charge density on the MNP surface is weakened, facilitating the disruption of mixed-hemimicelles. Among the aqueous solutions, NaOH demonstrated the highest desorption efficiency. Consequently, a basic NaOH solution (0.05 M) was chosen for further studies.
 |
| | Fig. 10 Comparison of different eluents for CPZH desorption from Fe3O4@CPTES. | |
3.8. Optimization of eluent volume and desorption time
The influence of eluent volume and desorption time on the extraction efficiency of CPZH was investigated. A basic solution of 0.05 M NaOH was used as the eluent, with volumes ranging from 0.1 to 1.0 mL. As illustrated in Fig. 11, the desorption efficiency of CPZH elevated with eluent volume up to 0.5 mL, beyond which peak areas decreased due to dilution effects. The optimization study determined 0.5 mL as the ideal eluent volume, as shown in Fig. 8. Additionally, the impact of desorption time was examined over a range of 1 to 15 minutes. Experimental results indicated that desorption time had negligible effect on CPZH desorption efficiency. To minimize overall analysis time, a desorption period of 1 minute was chosen for subsequent studies. These optimizations ensure efficient CPZH extraction while maintaining analytical expediency.
 |
| | Fig. 11 Effect of eluent volume on CPZH extraction efficiency. | |
3.9. Effect of sample volume
Processing larger sample volumes is essential for achieving higher enrichment factors. The use of Magnetic Carrier Technology (MCT) in MNPs mixed hemimicelles SPE offers key advantages in this regard. This approach eliminates time-consuming steps like column passing and filtration, demonstrating great potential for preconcentrating large volumes of water samples. The effect of sample volume on CPZH enrichment was studied by extracting 20 μg CPZH from aqueous solutions with volumes ranging from 25 to 250 mL. Using optimized conditions, CPZH recovery remained high (>94%) even with sample volumes reaching 200 mL. Although the data supported using volumes up to 150 mL, the researchers opted to proceed with 75 mL sample solutions for further studies. This decision was made due to the limited volumes typically available in biological samples. However, this finding is valuable as it can be applied to dilute biological samples, potentially reducing matrix effects in the analysis.
3.10. Assessment of analytical method performance
The performance of the proposed method is summarized in Table 1, which presents the linearity, enrichment factor (EF), limit of detection (LOD), sensitivity, precision, and limit of quantification (LOQ) for the extraction of CPZH from 75 mL of three distinct aqueous solutions.
Table 1 Relative standard deviation (RSD), extraction efficiency, and enrichment factors (EF) for CPZH analysis in different matrices
| Sample |
RSD%, (n = 5), [CPZH] = 25 ng mL−1 |
Extraction% |
EF |
| Inter day |
Intra day |
| CPZH tablet |
0.9 |
1.1 |
98 |
102 |
| Serum |
3.1 |
5.3 |
49 |
52 |
| Urine |
2.9 |
3.8 |
33 |
41 |
3.10.1. Linearity and calibration range. Under optimized conditions, the calibration curves were linear over the ranges of 0.25–300 ng mL−1, 5.0–300 ng mL−1, and 10.0–300 ng mL−1 for pharmaceutical solution, urine, and serum samples, respectively (n = 6). Recovery percentages were calculated using the formula:
where C and V represent analyte concentrations and volumes, respectively. enrichment factors (EF) were determined as the ratio of Celuate and Csample. While formula for EF was:
3.10.2. Enrichment factor (EF). The EF for CPZH was determined by comparing the calibration curve slopes obtained from preconcentrated samples versus direct injection of CPZH standards using HPLC. The above results showed that the proposed method provided EFs of 102, 52, and 33 for pharmaceutical solution, urine, and serum samples, respectively, reflecting its effectiveness in the enriching of CPZH from these matrices, as listed in Table 1.
3.10.3. Sensitivity and precision. The precision of the developed method was verified by calculating intra-day and inter-day RSDs. The RSDs of reproducibility and repeatability (at n = 5) for CPZH were below 7% at the 20 μg L−1 level, which reveals good precision. Furthermore, no significant differences in inter-day peak areas obtained for CPZH were determined using a one-way ANOVA test, demonstrating the excellent reproducibility of the developed method (See Table 1).
3.10.4. LOD and LOQ. The LODs were 0.1 ng mL−1 for pharmaceutical solutions, 0.5 ng mL−1 for urine samples, and 1.0 ng mL−1 for serum samples. This means that the assay could reliably detect concentrations of the analyte as low as these values. The LOQs were 0.25 ng mL−1, 1.0 ng mL−1, and 5.0 ng mL−1 for the respective sample types. This indicates the lowest concentration at which the analyte could be accurately quantified. The LODs were determined using a signal-to-noise ratio of 3.
3.11. The proposed mechanism of the MSPE-UV method
The electrostatic attraction between the positively charged amino group of CPZH and the negatively charged surface of the MSP NPs is thought to be the driving force behind the CPZH extraction mechanism. The MSPE-UV-based extraction of CPZH can be understood in light of its ionizable amino group (Fig. 12). With a pKa of 9.3, the ionization state of this amino group significantly influences the extraction process. In acidic conditions (pH < pKa), the amino group of CPZH becomes protonated, resulting in a positive charge. With this, the CPTES-coated MNPs are supposed to form mixed-hemimicelles at the surface, which would project a negatively charged interface on their surface. The positively charged CPZH molecules are consumed onto the negatively charged surface of the CPTES-coated MNPs in a very fast way. According to this, the main driving force for the extraction was an electrostatic attraction. Thus, under acidic conditions, the extraction of CPZH is predominantly driven by electrostatic interactions between the protonated CPZH and the mixed-hemimicelles on the CPTES-coated MNPs.
 |
| | Fig. 12 Proposed mechanism for CPZH extraction using Fe3O4@CPTES nanoparticles. | |
3.12. Application of MSEP method with real samples
To test the effectiveness of the new extraction system on complex matrices, we analyzed spiked samples of pharmaceutical solution, urine, and serum under optimized conditions of the method. As CPZH wasn't initially detected in these samples, known quantities were added (in ng mL−1) before proceeding with the extraction and analysis. Results from Table 2 show that our method yielded satisfactory outcomes across three replicate analyses for each sample type. The measured values closely matched the spiked amounts, with relative errors below 7.0%. Furthermore, the method demonstrated good reproducibility in real samples, with intra-day RSD% values ranging from 2.3% to 9.7%. Fig. 13 presents the chromatographic results from MSPE-HPLC-UV analysis, showing CPZH determination in diluted urine (A) and serum (B) matrices, with comparisons shown between the un-spiked (native) samples and those spiked with CPZH Table 3.
Table 2 Statistical parameters of the proposed method for CPZH determination
| Sample |
Linear range ng mL−1 |
LOD (ng mL−1) |
LOQ (ng mL−1) |
R2 |
| CPZH tablet |
0.15-400 |
0.08 |
0.25 |
0.9988 |
| Serum |
8.0-400 |
4.0 |
10.0 |
0.9989 |
| Urine |
3.0-400 |
0.9 |
3.0 |
0.9978 |
 |
| | Fig. 13 HPLC-UV chromatograms of CPZH in (A) urine and (B) serum samples before and after spiking. | |
Table 3 Recovery results for CPZH determination in real samples using the proposed MSPE-UV method
| Sample |
Added [CPZH], ng mL−1 |
Found [CPZH], ng mL−1 |
RSD%, n = 3 |
Rec% |
| CPZH tablet 1 |
2.0 |
2.1 |
1.2 |
105.00 |
| CPZH tablet 2 |
5.0 |
5.08 |
0.99 |
101.60 |
| Serum 1 |
20 |
20.2 |
5.2 |
101.00 |
| Serum 1 |
40 |
40.19 |
6.3 |
100.47 |
| Serum 1 |
60 |
60.25 |
4.9 |
100.41 |
| Urine 1 |
5.0 |
4.98 |
1.8 |
99.60 |
| Urine 2 |
15 |
14.88 |
3.2 |
99.20 |
| Urine 3 |
30 |
30.3 |
4.8 |
101.00 |
3.13. Comparison of the proposed method with existing techniques
The performance of the proposed method was evaluated and compared with other recently published methods for the preconcentration and determination of CPZH in different matrices. The key advantages of the proposed method are outlined in Table 4. Significantly, the proposed method provides a broad linear range and lower LODs, while maintaining RSDs that are competitive with or better than those of LLE methods. This is remarkable considering that LLE is often coupled with highly sensitive detection techniques like LC-ESI-MS/MS and chemiluminescence. Furthermore, the proposed method's use of MNPs enables rapid extraction dynamics and a large surface area, resulting in significantly shorter extraction times compared to other reported methods. This, in turn, allows for higher sample throughput, making the proposed method a more efficient option.
Table 4 Comparison of different analytical methods for CPZH determination
| Reported method |
Linear range (ng mL−1) |
LOD (ng mL−1) |
Matrix |
Ref. |
| MIP-electrochemical |
0.5-100 |
0.16 |
Pharmaceuticals |
36 |
| LC-MS/MS |
1.0-500 |
0.3 |
Blood/Urine |
37 |
| SPME-HPLC |
5.0-1000 |
1.5 |
Biological fluids |
38 |
| GC-MS |
10-1000 |
3.0 |
Biological samples |
39 |
| MSPE-UV |
0.15-400 |
0.08 |
Pharmaceuticals |
Present work |
| MSPE-UV |
3.0-400 |
0.9 |
Urine |
Present work |
| MSPE-UV |
8.0-400 |
4.0 |
Serum |
Present work |
4. Conclusion
A novel MSPE-UV method was developed using MNPs coated with CPTES to form mixed-hemimicelles. This method effectively preconcentrates CPZH in pharmaceutical solution, urine, and serum samples. The proposed method offers several key advantages that make it a robust and efficient analytical technique. It has a high extraction capacity with high preconcentration factors attained by the use of MNPs. Additionally, magnetic separation considerably improves the separation rate since column passing or filtration steps, which can be time-consuming, are avoided. High extraction efficiency and capacity is further obtained due to strong electrostatic interactions between protonated CPZH and mixed hemimicelles. Besides, the analyte can be easily desorbed by basic ethanol without any carry-over effects in subsequent analyses, which guarantees the accuracy and reliability of test results. Conclusion: all these advantages turn the proposed method into real help for CPZH analysis. The method demonstrated its effectiveness in concentrating trace amounts of CPZH in various samples before using either HPLC or UV analysis. Based on these results, this approach shows great potential for preconcentrating drugs from real samples in similar applications.
Data availability
All data supporting the findings of this study are included within the article. As all experimental data, results, and analyses have been fully incorporated into the manuscript, there are no additional datasets stored elsewhere that would require external access provisions.
Conflicts of interest
The authors confirm that there are no relevant financial or non-financial competing interests to report.
Acknowledgements
The authors are grateful to the University of Baghdad, College of Science for supporting this manuscript.
References
- A. A. Baumeister, The chlorpromazine enigma, J. Hist. Neurosci., 2013, 22, 14–29, DOI:10.1080/0964704X.2012.664087.
- T. A. Ban, Fifty years chlorpromazine: a historical perspective, Neuropsychiatric Dis. Treat., 2007, 3, 495–500 CAS.
- S. Narimani, N. Samadi and E. Delnavaz, Highly sensitive and novel dual-emission fluorescence nanosensor utilizing hybrid carbon dots–quantum dots for ratiometric determination of chlorpromazine, Anal. Sci., 2024, 1–8, DOI:10.1007/s44211-024-00591-x.
- M. K. Hammood, J. N. Jeber and M. A. Khalaf, Rapid colorimetric sensing of chlorpromazine HCl antipsychotic through in situ growth of gold nanoparticles, RSC Adv., 2024, 14, 2327–2339, DOI:10.1016/j.msec.2016.07.025.
- J. N. Jeber, R. F. Hassan, M. K. Hammood and O. H. R. Al-Jeilawi, Sensitive and simple colorimetric methods for visual detection and quantitative determination of semicarbazide in flour products using colorimetric reagents, Sensor. Actuator. B Chem., 2021, 341, 130009, DOI:10.1016/j.snb.2021.130009.
- N. S. Turkey and J. N. Jeber, A flow analysis system integrating an optoelectronic detector for the quantitative determination of active ingredients in pharmaceutical formulations, Microchem. J., 2021, 160, 105710, DOI:10.1016/j.microc.2020.105710.
- J. N. Jeber and N. S. Turkey, An optoelectronic flow-through detectors for active ingredients determination in the pharmaceutical formulations, J. Pharm. Biomed. Anal., 2021, 201, 114128, DOI:10.1016/j.jpba.2021.114128.
- S. Srinivasan, C. S. S. Narayanan, J. Kanimozhi, I. Suresh, B. M. Gunasekaran, M. Ezhilan, N. Nesakumar and R. Venkatachalam, Electrochemical Sensor Based on Electrodeposited Zinc-Aluminium Layered Double Hydroxide Modified Glassy Carbon Electrode for Chlorpromazine Sensing, J. Electrochem. Soc., 2024, 171, 037517, DOI:10.1149/1945-7111/ad2ef2.
- F. Martinez-Rojas, C. Espinosa-Bustos, G. Ramirez and F. Armijo, Electrochemical oxidation of chlorpromazine, characterisation of products by mass spectroscopy and determination in pharmaceutical samples, Electrochim. Acta, 2023, 443, 141873, DOI:10.1016/j.electacta.2023.141873.
- N. S. Turkey and J. N. Jeber, Turbidimetric Determination of Mebeverine Hydrochloride in Pharmaceutical Formulations Using Two Consecutive Detection Zones Under Continuous Flow Conditions, Chem. Chem. Technol., 2022, 16, 600–613, DOI:10.23939/chcht16.04.600.
- N. S. Turkey and J. N. Jeber, Light scattering detector based on light-emitting diodes-Solar cells for a flow analysis of Warfarin in pure form and pharmaceutical formulations, J. Phys.: Conf. Ser., 2021, 2063, 012006, DOI:10.1088/1742-6596/2063/1/012006.
- K. I. Alzand, H. R. Younis, B. Qasim and J. N. Jeber, Development and Validation of a Dispersive Liquid–Liquid Microextraction Method for Metoclopramide Analysis in Pharmaceuticals and Biological Samples, Russ. J. Gen. Chem., 2024, 94, 2517–2528, DOI:10.1134/S1070363224090287.
- A. Kul and O. Sagirli, A new method for the therapeutic drug monitoring of chlorpromazine in plasma by gas chromatography–mass spectrometry using dispersive liquid–liquid microextraction, Bioanalysis, 2023, 15, 1343–1354, DOI:10.4155/bio-2023-0176.
- J. Dai, H. Lin, Y. Pan, Y. Sun, Y. Wang, J.-q. Qiao, H.-z. Lian and C.-x. Xu, Determination of chlorpromazine and its metabolites in animal-derived foods using QuEChERS-based extraction, EMR-Lipid cleanup, and UHPLC-Q-Orbitrap MS analysis, Food Chem., 2023, 403, 134298, DOI:10.1016/j.foodchem.2022.134298.
- V. S. Kovalenko and S. I. Merzlikin, Determination of chlorpromazine metabolite in objects of biological origin by chromatography, J. Mod. Med., 2023, 12, 707–713, DOI:10.1002/bms.1200121207.
- Y. Yamini and M. Faraji, Extraction and determination of trace amounts of chlorpromazine in biological fluids using magnetic solid phase extraction followed by HPLC, J. Pharm. Anal., 2014, 4, 279–285, DOI:10.1016/j.jpha.2014.03.003.
- S. R. Fadhel and S. I. Khalil, Simultaneous estimation of chlorpromazine hydrochloride and carvedilol in bulk and pharmaceutical dosage forms using HPLC, J. Biochem. Technol., 2018, 9, 5–9 CAS.
- H. R. Sobhi, Y. Yamini and R. H. H. B. Abadi, Extraction and determination of trace amounts of chlorpromazine in biological fluids using hollow fiber liquid phase microextraction followed by high-performance liquid chromatography, J. Pharmaceut. Biomed. Anal., 2007, 45, 769–774, DOI:10.1016/j.jpba.2007.09.026.
- T. M. Pizzolato, M. J. L. de Alda and D. Barceló, LC-based analysis of drugs of abuse and their metabolites in urine, TrAC, Trends Anal. Chem., 2007, 26, 609–624 CrossRef CAS.
- L. Zhang, P. Wu, Y. Zhang, Q. Jin, D. Yang, L. Wang and J. Zhang, A GC/MS method for the simultaneous determination and quantification of chlorpromazine and diazepam in pork samples, Anal. Methods, 2014, 6, 503–508, 10.1039/C3AY41130C.
- Y. Liu, X. Hu, Y. Xia, F. Zhao and B. Zeng, A novel ratiometric electrochemical sensor based on dual-monomer molecularly imprinted polymer and Pt/Co3O4 for sensitive detection of chlorpromazine hydrochloride, Anal. Chim. Acta, 2022, 1190, 339245, DOI:10.1016/j.aca.2021.339245.
- J. Li, J. An and Y. Jiang, Development of a method of hollow fiber-based solid-phase microextraction followed by ultra performance liquid chromatography-tandem mass spectrometry for determination of five antipsychotics in human whole blood and urine, J. Chromatogr. A, 2020, 1620, 461000, DOI:10.1016/j.chroma.2020.461000.
- J. J. W. Broeders, B. J. Blaauboer and J. L. M. Hermens, Development of a negligible depletion-solid phase microextraction method to determine the free concentration of chlorpromazine in aqueous samples containing albumin, J. Chromatogr. A, 2011, 1218, 8529–8535, DOI:10.1016/j.chroma.2011.09.064.
- N. Jeber Jalal, F. Hassan Raed and K. Hammood Mohammad, Solid Phase Extraction of Theophylline in Aqueous Solutions by Modified Magnetic Iron Oxide Nanoparticles as an Extractor Material and Spectrophotometry Technique for the Determination, Res. J. Chem. Environ., 2019, 1, 94–100 Search PubMed.
- A. Al-Ani, J. Jeber and A. Elewi, Development of a nanostructured double-layer coated tablet based on polyethylene glycol/gelatin as a platform for hydrophobic molecules delivery, Egypt. J. Chem., 2021, 64, 1759–1767, DOI:10.21608/ejchem.2021.52019.3066.
- H. Serbest, Determination of Rhodium in Soil by Dispersive Solid-Phase Extraction (DSPE) with Manganese Ferrite Nanoparticles and Flame Atomic Absorption Spectrometry (FAAS), Anal. Lett., 2024, 57, 456–468, DOI:10.1080/00032719.2023.2212092.
- E. Pourbasheer, L. Malekpour, Z. Azari, V. H. Masand and M. R. Ganjali, Magnetic solid phase extraction of Sunitinib malate in urine samples assisted with mixed hemimicelle and spectrophotometric detection, Sci. Rep., 2023, 13, 3361 CrossRef CAS PubMed.
- S. Oshaghi, Nano-sized magnetic molecularly imprinted polymer solid-phase microextraction for highly selective recognition and enrichment of sulfamethoxazole from spiked water samples, J. Chromatogr. A, 2024, 1729, 465016, DOI:10.1016/j.chroma.2024.465016.
- M. Faraji, Y. Yamini, E. Tahmasebi, A. Saleh and F. Nourmohammadian, Cetyltrimethylammonium bromide-coated magnetite nanoparticles as highly efficient adsorbent for rapid removal of reactive dyes from the textile companies' wastewaters, J. Iran. Chem. Soc., 2010, 7, S130–S144, DOI:10.1007/BF03246192.
- M. Faraji, Y. Yamini and M. Rezaee, Extraction of trace amounts of mercury with sodium dodecyle sulphate-coated magnetite nanoparticles and its determination by flow injection inductively coupled plasma-optical emission spectrometry, Talanta, 2010, 81, 831–836, DOI:10.1016/j.talanta.2010.01.023.
- L. Sun, L. Chen, X. Sun, X. Du, Y. Yue, D. He, H. Xu, Q. Zeng, H. Wang and L. Ding, Analysis of sulfonamides in environmental water samples based on magnetic mixed hemimicelles solid-phase extraction coupled with HPLC–UV detection, Chemosphere, 2009, 77, 1306–1312, DOI:10.1016/j.chemosphere.2009.09.049.
- L. Sun, C. Zhang, L. Chen, J. Liu, H. Jin, H. Xu and L. Ding, Preparation of alumina-coated magnetite nanoparticle for extraction of trimethoprim from environmental water samples based on mixed hemimicelles solid-phase extraction, Anal. Chim. Acta, 2009, 638, 162–168, DOI:10.1016/j.aca.2009.02.039.
- M. H. Nouriyeh, M. R. A. Mogaddam, M. Nemati, M. A. Farajzadeh, A. Abbasalizadeh and A. S. Hojghan, Development of dispersive solid phase extraction based on magnetic metal organic framework for the extraction of sunitinib in biological samples and its determination by high performance liquid chromatography-tandem mass spectrometry, J. Chromatogr. B, 2024, 1239, 124109, DOI:10.1016/j.jchromb.2024.124109.
- A. Karataş, T. Oymak and A. Cel
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) k, Development of a new magnetic solid-phase extraction method prior to HPLC determination of naproxen in pharmaceutical products and water samples, J. Pharm. Biomed. Anal., 2024, 116336, DOI:10.1016/j.jpba.2024.116336.
k, Development of a new magnetic solid-phase extraction method prior to HPLC determination of naproxen in pharmaceutical products and water samples, J. Pharm. Biomed. Anal., 2024, 116336, DOI:10.1016/j.jpba.2024.116336. - B. Rezaei, P. Yari, S. M. Sanders, H. Wang, V. K. Chugh, S. Liang, S. Mostufa, K. Xu, J. P. Wang and J. Gómez-Pastora, Magnetic nanoparticles: a review on synthesis, characterization, functionalization, and biomedical applications, Small, 2024, 20, 2304848, DOI:10.1002/smll.202304848.
- S. Zeng, P. Zhu, D. Liu, Y. Hu, Q. Huang and H. Huang, A sensitive electrochemical sensor based on CoWO 4/multi-walled carbon nanotubes for the selective determination of chlorpromazine hydrochloride, Analyst, 2025, 150, 81–86, 10.1039/D4AN01298D.
- L. N. Rodda, M. Farley, S. Towler, T. Devincenzi and S. Pearring, Multi-class analysis of 57 drugs quantitatively in blood and qualitatively in urine by LC–MS–MS to complement comprehensive DFC, DUID, and postmortem testing, J. Anal. Toxicol., 2025, 49, 1–13, DOI:10.1093/jat/bkae077.
- Y. Yamini and M. Faraji, Extraction and determination of trace amounts of chlorpromazine in biological fluids using magnetic solid phase extraction followed by HPLC, J. Pharm. Anal., 2014, 4, 279–285, DOI:10.1016/j.jpha.2014.03.003.
- A. Kul and O. Sagirli, A new method for the therapeutic drug monitoring of chlorpromazine in plasma by gas chromatography–mass spectrometry using dispersive liquid–liquid microextraction, Bioanalysis, 2023, 15, 1343–1354, DOI:10.4155/bio-2023-0176.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hussein Fares Abd-Alrazackb,
Mohammad K. Hammooda and
Muhannad M. Abdc
*a,
Hussein Fares Abd-Alrazackb,
Mohammad K. Hammooda and
Muhannad M. Abdc