
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hyaluronic acid-coated capecitabine nanostructures for CD44 receptor-mediated targeting in breast cancer therapy†
Sruthi Laakshmi Mugundhan and
Mothilal Mohan*
and
Mothilal Mohan*
Department of Pharmaceutics, SRM College of Pharmacy, SRM Institute of Science and Technology, Kattankulathur-603203, Chengalpattu, Tamil Nadu, India. E-mail: mothipharma78@gmail.com
First published on 22nd April 2025
Abstract
Hyaluronic acid-coated capecitabine-loaded nanomicelles (HA-CAP-M) are synthesized to overcome the challenges associated with capecitabine (CAP) conventional delivery such as low permeability and systemic toxicity. Nanomicelles containing saponin, glycerol, and vitamin-E TPGS formulation of capecitabine were further encapsulated with hyaluronic acid (HA) for CD44 receptor-mediated targeting. Optimization of the formulation was carried out using a Box–Behnken design resulting in 17.8 nm particle size, 89.3% entrapment efficiency and a biphasic drug release profile. Characterization studies validated stability, spherical structure, and desirable encapsulation characteristics of the nanomicelles. Lowered critical micelle concentration (CMC) and acceptable drug release kinetics revealed improved thermodynamic stability and controlled drug release, as predicted by the Hixson–Crowell model. HA-CAP-M showed much higher permeability and cytotoxicity than the free CAP, with an IC50 of 2.964 μg mL−1 in in vitro experiments. AO/PI staining also demonstrated dose-dependent apoptosis in MCF-7 breast cancer cells and validated the highly effective active targeting of HA. In addition, the formulation demonstrated good stability during storage and dilution conditions, confirming its stability as a drug delivery platform. In conclusion, HA-functionalized nanomicelles provide a biocompatible and efficient system for the targeted breast cancer therapy, enhancing the therapeutic efficacy of capecitabine.
Introduction
Cancer is a condition characterised by unregulated/uncontrolled proliferation of cells with the failure to undergo programmed cell death, resulting in the destruction of adjacent tissues.1 Breast cancer (BC) was the most common cancer in women globally in 2022, as per the Global Cancer Statistics Report by WHO, 2024.2 Female BC ranked second in the most common cancer types among 36 cancers in 186 countries, with about 2.3 million cases covering 11.6% of the total with 670![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fatalities globally in the same year.3 Incidences of breast cancer rose by 130% from 2008 to 2020, increasing from 1.38 million new cases to 1.67 million in 2012, 2.1 million in 2018, and 2.3 million in 2020.4
000 fatalities globally in the same year.3 Incidences of breast cancer rose by 130% from 2008 to 2020, increasing from 1.38 million new cases to 1.67 million in 2012, 2.1 million in 2018, and 2.3 million in 2020.4
Despite advancements in imaging technology and therapeutic methods, BC continues to result in numerous fatalities.5 BC impacts both the quality of life and the survival probability of patients. BC patients have access to various treatment modalities, including medication, chemotherapy, and surgical interventions, based on the specific type and location of the cancer. Conventional drug therapies exhibit limitations due to the inefficiencies in anti-cancer drug delivery. Nanomedicines utilising nanocarriers present promising therapeutic agents for breast cancer, addressing the limitations of potential drugs.6,7
Nanomicelles are small colloidal structures that consist of molecules with two distinct regions: a core and a shell, which possess different affinities for water.8 Poorly water-soluble drugs are encapsulated in the hydrophobic core of nanomicelles, protected by a hydrophilic corona that stabilizes the micelles and minimizes recognition by the reticuloendothelial system (RES) in vivo.9 This unique structure enables nanomicelles to enhance drug solubility and bioavailability, protect drugs from degradation, and facilitate transport across biological barriers.10 Their ability to accumulate in diseased tissues via the enhanced permeability and retention (EPR) effect allows targeted drug delivery with reduced systemic toxicity, making them valuable for therapeutic and diagnostic applications.11
Nanomicelles offer thermodynamic and kinetic stability due to the intra-micellar polymer chain entanglement.12 Thermodynamic stability arises when nanomicelles exceed their critical micelle concentration (CMC), typically low (10−6 to 10−7 M), ensuring stability even after dilution. Kinetic stability allows nanomicelles to retain drugs under unpredictable release conditions, with slow disintegration below the CMC, preserving drug content until delivery to the target site, thus enhancing bioavailability.13
Nanomicelles improve treatment efficacy by enhancing drug selectivity and specificity, minimizing adverse effects, and reducing exposure to non-target tissues.14,15 Their enhanced permeability and retention (EPR) effect increases drug concentration in tumours, overcoming drug resistance.
Compared to liposomes, nanoparticles, and nanotubes, nanomicelles exhibit superior performance in cancer therapy due to their nanoscale size, high drug-loading capacity, stability, sustained release, and ability to penetrate tumour tissues effectively.16
Capecitabine (CAP) is an oral prodrug of Fluorouracil that is used to treat various cancers, including breast, colorectal, and gastric cancers.17 CAP is a BCS Class-III drug with high solubility but limited permeability and is rapidly absorbed from the gastrointestinal tract. With a short half-life (0–2 h) and a high daily dosage (2.5 g m−2), it requires a sustained-release formulation to prolong antitumour activity and reduce toxicity.18 However, its poor permeability limits efficient transport to target tissues.19
Nanocarriers improve the efficacy of nanomedicines for breast cancer treatment by mitigating adverse effects and delivering precise drug doses to primary and metastatic tumours, including the vasculature, stroma, cancer cells, and immune cells.20 To date, only ten nanomedicines have been approved for clinical treatment of breast cancer,21 while many others are under trial, including active and non-targeting formulations. However, active-targeting nanomedicines have yet to achieve the anticipated outcomes.22 Drugs in nanocarriers are more stable, circulate longer, and accumulate at specific tumour sites due to the EPR effect.23 Nanomicelles can passively accumulate in tumour tissues; however, most of the drug is released before receptor-mediated endocytosis.24 Recently developed drug delivery techniques target cancer cell surface receptors, presenting a viable alternative to passive targeting.25
Cluster determinant 44 (CD44) is a cell surface glycoprotein receptor highly expressed in tumours such as lung, pancreatic, and breast cancers, particularly known in promoting breast cancer metastasis to the liver.26 Targeting CD44 for anticancer therapy offers a promising approach. Hyaluronic acid (HA) is a natural and biocompatible polysaccharide that selectively binds to CD44, enabling intracellular drug delivery to cancer cells. HA-based systems are known for their biocompatibility, biodegradability, and non-toxicity, and are ideal platforms for chemotherapy.27
The HA-CD44 interaction plays a critical role in cancer proliferation, migration, and growth. HA-coated nanocarriers with robust core–shell structures enable precise drug delivery to tumour sites, minimizing premature drug release into the bloodstream. Thus, many investigations have analysed HA-coated nanocarriers for targeting CD44 over-expressing cancer cells.28–31
Moreover, biocompatible natural surfactants offer significant advantages in micelle drug delivery. Saponins are natural biosurfactants found in plants like Gypsophila, Quillaja, Saponaria, and Glycyrrhiza species that enhance the solubility and stability of hydrophobic drugs.32 As a primary surfactant, saponin promotes micelle formation and improves encapsulation efficiency.33,34 Glycerol acts as a co-surfactant to stabilize micelles by reducing interfacial tension, increasing stability, and enhancing drug loading.35 The combination of saponin and glycerol makes them ideal for developing micelles for targeted breast cancer therapy. In addition, vitamin-E TPGS stabilizes micelles by enhancing integrity and drug solubility, supporting efficient delivery in cancer therapy.36,37
A novel ligand-coated, receptor-driven nanosystem was designed to encapsulate capecitabine for improved breast cancer treatment. The system uses a saponin micelle loaded with CAP and coated with HA to target tumours via HA/CD44 receptor binding. The study used a design of experiments (DoE) approach to optimize formulation and processing parameters, evaluating their effects on particle size, entrapment efficiency, and drug release. The nanomicelle structure is expected to accumulate at tumour sites through EPR and be internalized by CD44-overexpressing breast cancer cells. This delivery system aims to induce apoptosis and suppress metastasis.
Materials
Chemicals
Capecitabine (CAS Number: 154361-50-9) was obtained from Dr Reddy's Laboratories (Hyderabad, India) as a gift sample. Saponin (ex. Gypsophila roots 25% sapogenin; CAS Number: 8047-15-2) and glycerol (CAS Number: 56-81-5) were purchased from Sisco Research Laboratories (SRL) Pvt. Ltd (Maharashtra, India). Vitamin-E TPGS (CAS Number: 9002-96-4) was received as a gift sample from Matrix Life Science Pvt. Ltd (Aurangabad, India). Hyaluronic acid (800 kDa) (CAS Number: 9004-61-9) was purchased from BFC Lab Pvt. Ltd (Bangalore, India). If not specified, distilled filtered water was used and all other organic solvents or reagents were of analytic grade and used as received.Cell culture
Human breast cancer cell line MCF-7 was purchased from the National Centre for Cell Science (NCCS), Pune, India. Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), and 1% penicillin–streptomycin was used to culture MCF-7 breast cancer cells. All cells were incubated at 37 °C in a humidified atmosphere with 5% CO2. Trypsin–EDTA, trypan blue, phosphate-buffered saline (PBS), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were used.Methods
Experimental design
Initially, preliminary experiments (one factor at a time approach) were performed to determine the main factors and the appropriate ranges in which the optima lie. Through preliminary screening, the most significant variables were identified, i.e., the effects of concentrations of surfactant (saponin) and co-surfactant (glycerol), and the sonication time as independent variables on the particle size (nm), entrapment efficiency (%) and in vitro drug release were tested after 4 h. Based on the preliminary trials, a 3-factor, 3-level Box–Behnken design (BBD) by a response surface methodology (RSM) was applied through the Design-Expert v12.0. Software (Stat-Ease Inc., Minnesota, U.S.A) to study the effect of each independent variable on the dependent variables. For each factor, natural values corresponding to the coded levels of −1, 0, and +1 were selected to cover a range of values of practical interest based on the results of preliminary experiments conducted to assess their effect on the responses (Table S1†). A 33 BBD requires 15 runs with a minimum of three central points to determine the experimental error and precision of the design using RSM. This design is suitable for exploring quadratic response surfaces and constructing second-order polynomial models. It consists of replicated centre points and the set of points lying at the midpoint of each edge of the multidimensional cube that defines the region of interest. The 15 runs were carried out for 3 factors, which were conducted randomly to minimize the effects of uncontrolled factors. Besides, the percentage error was calculated between the observed and expected data. Eventually, the optimized formulation was selected for the next steps.Preparation of capecitabine-loaded nanomicelles
The preparation of capecitabine-loaded nanomicelles (CAP-M) was done using the direct dissolution method.38 It is the simplest and most feasible method for preparing micelles and is especially appropriate for water-soluble molecules.39 This process involves rapid solubilisation of the surfactant and co-surfactant in the aqueous medium followed by steady sonication. Saponin ex. Gypsophila roots, glycerol, vitamin-E TPGS, and distilled water were all used for the preparation of nanomicelles. Fifteen formulations of CAP-M were prepared as suggested by the DoE software and were subjected to further characterization. The procedure involved in the preparation of CAP-loaded micelles is illustrated in Fig. 1. Following a preliminary trial, a 33 BBD with 15 formulations was suggested by the Design Expert software (Table 1). | ||
| Fig. 1 Schematic of the structure of CAP-M and HA-CAP-M nanomicelles and their method of preparation for the CD44 receptor-mediated endocytosis. | ||
| Run | Levels of independent variables | ||
|---|---|---|---|
| A: saponin (mmol) | B: glycerol (mmol) | C: sonication time (minutes) | |
| 1 | +1 | 0 | +1 |
| 2 | 0 | +1 | −1 |
| 3 | 0 | −1 | −1 |
| 4 | 0 | 0 | 0 |
| 5 | +1 | +1 | 0 |
| 6 | +1 | 0 | −1 |
| 7 | +1 | −1 | 0 |
| 8 | 0 | +1 | +1 |
| 9 | −1 | 0 | −1 |
| 10 | −1 | 0 | +1 |
| 11 | 0 | 0 | 0 |
| 12 | −1 | −1 | 0 |
| 13 | 0 | −1 | +1 |
| 14 | 0 | 0 | 0 |
| 15 | −1 | +1 | 0 |
Preparation and coating of hyaluronic acid on capecitabine-loaded micelles
The optimized CAP-M formulation was coated with hyaluronic acid (HA-CAP-M) by adding 10 mL of 0.1% (w/v) HA solution with stirring at room temperature for 1 h.Characterization of capecitabine-loaded micelles
where γsample (N m−1) and γwater (N m−1) denote the surface tension of the surfactant solution and water (at 25 °C), respectively, while msample (g) and mwater (g) represent the masses of the surfactant solution and that of water. CMC was computed from a plot of concentration vs. surface tension. The same approach was used to determine micelle CMC and compare it to the surfactant. A comparison of this CMC value demonstrates the efficacy and stability of micelles, which is essential for the development of a targeted drug delivery system.43
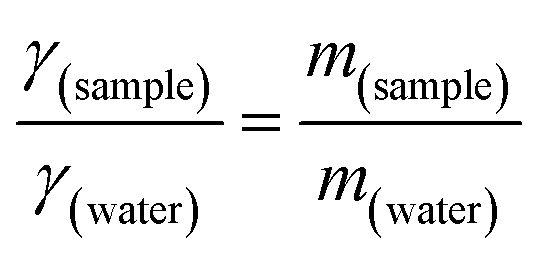 000 rpm for 1 h at 4 °C using a refrigerated centrifuge (REMI C-24, Maharashtra, India). The clear supernatant was carefully collected, diluted appropriately with phosphate buffer (pH 7.4), and analysed using a UV spectrophotometer (V-730 Double Beam, Jasco, Inc., Japan) at 304 nm in PBS pH 7.4.000 to 14000 Da; Dialysis Membrane-60, Himedia, Maharashtra, India), continuously agitated at 50 rpm and maintained at 37 °C in a 50 mL PBS solution (T = 37, pH = 7.4). Samples were withdrawn at 0, 1, 2, 4, 6, 8, 12, and 24 h and the amount of drug present was quantified using UV spectroscopy. The procedure was performed in triplicate. To analyse the drug release profile and mechanism, the release data were fitted to various linear kinetic models, including zero-order-, first-order-, Higuchi-, Hixson–Crowell-, and Korsmeyer–Peppas models.
000 rpm for 1 h at 4 °C using a refrigerated centrifuge (REMI C-24, Maharashtra, India). The clear supernatant was carefully collected, diluted appropriately with phosphate buffer (pH 7.4), and analysed using a UV spectrophotometer (V-730 Double Beam, Jasco, Inc., Japan) at 304 nm in PBS pH 7.4.000 to 14000 Da; Dialysis Membrane-60, Himedia, Maharashtra, India), continuously agitated at 50 rpm and maintained at 37 °C in a 50 mL PBS solution (T = 37, pH = 7.4). Samples were withdrawn at 0, 1, 2, 4, 6, 8, 12, and 24 h and the amount of drug present was quantified using UV spectroscopy. The procedure was performed in triplicate. To analyse the drug release profile and mechanism, the release data were fitted to various linear kinetic models, including zero-order-, first-order-, Higuchi-, Hixson–Crowell-, and Korsmeyer–Peppas models.Stability studies
Results
Pre-formulation studies
 | ||
| Fig. 2 (A) UV/vis spectrum and (B) calibration curve of capecitabine in PBS pH 7.4. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (ester and amide) at 1641.42 cm−1, aromatic CC stretching at 1454.33 cm−1, and C–F vibrations at 1078.21 cm−1. These significant peaks are preserved in the spectra of the formulations with different excipients, indicating that no chemical interaction or incompatibility occurred between CAP and the excipients. The slight variations in intensity are attributed to physical effects such as dilution or blending rather than chemical modification.
O stretching (ester and amide) at 1641.42 cm−1, aromatic CC stretching at 1454.33 cm−1, and C–F vibrations at 1078.21 cm−1. These significant peaks are preserved in the spectra of the formulations with different excipients, indicating that no chemical interaction or incompatibility occurred between CAP and the excipients. The slight variations in intensity are attributed to physical effects such as dilution or blending rather than chemical modification.
 | ||
| Fig. 3 Stacked FTIR spectra of capecitabine and excipients (HA, saponin, glycerol and vit-E TPGS). | ||
The absence of new peaks, disappearance of critical functional group peaks, or significant shifts confirms the compatibility between CAP and the tested excipients. Each formulation retains the structural integrity of CAP, as evidenced by the consistent presence of its key functional groups. This suggests that the excipients do not chemically react with CAP, supporting the stability of the drug in these formulations.
Characterization of capecitabine-loaded micelle formulation
 | ||
| Fig. 4 FTIR spectra of capecitabine, CAP-M and HA-CAP-M. | ||
The preservation of capecitabine's structural peaks in the micelle formulations confirms the stability and integrity of the drug. The formulations encapsulate the drug without inducing any chemical interaction, as evidenced by the consistent FTIR profiles. This suggests that the micelles are effective in maintaining the drug's chemical nature and stability during formulation. Fig. S1–S7 and Table S2† show the results and interpretation of the FTIR analysis.
 | ||
| Fig. 5 Tyndall effect exhibited by CAP-M and HA-CAP-M. | ||
 | ||
| Fig. 6 CMC of saponin, glycerol and CAP-M. | ||
Upon incorporating CAP into the micellar system, an approximately 2.5 decrease in the CMC was observed compared with the CMC of saponin. Comparable findings in other studies56,57 highlight the combined effect of the components on drug encapsulation and micelle formation. The drug–surfactant interactions likely enhance the thermodynamic stability of the micelles, facilitating their formation at much lower surfactant concentrations. The marked reduction in the CMC of the CAP-M demonstrates its superior surface activity and underscores the enhanced stability and compactness of the micelles. These characteristics are particularly advantageous in pharmaceutical formulations as they ensure efficient micellization and drug loading at reduced surfactant concentrations.58
| Run | Particle size (nm) | Entrapment efficiency (%) | Drug release after 4 h (%) |
|---|---|---|---|
| a Values are presented as means ± SD (n = 3). | |||
| 1 | 19.47 ± 1.43 | 89.3 ± 2.65 | 65.82 ± 4.53 |
| 2 | 25.89 ± 1.03 | 62.11 ± 3.22 | 60.36 ± 3.22 |
| 3 | 29.42 ± 0.53 | 56.2 ± 1.89 | 61.08 ± 2.64 |
| 4 | 17.81 ± 1.54 | 75.01 ± 4.53 | 66.52 ± 2.07 |
| 5 | 14.92 ± 2.03 | 87.97 ± 2.43 | 67.24 ± 3.04 |
| 6 | 23.82 ± 0.04 | 88.43 ± 2.11 | 64.07 ± 2.53 |
| 7 | 21.94 ± 0.42 | 85.24 ± 1.53 | 64.75 ± 1.53 |
| 8 | 22.69 ± 1.52 | 63.29 ± 1.11 | 62.48 ± 4.24 |
| 9 | 44.52 ± 1.68 | 28.19 ± 2.59 | 61.74 ± 2.67 |
| 10 | 35.41 ± 2.53 | 29.02 ± 2.31 | 58.94 ± 2.54 |
| 11 | 17.27 ± 2.11 | 76.92 ± 1.96 | 66.69 ± 3.25 |
| 12 | 42.08 ± 1.80 | 23.75 ± 1.04 | 55.45 ± 4.62 |
| 13 | 24.28 ± 0.98 | 55.45 ± 1.54 | 61.2 ± 2.34 |
| 14 | 17.35 ± 1.04 | 76.73 ± 2.45 | 66.87 ± 3.24 |
| 15 | 36.82 ± 1.66 | 32.93 ± 1.42 | 60.34 ± 2.89 |
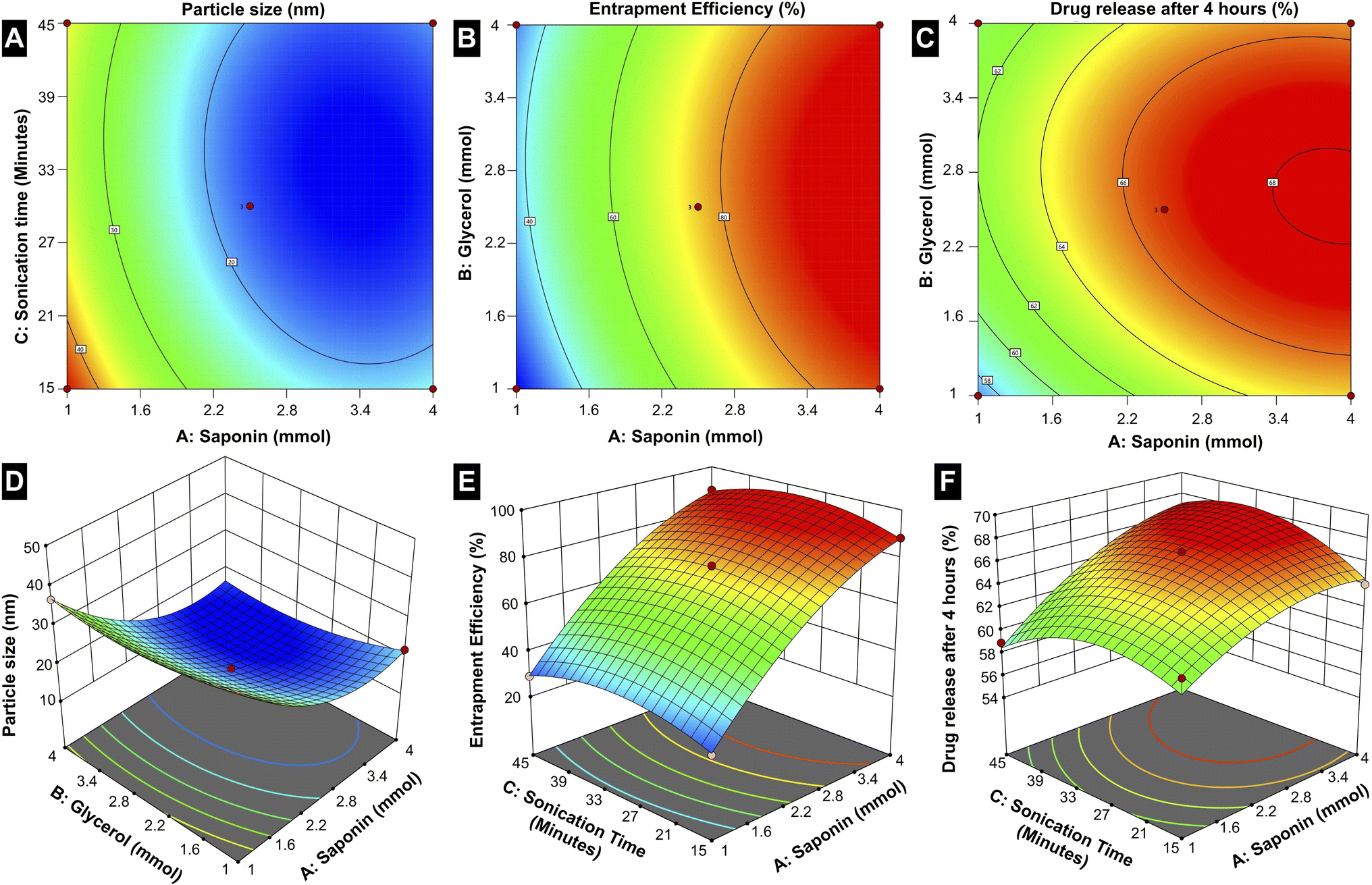 | ||
| Fig. 7 Box–Behnken design for (A and D) particle size, (B and E) entrapment efficiency and (C and F) in vitro drug release after 4 h as a function of the parameters (surfactant and co-surfactant concentration, and sonication time). | ||
The particle size is notably dependent on all three factors, with the contour and 3D surface plots demonstrating a reduction in size at optimized combinations of A, B, and C. This indicates that precise adjustments in the formulation components and processing parameters are essential to achieving smaller, more uniform micelles, which are critical for effective drug delivery. Entrapment efficiency is primarily governed by A and B, where increasing the saponin concentration and optimizing the glycerol concentration yield higher drug encapsulation. This suggests that the amphiphilic nature of saponin plays a crucial role in creating a stable micellar structure capable of efficiently entrapping the drug, while the co-surfactant glycerol facilitates the stabilization of the micelle. Interestingly, sonication time (C) has less influence on the EE, suggesting that encapsulation is mainly determined by the surfactant-co-surfactant interactions rather than the mechanical energy imparted during sonication.
The drug release behaviour is distinctly influenced by A and C; the higher concentration of saponin changes the drug release rates, which is because of a favourable influence on solubilizing the drug in the micelles, while optimized sonication time allows the proper formation of the micelles and stability. Thus, the interplay of these conditions and formulation components outlines the complexities encountered when achieving drug release control. While saponin concentration (A) exerts the most pervasive influence on all three responses among the three factors, glycerol concentration (B) mostly affects particle size and EE. On the other hand, sonication time (C) is crucial for optimizing particle size and drug release, underscoring its role in the physical structuring and functional performance of the micelles.
The statistical evaluation further supports these findings, with R-squared and adjusted R-squared values closely aligning, indicating a reliable model capable of accurately predicting the responses within the design space (Table S5†). The adequate precision values that measure the signal-to-noise ratio exceed the favourable threshold of 4 for all responses, confirming the robustness of the model. Further, 2.5 mmol of surfactant, 2.5 mmol of co-surfactant, and a sonication time of 30 minutes yields optimum formulations (CAP-M) in terms of particle size, EE, and CDR after 4 h. The values of optimized responses generated from the RSM method and their experimental data under optimal conditions are given in Table S6.†
As seen in the plots, increasing saponin concentration (A) from 1 to 4 mmol initially reduces particle size and then slightly increases it beyond the optimized level. Entrapment efficiency (EE%) follows a similar trend, peaking at intermediate concentrations, while drug release consistently increases with higher saponin levels. Glycerol concentration (B) stabilizes particle size and EE at optimized levels but plateaus at higher values. Meanwhile, sonication time (C) will significantly reduce the particle size with longer durations and facilitate drug release by enhancing micelle stability and uniformity. Moreover, trends relating to sonication and micelle performance emphasize the position of formulation variables. These results describe the need for an optimized formulation, in which a careful balance between the factors is essential to provide the desired responses. Such an in-depth understanding of factor-response relationships forms a sound basis for the rational design of micellar drug delivery systems.
 | ||
| Fig. 8 Graphical presentation of the size distribution of plain micelles, CAP-M and HA-CAP-M (mean ± SD (n = 3)). | ||
CAP-M micelles exhibit an average zeta potential of about −15.2 mV (Fig. 9), indicating moderate stability, while the HA-CAP-M micelles have a more negative average zeta potential of −26.8 mV, suggesting enhanced stability due to stronger electrostatic repulsion. The shift in zeta potential reflects surface modifications induced by HA coating. The particle size range aligns with reported ranges, supported by literature attributing them to the core–shell structure and HA coating of the micelles.60,61
 | ||
| Fig. 9 Graphical presentation of the zeta potential distribution of CAP-M and HA-CAP-M (mean ± SD (n = 3)). | ||
 | ||
| Fig. 10 TEM images of (A) CAP-M and (B) HA-CAP-M. | ||
Additionally, TEM results highlighted the spherical shape and uniformity of the micelles. The results were consistent with the findings of TEM analysis in previously reported micelle formulations.62 The findings emphasize that HA coating significantly increases the size of the micelles while maintaining their spherical structure and uniform distribution.
 | ||
| Fig. 11 In vitro drug release profile of the optimized CAP-M (n = 3). | ||
Release kinetics were analysed by fitting the formulations to various mathematical models. Regression coefficient values are summarized in Table 3. The highest value of the coefficient of determination (R2 = 0.9179) was given by the Hixson–Crowell model, indicating that this model was the most suitable to describe the drug release because it is predominantly controlled by surface area and/or particle size change of the micelles. The Korsmeyer–Peppas model yields an R2 value of 0.8655, with release exponent (n = 0.49) indicating that drug release is primarily governed by anomalous diffusion, which incorporates both diffusion and erosion. Thus, it seems that the actual release mechanism is complex and involves a combination of diffusion and surface erosion processes as characterized by the Hixson–Crowell and anomalous diffusion models.
| Model | Equation | R2 |
|---|---|---|
| Zero-order | Ct = C0 − K0·t | 0.5907 |
| First order | lnCt/C0 = −k1·t |
0.8857 |
| Higuchi | Mt/M∞ = Ktn | 0.8534 |
| Hixson–Crowell | W01/3 − Wt1/3 = kt·t | 0.9179 |
| Korsmeyer–Peppas | C = KH·√t | 0.8655 (n = 0.49) |
These findings are consistent with previous studies. For instance, the Hixson–Crowell model describes drug release from systems where there is a change in surface area and diameter of particles or tablets, which aligns with the observed release behaviour in micellar systems.63 Similarly, the Korsmeyer–Peppas model has been utilized to study drug release from liposomes, indicating that drug release is primarily governed by anomalous diffusion, which incorporates diffusion and erosion mechanisms.64 Therefore, the observed biphasic release pattern and the applicability of these models to describe the release kinetics are well-supported by existing literature.
 | ||
| Fig. 12 In vitro permeation profile of capecitabine and CAP-M formulation. Values are presented as means ± SD (n = 3). | ||
| Formulation | Jss or flux (μg cm−2 h−1) | Kp (cm h−1) |
|---|---|---|
| a Values are presented as means ± SD (n = 3). | ||
| Pure drug capecitabine | 0.075 ± 0.015 | 0.0100 ± 0.0020 |
| Optimized Cap-M | 0.391 ± 0.017 | 0.0521 ± 0.0023 |
The in vitro permeation profile of the optimized CAP-M formulation, comprising saponin and glycerol micelles, demonstrated a significant enhancement in CAP permeation compared to the pure drug CAP. This finding aligns with previous research indicating that saponin-based micelles can effectively increase the solubility and permeability of poorly soluble drugs. For instance, a study utilizing saponin micelles for budesonide delivery reported improved mucosal permeation and a faster onset of action, highlighting the potential of saponin micelles in enhancing drug absorption.65 This improvement can be attributed to the ability of micelles to solubilize hydrophobic drugs and modify the barrier properties of biological membranes, thereby enhancing drug transport.
Stability studies
 | ||
| Fig. 13 Storage stability of CAP-M and HA-CAP-M stored at 4 °C and 25 °C for 2 months. (A) Particle size (nm). (B) PDI. (C) EE (%). Values are presented as means ± SD (n = 3). | ||
| Formulation | Particle size (nm) | ||
|---|---|---|---|
| Dilution volume 5 mL | Dilution volume 10 mL | Dilution volume 100 mL | |
| a Values are presented as means ± SD (n = 3). | |||
| CAP-M | 18.01 ± 0.32 | 22.15 ± 0.96 | 25.32 ± 1.42 |
| HA-CAP-M | 208.73 ± 0.63 | 212.67 ± 0.55 | 214.11 ± 0.05 |
 | ||
| Fig. 14 Cytotoxicity of CAP, CAP-M and HA-CAP-M against the MCF-7 cell line by MTT assays. IC50 values and % cell viability of (A and D) capecitabine, (B and E) CAP-M and (C and F) HA-CAP-M. | ||
Free CAP exhibited an IC50 value of 5.929 μg mL−1, reflecting its potent anti-cancer activity. However, this also highlights its limitations, including poor permeability and limited intracellular uptake. The CAP-M micellar formulation demonstrated improved cytotoxicity with an IC50 value of 4.209 μg mL−1, indicating its ability to enhance drug solubility and stability. This improvement is attributed to the micelles' capacity to facilitate intracellular delivery via passive targeting mechanisms, such as the EPR effect. The most significant enhancement in cytotoxicity was observed with the HA-CAP-M formulation, which displayed an IC50 of 2.964 μg mL−1, the lowest among the three formulations. The superior efficacy of HA-CAP-M can be attributed to the active targeting properties provided by the HA coating. HA selectively binds to CD44 receptors, which are overexpressed on breast cancer cells, enabling receptor-mediated endocytosis and improving intracellular drug accumulation. This targeted delivery combines the benefits of micelle-mediated solubilization and stabilization with active receptor targeting.
HA-conjugated nanocarriers can improve drug accumulation in tumour cells while reducing off-target effects, as demonstrated in breast cancer models.66,67 Additionally, micelle-based formulations enhance the solubility and stability of poorly soluble drugs and exploit the EPR effect for passive tumour targeting.68 The observed reduction in IC50 values for HA-functionalized micelles aligns with previous studies, confirming the synergistic benefits of combining passive micellar delivery with active receptor targeting for effective and selective cancer therapy. Overall, CAP-HA-M exhibited the most improved cytotoxicity compared to CAP and CAP-M, combining enhanced solubility, passive EPR targeting, and active CD44 receptor-mediated targeting. These findings underscore the potential of HA-functionalized micelles as a promising strategy for targeted breast cancer therapy, offering a more effective and selective delivery system for capecitabine.
Fig. 15 shows the AO/PI staining fluorescence micrographs of MCF-7 cells. The control group (Fig. 15A) showed predominantly green fluorescence, indicating healthy, viable cells with intact membranes. A slight increase in orange/red fluorescence was observed at 6.25 μg mL−1 (Fig. 15B), suggesting limited apoptosis or necrosis. As the concentration increased to 12.5 and 25 μg mL−1 (Fig. 15C and D), a higher proportion of apoptotic and necrotic cells was evident, with a noticeable decline in green fluorescence. Red fluorescence dominated at the highest concentration (50 μg mL−1, Fig. 15E), indicating extensive cell death through late apoptosis and necrosis.
 | ||
| Fig. 15 Fluorescence micrographs of MCF-7 cells double-stained with AO/PI. Cells were treated with (A) control and (B–E) HA-CAP-M at 6.25, 12.5, 25, and 50 μg mL−1, respectively. | ||
These findings highlight the potent, dose-dependent cytotoxicity of HA-CAP-M, likely due to its ability to target CD44 receptors on MCF-7 cells, thereby enhancing drug delivery and inducing cell death effectively. The literature strongly supports the findings of dose-dependent cytotoxicity observed with HA-CAP-M against MCF-7 cells using AO/PI staining. Hyaluronic acid-functionalized drug delivery systems have been well-documented to effectively target CD44 receptors, which are overexpressed on breast cancer cells, enhancing intracellular drug delivery via receptor-mediated endocytosis.69
The differential staining observed in AO/PI assays is a widely recognized method for distinguishing live-, apoptotic-, and necrotic cells, with studies reporting similar fluorescence patterns in dose-dependent cytotoxicity assessments for HA-conjugated formulations.70 Furthermore, the increased induction of apoptosis and necrosis at higher concentrations aligns with previous findings demonstrating that HA-functionalized micelles enhance therapeutic efficacy by promoting targeted delivery and inducing significant cancer cell death.71,72 These results reinforce the potential of HA-CAP-M as an effective, receptor-targeted therapeutic for breast cancer treatment.
Discussion
This study focuses on the development and evaluation of hyaluronic acid-coated capecitabine-loaded nanomicelles for CD44 receptor-mediated breast cancer therapy. capecitabine is extensively used for treating numerous cancers including breast cancer. The intrinsic limitations of CAP, such as low permeability, short half-life, and high dosage requirements, necessitate developing a novel delivery system. To overcome these limitations, the current study aims to increase the therapeutic effectiveness and bioavailability of CAP by utilizing nanotechnology and active targeting mechanisms. Nanomicelles have been extensively reported as effective drug carriers, offering enhanced solubility, stability, and tumour-targeted delivery via the EPR effect.73 The study employed a design of experiments (DoE) approach using a Box–Behnken design to optimize the formulation parameters. The micelles were prepared using saponin as the primary surfactant, glycerol as the co-surfactant, and vitamin-E TPGS as the stabilizing agent. Hyaluronic acid, a natural polysaccharide known for its affinity to CD44 receptors, was used to coat the optimized micelles, enabling active targeting of breast cancer cells. The use of HA as a targeting ligand is well-documented for its specific binding to CD44 receptors, which are overexpressed in many cancer cells, promoting receptor-mediated endocytosis and increasing intracellular drug accumulation. HA-coated nanocarriers improve therapeutic efficacy, reduce toxicity, and enhance drug bioavailability.74The key findings of this study are the successful development of HA-CAP-M. Through DoE, the independent variables (concentration of saponin and glycerol, and sonication time) were optimized to obtain the desired particle size, entrapment efficiency (EE), and drug release profiles. The optimized formulation exhibited an average particle size of 17.8 nm, an EE of 89.3%, and a drug release of 67.24% after 4 h. ATR-FTIR confirmed the absence of chemical interactions between CAP and the excipients, indicating formulation stability, while the Tyndall effect confirmed the successful formation of nanomicelles. TEM analysis indicated spherical and uniformly dispersed micelles with HA-coated micelles having a larger size as a result of the HA layer. It is critical to clarify that the increase in hydrodynamic diameter of HA-coated micelles (HA-CAP-M) is primarily due to the hydration shell and surface coating of high molecular weight hyaluronic acid (HA), not an actual shift in core structural size into the microscale domain as measured by DLS. Furthermore, although the IUPAC Recommendations 2011 provide particular classifications separating “nano” from “micro” depending on particle size, such recommendations are precisely meant for polymer-based dispersion systems. In contrast, the micelles developed in this work are based on non-polymeric surfactants and co-surfactants, so they are beyond the direct scope of polymer colloidal nomenclature. Moreover, in line with current usage in nanomedicine literature,75–77 formulations displaying nanoscale core architecture and functional behaviour (such as targeted receptor-mediated delivery) are generally referred to as “nanomicelles”, or “nanostructures”, even if their hydrodynamic size slightly exceeds 100 nm due to ligand corona. Thus, the use of the word “nanomicelles” for CAP-M and HA-CAP-M is in line with the accepted scientific standard and adequately captures the intended usage and formulation design. The critical micelle concentration (CMC) of CAP-loaded micelles was markedly decreased from that of the surfactants, suggesting increased thermodynamic stability and efficient micelle formation with lower surfactant concentrations. Drug release studies confirmed a biphasic release profile, including a rapid release phase and a sustained release phase. The Hixson–Crowell model fits the release kinetics best, indicating surface area and particle size changes as governing factors. HA-CAP-M presented a 5-fold increase in flux (0.391 μg cm−2 h−1) compared to that of CAP (0.075 μg cm−2 h−1) and a permeability coefficient (0.0521 cm h−1) with an increase in permeability compared to free CAP (0.01 cm h−1), reflecting improved permeability. The formulation presented high stability during storage and dilution conditions and HA-CAP-M presented higher stability than uncoated micelles. MTT assay data showed dose-dependent cytotoxicity, with the HA-CAP-M resulting in the lowest IC50 value (2.964 μg mL−1) as a result of CD44 receptor-mediated endocytosis compared with pure drug CAP (5.929 μg mL−1). These results clearly show that the ligand-coated carrier improved the cytotoxicity potential by effective targeting of cancer cells. In addition, AO/PI staining showed that MCF-7 cells treated with HA-CAP-M exhibited significant apoptosis, further supporting its potent anticancer activity and CD44-targeting efficiency.
The hydrodynamic diameter of HA-coated micelles (HA-CAP-M) was approximately 205 nm, as determined by dynamic light scattering (DLS). Transmission electron microscopy (TEM) analysis revealed a core size distribution ranging from 180 to 360 nm, primarily due to the hydration shell formed around the high molecular weight (800 kDa) hyaluronic acid, despite the fact that this size may appear larger than typical nanocarriers. The negative zeta potential value (−26.8 mV), low polydispersity index, and consistent results from dilution stability experiments help to further explain this increase in size by hydration effects instead of aggregation or formulation instability. Notably, recent research has demonstrated that hyaluronic acid-coated nanostructures with sizes ranging from 150 to 250 nm can still achieve enhanced tumour accumulation through the enhanced permeability and retention (EPR) effect, while also enabling active targeting through CD44 receptor-mediated uptake.78,79 Moreover, the HA-CAP-M formulation showed effective cytotoxic ability and cellular intake, as indicated by the low IC50 value (∼2.96 μg mL−1) and unambiguous proof of dead cells seen in AO/PI dual staining experiments. These results show that the slightly larger particle size has no negative effect on biological performance. Preliminary optimisation steps include modification of HA concentration and coating kinetics and lowering the average particle size to ∼160–170 nm without compromising target effectiveness or colloidal stability.
Compared to previous studies, the development of HA-CAP-M in this study demonstrates enhanced drug delivery and therapeutic efficacy through active targeting of CD44 receptors. This strategy improves tumour specificity and reduces systemic toxicity.80 Our results are consistent with previous studies on cancer cells in the literature. For instance, studies by Liu et al. (2017) and Dartora et al. (2020) also highlight the use of HA-coated nanocarriers for targeted cancer therapy, showing improved therapeutic outcomes and reduced side effects compared to conventional drug formulations.81,82 Additionally, the findings of the current study showing improved drug release profiles, enhanced permeability, and increased cytotoxicity support the growing body of evidence that receptor-mediated drug delivery via HA-functionalized carriers offers substantial advantages over non-targeted therapies83 These results are consistent with the literature, where HA-coated formulations increase cellular uptake and enhance anticancer activity, demonstrating the potential of HA-based nanocarriers for efficient cancer treatment.
Conclusion
This study successfully engineered and optimized HA-coated capecitabine-encapsulated nanomicelles as a novel targeted breast cancer therapy platform. The nanomicelles showed enhanced encapsulation efficiency, stability, and release profile with controlled release. The HA coating facilitated active targeting properties through the specific binding of CD44 receptors, thereby improving the therapeutic effects and selectivity of capecitabine. The capacity of the formulation to circumvent the limitations of traditional CAP delivery (e.g., low permeability and systemic toxicity) was demonstrated. The synergic effect of saponin and glycerol for the formation of such a stable micelle structure with a high loading capacity of the drug was the key. Active targeting using HA enabled higher intracellular delivery and decreased off-target effects. The ability of HA-CAP-M to cause apoptosis was apparent, as shown through in vitro cytotoxicity and AO/PI staining. The results highlight the feasibility of HA-functionalized nanomicelles as a stable, biocompatible, and effective cancer drug delivery system for breast cancer. Future directions should encompass in vivo experiments to confirm the therapeutic efficacy, determine the pharmacokinetics and biodistribution of HA-CAP-M, and scale up the production of the formulation for clinical use, which can serve as an alternative to conventional chemotherapy to improve patient outcomes and quality of life. By integrating nanotechnology and receptor-mediated targeting, this study provides a significant step forward in advancing personalized and precision medicine for cancer therapy.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Sruthi Laakshmi Mugundhan: writing – original draft, review and editing, conceptualization, data curation, and methodology. Mothilal Mohan: writing – review and editing, validation, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely thank the management of SRM College of Pharmacy and the SRM Institute of Science and Technology, Kattankulathur, Tamil Nadu, for providing all the necessary facilities and support to carry out this research work.References
- B. K. Chagaleti, V. Saravanan, C. Vellapandian and M. K. Kathiravan, RSC Adv., 2023, 13, 33770–33785 RSC.
- Breast Cancer WHO, 2024, Global Cancer statistics Report by WHO, https://www.who.int/news-room/fact-sheets/detail/breast-cancer, (accessed 12 November 2024) Search PubMed.
- IARC-WHO, https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services, (accessed 12 November 2024).
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- R. A. Leon-Ferre and M. P. Goetz, BMJ, 2023, 381, e071674 CrossRef CAS PubMed.
- D. Wu, M. Si, H. Y. Xue and H. L. Wong, Int. J. Nanomed., 2017, 12, 5879–5892 CrossRef CAS PubMed.
- Y. Jiang, Z. Jiang, M. Wang and L. Ma, Adv. Drug Delivery Rev., 2022, 180, 114034 CrossRef CAS PubMed.
- H. M. Trinh, M. Joseph, K. Cholkar, R. Mitra and A. K. Mitra, Emerging Nanotechnologies for Diagnostics, Drug Delivery and Medical Devices, 2017, pp. 45–58 Search PubMed.
- S. M. Tawfik, S. Azizov, M. R. Elmasry, M. Sharipov and Y. I. Lee, Nanomaterials, 2021, 11, 70 CrossRef CAS PubMed.
- S. Khatun, S. Bonala, S. V. Pogu and A. K. Rengan, Functional Biomaterials: Drug Delivery and Biomedical Applications, 2022, pp. 1–36 Search PubMed.
- H. Maeda, J. Drug Targeting, 2017, 25, 781–785 CrossRef CAS PubMed.
- A. Bose, D. R. Burman, B. Sikdar and P. Patra, IET Nanobiotechnol., 2021, 15, 19–27 CrossRef PubMed.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11, 4985–4998 CrossRef PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9(1), 1–12 CrossRef CAS PubMed.
- P. M. Giri, A. Banerjee and B. Layek, Cancers, 2023, 15, 2256 CrossRef CAS PubMed.
- S. L. Mugundhan and M. Mohan, RSC Adv., 2024, 14, 14017–14040 RSC.
- M. Malet-Martino and R. Martino, Oncologist, 2002, 7, 288–323 CrossRef CAS PubMed.
- G. V. Koukourakis, V. Kouloulias, M. J. Koukourakis, G. A. Zacharias, H. Zabatis and J. Kouvaris, Molecules, 2008, 13, 1897–1922 CrossRef CAS.
- L. S. L. Janardhanam, A. S. Deokar, S. R. Bollareddy and V. V. K. Venuganti, AAPS PharmSciTech, 2022, 23, 1–13 CrossRef.
- M. Chehelgerdi, M. Chehelgerdi, O. Q. B. Allela, R. D. C. Pecho, N. Jayasankar, D. P. Rao, T. Thamaraikani, M. Vasanthan, P. Viktor, N. Lakshmaiya, M. J. Saadh, A. Amajd, M. A. Abo-Zaid, R. Y. Castillo-Acobo, A. H. Ismail, A. H. Amin and R. Akhavan-Sigari, Mol. Cancer, 2023, 22, 1–103 Search PubMed.
- B. Wang, S. Hu, Y. Teng, J. Chen, H. Wang, Y. Xu, K. Wang, J. Xu, Y. Cheng and X. Gao, Signal Transduction Targeted Ther., 2024, 9(1), 1–65 CrossRef.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- E. Blanco and M. Ferrari, Breast, 2014, 23, 10–18 CrossRef PubMed.
- S. K. S. S. Pindiprolu, P. T. Krishnamurthy, P. K. Chintamaneni and V. V. S. R. Karri, Artif. Cells, Nanomed., Biotechnol., 2017, 46, 885–898 CrossRef.
- L. Hong, W. Li, Y. Li and S. Yin, RSC Adv., 2023, 13(31), 21365–21382 RSC.
- E. Chiesa, A. Greco, F. Riva, R. Dorati, B. Conti, T. Modena and I. Genta, Pharmaceuticals, 2022, 15(1), 103 CrossRef CAS.
- R. J. Babu, M. Annaji, R. D. Arnold and A. Alsaqr, Polymeric Nanoparticles as a Promising Tool for Anti-cancer Therapeutics, 2019, 319–341 Search PubMed.
- C. Yang, Y. He, H. Zhang, Y. Liu, W. Wang, Y. Du, F. Gao, C. Yang, Y. He, H. Zhang, Y. Liu, W. Wang, Y. Du and F. Gao, Oncotarget, 2015, 6, 15283–15296 CrossRef.
- G. Nabil, R. Alzhrani, H. O. Alsaab, M. Atef, S. Sau, A. K. Iyer and H. El Banna, Cancers, 2021, 13, 898 CrossRef CAS PubMed.
- Z. Hussain, A. H. Akbari, S. H. Barbuor, D. S. Dawood Alshetiwi, I. S. Ahmed and M. Rawas-Qalaji, J. Drug Delivery Sci. Technol., 2024, 101, 106183 CrossRef CAS.
- A. Tirella, K. Kloc-Muniak, L. Good, J. Ridden, M. Ashford, S. Puri and N. Tirelli, Int. J. Pharm., 2019, 561, 114–123 CrossRef CAS PubMed.
- I. M. Tucker, A. Burley, R. E. Petkova, S. L. Hosking, J. P. Webster, P. X. Li, K. Ma, J. Doutch, J. Penfold and R. K. Thomas, Colloids Surf., A, 2022, 633, 127854 CrossRef CAS.
- Y. Liao, Z. Li, Q. Zhou, M. Sheng, Q. Qu, Y. Shi, J. Yang, L. Lv, X. Dai and X. Shi, Int. J. Pharm., 2021, 603, 120709 CrossRef CAS.
- B. Kołodziej, Ł. Sęczyk, D. Sugier, B. Kędzia, M. Chernetskyy, R. Gevrenova and M. Henry, Ind. Crops Prod., 2019, 138, 111422 CrossRef.
- N. J. Kale and L. V. Allen, Int. J. Pharm., 1989, 57, 87–93 CrossRef CAS.
- Y. Guo, J. Luo, S. Tan, B. O. Otieno and Z. Zhang, Eur. J. Pharm. Sci., 2013, 49, 175–186 CrossRef CAS PubMed.
- C. Yang, T. Wu, Y. Qi and Z. Zhang, Theranostics, 2018, 8, 464 CrossRef CAS PubMed.
- B. Hu, F. Pei, X. Sun, Y. Liang, Z. He, L. Zhang and J. Li, New J. Chem., 2018, 42, 19600–19607 RSC.
- X. Bu, N. Ji, L. Dai, X. Dong, M. Chen, L. Xiong and Q. Sun, Trends Food Sci. Technol., 2021, 114, 386–398 CrossRef CAS.
- R. shaikh, S. Bhattacharya and S. D. Saoji, Heliyon, 2024, 10, e39632 CrossRef CAS PubMed.
- H. P. Nijhawan, P. Shyamsundar, B. Prabhakar and K. S. Yadav, AAPS PharmSciTech, 2024, 25, 1–16 CrossRef PubMed.
- G. You, T. Feng, G. Zhang, M. Chen, F. Liu, L. Sun, M. Wang and X. Ren, Int. J. Pharm., 2021, 601, 120546 CrossRef CAS PubMed.
- E. İspir, M. İnal, Z. Gün Gök and M. Yiğitoğlu, Polym. Bull., 2024, 81, 6801–6822 CrossRef.
- A. A. Aljohani, M. A. Alanazi, L. A. Munahhi, J. D. Hamroon, Y. Mortagi, M. Qushawy and G. M. Soliman, OpenNano, 2023, 11, 100145 CrossRef CAS.
- E. Haladjova, M. Kyulavska, J. Doumanov, T. Topouzova-Hristova and P. Petrov, Colloid Polym. Sci., 2017, 295, 2197–2205 CrossRef CAS.
- H. M. Aboud, S. F. El Menshawe, N. H. Mohammed, A. S. Tulbah and A. A. Ali, Pharmaceuticals, 2024, 17, 1275 CrossRef CAS PubMed.
- A. Mansoori-Kermani, S. Khalighi, I. Akbarzadeh, F. R. Niavol, H. Motasadizadeh, A. Mahdieh, V. Jahed, M. Abdinezhad, N. Rahbariasr, M. Hosseini, N. Ahmadkhani, B. Panahi, Y. Fatahi, M. Mozafari, A. P. Kumar and E. Mostafavi, Mater. Today Bio, 2022, 16, 100349 CrossRef CAS PubMed.
- A. Simon, M. I. Amaro, A. M. Healy, L. M. Cabral and V. P. de Sousa, Int. J. Pharm., 2016, 512, 234–241 CrossRef CAS PubMed.
- S. Mordon, A. B. Eldeen Yassin, S. Massadeh, A. A. Alshwaimi, R. H. Kittaneh, M. E. Omer, D. Ahmad, A. Hassan Aodah, F. Shakeel, M. Halwani, S. A. Alanazi and P. Alam, Pharmaceuticals, 2023, 2024, 19 Search PubMed.
- O. Inal, G. Amasya, Z. Sezgin Bayindir and N. Yuksel, Int. J. Biol. Macromol., 2023, 241, 124651 CrossRef CAS PubMed.
- N. M. Gaikwad, P. D. Chaudhari, K. S. Shaikh, S. Y. Chaudhari, S. S. Pathare, A. A. Shaikh, N. H. Aljarba, A. Kumer and B. Dhara, J. Cell. Mol. Med., 2024, 28, e18389 CrossRef CAS PubMed.
- G. Singh, D. Singh, M. Choudhari, S. D. Kaur, S. K. Dubey, S. Arora and N. Bedi, J. Pharm. Invest., 2021, 51, 701–714 CrossRef CAS.
- P. S. Chary, A. Bansode, N. Rajana, V. Bhavana, S. Singothu, A. Sharma, S. K. Guru, V. Bhandari and N. K. Mehra, Int. J. Pharm., 2024, 657, 124109 CrossRef CAS PubMed.
- H. Ameli and N. Alizadeh, RSC Adv., 2022, 12, 4681–4691 RSC.
- P. V. Ramana, Theor. Chem. Acc., 2023, 142, 1–20 Search PubMed.
- R. G. Alargova, I. I. Kochijashky, M. L. Sierra, K. Kwetkat and R. Zana, J. Colloid Interface Sci., 2001, 235, 119–129 CrossRef CAS PubMed.
- X. Cui, S. Mao, M. Liu, H. Yuan and Y. Du, Langmuir, 2008, 24, 10771–10775 CrossRef CAS PubMed.
- R. Wang, H. Wang, Y. Yao and Y. Chai, RSC Adv., 2018, 8, 36879–36885 RSC.
- A. M. S. Villar, B. C. Naveros, A. C. C. Campmany, M. A. Trenchs, C. B. Rocabert and L. H. Bellowa, Int. J. Pharm., 2012, 431, 161–175 CrossRef CAS PubMed.
- W. Xu, H. Wang, L. Dong, P. Zhang, Y. Mu, X. Cui, J. Zhou, M. Huo and T. Yin, Int. J. Nanomed., 2019, 14, 4649–4666 CrossRef CAS PubMed.
- K. Suzuki, Y. Yoshizaki, K. Horii, N. Murase, A. Kuzuya and Y. Ohya, Biomater. Sci., 2022, 10, 1920–1928 RSC.
- T. Q. M. Tran, M. F. Hsieh, K. L. Chang, Q. H. Pho, V. C. Nguyen, C. Y. Cheng and C. M. Huang, Polymers, 2016, 8, 321 CrossRef.
- S. Liu, L. Li, X. Zhang and Q. Meng, Smart Mater. Med., 2023, 4, 183–198 Search PubMed.
- I. Y. Wu, S. Bala, N. Škalko-Basnet and M. P. di Cagno, Eur. J. Pharm. Sci., 2019, 138(1), 105026 CrossRef CAS PubMed.
- S. Nakowitsch, C. Koller, J. M. Seifert, M. König-Schuster, N. Unger-Manhart, C. Siegl, N. Kirchoff, E. Foglar, C. Graf, M. Morokutti-Kurz, M. Neurath, S. Sladek, C. Knecht, W. Sipos, E. Prieschl-Grassauer and A. Grassauer, Pharmaceutics, 2020, 12, 847 CrossRef CAS PubMed.
- C. Xu, Y. Ding, J. Ni, L. Yin, J. Zhou and J. Yao, RSC Adv., 2016, 6, 27542–27556 RSC.
- Q. Xiong, M. Cui, Y. Bai, Y. Liu, D. Liu and T. Song, Smart Mater. Med., 2017, 155, 93–103 CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS PubMed.
- X. Wei, T. H. Senanayake, G. Warren and S. V. Vinogradov, Bioconjugate Chem., 2013, 24, 658–668 CrossRef CAS PubMed.
- S. Kari, K. Subramanian, I. A. Altomonte, A. Murugesan, O. Yli-Harja and M. Kandhavelu, Apoptosis, 2022, 27(7), 482–508 CrossRef PubMed.
- N. B. Naeeni, M. H. Tabrizi, E. Karimi and H. Ghafaripour, Polym. Bull., 2024, 81, 2671–2683 CrossRef CAS.
- V. R. Ranjitha and V. Ravishankar Rai, Bionanoscience, 2021, 11, 371–379 CrossRef.
- V. P. Torchilin, Pharm. Res., 2006, 24(1), 1–16 CrossRef PubMed.
- Z. J. Deng, S. W. Morton, E. Ben-Akiva, E. C. Dreaden, K. E. Shopsowitz and P. T. Hammond, ACS Nano, 2013, 7, 9571–9584 CrossRef CAS PubMed.
- D. Xin, Y. Wang and J. Xiang, Pharm. Res., 2010, 27, 380–389 CrossRef CAS PubMed.
- C. Xu, W. He, Y. Lv, C. Qin, L. Shen and L. Yin, Int. J. Pharm., 2015, 493, 172–181 CrossRef CAS PubMed.
- J. Shi, Y. Ren, J. Ma, X. Luo, J. Li, Y. Wu, H. Gu, C. Fu, Z. Cao and J. Zhang, J. Nanobiotechnol., 2021, 19, 1–22 CrossRef PubMed.
- V. F. C. Dartora, G. C. Salata, J. S. Passos, P. C. Branco, E. Silveira, A. A. Steiner, L. V. Costa-Lotufo and L. B. Lopes, Int. J. Biol. Macromol., 2022, 219, 84–95 CrossRef CAS PubMed.
- J. Zhang, M. Deng, C. Xu, D. Li, X. Yan, Y. Gu, M. Zhong, H. Gao, Y. Liu, J. Zhang, X. Qu and J. Zhang, ACS Appl. Mater. Interfaces, 2024, 16(38), 50459–50473 CrossRef CAS PubMed.
- C. Hepokur, İ. A. Kariper, S. Mısır, E. Ay, S. Tunoğlu, M. S. Ersez, Ü. Zeybek, S. E. Kuruca and İ. Yaylım, Toxicol. in Vitro, 2019, 61, 104600 CrossRef CAS PubMed.
- V. F. C. Dartora, G. C. Salata, J. S. Passos, P. C. Branco, E. Silveira, A. A. Steiner, L. V. Costa-Lotufo and L. B. Lopes, Int. J. Biol. Macromol., 2022, 219, 84–95 CrossRef CAS PubMed.
- K. Liu, Z. Q. Wang, S. J. Wang, P. Liu, Y. H. Qin, Y. Ma, X. C. Li and Z. J. Huo, Int. J. Nanomed., 2015, 10, 6445–6454 CAS.
- J. Li, M. Li, L. Tian, Y. Qiu, Q. Yu, X. Wang, R. Guo and Q. He, Int. J. Pharm., 2020, 578, 119122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01275a |
| This journal is © The Royal Society of Chemistry 2025 |