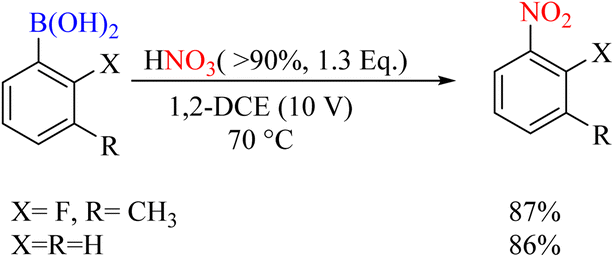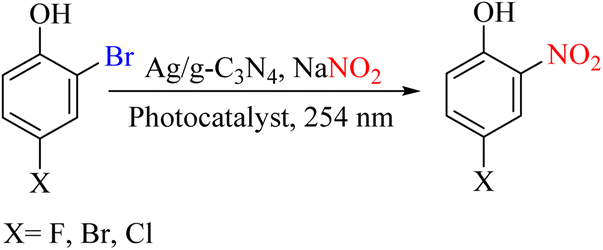DOI:
10.1039/D5RA01261A
(Review Article)
RSC Adv., 2025,
15, 23499-23558
Ipso nitration in organic synthesis
Received
20th February 2025
, Accepted 30th May 2025
First published on 8th July 2025
Abstract
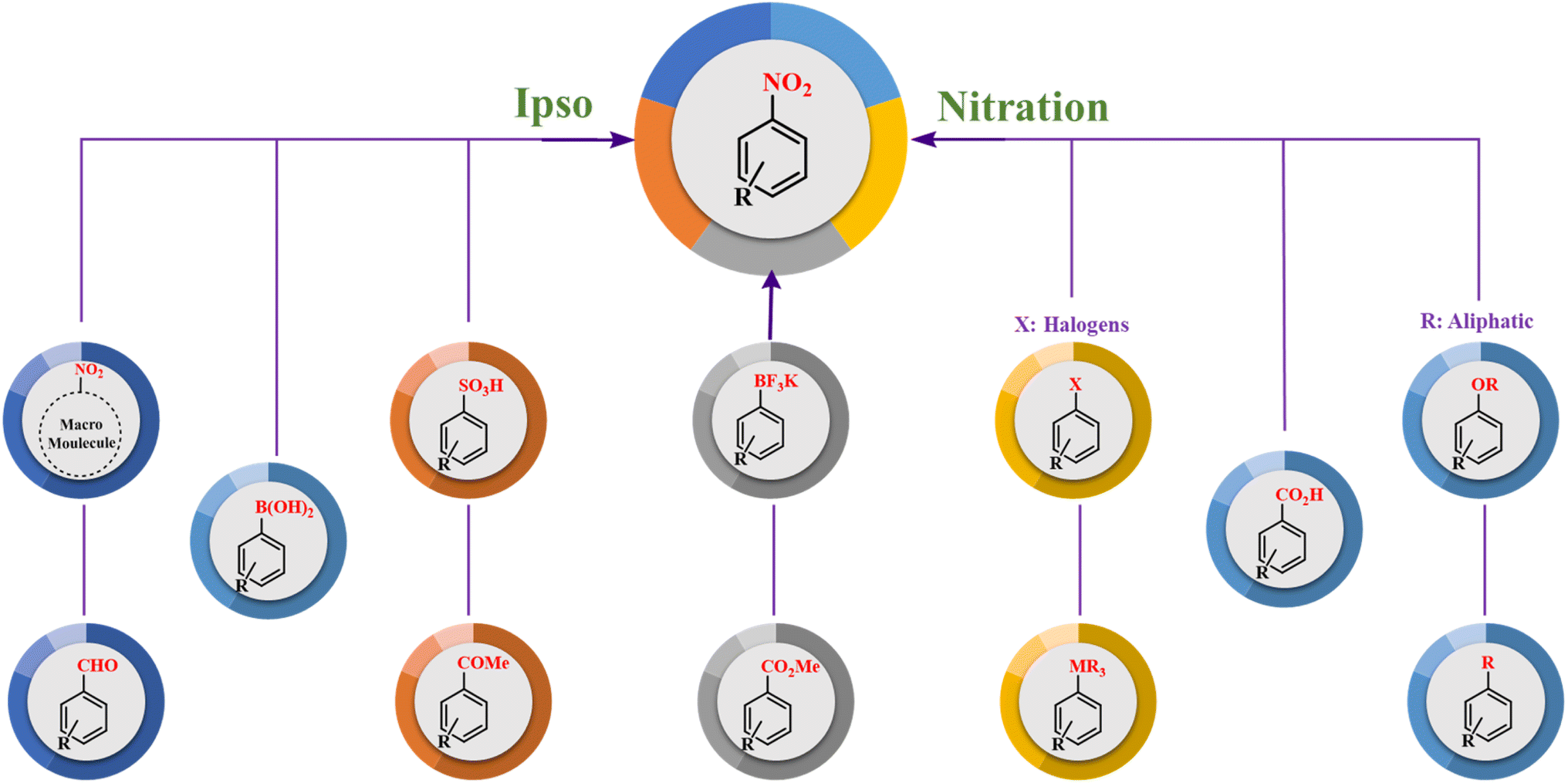
Ipso reaction is a class of nitration reactions that involve the concerted movement of multiple atoms in a transition state. Nitro structures are vital intermediates in synthetic organic chemistry and related industries. This method has been introduced in the preparation of NO2-containing structures with a wide range of leaving groups such as –B(OH)2, –CO2H, –SO3H, –OR, –R, –X, –CHO, –COMe, –CO2Me, –MR3 and –BF3K. Ipso nitration reactions have an essential impact on the synthesis of complex organic molecules and have been widely studied and utilized in organic chemistry. This review provides a comprehensive overview of the fundamental concepts and mechanisms underlying ipso reactions, highlighting their significance in contemporary organic chemistry.
 Hassan Sepehrmansourie | Hassan Sepehrmansourie was born in Hamedan, Iran, in 1994. He completed his BSc in Applied Chemistry (2017) from Bu-Ali Sina University, Iran, and his M.Sc. in Organic Chemistry (2019) under the supervision of Prof. Mohammad Ali Zolfigol. In addition, he was accepted for a PhD in organic chemistry at the Bu-Ali Sina University in the same year. He graduated with a PhD course in organic chemistry from Bu-Ali Sina University, Iran (2023). His research interests include the synthesis, characterization and applications of homogeneous and heterogeneous reagents and catalysts in organic synthesis. In recent years, he has also been involved in photocatalytic activities. He is currently an assistant professor of organic chemistry at the University of Qom, Iran. |
 Mahmoud Zarei | Mahmoud Zarei was born in Hamedan, Iran, in 1986. He completed his BSc in Pure Chemistry (2010) and M.Sc. in Organic Chemistry (2013). Additionally, he received his PhD in Organic Chemistry (2017) under the supervision of Prof. Mohammad Ali Zolfigol. His research interests include the synthesis, characterization and applications of homogeneous and heterogeneous reagents and catalysts in organic synthesis. He has been studying nanotechnology and nanoscience in chemistry. His current research interests include metal nanoparticles, metal–organic framework (MOF) and organic synthesis. He is currently an assistant professor of organic chemistry at the University of Qom, Iran. |
 Professor Shadpour Mallakpour | Professor Shadpour Mallakpour, an organic polymer chemist, graduated from the Chemistry Department of the University of Florida (UF), Gainesville, Florida, U.S.A. in 1984. He spent two years as a post-doc at UF. He joined the Department of Chemistry at Isfahan University of Technology (IUT), Iran, in 1986. He held several positions such as chairman of the Department of Chemistry and deputy of research in the Department of Chemistry at IUT. He worked as a visiting professor at the University of Mainz, Germany, from 1994 to 1995 as well as a visiting professor at Virginia Tech, Blacksburg, USA, from 2003 to 2004. He has published more than 890 journal papers and book chapters and more than 440 conference papers and received more than 40 items of awards. The most important award given to him was the selection of the first laureate of fundamental research at the 21st Khwarizmi International Award in 2008. He has been listed among the top 1% of scientists in chemistry in the ISI Essential Science Indicators since 2003. He was selected as an academic guest of the 59th Meeting of Nobel Prize Winners in Chemistry, 2009, at Lindau, Germany. He presented many lectures as an invited or keynote speaker at different national and international conferences or universities. He has served as a member of organizing and scientific committees for many national and international conferences. He was also the chairperson of many national and international meetings. In recent years, he has focused on the preparation and characterization of polymers containing chiral amino acid moieties under green conditions using ionic liquids and microwave irradiation as new technology, bringing these aspects towards nanotechnology for the preparation of novel chiral bionanocomposite polymers as well as polymer nanocomposites to develop hazardous material removal technologies https://scholar.google.com/citations?hl=en%26user=aZ-gAxgAAAAJ%26view_op=list_works. |
 Farbod Tabesh | Farbod Tabesh received his PhD in Organic Polymer Chemistry in 2019, with his research focusing on developing polysaccharide-based hydrogel nanocomposites for water pollutant removal. Farbod is a researcher at the Stanford University School of Medicine, supported by a prestigious NIH fellowship, with research interests at the intersection of nanotechnology, molecular imaging, and gene therapy. He specializes in targeted gene delivery using polymeric nanocomposites, including CRISPR-Cas9 systems, for applications in cancer treatment, immunotherapy, and development of anticancer vaccines. Farbod is also advancing innovative approaches for brain-targeted therapeutic delivery, such as using nanocomposites for intranasal administration in neurodegenerative diseases such as Parkinson's disease. With expertise in ultrasound imaging for molecular diagnostics and experience in creating microbubble systems for endothelial PD-L1 imaging, Farbod combines cutting-edge material science with translational biomedical applications to address critical challenges in precision medicine. |
 Mohammad Ali Zolfigol | Mohammad Ali Zolfigol was born in 1966 in Salehabad (Ashtian) and completed his BSc from Arak University, his M.Sc. from Isfahan University of Technology, with Prof. Shadpour Mallakpour as the supervisor, and his PhD from Shiraz University under the supervision of Prof. Nasser Iranpoor. He has been a faculty member at Bu-Ali Sina University since 1997, and he was promoted to Professor in 2005. His name is listed among the top 1% ESI scientists since 2004. He won the 21st International Khwarizmi Festival and the COMSTEC awards in 2008. He has been the General Secretary of the Chemical Society of Iran (2007–2021), president of the Bu-Ali Sina University (2008–2014) and Minister of Science, Research and Technology (2021–2024). Zolfigol's research involves the development of catalysts, basic concepts and new methodologies in organic chemistry. |
1. Introduction: ipso-nitration reaction (C–N bond formation)
Nitro compounds play important roles in organic chemistry, e.g., in the preparation of chemical intermediates in the perfume industry, dyes, plastics, pharmaceuticals, and energetic materials.1–5 The electrophilic nitration of organic molecules is a well-studied organic reaction involving the use of HNO3/H2SO4 as a classical electrophilic nitration reaction under harsh conditions. This method faces the challenge of regioselectivity, producing a mixture of isomers alongside oxidation by-products. Moreover, several reported methodologies suffer from numerous drawbacks, such as low yields, extended reaction duration, excessive nitration, high catalyst loading, poor regioselectivity, reagent oxidation, and safety concerns. In pursuing regioselective nitration, research endeavors have uncovered the ipso and Hunsdiecker reactions as promising alternatives.6–12 Olah and coworkers reported the first ipso-nitration from aryl boronic acids using Crivello's reagent (ammonium nitrate/trifluoro acetic anhydride).13
The ipso-nitration method involves the introduction of a nitro functional group into a compound using a nitrating agent. This process entails replacing a good leaving group (L.G) with the compound's nitro group. However, if the reagent is ineffective or there is no suitable leaving group in the compound, adverse reactions may occur, and ipso nitration may not occur as intended. Many groups, such as –B(OH)2, –CO2H, –SO3H, –OR, –R, –X, –CHO, –COMe, –CO2Me, –MR3, and –BF3K, can be replaced by a nitro group (Scheme 1). The high efficiency, general application, and broader range of substrates, including heterocycles and functional groups, make this method useful.14–20 Optimal selection is one of the most important aspects of nitration reaction because the reaction compounds can lead to different products, some of which are pollutants and others are just a waste of capital.21–23 Therefore, it is preferable to design the reactions in the best way in advance, and the ipso-nitration replacement reaction is one of the best methods. Performing ipso-nitration substitution reactions requires appropriate reagents. The most important reagents for conducting the nitration reaction are NaNO2, HNO3/H2SO4, and nitrite salt.24–28 Dinitrogen tetroxide and its derivatives are also used in various nitration reactions.29
 |
| | Scheme 1 Most important leaving groups in the ipso–nitration reaction. | |
Yan and Yang comprehensively reviewed the synthesis of aromatic nitro via the nitration of aromatic hydrocarbons, aryl boronic acids, aryl halides and pseudohalides, aryl carboxylic acids, and aryl primary amines and azides.9 Recently, various types of ipso-nitration reactions, particularly advances in ipso-nitration reactions that have been reported since 2000–2015, have been comprehensively discussed.14 Continuing our experiences related to nitration reaction,5,30–38 we reviewed the ipso nitration of organic compounds. This study addresses the issue of purpose with greater insight.
2. Decarboxylative ipso-nitration [ipso-CO2H]
Carboxylic acids (R-COOH) are abundant in nature, commercially available, exhibit high structural diversity, are easy to work with and maintain, and can be synthesized using readily available methods. The decarboxylative group plays an important role in various syntheses.39 One of the most significant reactions involving carboxylic acid groups is the ipso-nitration reaction, which is facilitated by the excellent leaving-group property of the –COOH moiety.40,41 Recently, the decarboxylation–nitration reaction has attracted much attention because it opens a new path for forming the carbon–nitrogen bond. Many important reagents and methods have been introduced to perform this reaction.
In 1987, Peterson et al. conducted an electrophilic reaction to suppress ipso-nitration in cinnamic acid compounds. They used cerium(IV) ammonium nitrate (CAN) as the nitro source. Cinnamic acid compounds are known as unsaturated carboxylic acid compounds. This report proposes an electrophile-based mechanism for the internal nitration of cinnamic acids (Scheme 2).42
 |
| | Scheme 2 Ipso-nitration reaction of cinnamic acids using cerium(IV) ammonium nitrate (CAN) as a nitro source. | |
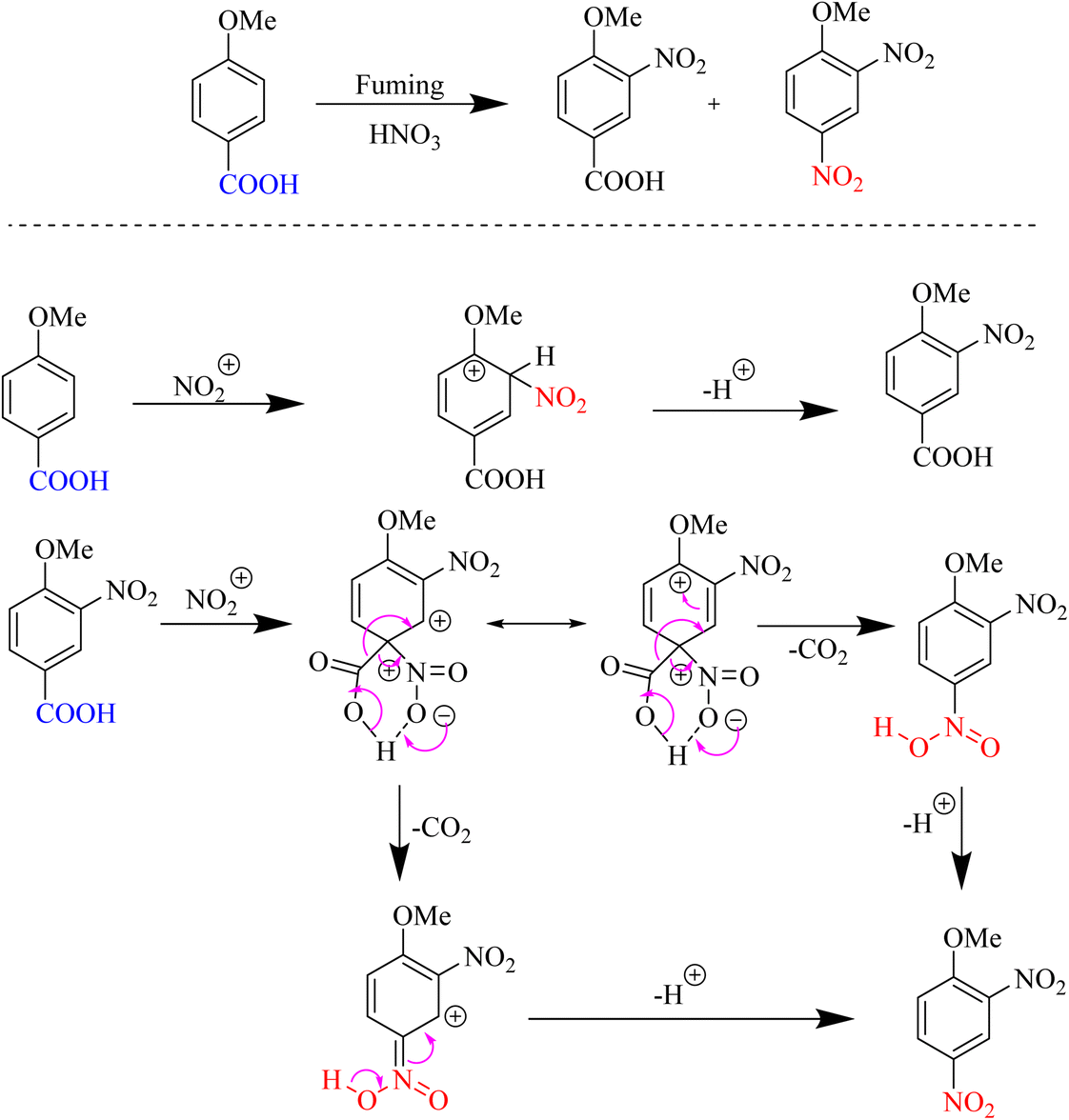
In 1993, the decarboxylation nitration of aromatic carboxylic acids was introduced using a clay catalyst and HNO3 as a nitro source. The nitration of a p-anisic acid compound produces 2,4-dinitroanisole with a better yield than conventional nitration, 3-nitro-4-methoxybenzoic acid and the efficiency and selectivity of natural montmorillonite clays, as well as cation exchange in the ipso-nitration reaction, are highlighted in the nitration of aromatic carboxylic acids (Scheme 3).43
 |
| | Scheme 3 Ipso-nitration reaction of aromatic acids using clay as a catalyst and HNO3 as a nitro source. | |
Another report described the decarboxylation nitration of unsaturated aromatic carboxylic acids and ring-activated benzoic acids using nitric acid. In this report, α,α′-azoisobutyronitrile (AIBN) was used to generate the radical required for the reaction. Due to the radical nature of the reaction pathway, the ipso-nitration reaction described in this paper is called the nitro–Hunsdiecker reaction (Scheme 4).44
 |
| | Scheme 4 Nitro–Hunsdiecker reaction using HNO3/AIBN. | |
Internal replacement of a vinyl carboxyl group with a nitro group by ipso-nitration is one of the main goals of scientists. In a 2004 study, the abovementioned reaction was performed using diluted nitric acid and acetic acid under microwave irradiation. The desired nitroproducts were obtained along with a minor amount of another crystalline compound—technique (Scheme 5).45
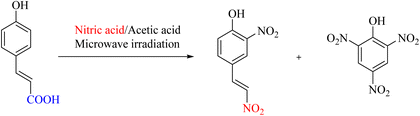 |
| | Scheme 5 Microwave-assisted synthesis of an unusual dinitro phytochemical. | |
In 2004, the Fiorentino research team reported the nitration of cinnamic acids using CAN. In this method, nitration and ipso-nitration reactions were conducted by introducing the substrate onto the silica substrate. The ipso reaction mechanism occurs when the α-position of the cinnamic acid compounds is electron-rich (Scheme 6).46
 |
| | Scheme 6 Nitration of cinnamic acids using cerium(IV) ammonium nitrate immobilized on silica. | |
Another study on the ipso-nitration reaction using cerium nitrate reagent reported an efficient method for synthesizing conjugated nitro-olefins from unsaturated alpha and beta acids under mild conditions. This method uses CAN at room temperature in acetonitrile, yielding moderate to well-described yields (Scheme 7).47
 |
| | Scheme 7 Efficient synthesis of conjugated nitro-olefins using cerium(IV) ammonium nitrate. | |
Another report proposed a new experimental method for chemical reactions: mixing and freezing reagents to sub-zero temperatures such as −30 °C. After freezing, the mixture was briefly exposed to microwave radiation. In this report, the ipso-nitration reaction was performed under these conditions, and the unsaturated (E)-3-(4-hydroxyphenyl) acrylic was substituted with a nitro group (Scheme 8).48
 |
| | Scheme 8 Cold microwave chemistry: synthesis using pre-cooled reagents. | |
Performing organic reactions under solvent-free conditions can provide favorable conditions. Performing an ipso-nitration reaction in the absence of a solvent can be a dream. In 2007, Rajanna et al. performed the ipso Hunsdiecker reaction under solvent-free conditions using metal nitrate salts on unsaturated carboxylic acids. The results obtained in this methodology show mild reaction conditions with high yields of the desired products (Scheme 9).49
 |
| | Scheme 9 Metal nitrate-driven nitro–Hunsdiecker reaction with unsaturated carboxylic acids under solvent-free conditions. | |
The importance of CAN was demonstrated in organic reactions in 2008. It is one of the most widely used oxidants in organic chemistry. It serves as a mediator in forming carbon–heteroatom bonds and initiates the radical addition of functional compounds with activated C–N bonds. In this report, this oxidant, along with sodium nitrate salt, was employed to introduce a new approach to the ipso-nitration reaction of unsaturated acids (Scheme 10).50
 |
| | Scheme 10 Solid-phase oxidative decarboxylation of unsaturated acids using a ceric ammonium nitrate-alkali halide system. | |
The synthesis of pure medicinal compounds is one of the main goals of scientists. Changing the functional group can be very useful for synthesizing drug compounds to perform the reaction and obtain the main product. One of these changes involves replacing the nitro group with the acid group. In a previous report, different reagents were used to synthesize 5,6,7-trioxygenated dihydroflavonol derivatives in different stages. In one of these steps, the ipso-nitration reaction was performed using a nitric/acetic acid reagent at room temperature. This report illustrates the importance of ipso reactions and factor functional group change well (Scheme 11).51
 |
| | Scheme 11 Preparation of 5,6,7-trioxygenated dihydroflavonol derivatives as free radical scavengers and neuronal cell protectors to oxidative damage. | |
Tert-butyl nitrite (TBN) is a nitrating agent that acts under acid-free conditions. Due to the absence of acid usage, this method is safe and offers preferential ipso-nitration reactions of cinnamic acids with excellent performance under classical conditions. In 2013, a study reported using TBN for the nitration of unsaturated acids using three methods: ultrasonic, conventional, and microwave. The application of ultrasonics and microwaves notably reduced reaction times and enhanced efficiency from good to excellent, as stated in our previous report (Scheme 12).52
 |
| | Scheme 12 Ultrasonic and microwave-assisted synthesis of β-nitro styrenes with tert-butyl nitrite (TBN) under acid-free conditions. | |
In 2013, the Prabhu research group reported a new, gentle, and convenient method for the nitrocarboxylation of cinnamic acid derivatives substituted for their nitroolefins using TBN as a nitrating agent and a catalytic amount of CuCl. In this method, different aromatic derivatives were nitrated (Scheme 13). The presence of CuCl significantly altered the reaction mechanism (Scheme 14). This offers a valuable approach for synthesizing nitroolefins, which have traditionally been more challenging to obtain using conventional methods. Another noteworthy aspect of this method is that, unlike other approaches, it does not require metal nitrates or HNO3 for nitration.53
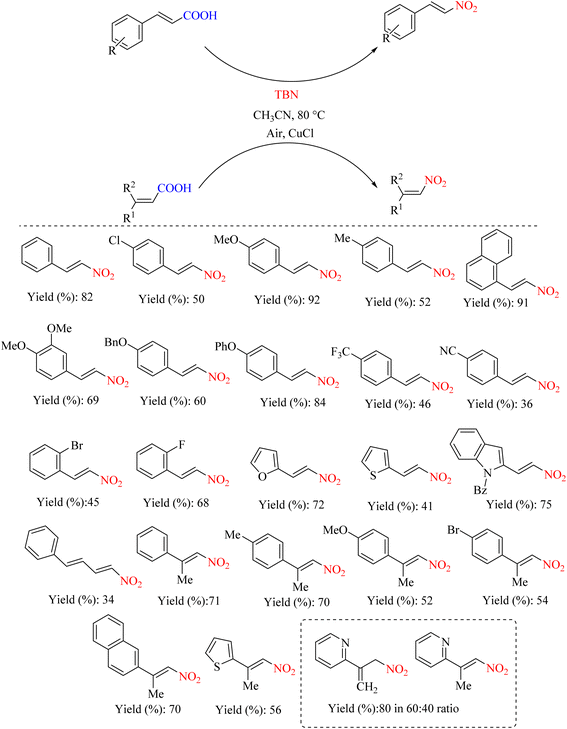 |
| | Scheme 13 Synthesis of substituted nitroolefins: copper-catalyzed nitro decarboxylation of unsaturated carboxylic acids. | |
 |
| | Scheme 14 Possible mechanism underlying the ipso-nitration of substituted nitroolefins: copper-catalyzed nitro decarboxylation of unsaturated carboxylic acids. | |
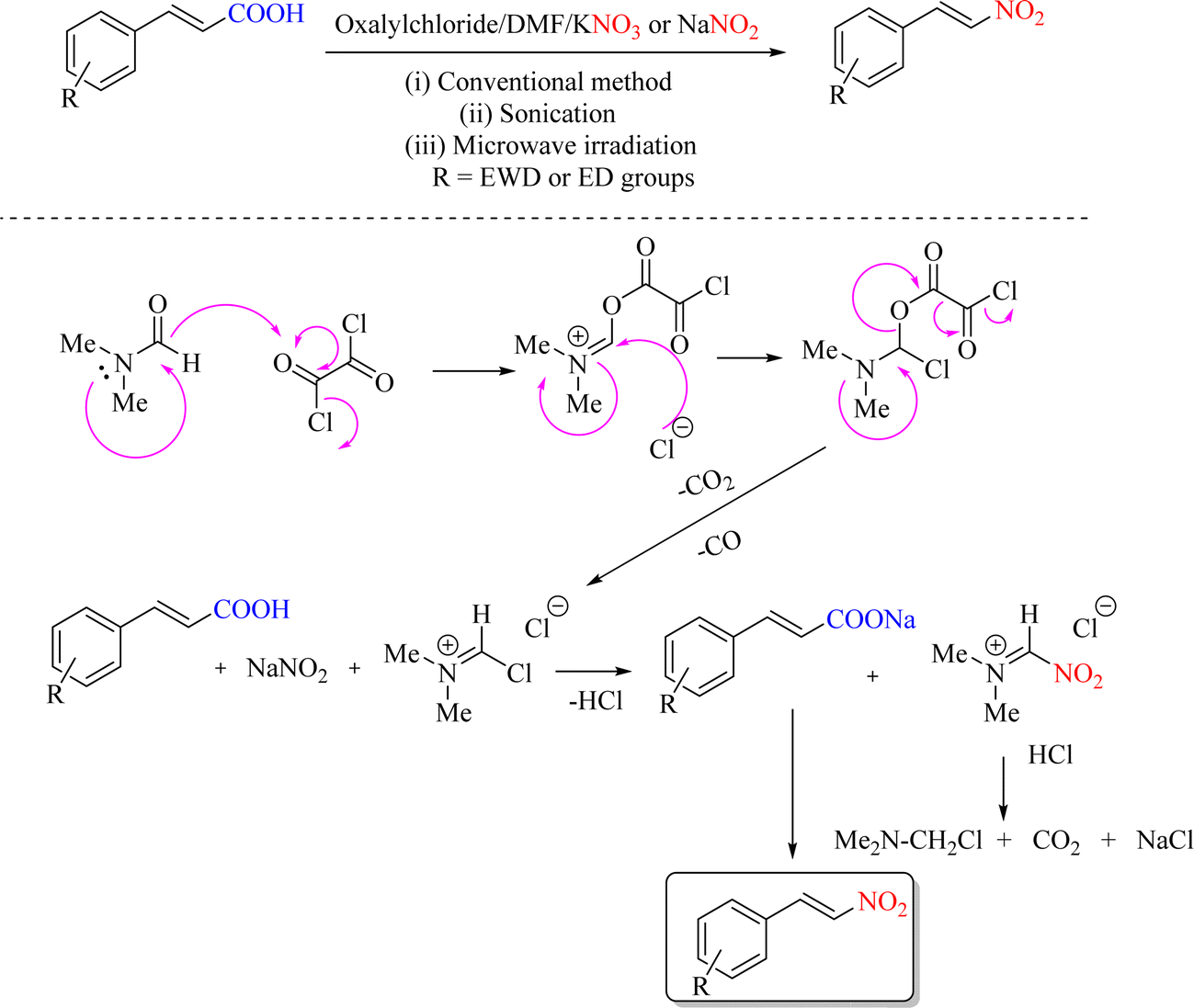
An ipso-nitration reaction using a new method can be fascinating. In a 2013 report, the nitration of aromatic compounds and cinnamic acids with oxalylchloride/DMF of the corresponding nitro derivatives in the presence of KNO3 or NaNO2 was obtained under normal and unconventional conditions. The current method boasts several advantages over other common nitration methods for various compounds, including excellent efficiency, straightforward operation, and short reaction times. The report indicates that the efficiencies achieved using the present methodology are comparable to those obtained from other systems (POCl3/DMF/KNO3 or NaNO2) and (SOCl2/DMF/KNO3 or NaNO2), with shorter reaction times. The reaction time under ultrasound and microwave conditions was much shorter than under other conditions (Scheme 15).54
 |
| | Scheme 15 Mechanism of the decarboxylation nitration of cinnamic acid with NaNO2 or KNO3 in the presence of oxalylchloride/DMF. | |
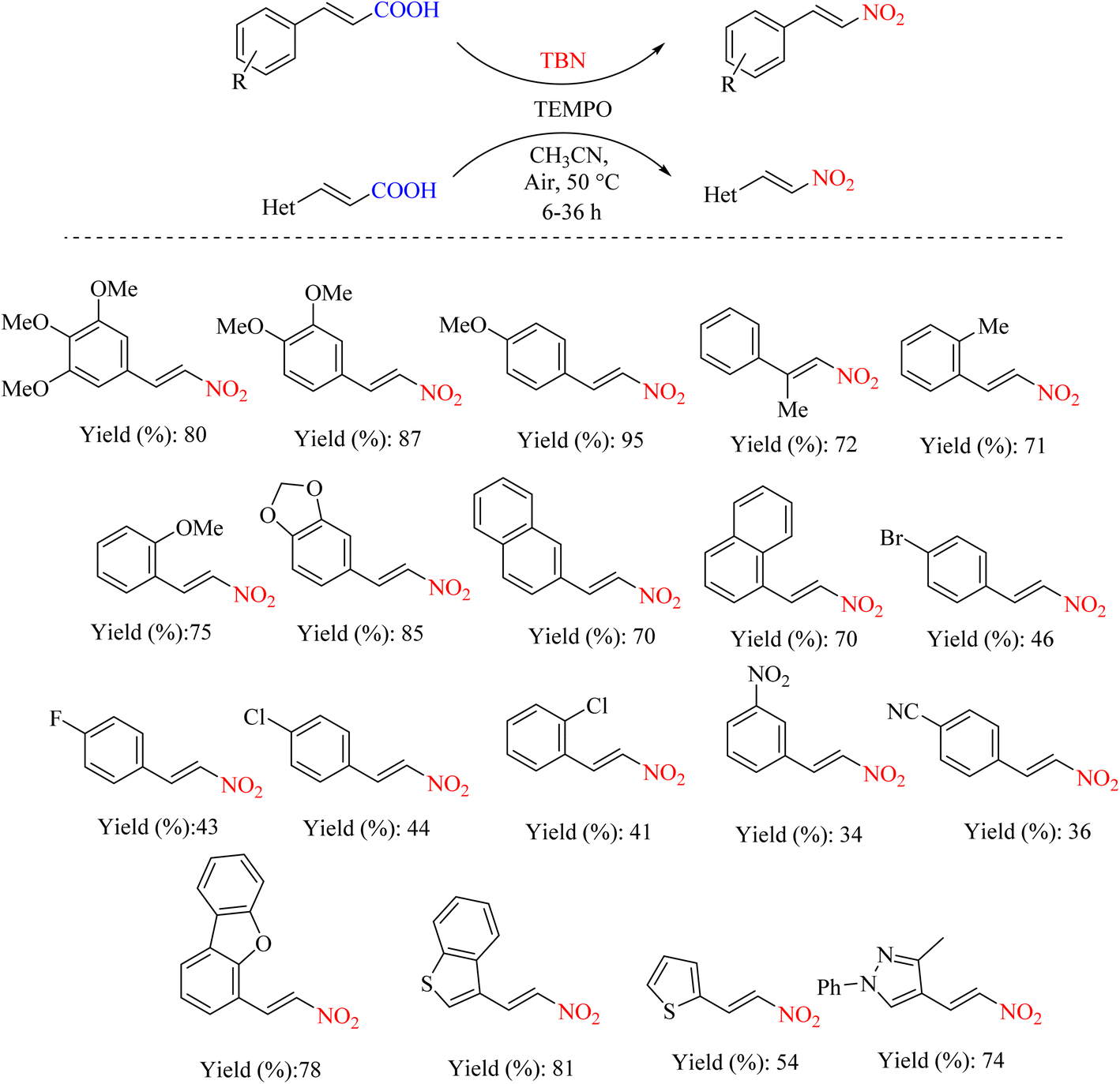
As mentioned in the previous sections, the nitration reaction can proceed via a radical mechanism. Providing reagents that can perform under these conditions with high efficiency is important. A research study reported a metal-free decarboxylate nitration protocol for preparing nitroolefin from unsaturated carboxylic acids a and b using TBN and TEMPO (Scheme 16).55 The presence of TEMPO provides a radical-based pathway for the carboxylation–nitration reaction (Scheme 17).
 |
| | Scheme 16 Synthesis of (E)-nitroolefins via decarboxylative nitration using TBN and TEMPO. | |
 |
| | Scheme 17 Mechanism of nitro decarboxylation during the synthesis of (E)-nitroolefins using TBN and TEMPO. | |
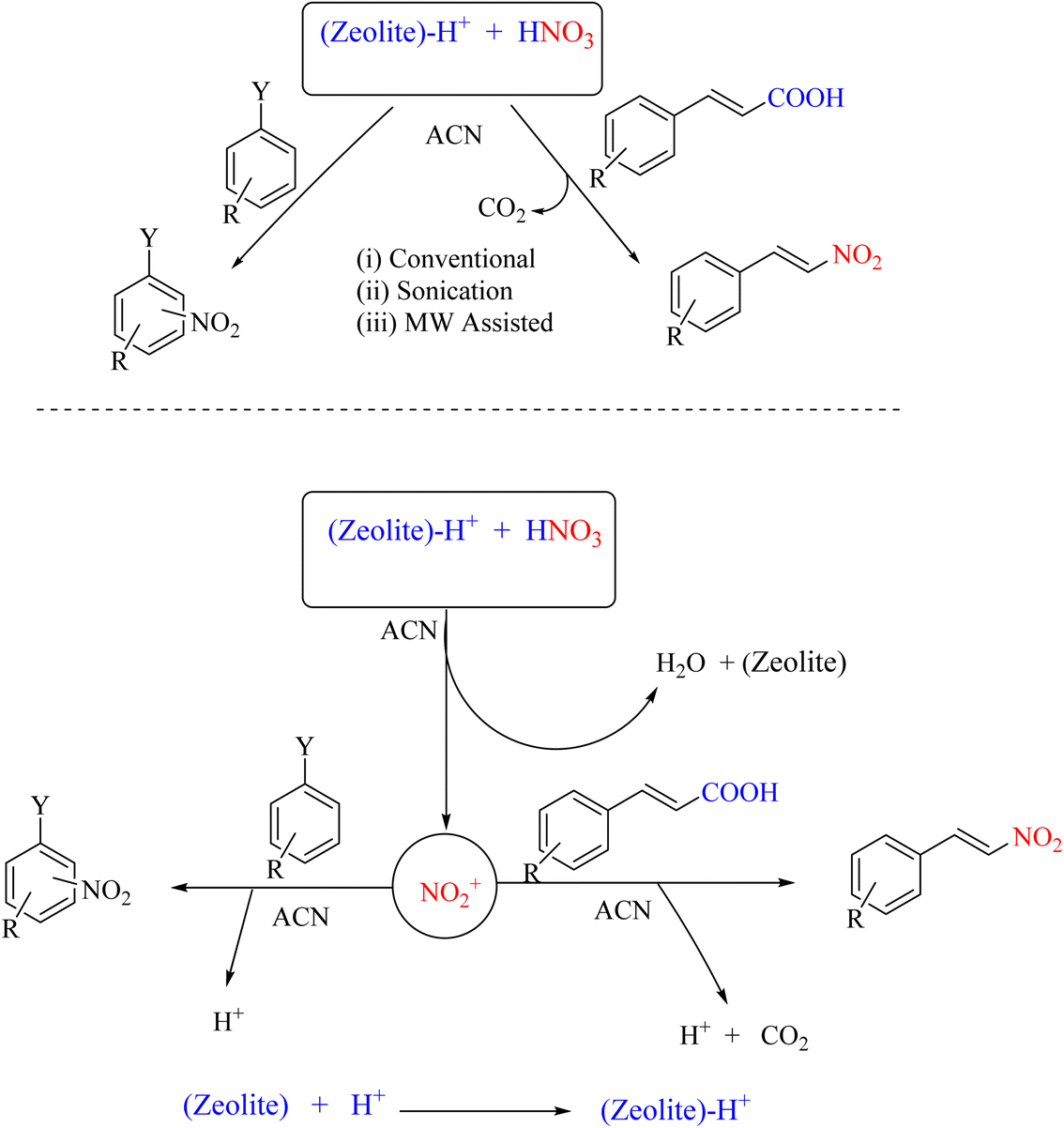
Zeolites, primarily composed of aluminosilicate, are minerals widely used in various industrial applications, with the main commercial use being as surface adsorbents. They are extensively used in water treatment, catalysis, and the production of advanced materials in various industries. In a 2015 study, zeolite Y was employed as an effective catalyst for the facile nitration of aromatic compounds in the presence of small quantities of HNO3 in acetonitrile solvent under ambient temperature conditions. These reaction conditions yielded mono-nitro derivatives of aromatic compounds within approximately three hours under stirred conditions with high regioselectivity and yields. The ipso-nitration reaction was conducted under ultrasonic and microwave irradiation, which significantly reduced the reaction time. Furthermore, the zeolite could be recovered after completing the reaction and recycled three to four times without any issues (Scheme 18).56
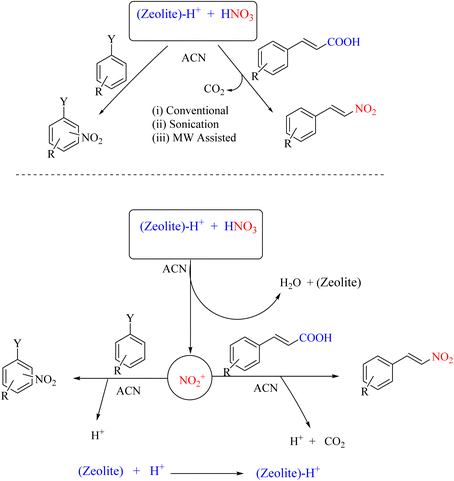 |
| | Scheme 18 Zeolite Y-mediated nitration and nitro decarboxylation. | |
The conversion of functional groups into a single step is important for the synthesis of biological compounds. One of these conversions is the conversion of carboxylic acid groups to nitro, which should provide the conditions for high reaction yields and the production of few by-products. In one of these methods for synthesizing and evaluating the biological activity of 3-alkyl-1,5-diaryl-1H-pyrazoles as rigid analogs of combretastatin A-4 with potent antiproliferative activity, acetic acid and nitric acid were used in one of the synthesis steps of these compounds (Scheme 19).57
 |
| | Scheme 19 Part of the synthesis and evaluation of a drug compound with strong anti-proliferative activity. | |
An efficient and safe method for the nitration of aromatic compounds and decarboxylation nitration was developed using trichloroisocyanuric acid (TCICA)/DMF in the presence of NaNO2. This reaction is beneficial both economically and environmentally. The advantages of this method include the simplicity of operation, mild reaction conditions, the avoidance of organic solvents and toxic and expensive reagents, a short reaction time, and high product yield (Scheme 20).58 The presence of ultrasonic waves in this method has increased the rate of nitration. The reaction mechanism is shown in Scheme 21.
 |
| | Scheme 20 Nitro decarboxylation and nitration of trichloroisocyanuric acid (TCICA)/dimethylformamide (DMF) in the presence of NaNO2. | |
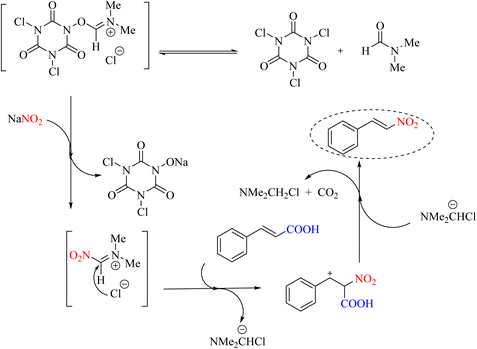 |
| | Scheme 21 Mechanism of nitro decarboxylation and nitration of trichloroisocyanuric acid (TCICA)/dimethylformamide (DMF) in the presence of NaNO2. | |
In 2015, a new and efficient method for the nitration of selected regions of aliphatic and aromatic carboxylic acid compounds to their corresponding nitro compounds was reported using the reagent of tetrafluoroborate nitronium and silver carbonate in dimethyl acetamide (Scheme 22).59 This conversion is believed to occur via an alkyl-silver or aryl-silver intermediate, which subsequently reacts with the nitronium ion to form nitro materials (Scheme 23).
 |
| | Scheme 22 Silver(I)-induced ipso-nitration of carboxylic acids by nitronium tetrafluoroborate. | |
 |
| | Scheme 23 Proposed reaction mechanism underlying the decarboxylative nitration reaction. | |
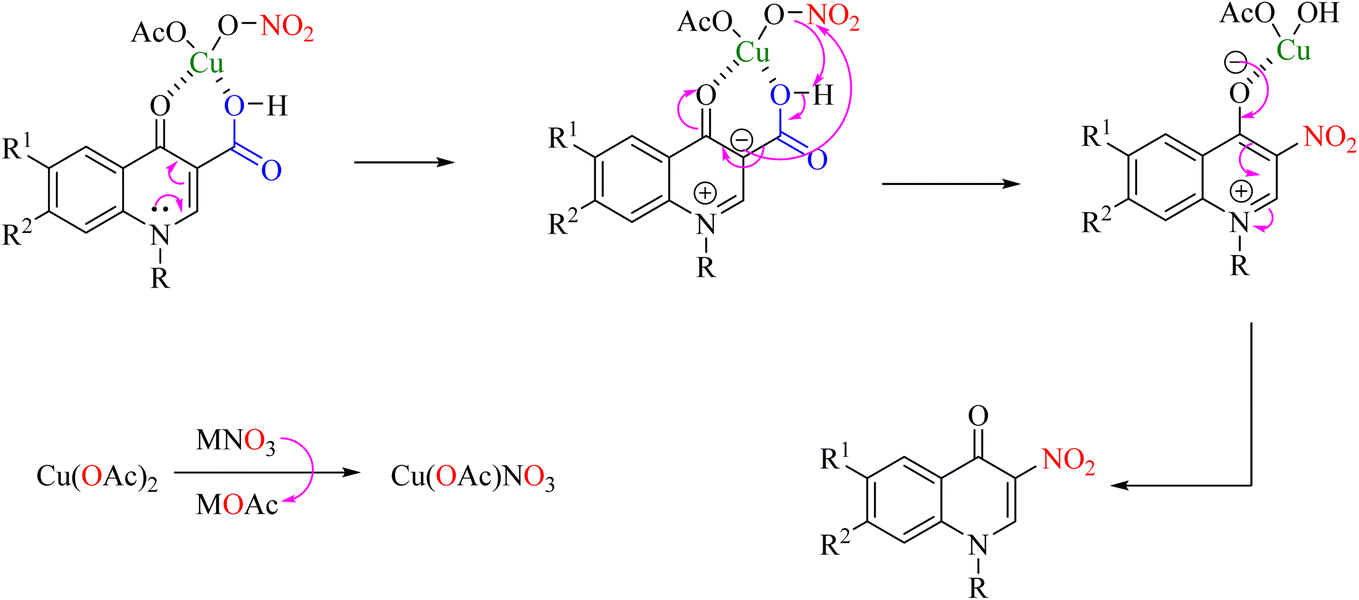
An efficient, cost-effective, and green method for ipso-nitration has been reported for synthesizing 3-nitro derivatives of 3-carboxy-quinolones using copper acetate and silver nitrate in water as solvent (Scheme 24).60 In the mechanism of this reaction, two carbonyl groups chelate with copper(II), containing nitrate ions formed by anion exchange between Cu(OAc)2 and a nitrate salt. Decarboxylation nitration is then performed through a coordinated mechanism in which NO2 is attacked by a charge created on carbon C-3 and a protonable acid ionized by Cu–O to form Cu(OH)OAc (Scheme 25).
 |
| | Scheme 24 Operative conversion of 3-carboxy-4-quinolones into 3-nitro-4-quinolones via ipso-nitration. | |
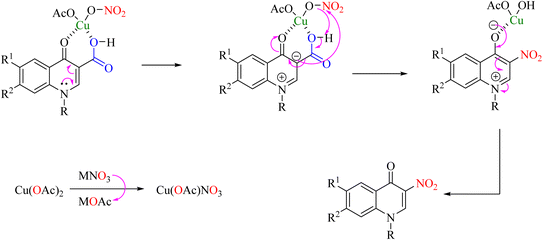 |
| | Scheme 25 Plausible reaction mechanism underlying the operative conversion of 3-carboxy-4-quinolones into 3-nitro-4-quinolones via ipso-nitration. | |
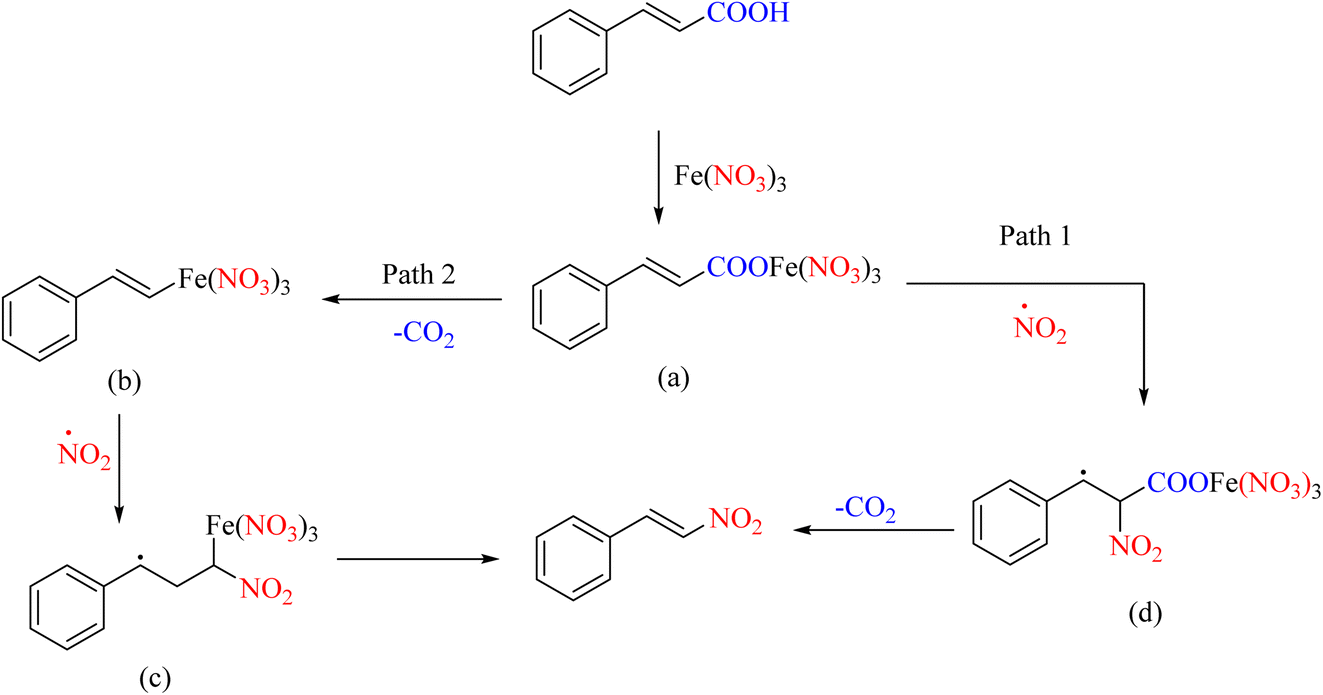
Many nitration–substitution reactions proceed through a radical mechanism. A report has shown that iron nitrate salts can perform the ipso-nitration reaction well, decarboxylating many unsaturated acids. The effect of the iron(III)/pyridine-mediated decarboxylate nitration reaction on the selectivity of nitroolefin has also been proposed, which is an interesting topic (Scheme 26).61 The radical mechanism of this reaction is presented in Scheme 27.
 |
| | Scheme 26 Decarboxylative nitration of α,β-unsaturated acids with nitrate ions. | |
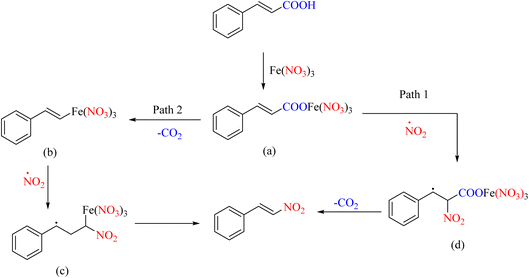 |
| | Scheme 27 Proposed mechanism underlying the decarboxylative nitration process. | |
Many heterogeneous catalysts are synthesized from metals. One of these widely used metals is copper. In a report on the placement of copper particles on chitosan as a suitable substrate, heterogeneous catalysts were used to synthesize 3-nitro-4-quinolones from 3-carboxy-4-quinolones by ipso-nitration (Scheme 28).62 The reduction of the copper composition by NaBH4 produces copper nanoparticles that efficiently perform the decarboxylation reaction and provide nitration positions (Scheme 29).
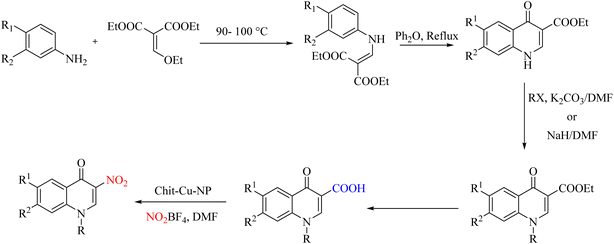 |
| | Scheme 28 Ipso-nitration using polysaccharide-supported copper nanoparticles. | |
 |
| | Scheme 29 Probable reaction mechanism underlying ipso-nitration using polysaccharide-supported copper nanoparticles. | |
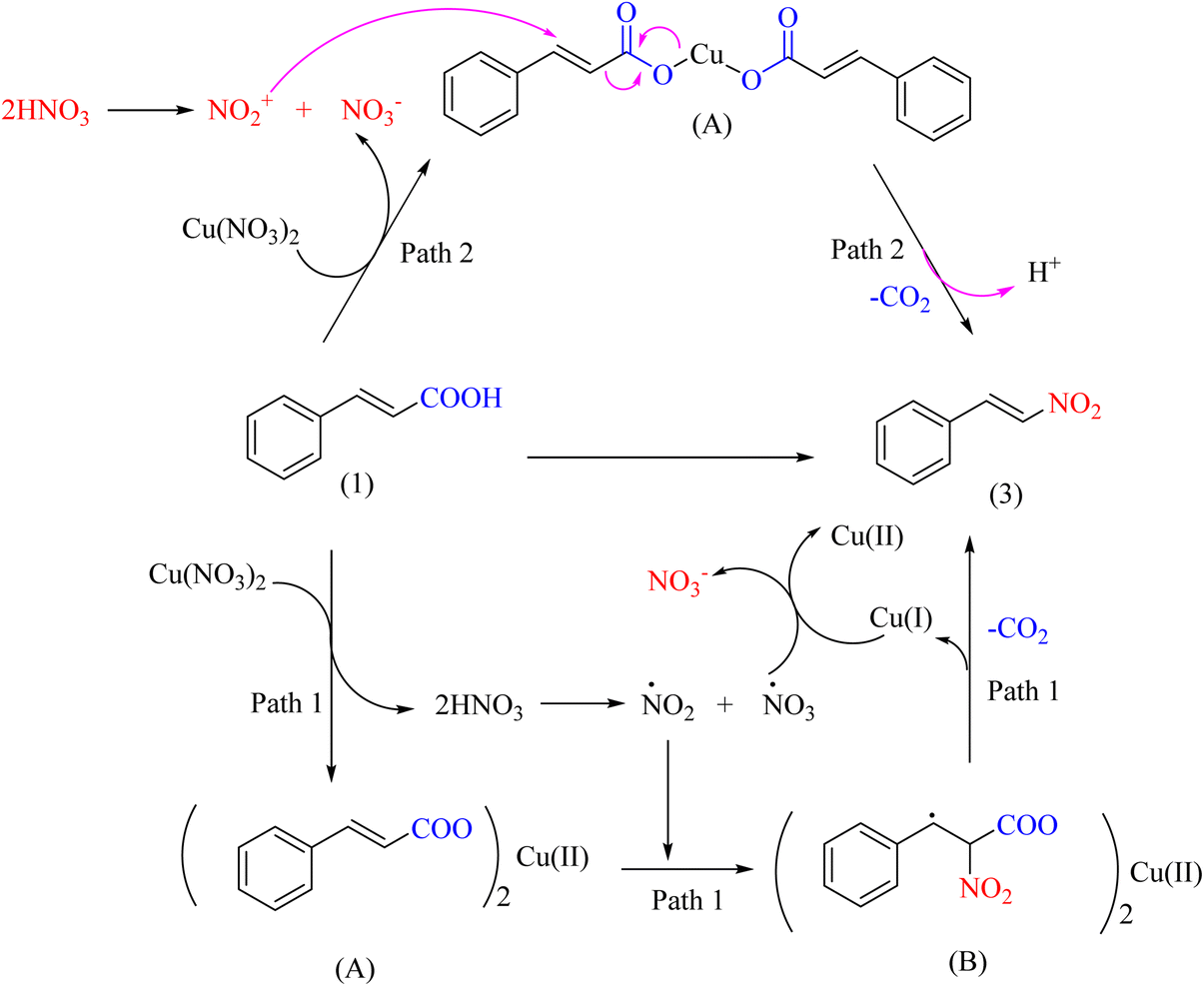
The importance of copper metal in performing chemical reactions as a catalyst is well demonstrated. Copper metal can participate in the ipso-nitration reaction to produce nitro radicals in the environment through its reduction. A previous report showed that a copper nitrate salt was used for the ipso-nitration reaction. This efficient method converts many carboxylic acid derivatives to nitro compounds (Scheme 30).63
 |
| | Scheme 30 Proposed mechanism underlying the Cu(NO3)2-catalyzed decarboxylation nitration of unsaturated carboxylic acids. | |
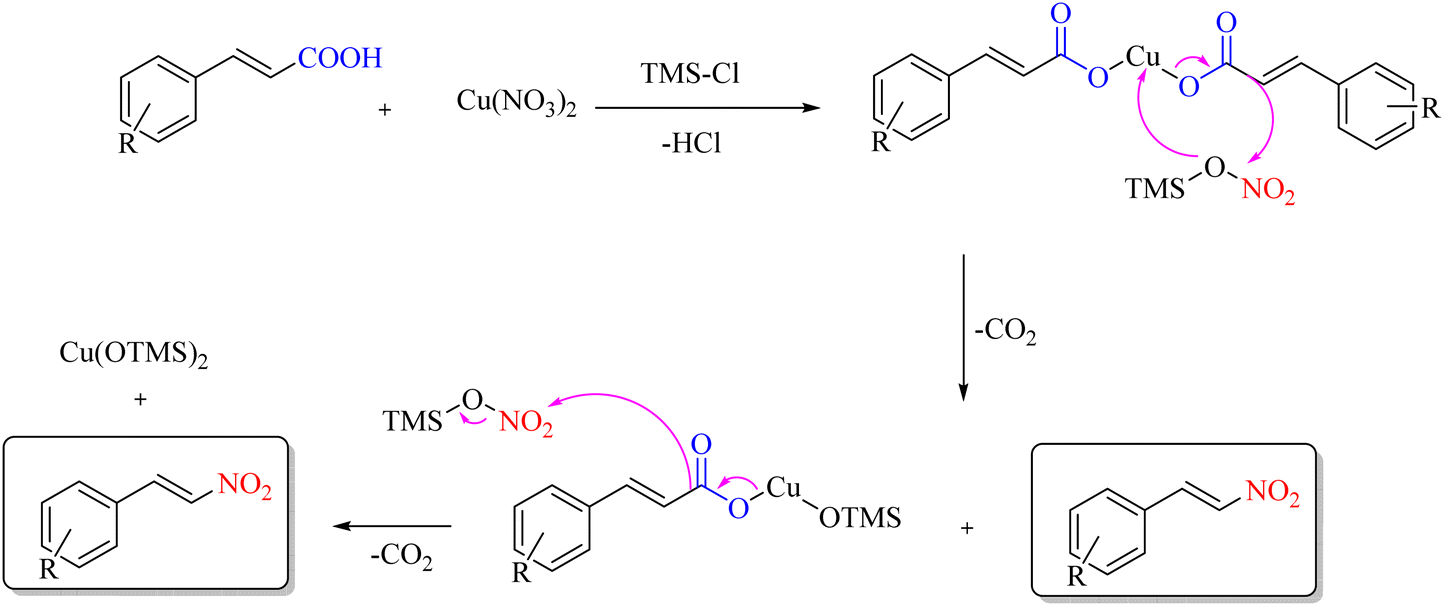
The halotrimethylsilane nitrate salt has been proven to be of greater importance. A mixture of the TMSX-Cu(NO3)2·3H2O system has been shown to be an efficient reagent system for ipso-nitration when X = Cl and the debromination of cinnamic acids when X = Br under mild conditions. These reactions are benign and straightforward, allowing avoidance of hazardous and toxic nitrating systems like acetyl nitrate and brominating agents such as molecular bromine (Scheme 31).64 The reaction mechanism is illustrated in Scheme 32.
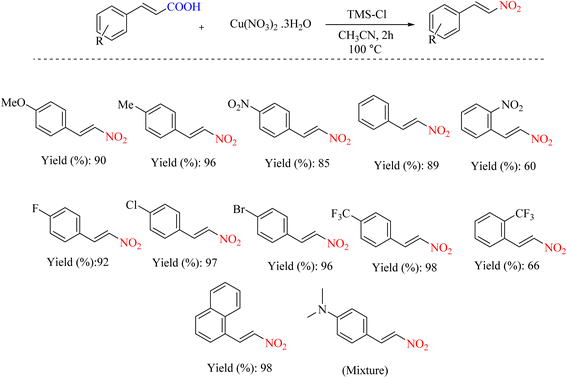 |
| | Scheme 31 Decarboxylative ipso-nitration using halotrimethylsilane-nitrate salt. | |
 |
| | Scheme 32 Plausible mechanism underlying decarboxylative ipso-nitration. | |
Switching between different groups has always been inevitable in synthesizing drug compounds. Among the types of switching groups, the placement of nitro groups instead of acid groups, which is the reaction of ipso-nitration, has been widely used in the synthesis of medicinal compounds. Sulfonyl amides are newly synthesized drug compounds, and their effects on melanin synthesis in mouse B16 cells have been investigated. All target sulfonamides were synthesized by an ipso-nitration reaction (Scheme 33).65
 |
| | Scheme 33 Synthesis of sulfonyl amides via the ipso-nitration reaction. | |
Numerous chemical reactions proceed via radical mechanisms. While some radicals can form spontaneously, many reactions require a radical initiator to generate the radicals. K2S2O8 is one of the most prevalent radical initiators. This compound can react with nitrate salts to generate nitro radicals, creating conditions for the ipso-nitration reaction (Scheme 34).66
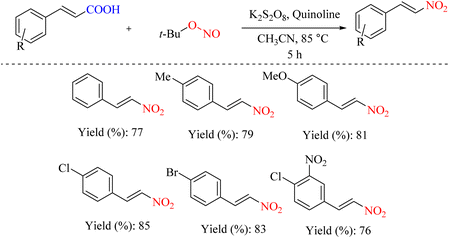 |
| | Scheme 34 Ipso-nitration reaction using TBN and K2S2O8. | |
An environmentally friendly and straightforward ipso nitration protocol has been reported for aliphatic and (hetero) aromatic carboxylic acids using ionic liquids. Although this protocol contradicts recently established ipso-nitration decarboxylation strategies for preparing nitro compounds, it does not require a toxic metal catalyst or harmful volatile organic solvents. In addition, this method is excellent for various functional groups and provides exclusively nitrated products in moderate to good yields (Scheme 35).67
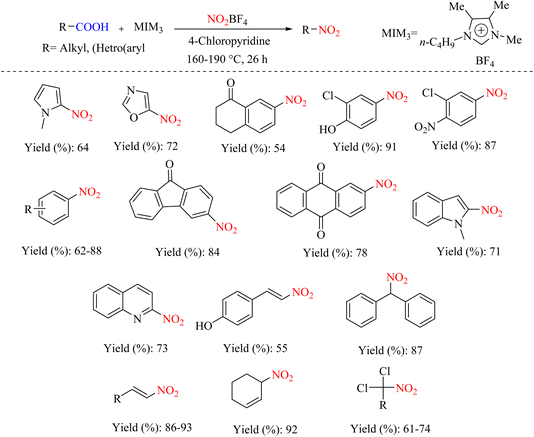 |
| | Scheme 35 Substrate scope for the decarboxylative nitration of alkyl and (hetero)aryl carboxylic acids to nitro compounds. | |
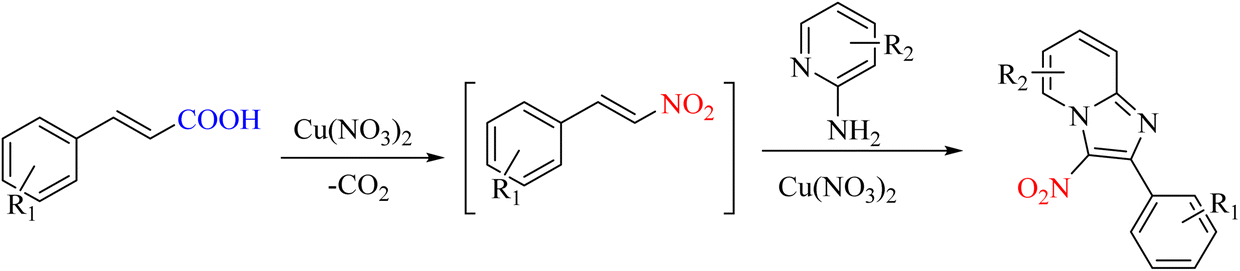
In organic reactions, products can be used and produced as intermediates to create the main products. Ipso-nitration products can be used to access future products. This issue has been studied in a report. The compounds imidazo[1, 2-a]pyridines are formed as intermediates of ipso-nitration. Due to its good performance in copper, this metal has been used in ipso-nitration reactions (Scheme 36).68
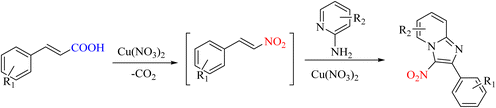 |
| | Scheme 36 Novel approach in the synthesis of imidazo[1, 2-a]pyridine from phenyl acrylic acids. | |
3. Ipso-nitration of boronic acids [ipso-B(OH)2]
Organic boronic acids are widely used in organic synthesis because of their commercial availability and excellent stability against air and moisture. The boronic acid functional group is reputed to have low inherent toxicity. Boronic acids act as Lewis acids,69 and the presence of two hydroxy groups on the boron element makes these compounds good leaving groups in many substitution reactions. There are many examples of the ipso-nitration of aryl boronic acids in articles. This report highlights the most important methods of ipso-nitration using organic boronic acids.70,71 Many reagents and methods have been introduced to perform this reaction (Scheme 37).
 |
| | Scheme 37 Ipso-nitration of boronic acids [ipso-B(OH)2]. | |
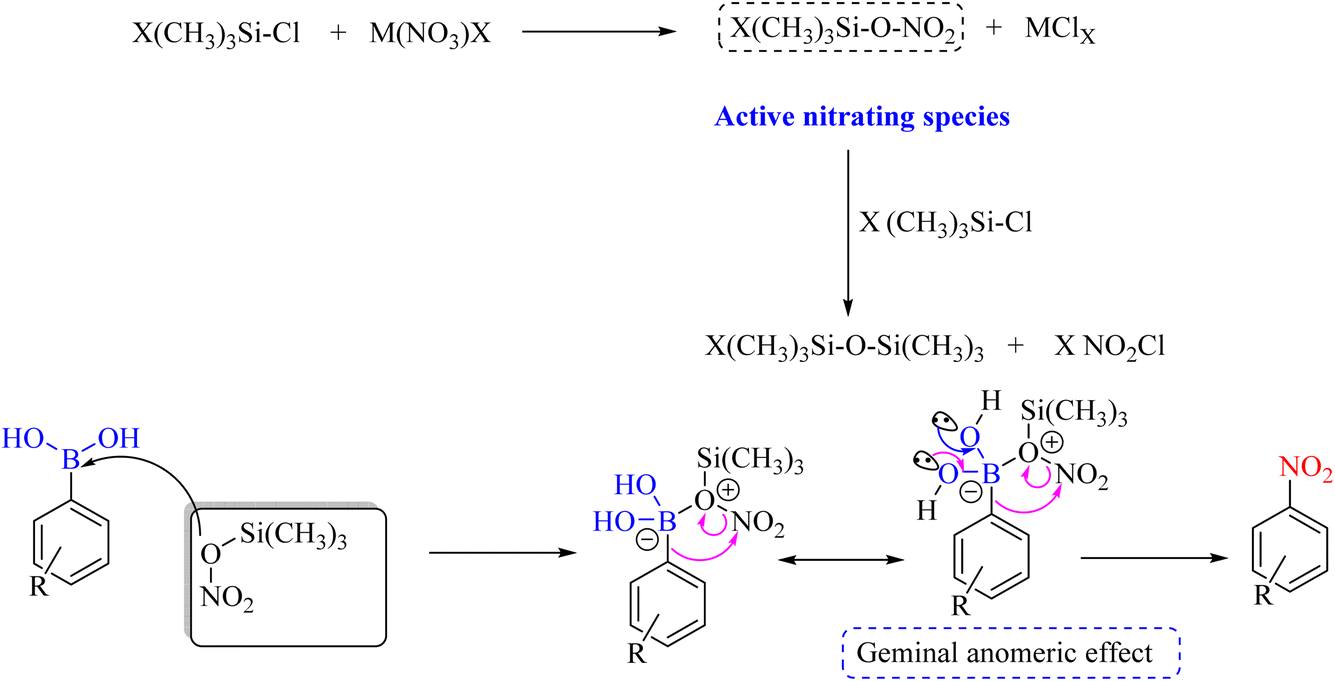
In 2000, Olah et al. used the regioselective electrophilic method and NH4NO3 reagent to perform ipso-nitration with raw aryl boronic acid materials.13 With this method, the authors showed that active and inactive aromatic compounds can be nitrated to obtain mono- and dinitro products with good efficiency (Scheme 38). The reaction mechanism is described in Scheme 39. Recently, our research group has reviewed the role of stereoelectronic effects as a fundamental concept in organic reactions.72,73 We considered the anomeric effect and modified related reported mechanisms in Scheme 39. These concepts are applicable to the ipso nitration of borane organic compounds. Our recently reported educational synthetic organic methodology is also applicable for modifying the abovementioned nitration reaction.74
 |
| | Scheme 38 Ipso-nitration of the B(OH)2 leaving group via the regioselective electrophilic method. | |
 |
| | Scheme 39 Reaction mechanism of the B(OH)2 leaving group via the regioselective electrophilic method. | |
In 2004, Parkash et al. introduced the synthesis of nitro compounds as an efficient method using a mixture of nitrate salt and chlorotrimethylsilane as efficient regioselective nitrating agents (Scheme 40).75 The reaction mechanism indicates that the reaction proceeded via an increase in electrophilicity (Scheme 41).
 |
| | Scheme 40 Ipso-nitration of the B(OH)2 leaving group using nitrate salt and chlorotrimethylsilane. | |
 |
| | Scheme 41 Reaction mechanism of the B(OH)2 leaving group using nitrate salt and chlorotrimethylsilane. | |
This report describes a non-catalytic nitration of aryl boronic acids using bismuth(III) nitrate as the nitrating agent. The reaction proceeded with shorter reaction times with moderate to excellent efficiency. This method is operationally simple, exhibits regional selectivity, and is compatible with various functional groups, enabling the synthesis of nitroarenes (Scheme 42).76 Electrophilic bismuth(III) nitrate promotes ipso-nitration (Scheme 43).
 |
| | Scheme 42 Catalyst-free ipso-nitration of aryl boronic acids using bismuth(III) nitrate. | |
 |
| | Scheme 43 Proposed mechanism underlying the ipso-nitration of aryl boronic acids by Bi(NO3)3·5H2O. | |
Changing functional groups is essential in some chemical reactions. In the synthesis of organic matter, we may have to perform the ipso-reaction at one stage. In 2007, it was reported that in one of the steps of synthesizing benzopyran derivatives, an ipso-nitration reaction was performed on boronic acid (Scheme 44).77
 |
| | Scheme 44 Ipso-nitration reaction of boronic acid in the synthesis of benzopyran derivatives. | |
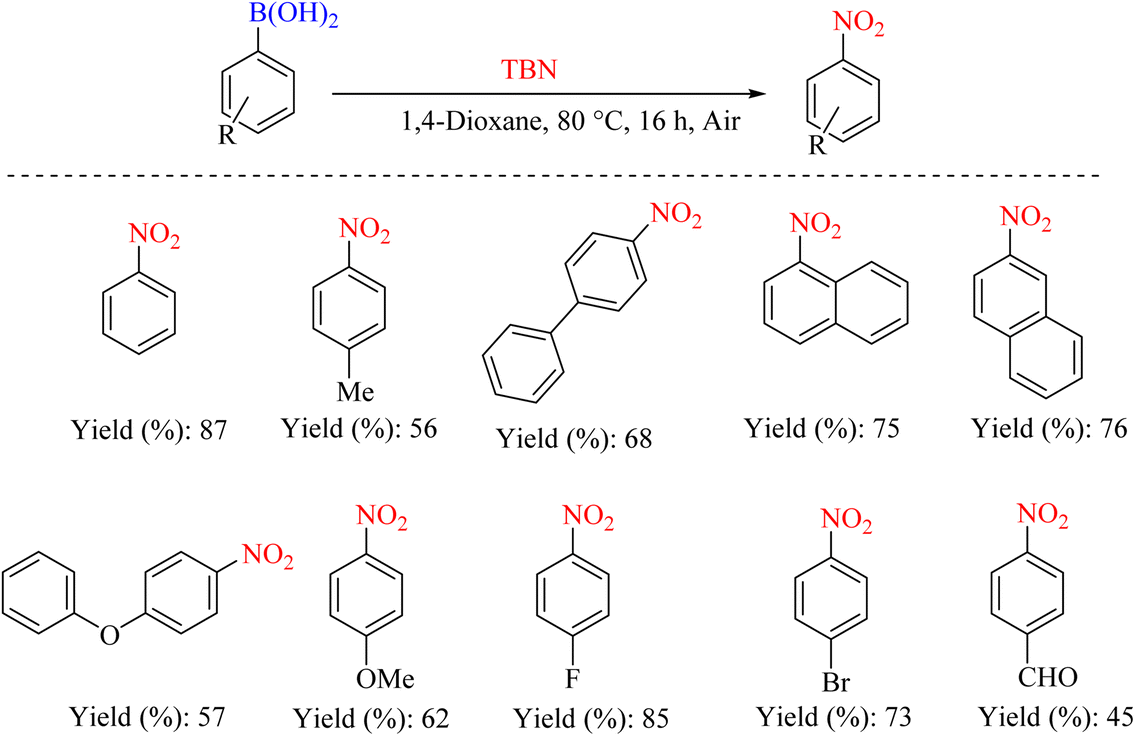
In 2011, Wu et al. reported a new method for nitrating various aryl boronic acids. The report indicates that various nitroarenes with moderate to good yields can be produced using low-cost TBN. This method offers several advantages, including ease of operation and the absence of the need for expensive catalyst systems (Scheme 45).78
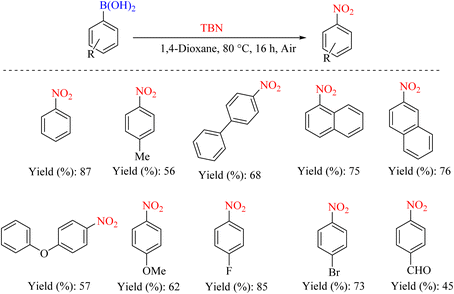 |
| | Scheme 45 Convenient and mild synthesis of nitroarenes via metal-free nitration of aryl boronic acids. | |
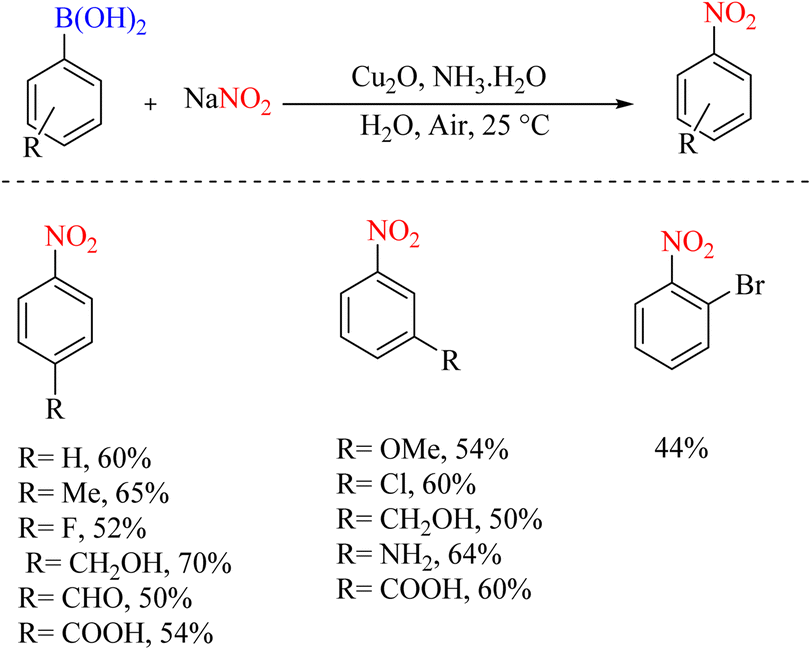
The conversion of copper-catalyzed functional groups from aryl boronic acids in water was reported in 2011. In a certain part of this change in the functional group, the ipso-nitration reaction on boronic acids was performed this way (Scheme 46).79 The very inexpensive Cu2O/NH3 system has been used as a catalyst, environmentally friendly water as a solvent, and readily available and affordable NaNO2 as a reagent.
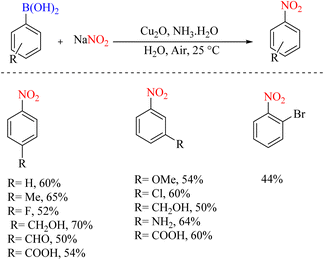 |
| | Scheme 46 Convenient and mild synthesis of nitroarenes by copper-catalyzed and NaNO2. | |
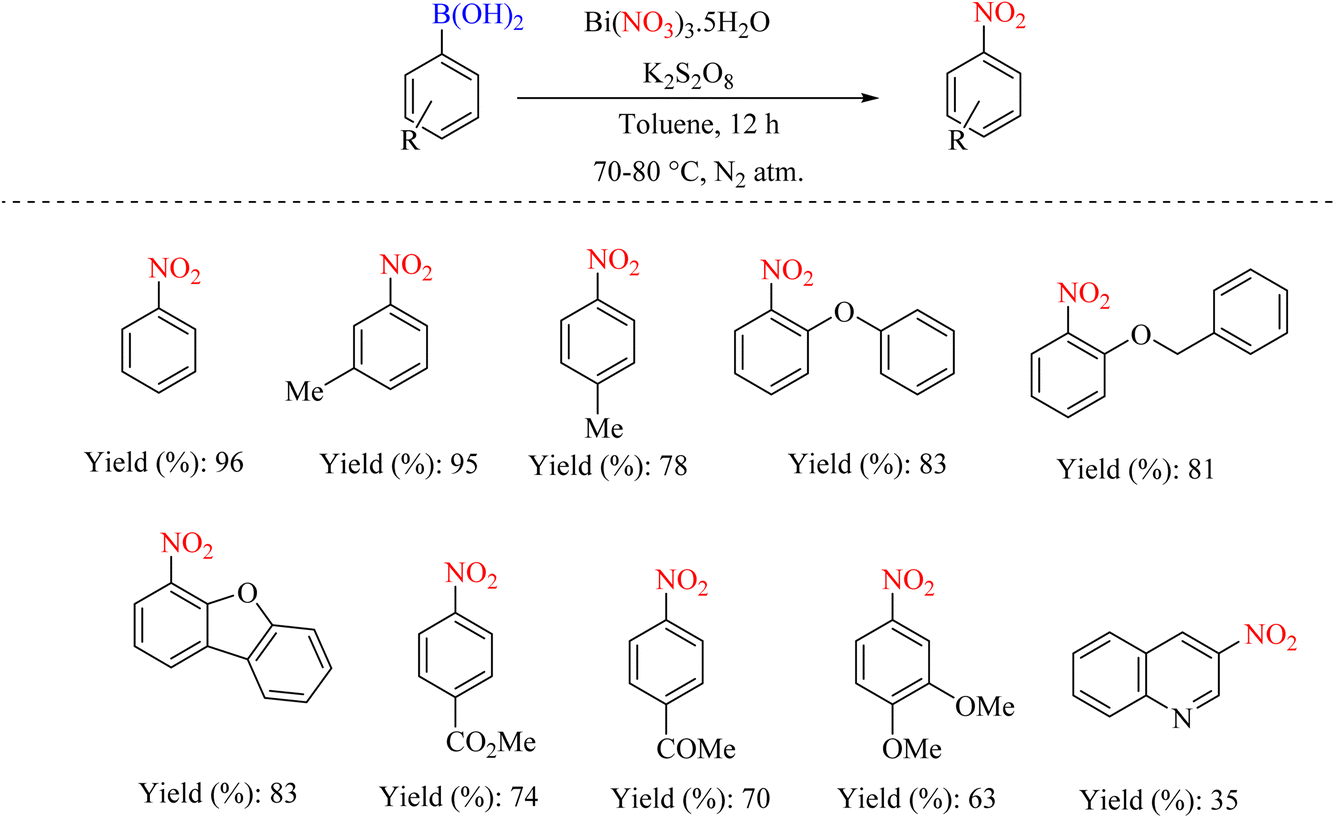
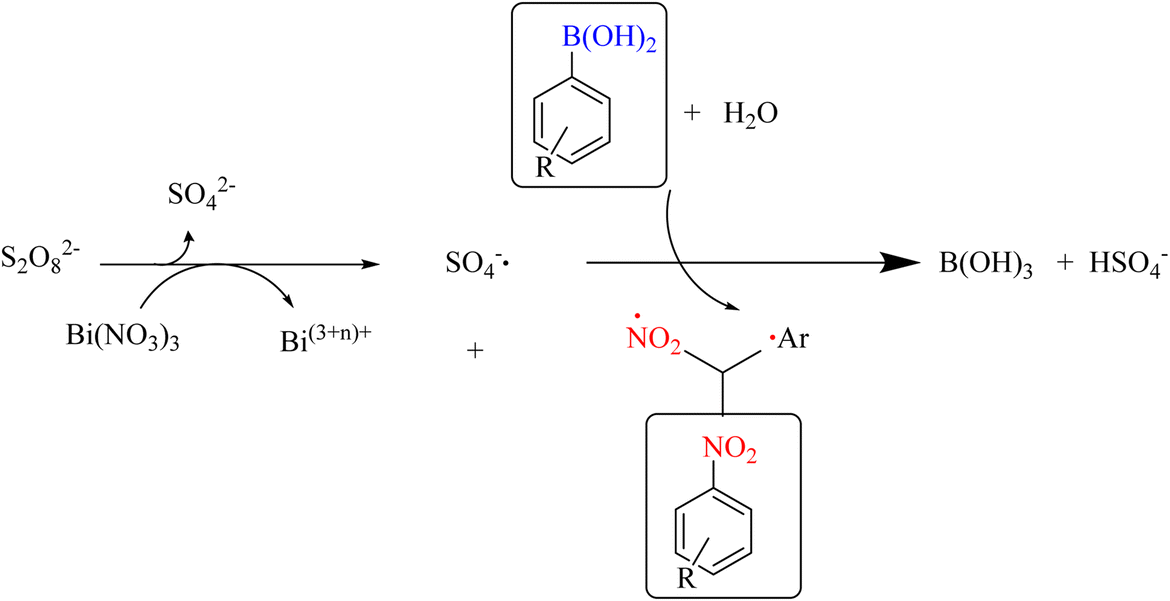
In 2012, Maiti et al. reported an efficient synthetic method for the one-pot internal nitration of aryl boronic acids. The high efficiency and broader substrate scope, which encompasses heterocycles and functional groups, render this method highly valuable (Scheme 47). In a radical mechanism, this method performs an ipso-nitration reaction using Bi(NO3)3·5H2O/K2S2O8. The team showed that the nitration reaction can be performed in a way other than the electrophilic method (Scheme 48).80
 |
| | Scheme 47 Ipso-nitration of arylboronic acids using bismuth nitrate and perdisulfate. | |
 |
| | Scheme 48 Proposed mechanism underlying the ipso-nitration of aryl boronic acids by Bi(NO3)3·5H2O/K2S2O8. | |
Metal-free nitration and a controllable green method for the synthesis of aryl boronic acids were introduced in 2012. Yan et al. used TBN as the nitrating agent. They performed an ipso-nitration reaction on boronic acids under mild conditions (Schemes 49, 50).81
 |
| | Scheme 49 Ipso-nitration of boronic acids using TBN as a nitrating agent. | |
 |
| | Scheme 50 Proposed mechanism underlying the ipso-nitration of boronic acids using TBN as a nitrating agent. | |
In 2012, Jan et al. developed the nitration of copper-catalyzed aryl boronic acids using nitrite salts as a nitrating agent under mild conditions. Due to the simple experimental method and the availability of a suitable and inexpensive copper catalyst, this process is an efficient and practical method for synthesizing nitro aromatics (Scheme 51).82
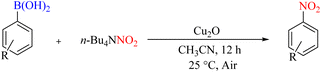 |
| | Scheme 51 Copper-catalyzed nitration of aryl boronic acids using nitrite salts under mild conditions. | |
In 2013, a brief synthesis of C and E marinoquinolines was reported. The synthetic pathway involves the internal nitration of an electron-deficient boronic acid, the first example of Bartoli indolization on nitroquinoline and Suzuki paired between 2-chloropyrroloquinoline and two separate MIDA boronates (Scheme 52).83
 |
| | Scheme 52 Ipso-nitration of boronic acid in the retrosynthetic analysis of marinoquinolines. | |
In another exploration, a simple and efficient nitration of aryl boronic acids with iron nitrate was proposed. The protocol uses aryl boronic acids as raw materials with low-cost iron nitrate as a nitro source without additives, and the corresponding nitroarenes are obtained with good to excellent efficiency (Scheme 53). The reaction mechanism indicates the radical progression of the reaction (Scheme 54).84
 |
| | Scheme 53 Efficient ipso-nitration of arylboronic acids using iron nitrate as a nitro source. | |
 |
| | Scheme 54 Possible mechanism underlying the ipso-nitration of aryl boronic acids using iron nitrate. | |
Nitroso compounds are versatile reagents in synthetic organic chemistry. A practical protocol for the arbitrary nitrification of aryl boronic acids using chlorotrimethylsilane and sodium nitrite Unison as a nitrosation reagent system was reported. During this reaction, ipso-nitration products are generated as by-products. This observation is similar to many other direct nitrosation reactions in which nitrosoarenes are oxidized in situ and converted to nitro groups (Scheme 55).85
 |
| | Scheme 55 Ipso-nitration of aryl boronic acids using chlorotrimethylsilane and sodium nitrite. | |
In 2015, Goswami et al. performed the ipso-nitration reaction of boronic acids using [bis-(trifluoroacetoxy)-iodo]benzene (PIFA). No metal was used in this reaction. The presence of N-bromosuccinimide (NBS) makes the reaction mechanism radical, and sodium nitrite is the nitro source (Scheme 56).86
 |
| | Scheme 56 PIFA-mediated ipso-nitration of aryl and alkylboronic acids. | |
The reaction proceeded through the in situ production of NO2 and O-centered boronic acid radicals, followed by the formation of an O–N bond through the combination of these radicals. Finally, the transfer of the NO2 group to the aryl fraction was accomplished via the migration of 1,3-aryl to the preparation of nitroarenes (Scheme 57).
 |
| | Scheme 57 Possible mechanism underlying the PIFA-mediated ipso-nitration of aryl and alkylboronic acids. | |
Nitration of electrophilic arenas is a classic method that involves a mixture of nitric acid and sulfuric acid or nitrogen pentoxide. In another report, an economic protocol was proposed in terms of simple operation and regional selectivity for the synthesis of nitro-aromatic compounds by direct nitration of aryl-boronic acids using trifluoroacetic acid and an aqueous solution of commercially available 68% nitric acid (Scheme 58).87
 |
| | Scheme 58 Convenient metal-free ipso-nitration of aryl boronic acids using nitric acid and trifluoroacetic acid. | |
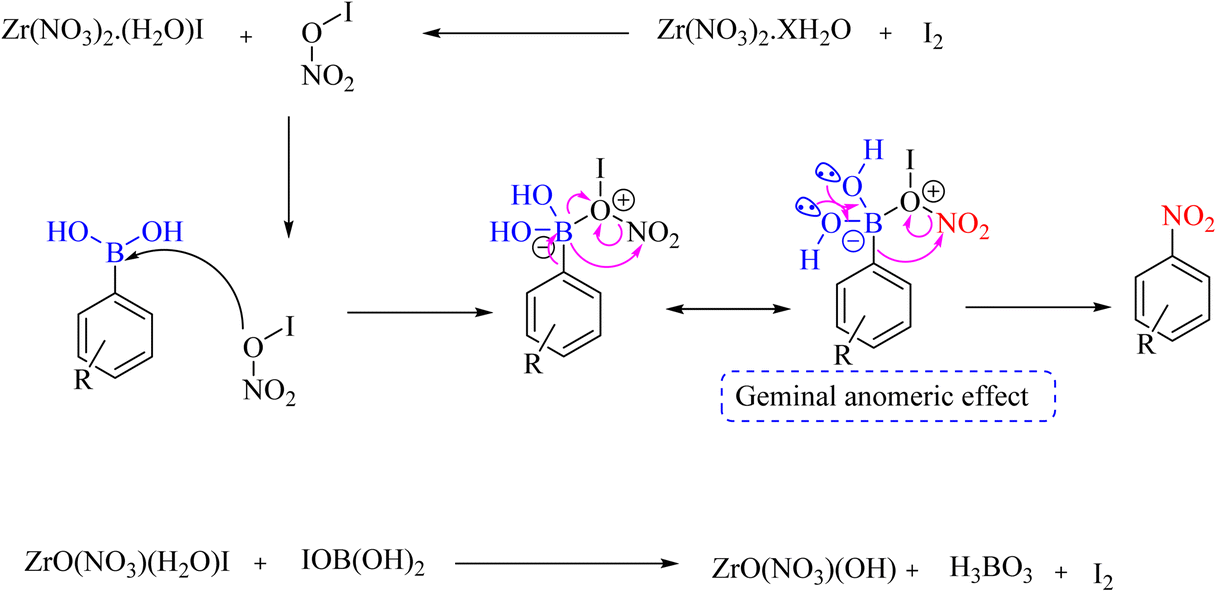
In another exploration, the ipso-nitration reaction of the aryl boronic acid group was catalyzed by iodine and zirconium oxynitrate as the nitrating species. The protocol applies to electronically diverse aryl- and hetero-aryl boronic acid moieties under mild reaction conditions with good to excellent isolated yields (Scheme 59).88 The reaction mechanism is shown in Scheme 60.
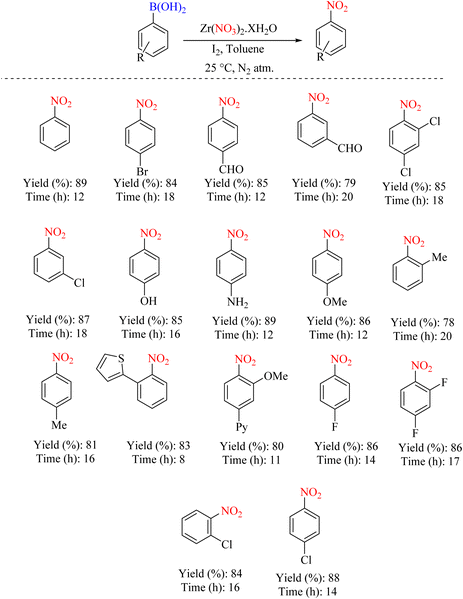 |
| | Scheme 59 Ipso-nitration reaction of the aryl boronic acid group catalyzed by iodine and zirconium oxynitrate. | |
 |
| | Scheme 60 Possible mechanism underlying the ipso-nitration reaction of the aryl boronic acid group catalyzed by iodine and zirconium oxynitrate. | |
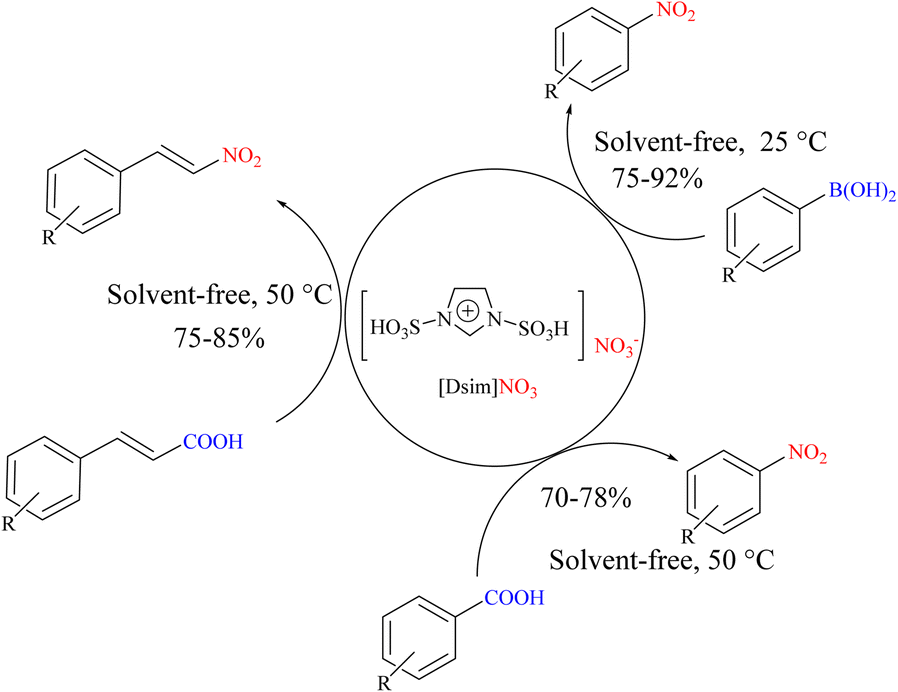
One of the ionic liquids that has the potential to design and synthesize various ILs and MSs is ionic liquids functionalized with N-sulfonic acid(s). In 2018, Zolfigol's research team introduced 1,3-disulfonic acid imidazolium nitrate ([Dsim]NO3) as a new ionic liquid and nitrating agent for the ipso-nitration of various aryl boronic acids and the nitro-Hunsdiecker reaction of different α,β-unsaturated carboxylic acids and benzoic acid derivatives (Scheme 61).89 In this section, the replacement of ipso-nitration of boronic acids is described. The mechanism of the reaction indicates the radical nature of the reaction, as shown in Scheme 62.
 |
| | Scheme 61 Ipso-nitration reaction of arylboronic acids using {[Dsim]NO3} as a new ionic liquid and nitrating agent. | |
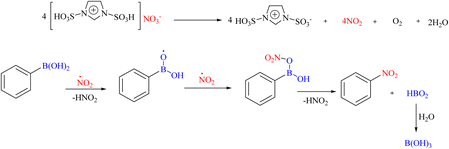 |
| | Scheme 62 Possible mechanism underlying the ipso-nitration reaction of aryl boronic acids using [Dsim]NO3 as a new ionic liquid and nitrating agent. | |
Another study reported the highly efficient nitration of aryl boronic acids using vaporized nitric acid. This report's significance lies in using vaporized nitric acid as the nitro source, enabling the process to be scalable for industrial applications. The report also provides detailed product yields, as mentioned above Scheme 63.90
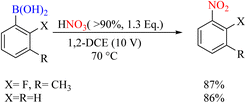 |
| | Scheme 63 Ipso-nitration of boronic acid using HNO3. | |
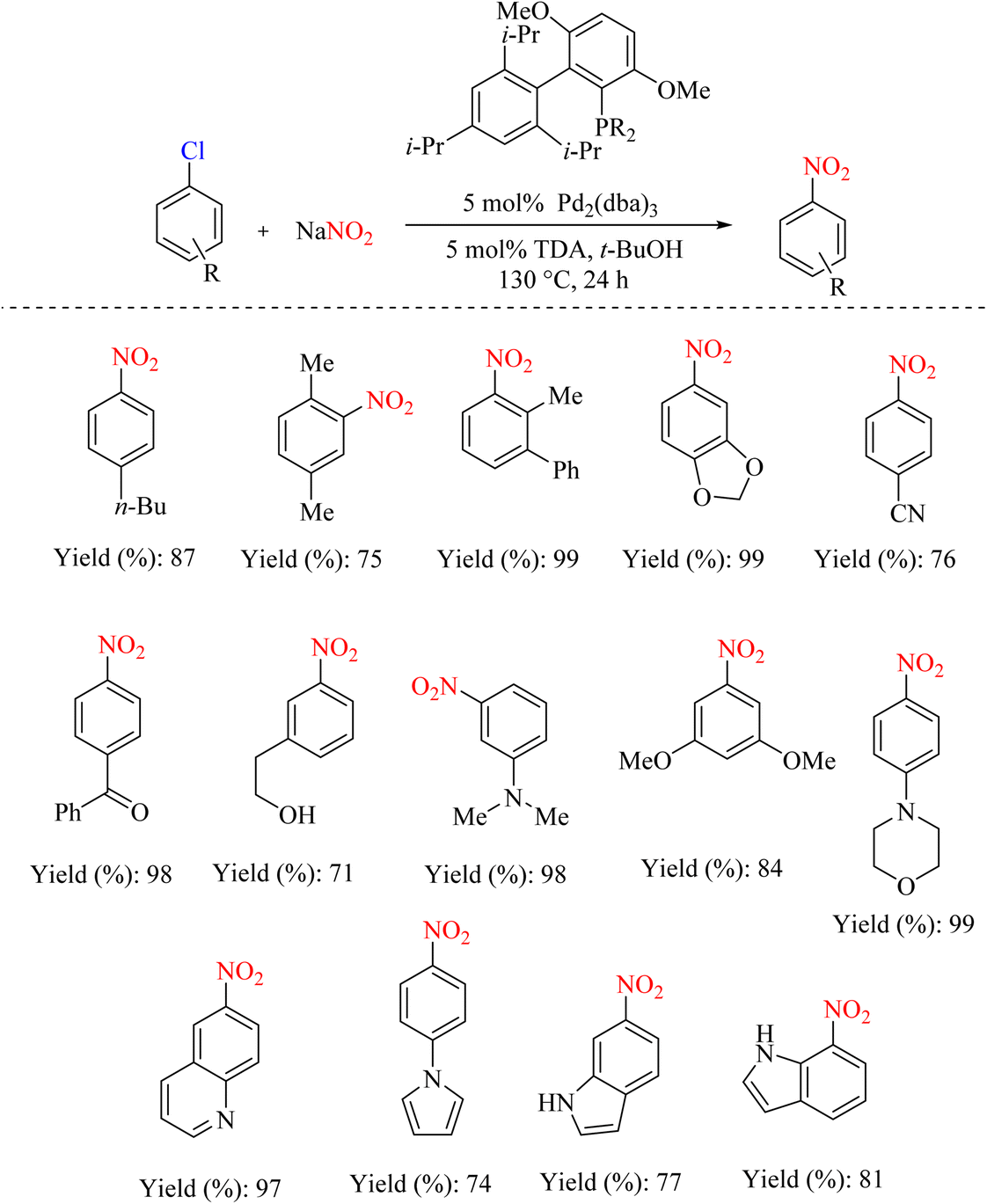
Potassium organotrifluoroborates are unique solid compounds. The use of these compounds in organic conversion has become increasingly important. Due to the good leaving nature, many other organic compounds can be substituted for this group. Instead of using dangerous reagents, nitrate salts can remove the potassium organotrifluoroborate group and perform an ipso-nitration reaction. Based on this method, the coupling reaction was investigated by microwave radiation in the presence of palladium as a catalyst. This cross-coupling reaction is an interesting development that can lead to the development of new nitration protocols (Scheme 64).91 The reaction mechanism and the role of Pd are illustrated in Scheme 65.
 |
| | Scheme 64 A novel route to obtain organonitrites via Pd-catalyzed cross-coupling of sodium nitrite and potassium organotrifluoroborates. | |
 |
| | Scheme 65 Mechanism underlying the cross-coupling of sodium nitrite and potassium organotrifluoroborates. | |
The ipso nitration of arylboronic acid derivatives using nitric acid as the nitrating agent was reported by Murray et al. in 2022. The reaction is driven by the presence and generation of nitro radicals, leading to faster product formation. This method provides easy, efficient, and chemoselective access to various aromatic nitro compounds. The report suggests that these processes likely produce a common active reagent, anhydrous HNO3 (Scheme 66).11
 |
| | Scheme 66 Ipso-nitration of aryl boronic acids using fuming nitric acid. | |
4. Ipso-nitration of halogens [ipso-X]
The leaving property of a halogen increases with its atomic radius as I > Br > Cl > F. Halogens are the best choices for ipso reactions, especially ipso-nitration. The nitro group replaces halogens under suitable conditions, facilitating the preparation of nitro compounds using this method. Therefore, the anionic mechanism for their exit is predicted (Scheme 67). Most metals, such as palladium and copper, contribute to the better release of halogens, some of which are described below.
 |
| | Scheme 67 General methods for the ipso-nitration of halogens [X] as leaving groups. | |
In 2005, research was conducted on the ipso-nitration of aryl halides. In this study, it was shown that for the removal of the halogen group from the copper catalyst as well as the N,N-dimethyl ethylenediamine in the presence of sodium nitrite-18-crown-6 or tetra-n-butylammonium nitrite (n-Bu4NNO2) as a nitration reagent was used, and ipso-nitration nitration operation was performed under these conditions (Scheme 68).92
 |
| | Scheme 68 Copper-catalyzed coupling of aryl halides and nitrite salts. | |
Under these conditions, Cu(I) is converted to Cu(0) and is placed between the benzene and the halogen. The halogen–nitrite exchange reaction and the reduction of copper species are performed. Soluble ammonium salts must effectively supply nitrite ions. This action produces nitro compounds, and it performs the ipso-nitration reaction (Scheme 69). This reaction was conducted under microwave conditions, with copper powder or CuI as the catalyst and NaNO2 or n-Bu4NNO2 as the NO2 source (Schemes 70 and 71).93,94
 |
| | Scheme 69 Mechanism underlying the copper-catalyzed coupling of aryl halides and nitrite salts. | |
 |
| | Scheme 70 Microwave-accelerated fluorodenitrations and nitrodehalogenations. | |
 |
| | Scheme 71 Rapid microwave-assisted copper-catalyzed nitration of aromatic halides using nitrite salt. | |
In another similar study, palladium metal was used instead of copper for this type of reaction. Palladium is a metal that must be activated for better performance. A 2009 report used a specific ligand to activate palladium. Different leaving groups have been used in this research, but different halo-benzenes have been used in some, as shown in Scheme 72.95
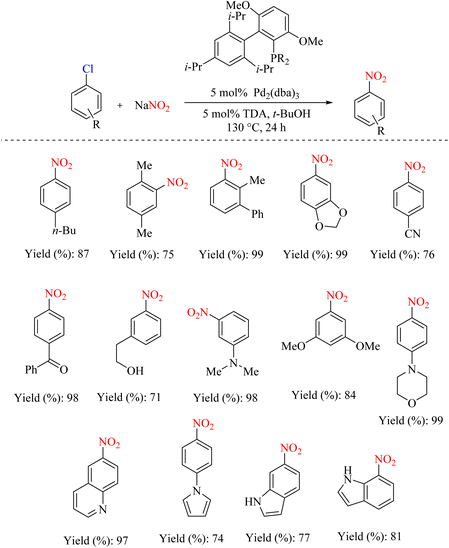 |
| | Scheme 72 Pd-catalyzed conversion of aryl chlorides to nitroaromatics. | |
In 2012, a suitable catalytic method was proposed for converting haloarenes to their corresponding nitroarenes using copper salts under conditions without any ligands. In this method, the internal nitration of a wide variety of haloes, including iodoarenes, bromoarenes, and heterocyclic haloarenes, is performed under mild conditions with a high-yield product (Scheme 73).96
 |
| | Scheme 73 Copper-catalyzed ipso-nitration of iodoarenes, bromoarenes, and heterocyclic haloarenes under ligand-free conditions. | |
In some cases, nucleophilic reactions can be performed in the absence of metals. A transition metal-free approach was reported for the facile synthesis of thiocyanate and nitroarenes from KSCN or NaNO2 with diaryliodonium salts in good yields under mild conditions (Scheme 74).97 Because of the reaction properties of iodonium salts, the reaction proceeded under ideal conditions, and the side products were avoided. This method is one of the best conditions for conducting ipso-nitration reactions in metal-free conditions. Nitroarenes are versatile building blocks for synthesizing highly complex organic molecules of higher complexity. These potential scaffolds are often found in natural products and drugs, and they commonly exhibit biological activities. Therefore, it is important to expand their simple synthesis methods.
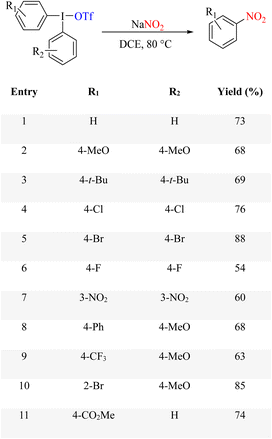 |
| | Scheme 74 Transition metal-free synthesis of thiocyanate- or nitro-arenes using diaryliodonium salts. | |
A similar review described the transition-metal-free, mild, and highly regioselective synthesis of nitroarenes from arenes. This report demonstrates that the products are synthesized through a sequential one-pot reaction, where iodine(III) reagents are nitrated, with two carbon ligands being generated in situ from iodine(I). In practice, this new concept has been extended to the formation of aryl azides and is an important step towards catalytic reactions using these multivalent iodide reagents. To carry out this reaction, diaryliodonium salts with different leaving groups are first synthesized through the C–H activation reaction, and then, using sodium nitrite salt, nitroarenes are synthesized through the ipso-nitration reaction (Scheme 75).98
 |
| | Scheme 75 One-pot C–H functionalization of arenes using diaryliodonium salts. | |
The chemical reaction of ipso-nitration on halogens continues to expand using new methods in recent years. In 2017, research was conducted on 2-halothiophenes. The reaction was catalyzed without a second metal such as Cu or Pd. In this reaction, a silver nitrate salt was used, showing that its silver metal helps to remove the halogen group, and its nitrate anion is used as a source of NO2 to perform the ipso-nitration. This attitude showed that instead of using several reagents and catalysts for ipso-nitration, one reagent with two different roles can be used (Schemes 76 and 77).99
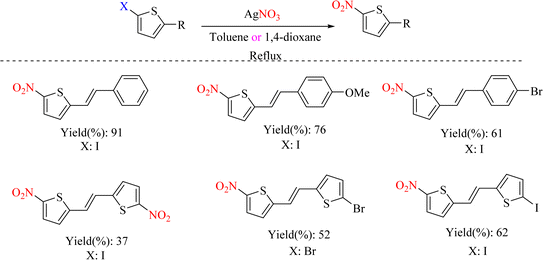 |
| | Scheme 76 Ipso-nitration of 2-halothiophenes using silver nitrate. | |
 |
| | Scheme 77 Proposed mechanism underlying the ipso nitration of 2-halothiophenes using silver nitrate. | |
A new selective nitration method was reported in 2022 in which MOFs were used as substrates, and palladium was deposited on the substrate using a post-modification method. In the following, sodium nitrite is used as a source of nitro. A wide range of compounds, such as boronic acids, aryl halides, aryl trifluoromethanesulfonate, and aroyl chlorides, have been converted into nitro products in the presence of this designed and synthesized system. The described method provides an advantageous and convenient strategy for the chemotherapeutic and co-selective ipso-nitration reaction. Unlike previously reported methods, nitric acid (HNO3) was not used during the nitration reaction (Scheme 78).100
 |
| | Scheme 78 Selective nitration reaction using MIL-101(Cr)–N(PC)–PdCl2/NaNO2 as a new nitrating agent. | |
An acceptable catalytic cycle is proposed as a targeted mechanism for the internal nitration reaction. In the first step, Pd(0) is prepared from a palladium precursor and reacted with Ar-X to form intermediate (I) via oxidative addition, followed by an X-nitrite exchange reaction to provide intermediate (II). In the next step, the reductive elimination of intermediate (II) yields the desired products (Scheme 79).
 |
| | Scheme 79 Plausible mechanism underlying the preparation of selective nitro compounds using MIL-101(Cr)–N(PC)–PdCl2/NaNO2. | |
5. Photoinduced ipso-nitration
Photocatalytic systems have attracted much attention in recent years. Such systems are important because they are environmentally friendly and use light sources such as sunlight, thus reducing energy consumption. The reaction proceeds via a radical mechanism in photocatalytic systems by creating an electron–hole system. Photoinduced radical reactions are a new mechanism that allows us to explain the formation of organic products through photocatalytic systems.101,102
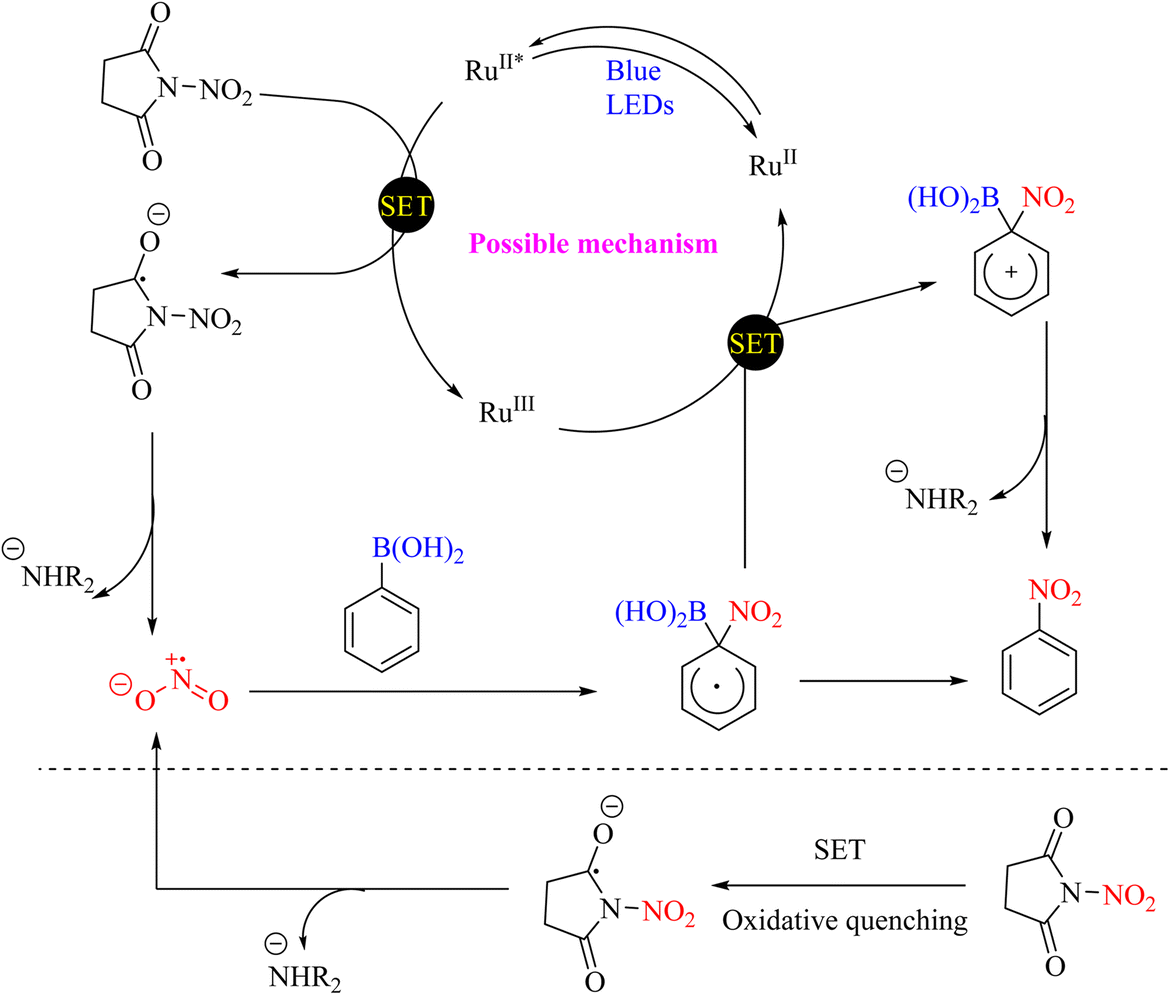
In 2020, photocatalytic and metal-free protocols were used to access various nitroaromatic and heteroaromatic compounds via internal ipso-nitration of boronic acid derivatives. In this method, non-metallic, stable and recyclable nitrating reagents were developed. The proposed methodologies are simple, gentle, and regionally selective in operation, compatible with various functional groups, and provide the desired products with high efficiency (Scheme 80).103 The reaction mechanism is shown in Scheme 81.
 |
| | Scheme 80 Photocatalytic protocols for accessing the ipso-nitration of boronic acid derivatives. | |
 |
| | Scheme 81 Possible mechanism underlying the photocatalytic and metal-free protocols for accessing the ipso-nitration of boronic acid derivatives. | |
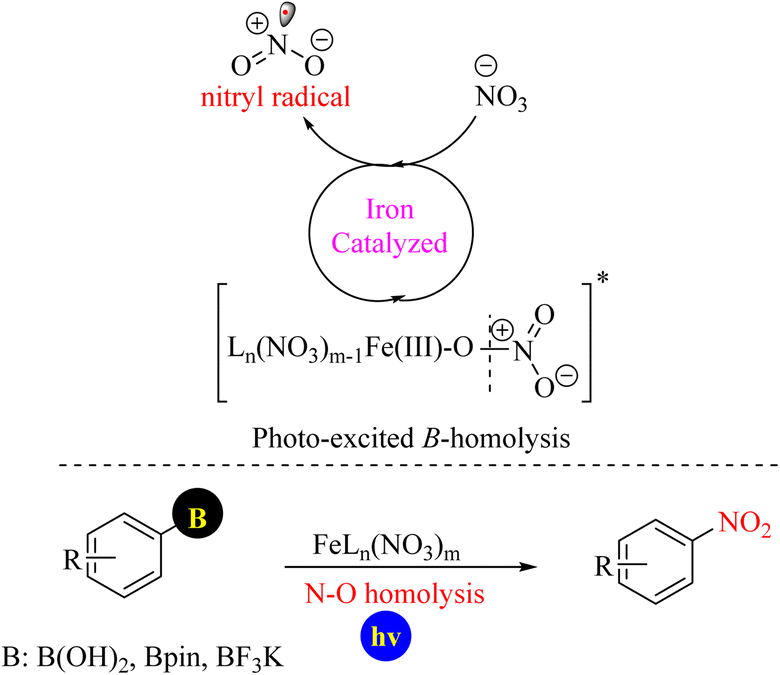
In 2025, a practical method for realizing the internal nitration of boronic acids, their corresponding pinacol esters, and trifluoroborate salts with the alkali metal salt NaNO3 as a nitrating reagent using a photocatalytic method with an Fe(III)/Fe(IV) catalytic cycle was reported (Scheme 82).104
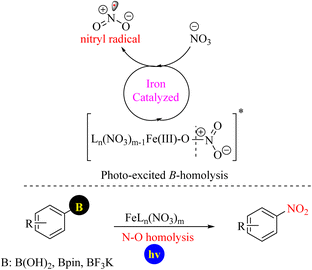 |
| | Scheme 82 Photocatalytic protocols for accessing the ipso-nitration of boronic acid derivatives using Fe(III)/Fe(IV) catalytic cycle. | |
In another recent report, a facile and generally applicable procedure for the nitration of boronic ipso esters by Fe(NO3)3·9H2O in hexafluoroisopropanol (HFIP) was reported. Nitroarene products were obtained under better and faster conditions than those obtained using the electrophilic C–H nitration method. This new protocol has shown that organoboron reagents with less reactivity and greater stability can be used to produce a wide range of these compounds (Scheme 83).105
 |
| | Scheme 83 Ipso-nitration of boronic esters enabled by Fe(NO3)3·9H2O in HFIP. | |
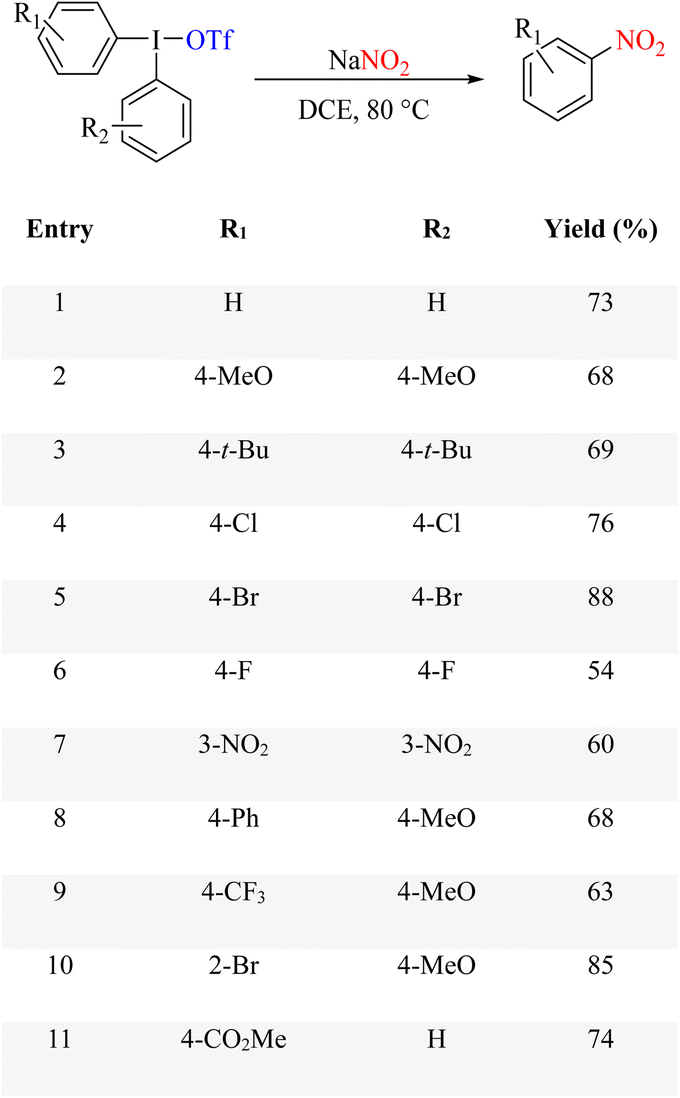
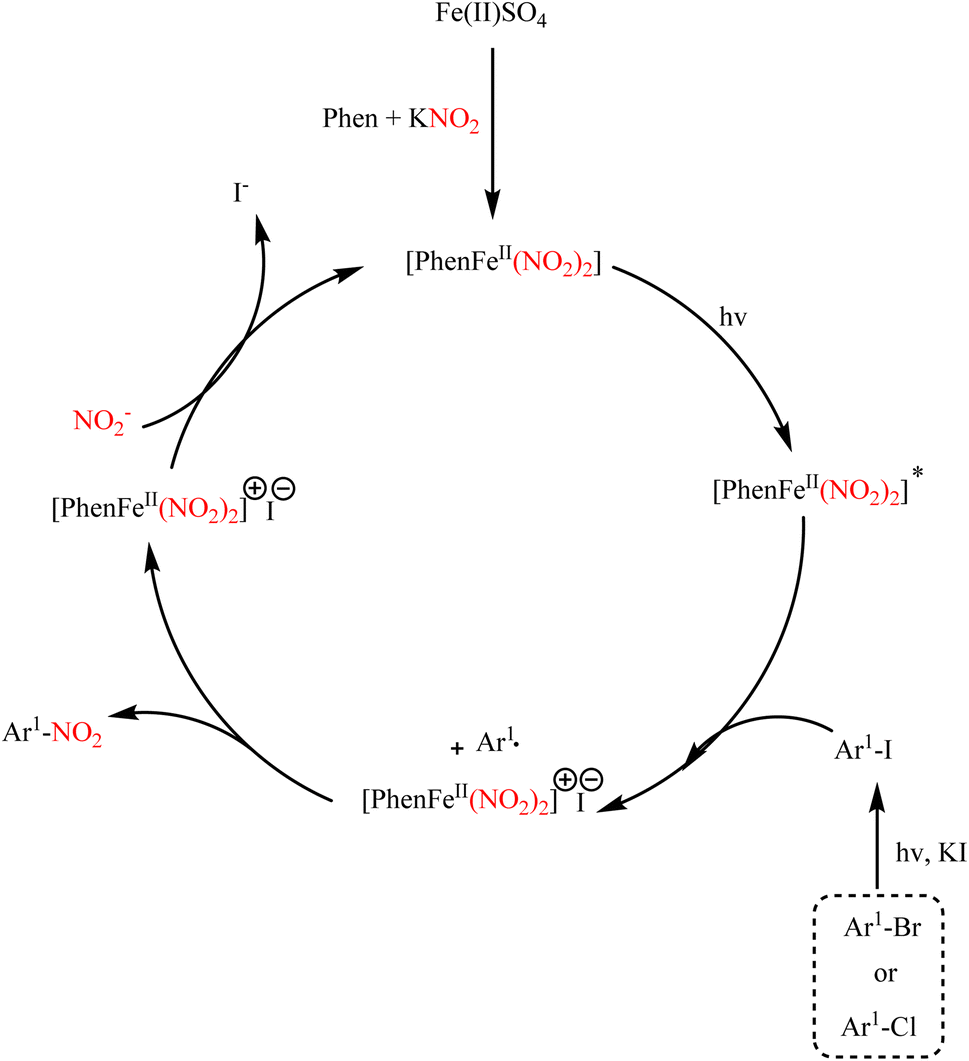
Ipso-nitration of various aryl halides in the presence of iron catalysis with KNO2 has been established under photocatalytic conditions, where aryl iodides, bromides, and some aryl chlorides have been converted to their nitro products. In this report, it is pointed out that the iron complex at the site formed by FeSO4, KNO2, and 1,10-phenanthroline acts as a photocatalyst, and the reaction undergoes a light-induced single-electron transfer (SET). The reaction conditions and progression run are exemplary of light-induced iron-catalyzed Ullmann-type couplings (Scheme 84).106 The reaction mechanism is shown in Scheme 85.
 |
| | Scheme 84 Ipso nitration of various aryl halides in the presence of iron catalysis. | |
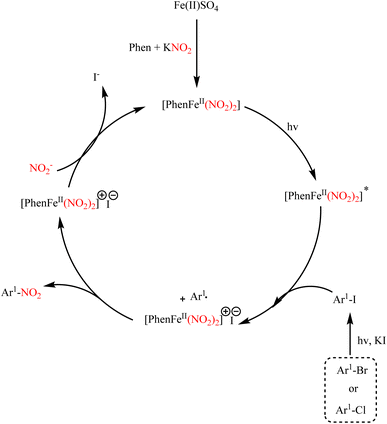 |
| | Scheme 85 Proposed mechanism underlying the ipso nitration of various aryl halides in the presence of iron catalysis. | |
As mentioned above, different systems are available for performing the ipso-nitration of halogen compounds. One of the best-known systems is photocatalytic systems, for which several examples have been presented. Another report used an Ag/g-C3N4 photocatalytic system to perform this process. The source of the nitro group in this report was sodium nitrite. In this case, the compounds of bromophenol are involved in the ipso-nitration reaction. The presence of g-C3N4 increased the electron transfer and product yield (Scheme 86).107
 |
| | Scheme 86 Ipso nitration of bromophenols in a photocatalytic system. | |
6. Ipso-nitration of aldehydes [ipso-CHO] and ketones [ipso-COMe]
Aldehyde and ketone groups form a large and important group of organic compounds. This group of factors is present in the structures of many organic and medicinal compounds. Many activities of organic compounds are influenced by aldehydes and ketones. Aldehydes and ketones typically play the role of electrophiles in their structures because of their carbonyl groups, which react with nucleophiles. These compounds can perform ipso–substitution reactions well. One of these reactions is the ipso-nitration substitution reaction, in which the aldehyde and ketone groups are removed from the compound structure and replaced by a nitro group. This activity shows that this compound can be a very good group leaving. This property of aldehydes and ketones can be exploited to prepare many nitro compounds using this method. Examples of the internal nitration of aldehyde and ketone groups are provided in the articles. This report highlights the most important internal nitration methods, such as aldehyde and ketone removal. Reports have indicated that the ipso-nitration reaction on aldehyde and ketone often occurs through a cationic mechanism in the presence of nitric acid (Scheme 87).
 |
| | Scheme 87 General methods for the ipso-nitration of CHO and COMe as leaving groups. | |
In 1974 and 1980, various research groups designed electrophilic reactions to perform ipso–substitution reactions using heterocyclic aldehydes such as thiophene aldehydes. They used nitric acid and sulfuric acid or acetic anhydride as nitro sources. The mechanism of this reaction shows that by adding nitric acid to the raw materials, the aldehyde group is removed from the system, and the nitro group is added to the system through a cationic reaction, which is the same as the ipso-nitration reaction (Scheme 88).108,109
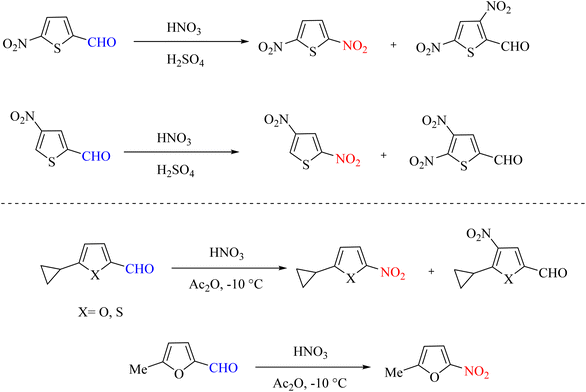 |
| | Scheme 88 Ipso-nitration reaction in aldehyde-containing heterocycles. | |
Another report investigated the synthesis of benzothiazole via the internal replacement of ortho-methoxy thiobenzamide. In one of the synthesis steps of these compounds, nitric acid was used for the ipso-nitration reaction on the aldehydes. This underscores the importance of such reactions for switching agent groups to reach the target molecules. This result also shows that CHO groups can be good leaving groups (Scheme 89).110
 |
| | Scheme 89 Synthesis of benzothiazoles via ipso substitution of ortho-methoxy thiobenzamides. | |
In independent studies, an electrophilic reaction involving acetophenones has been reported. These reports indicate that the addition of nitric acid, along with sulfuric acid, among others, generates nitro-cations. This subsequently reacts with the carbonyl ketone group, leading to the elimination of the ketone group. Such reactions present a novel approach to replacing ipso-nitration (Schemes 90 and 91).111
 |
| | Scheme 90 Electrophilic nitration of electron-rich acetophenones. | |
 |
| | Scheme 91 Electrophilic nitration of electron-rich acetophenones. | |
7. Ipso-nitration of esters [ipso-CO2Me]
An ester is a chemical compound derived from a carboxylic acid in which an alkoxy (RO) group replaces a hydroxyl (OH) group. They are important in biology because they are one of the major classes of lipids and comprise the bulk of animal fats and vegetable oils. Esters generally possess a pleasant aroma; those with low molecular weights are frequently employed as fragrances and are present in essential oils and pheromones. This group of factors is present in the structures of many organic and medicinal compounds. Esters commonly act as electrophiles in their structure owing to their carbonyl groups and readily react with nucleophiles. They exhibit a propensity to perform ipso substitution reactions effectively. One such reaction is the ipso-nitration substitution reaction, in which the ester group is displaced from the compound structure and replaced by a nitro group. Reports suggest that the ipso-nitration reaction on esters often proceeds via a cationic mechanism in the presence of nitric acid (Scheme 92).
 |
| | Scheme 92 General methods for the ipso-nitration of CO2Me as a leaving group. | |
In the quantum chemical studies performed on pyrimidine, nitric acid was used to synthesize the compounds to perform the ipso-nitration reaction. During this reaction, nitro cations are produced by adding nitric acid and sulfuric acid, which replace the leaving group. The leaving group in these studies was ester. This suggests that ester groups can be converted to good leaving to perform the ipso reaction (Scheme 93).112
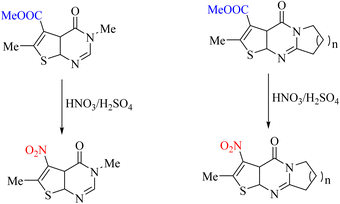 |
| | Scheme 93 Ipso-nitration reaction of an ester as a leaving group. | |
8. Ipso-nitration of [MR3]
The importance of performing ipso reactions has been well demonstrated. To perform the ipso reaction, it is necessary to have a suitable leaving group in the composition structure. The alkyl groups on the structure do not have good leaving power, so we need to convert them into good leaving compounds. The importance of Ge and Si in this case becomes more evident. Ge and Si were placed between the two elements to allow the alkyl groups to leave. Like carbonyl groups, the reaction mechanism proceeds through a cationic intermediate (Scheme 94).
 |
| | Scheme 94 General methods for the ipso-nitration of MR3 as a leaving group. | |
This survey was conducted in 1988. In this report, nitrate salt was subjected to special temperature conditions to perform the electrophilic substitution reaction of ipso-nitration on thiophene derivatives. Under these conditions, the ipso-nitration product is created as the main product (Scheme 95).113
 |
| | Scheme 95 Ipso-nitration of [MR3] as a leaving group using nitrate salt. | |
This type of ipso-nitration reaction can also be conducted using nitric acid instead of nitrate salts. The importance of ipso-nitration has been demonstrated in this reaction, and the main products obtained from the replacement of the ipso reaction have been demonstrated (Scheme 96).114
 |
| | Scheme 96 Ipso-nitration of [MR3] as a leaving group using nitric acid. | |
The development of nitroaromatic compounds is of great interest due to their importance for the synthesis of intermediates, preparation of pharmaceutical compounds, and production of materials on laboratory and industrial scales. In 2023, Mosiagin and co-workers reported an efficient, mild, and catalytic ipsonitration of organotrimethylsilane compounds. In this report, N-nitrosaccharin reagents were used to play a key role. This method is orthogonal to other common functionalizations and allows for the programming of molecular complexity through sequential transformations or final-step nitration. In this report, it is demonstrated that the mechanism proceeds via classical aromatic electrophilic substitution with a suitable and orderly transition state. In this study, the claimed cases were experimentally and theoretically proven to demonstrate the importance of the proposed method. In this report, various arylsilanes, as well as various reagents, were used, the efficiency of which indicated that N-nitrosaccharin had better conditions for the reaction to progress (Scheme 97).115
 |
| | Scheme 97 Catalytic ipso-nitration of organosilanes using an electrophilic N-nitrosaccharin reagent. | |
9. Ipso-nitration of trifluoromethane sulfonate [ipso-OTf]
The hydroxy functional group is crucial to the structure of organic and medicinal compounds. Its presence is often challenging to eliminate due to resonance effects with aromatic rings. Structural modifications are necessary to facilitate its removal. An effective approach involves the conversion of the hydroxy group to the trifluoromethane sulfonate group. Following this conversion, the resulting OTf group exhibits high stability and can be readily removed from the structure. This method creates suitable conditions for the ipso-nitration reaction, facilitating the production of nitro compounds. The anionic reaction mechanism can be carried out using nitrate salts (Scheme 98).
 |
| | Scheme 98 General methods for the ipso-nitration of OTf as a leaving group. | |
In a study conducted in 2013, researchers investigated the asymmetric catalytic benzidine rearrangement. In this method, the initial step involved converting the hydroxy groups to nitro groups to prepare the target compounds. Initially, the hydroxy group was converted to an OTf group to facilitate its removal. Subsequently, the ipso-nitration reaction was carried out using palladium metal and sodium nitrite salt (Scheme 99).116
 |
| | Scheme 99 Ipso-nitration of OTf as a leaving group in the catalytic asymmetric benzidine rearrangement. | |
10. Ipso-nitration of sulfonic acids [ipso-SO3H]
Sulfonic acid refers to a group of sulfur organic compounds with the general formula RSO3H, in which R is an alkyl or aryl organic group and the SO3H group is a sulfonyl hydroxide. A sulfonic acid can be considered a sulfuric acid in which an organic substituent replaces a hydroxyl group. The sulfonic functional group is important for the structure of these acids. These compounds can be considered for ipso reactions because the SO3H group exits relatively well. The cationic reaction mechanism can proceed with nitric acid (Scheme 100).
 |
| | Scheme 100 General methods for the ipso-nitration of sulfonic acid [SO3H] as a leaving group. | |
Numerous studies have been conducted in this area. One such study focused on synthesizing various heterocyclic compounds using nitric acid and employing a cationic mechanism, and the SO3H group was converted to a nitro group (Schemes 101–103).117–119
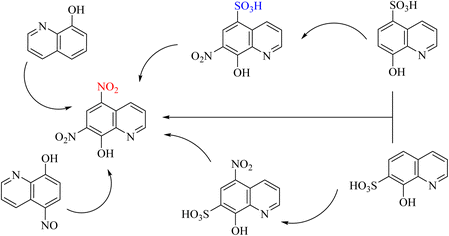 |
| | Scheme 101 Ipso-nitration of sulfonic acid [SO3H] as a leaving group in the thermal fragmentation of nitrated 8-quinolines. | |
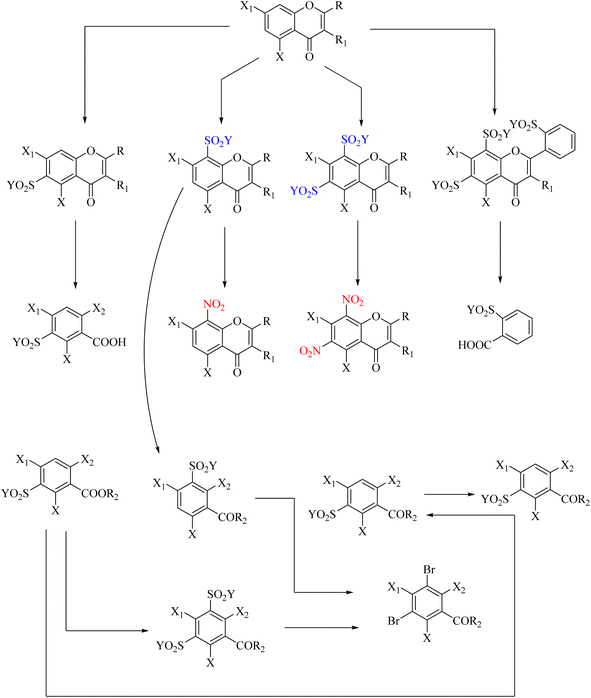 |
| | Scheme 102 Ipso-nitration of sulfonic acid [SO3H] as a leaving group in the substitution reaction of benzopyrone. | |
 |
| | Scheme 103 Ipso-nitration of sulfonic acid [SO3H] as the leaving group in synthesis and aldose reductase-inhibitory activity of benzo[b]furan derivatives. | |
11. Ipso-nitration of alkyl [Ipso-R]
The substitution of the nitro group with different functional groups has been explored. In most ipso-nitration reactions, various metals are required to convert functional groups into suitable leaving groups. Alkyl groups in the structure typically have poor leaving ability, but under appropriate conditions, such as the presence of concentrated nitric acid, the ipso-nitration reaction can occur. In this reaction type, the alkyl group replaced the nitro cation (Scheme 104). This reaction can occur in the structures of normal molecules and macromolecules.
 |
| | Scheme 104 General methods for the ipso-nitration of alkyl [R] as a leaving group. | |
11.1. Macromolecule
In 1977, Zheng's research group synthesized several calix[4] crowns using potassium iodide and potassium carbonate salts in acetone and toluene solvents. They further performed the ipso-nitration reaction on these compounds using nitric acid and acetic acid in a dichloromethane solvent and replaced the nitro group with a tert-butyl group (Scheme 105).120
 |
| | Scheme 105 Ipso-nitration of calix[4] crowns. | |
The ipso-nitration of p-tert-butylcalix[4] arenes was reviewed in 1992. In this research, an ipso-nitration reaction was performed using concentrated nitric acid. Reactions are often reported in high yields (Scheme 106).121
 |
| | Scheme 106 Ipso-nitration of p-tert-butylcalix[4] arenes. | |
In 1998, research was conducted on the nitration of tert-butylcalix[4] arene tetraethers and their subsequent hydrogenation (Scheme 107).122 In this reaction, the reaction with di- and tri-acid chlorides leads to different di- and tri-calix arenes. The ipso-nitration stage of this reaction was carried out in the presence of nitric acid and acetic acid in dichloromethane solvent at ambient temperature.
 |
| | Scheme 107 Ipso-nitration of t-butyl calix[4] arene. | |
In a similar report, the synthesis and nitration of calix[41](aza)crowns was conducted in 2000 (Scheme 108).123 In this reaction, HNO3 (65%) and AcOH perform the ipso-nitration reaction. During the nitration reaction, the nitro group replaced the alkyl group. Some products include two nitro groups, whereas others include only one nitro group. As a result, various products have been reported during this reaction, but the ipso-nitration products are well-formed.
 |
| | Scheme 108 Synthesis and nitration of calix[41](aza)crowns. | |
The synthesis of p-tert-butylcalix[4] arene containing polymerizable groups was reported by Vocanson et al. During this reaction, a calixarene monomer with a methacryloyloxy group on the phenolic rim is copolymerized with methyl methacrylate, resulting in linear polymers with pendant calixarene groups in a conical composition. Subsequently, the chemical modification of these macrocycles through ipso-nitration is described. This report demonstrated that nitration can also be conducted on compounds containing polymer groups (Scheme 109).124
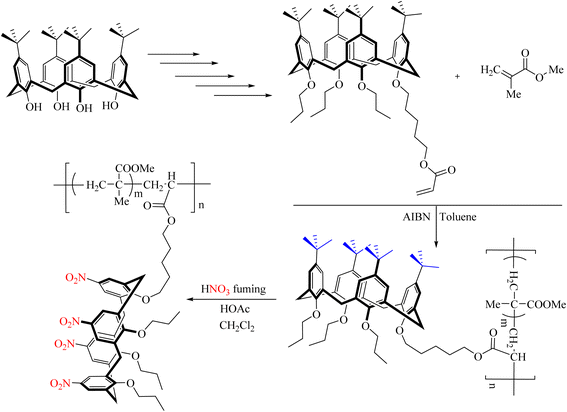 |
| | Scheme 109 Ipso-nitration in new calixarene copolymer. | |
In another study on the continuation of ipso-nitration reactions in macromolecules, in 2004, a report was presented by Daniela et al. By performing the ipso-nitration reaction, they prepared the compounds for other reactions. Nitro compounds can be reduced, and by converting the nitro group to an amine, new structures can be synthesized to create many macromolecule compounds for various synthetic purposes (Schemes 110 and 111).125
 |
| | Scheme 110 Ipso-nitration of 1,3-alternate calix[4] arenes. | |
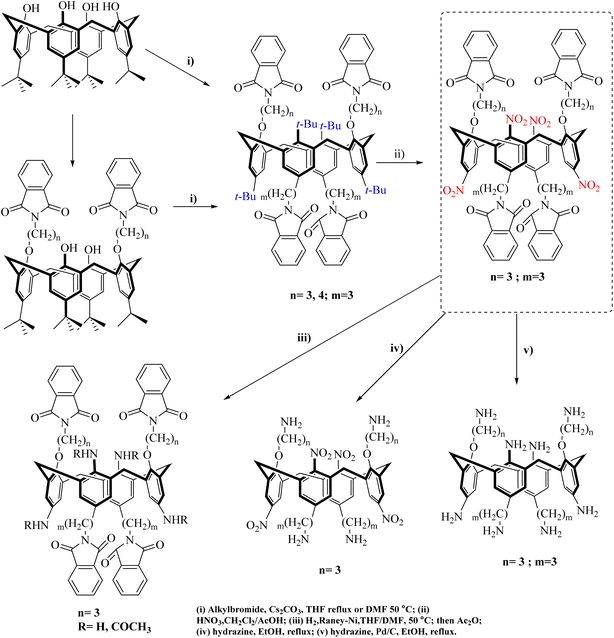 |
| | Scheme 111 Ipso-nitration of 1,3-alternate calix[4] arenes. | |
Jabin et al. prepared the first C3v and D3h-symmetrical triply bridged calix[6] azatubes in good yields from 1,3,5-tris-methylated calix6 arene via an efficient [1 + 1] macrocyclization reaction. They showed that such a reaction occurs during a hexa-ipso-nitration reaction that is alternately replaced at the wide rim in an alternate position by tert-butyl and nitro groups (Scheme 112).126
 |
| | Scheme 112 Synthesis and conformational study of the first triply bridged calix[6] azatubes. | |
In 2012, a new protection/deprotection method on calix[4] arene compounds was described. Introduction of nosyl (p-nitrobenzene sulfonyl) groups to the lower rim of the tert-butylcalix[4] arenes subsequently led to the selective nitration of the alkylated phenol rings, whereas the protecting groups could be easily removed in a subsequent step. The above substitution pattern is complementary to the isomers known so far in calix[4] arene chemistry and can be used in the design of a new type of calixarene-based receptors (Scheme 113).127
 |
| | Scheme 113 Regioselective ipso-nitration of calix[4] arenes. | |
The ipso-nitration of molecular receptors based on calix6 arene compounds is an important route for developing more complex systems. This reaction was studied by Reinaud et al. for various capped calixarenes in 2012. They showed a general trend for regioselective nitration with medium to high efficiency. This selectivity was partially attributed to the electronic coupling between the protonated cap on the small rim and the reactive sites on the large rim (Scheme 114).128
 |
| | Scheme 114 Ipso-nitration of molecular receptors based on calix[6] arene compounds. | |
In recent reports, many ipso-nitration of macromolecular compounds has caused the conversion of some alkyl groups into nitro groups. In 2014, a report showed that the ipso-nitration reaction can be performed on all alkyl groups, and substituting nitro with six bulky tert-butyl groups greatly influences the behavior of the obtained structure (Scheme 115).129
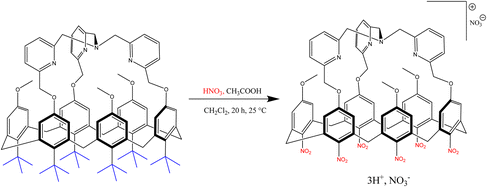 |
| | Scheme 115 A mechanical pivoting process of aromatic walls in the host-guest chemistry of calix[6] arene aza-cryptands. | |
A similar reaction was reported in 2014 by Yamato et al., who used nitric acid and acetic acid to perform an ipso-nitration reaction (Scheme 116).130 All alkyl groups were removed from the structure in this reaction, and the nitro groups were replaced.
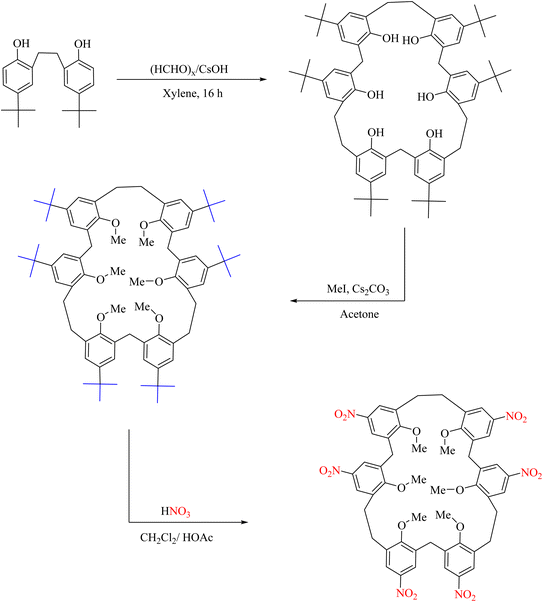 |
| | Scheme 116 Ipso-nitration of hexamethoxy[(2.1)3]metacyclophane. | |
11.2. Alkyl as the leaving group
In 1977, Yamato's research group used various nitrating reagents to perform the ipso-nitration reaction on compounds 5,13-di-tert-butyl-8,16-dimethoxy[2.2]MCP. In this reaction, the tert-butyl group is removed from the structure using nitro cations, and in fact, the tert-butyl group becomes a good leaving group (Scheme 117).131
 |
| | Scheme 117 Ipso-nitration of 5,13-di-tert-butyl-8,16-dimethoxy[2.2]MCP. | |
Nitrodealkylation, which is the electrophilic replacement of an alkyl group in an aromatic ring with a nitro group, is often associated with the nitration of para- and ortho-dialkylbenzenes. This process is usually a side reaction. Nitration reactions often occur in dialkylbenzenes containing isopropyl or tert-butyl groups. In 2000, research was conducted on replacing the nitro group with cyclopropylmethyl in para-substituted benzylcyclopropanes, in which the nitro group was replaced with a nitric acid reagent in acetic anhydride at −50 °C (Scheme 118).132
 |
| | Scheme 118 Uncommonly easy nitrodealkylation during the nitration of para-substituted benzylcyclopropanes. | |
Compounds [3.3] and [3.3.3] metacyclophanes are a structure in which various tert-butyl groups are present. In ipso-nitration, which is performed on this structure, it is often possible to replace tert-butyl groups with nitro groups using different nitrating reagents. The number of these substitutions can be controlled by selecting an appropriate reagent (Scheme 119).133
 |
| | Scheme 119 Nitration of [3.3] and [3.3.3]metacyclophanes. | |
The electrophilic reactions of tert-butylcalix6 arene were studied in 2003. In this study, it was demonstrated that ipso-nitration occurs selectively in calixarene anisole units. The process selectivity is related to the presence of protonable sites. The corresponding protonated heteroatom (N for amines, O for amides, and carboxylic acid) deactivates the corresponding aromatic ring by reducing electron density through intramolecular hydrogen bonding (Scheme 120).134
 |
| | Scheme 120 Unprecedented selective ipso-nitration of calixarenes monitored by O-substituents. | |
The selective ipso-nitration of tert-butylthiacalix[4] arene-tetrasulfone to produce thiacalixarene derivatives bearing one or two arylureido functions on the upper rim was reported in 2006. In this study, it was shown that the pre-organization of ureido units using thiacalix[4] arene moiety as a molecular scaffold led to new anion receptors with good complexation ability towards selective anions with different geometries (Scheme 121).135
 |
| | Scheme 121 Ipso-nitration in anion receptors based on ureido-thiacalix[4] arenes. | |
When preparing target compounds, accessing precursors from which the product can be derived in organic synthesis is often challenging. In such cases, substitution reactions play a crucial role. In a 2011 report, for synthesizing the monomer 2,2-diamino-7-tert-butyl-9,9-spirobifluorene, the ipso-nitration reaction was employed as one of the reaction stages. Subsequently, the target monomer was obtained by reducing the number of nitro groups. This highlights the significance of ipso-nitration in synthetic pathways (Scheme 122).136
 |
| | Scheme 122 Ipso-nitration in the synthesis of monomer 2,2-diamino-7-tert-butyl-9,9-spirobifluorene. | |
12. Conclusion
The ipso strategy for selective nitration is a well-established method for synthesizing valuable nitro aromatics and olefins with specific chemo- and regioselectivity. This approach uses a nitrating agent to replace a preferred group leaving within the molecule. Researchers have explored various new nitrating agents, including NaNO2, HNO3/H2SO4, and nitrite salt, to enhance the nitration of organic compounds. To address these issues, innovative ipso-nitration protocols have been developed, such as the nitro demetallation of aryl boronic acids, nitro decarboxylation of aryl carboxylic acids, and transition-metal-catalyzed transformations involving aryl halides and pseudohalides like –SO3H, –OR, –R, –X, –CHO, –COMe, –CO2Me, –MR3 and –BF3K. In addition, direct nitration of aromatic C–H bonds using transition-metal catalysts offers a promising alternative to traditional nitration methods. If the reagent used is not efficient and does not effectively perform the desired reaction, or if the leaving group is difficult to remove from the structure, undesirable reactions may occur and internal nitration may be hindered. Given the importance of nitro compounds due to their corresponding energetic properties, safety precautions are essential during their synthesis and handling. Currently, with the use of safer nitrate reagents, this has become increasingly important, and various methods for synthesizing these compounds have been developed. Furthermore, in industries, efforts to industrialize these compounds have intensified because of the growing emphasis on environmental protection and energy consumption as well as the need for safe and mild reagents.
Data availability
Data are available upon request from the authors.
Author contributions
H. S. and M. Z: investigation and writing of the original draft. M. A. Z.: supervision, resources, project administration, funding acquisition, conceptualization, and writing – review. S. M. and F. T.: final sorting, rewriting and editing of the text.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
We thank the Bu-Ali Sina University and the University of Qom for providing financial support for this review.
References
- R. Parry, S. Nishino and J. Spain, Nat. Prod. Rep., 2011, 28, 152–167 RSC.
- R. Winkler and C. Hertweck, ChemBioChem, 2007, 8, 973–977 CrossRef.
-
(a) H. Sepehrmansourie, M. Zarei, M. A. Zolfigol, A. Mehrzad and H. R. Hafizi-Atabak, RSC Adv., 2021, 11, 8367–8374 RSC;
(b) J. P. Agrawal, Organic Chemistry of Explosives, John Wiley & Sons, vol. 302, 2007 Search PubMed.
- H. Ghaderi, M. A. Zolfigol, Y. Bayat, M. Zarei and E. Noroozizadeh, Synlett, 2016, 27, 2246–2250 CrossRef.
- M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare, A. Zare, H. G. Kruger, Z. Asgari, V. Khakyzadeh and M. Kazem-Rostami, J. Org. Chem., 2012, 77, 3640–3645 CrossRef.
- G. Yan, A. J. Borah and L. Wang, Org. Biomol. Chem., 2014, 12, 6049–6058 RSC.
- C. E. Barnes and P. C. Myhre, J. Am. Chem. Soc., 1978, 100, 973–975 CrossRef CAS.
- P. Li and X. Jia, Synthesis, 2018, 50, 711–722 CrossRef CAS.
- G. Yan and M. Yang, Org. Biomol. Chem., 2013, 11, 2554–2566 RSC.
- S. Patra, I. Mosiagin, R. Giri and D. Katayev, Synthesis, 2022, 54, 3432–3472 CrossRef CAS.
- J. I. Murray, M. V. Silva Elipe, K. D. Baucom, D. B. Brown, K. Quasdorf and S. Caille, J. Org. Chem., 2021, 87, 1977–1985 CrossRef PubMed.
- D. M. Badgujar, M. B. Talawar and P. P. Mahulikar, Propellants, Explos., Pyrotech., 2016, 41, 24–34 CrossRef CAS.
- S. Salzbrunn, J. Simon, G. S. Prakash, N. A. Petasis and G. A. Olah, Synlett, 2000, 10, 1485–1487 Search PubMed.
- K. Bozorov, J. Y. Zhao and H. A. Aisa, ARKIVOC: The Free Internet Journal for Organic Chemistry, 2017, I, 41–66 Search PubMed.
-
(a) S. Agasti, S. Maiti, S. Maity, M. Anniyappan, M. B. Talawar and D. Maiti, Polyhedron, 2019, 172, 120–124 CrossRef CAS;
(b) Z. Q. Wang, M. P. Guo, Y. J. Wen, X. L. Shen, M. Y. Lv and X. L. Zhou, Lett. Org. Chem., 2018, 15, 891–894 CrossRef CAS.
- A. Fischer, D. L. Fyles and G. N. Henderson, J. Chem. Soc. Chem. Commun., 1980, 11, 513–514 RSC.
-
(a) M. R. Amin, L. Dekker, D. B. Hibbert, J. H. Ridd and J. P. Sandall, J. Chem. Soc. Chem. Commun., 1986, 9, 658–659 RSC;
(b) R. G. Clewley, A. Fischer and G. N. Henderson, Can. J. Chem., 1989, 67, 1472–1479 CrossRef CAS.
-
(a) N. Chatterjee and A. Goswami, Adv. Synth. Catal., 2017, 359, 358–371 CrossRef CAS;
(b) Z. Q. Wang, M. P. Guo, Y. J. Wen, X. L. Shen, M. Y. Lv and X. L. Zhou, Lett. Org. Chem., 2018, 15, 891–894 CrossRef CAS.
-
(a) C. E. Barnes and P. C. Myhre, J. Am. Chem. Soc., 1978, 100, 975–976 CrossRef CAS;
(b) J. T. Geppert, M. W. Johnson, P. C. Myhre and S. P. Woods, J. Am. Chem. Soc., 1981, 103, 2057–2062 CrossRef CAS.
-
(a) F. Al-Omran, K. Fujiwara, J. C. Giffney, J. H. Ridd and S. R. Robinson, J. Chem. Soc., Perkin Trans., 1981, 2, 518–525 RSC;
(b) S. Salzbrunn, J. Simon, G. S. Prakash, N. A. Petasis and G. A. Olah, Synlett, 2000, 10, 1485–1487 Search PubMed.
-
(a) C. E. Barnes, K. S. Feldman, M. W. Johnson, H. W. H. Lee and P. C. Myhre, J. Org. Chem., 1979, 44, 3925–3930 CrossRef CAS;
(b) P. Helsby and J. H. Ridd, J. Chem. Soc., Perkin Trans., 1983, 2, 311–315 RSC.
-
(a) W. Verboom, A. Durie, R. J. Egberink, Z. Asfari and D. N. Reinhoudt, J. Org. Chem., 1992, 57, 1313–1316 CrossRef CAS;
(b) A. M. Dimiev and Y. M. Kargin, Russ. J. Gen. Chem., 1996, 66, 1831–1836 Search PubMed.
-
(a) P. Helsby and J. H. Ridd, J. Chem. Soc. Chem. Commun., 1980, 19, 926–927 RSC;
(b) S. U. Paik and M. G. Jung, Bull. Korean Chem. Soc., 2012, 33, 689–691 CrossRef CAS.
-
(a) A. H. Clemens, J. H. Ridd and J. P. Sandall, J. Chem. Soc. Chem. Commun., 1983, 7, 343–344 RSC;
(b) A. Fischer, G. N. Henderson, T. A. Smyth, F. W. Einstein and R. E. Cobbledick, Can. J. Chem., 1981, 59, 584–590 CrossRef CAS.
-
(a) S. Salzbrunn, J. Simon, G. S. Prakash, N. A. Petasis and G. A. Olah, Synlett, 2000, 10, 1485–1487 Search PubMed;
(b) S. Mochalov, M. Smirnova, V. Karpova and Y. S. Shabarov, Chem. Informationsdienst, 1985, 16 Search PubMed.
- G. S. Bapat, A. Fischer, G. N. Henderson and S. Raymahasay, J. Chem. Soc. Chem. Commun., 1983, 3, 119–120 RSC.
-
(a) A. Zweig, K. R. Huffman and G. W. Nachtigall, J. Org. Chem., 1977, 42, 4049–4052 CrossRef CAS;
(b) T. Yamato, K. Maeda, H. Kamimura, K. Noda and M. Tashiro, ChemInform, 1995, 26 Search PubMed.
- K. S. Feldman and P. C. Myhre, J. Am. Chem. Soc., 1979, 101, 4768–4769 CrossRef CAS.
- M. Shiri, M. Ali Zolfigol, H. Gerhardus Kruger and Z. Tanbakouchian, Tetrahedron, 2010, 66, 9077–9106 CrossRef CAS.
- M. A. Zolfigol, E. Madrakian, E. Ghaemi and K. Niknam, Synth. Commun., 2008, 38, 3366–3374 CrossRef CAS.
- M. A. Zolfigol, B. F. Mirjalili, A. Bamoniri, M. A. K. Zarchi, A. Zarei, L. Khazdooz and J. Noei, Bull. Kor. Chem. Soc., 2004, 25, 1414–1416 CrossRef CAS.
- M. A. Zolfigol, F. Shirini and A. G. Chogamarani, Phosphorus, Sulfur Silicon, 2003, 178, 2019–2025 CrossRef CAS.
- M. A. Zolfigol, E. Madrakian and E. Ghaemi, Synlett, 2003, 14, 2222–2224 CrossRef.
- M. A. Zolfigol, E. Ghaemi and E. Madrakian, Synlett, 2003, 2, 0191–0194 CrossRef.
- M. A. Zolfigol, E. Madrakian and E. Ghaemi, Molecules, 2002, 7, 734–742 CrossRef CAS.
- M. A. Zolfigol, E. Ghaemi and E. Madrakian, Molecules, 2001, 6, 614–620 CrossRef CAS.
- M. A. Zolfigol, E. Ghaemi and E. Madrakian, Synth. Commun., 2000, 30, 1689–1694 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and M. A. Zolfigol, Synth. Commun., 1997, 27, 3301–3311 CrossRef CAS.
-
(a) Z. Zeng, A. Feceu, N. Sivendran and L. J. Gooben, Adv. Synth. Catal., 2021, 363, 2678–2722 CrossRef CAS;
(b) V. Ramadoss, Y. Zheng, X. Shao, L. Tian and Y. Wang, Chem. –Eur. J., 2021, 27, 3213–3228 CrossRef CAS;
(c) J. L. Tu, Z. Shen and B. Huang, Adv. Synth. Catal., 2024, 366, 4263–4273 CrossRef CAS.
- J. Shi, X. Y. Huang, J. P. Wang and R. Li, J. Phys. Chem., 2010, 114, 6263–6272 CrossRef CAS.
- E. T. Denisov and A. F. Shestakov, Kinet. Catal., 2013, 54, 22–33 CrossRef CAS.
- J. R. Peterson, H. D. Do and A. J. Dunham, Can. J. Chem., 1988, 66, 1670–1674 CrossRef CAS.
- K. Pitchumani, P. Baskar and C. Venkatachalapathy, Catal. Lett., 1993, 21, 157–163 CrossRef CAS.
- J. P. Das, P. Sinha and S. Roy, Org. Lett., 2002, 4, 3055–3058 CrossRef CAS PubMed.
- A. K. Bose, S. N. Ganguly, M. S. Manhas, V. Srirajan, A. Bhattacharjee, S. Rumthao and A. H. Sharma, Tetrahedron Lett., 2004, 45, 1179–1181 CrossRef CAS.
- A. Messere, A. Gentili, I. Garella, F. Temussi, B. Di Blasio and A. Fiorentino, Synth. Commun., 2004, 34, 3317–3324 CrossRef CAS.
- A. S. Rao, P. V. Srinivas, K. S. Babu and J. M. Rao, Tetrahedron Lett., 2005, 46, 8141–8143 CrossRef CAS.
- A. K. Bose, S. N. Ganguly, M. S. Manhas, W. He and J. Speck, Tetrahedron Lett., 2006, 47, 3213–3215 CrossRef CAS.
- S. Ramgopal, K. Ramesh, A. Chakradhar, N. M. Reddy and K. C. Rajanna, Tetrahedron Lett., 2007, 48, 4043–4045 CrossRef CAS.
- G. I. Nikishin, L. L. Sokova, V. D. Makhaev and N. I. Kapustina, Russ. Chem. Bull., 2008, 57, 118–123 CrossRef CAS.
- J. Gong, K. Huang, F. Wang, L. Yang, Y. Feng, H. Li, X. Li, S. Zeng, X. Wu, J. Stockigt and Y. Zhao, Bioorg. Med. Chem., 2009, 17, 3414–3425 CrossRef CAS PubMed.
- M. S. Kumar, K. C. Rajanna, K. R. Reddy, M. Venkateswarlu and P. Venkanna, Synth. Commun., 2013, 43, 2672–2677 CrossRef CAS.
- B. V. Rokade and K. R. Prabhu, Org. Biomol. Chem., 2013, 11, 6713–6716 RSC.
- M. S. Kumar, K. R. Reddy, K. C. Rajanna, P. Venkanna and G. Krishnaiah, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 977–983 CrossRef CAS.
- S. Manna, S. Jana, T. Saboo, A. Maji and D. Maiti, Chem. Commun., 2013, 49, 5286–5288 RSC.
- V. S. Chary, K. C. Rajanna, G. Krishnaiah and P. Srinivas, Catal. Sci. Technol., 2016, 6, 1430–1434 RSC.
- Q. Xu, H. Qi, M. Sun, D. Zuo, X. Jiang, Z. Wen, Z. Wang, Y. Wu and W. Zhang, PLoS One, 2015, 10, e0128710 CrossRef PubMed.
- M. Satish Kumar, K. C. Rajanna, M. Venkateswarlu, P. Venkanna and P. K. Saiprakash, Synth. Commun., 2015, 4, 2251–2258 CrossRef.
- P. Natarajan, R. Chaudhary and P. Venugopalan, J. Org. Chem., 2015, 80, 10498–10504 CrossRef CAS.
- C. S. Azad, V. M. Balaramnavar, I. A. Khan, P. K. Doharey, J. K. Saxena and A. K. Saxena, RSC Adv., 2015, 5, 82208–82214 RSC.
- Z. Yang, J. Li, J. Hua, T. Yang, J. Yi and C. Zhou, Synlett, 2017, 28, 1079–1082 CrossRef CAS.
- C. S. Azad and A. K. Narula, RSC Adv., 2016, 6, 19052–19059 RSC.
- Z. G. Luo, F. Xu, Y. Y. Fang, P. Liu, X. M. Xu, C. T. Feng, Z. Li and J. He, Res. Chem. Intermed., 2016, 42, 6079–6087 CrossRef CAS.
- S. Roshandel, L. Gurung, T. Mathew and G. S. Prakash, Tetrahedron Lett., 2017, 58, 2842–2845 CrossRef CAS.
- L. F. Nie, K. Bozorov, G. Huang, J. Zhao, C. Niu and H. A. Aisa, Phosphorus Sulfur Silicon Relat. Elem., 2018, 193, 656–667 CrossRef CAS.
- B. A. Mir, S. J. Singh, R. Kumar and B. K. Patel, Adv. Synth. Catal., 2018, 360, 3801–3809 CrossRef CAS.
- P. Natarajan, R. Chaudhary and P. Venugopalan, Tetrahedron Lett., 2019, 60, 1720–1723 CrossRef CAS.
- N. Mutkule, N. Bugad, S. Mokale, V. Choudhari and M. Ubale, J. Heterocycl. Chem., 2020, 57, 3186–3192 CrossRef CAS.
- Hall, D. G., Ed. Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, John Wiley & Sons, ( 2006) Search PubMed.
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC.
- T. Mathew, L. Gurung, S. Roshandel, S. B. Munoz and G. S. Prakash, Synlett, 2019, 30, 1037–1047 CrossRef CAS.
- I. V. Alabugin, L. Kuhn, M. G. Medvedev, N. V. Krivoshchapov, V. A. Vil, I. A. Yaremenko, P. Mehaffy, M. Yarie, A. O. Terent’ev and M. A. Zolfigol, Chem. Soc. Rev., 2021, 50, 10253–10345 RSC.
- I. V. Alabugin, L. Kuhn, N. V. Krivoshchapov, P. Mehaffy and M. G. Medvedev, Chem. Soc. Rev., 2021, 50, 10212–10252 RSC.
- M. A. Zolfigol, S. Azizian, M. Torabi, M. Yarie and B. Notash, J. Chem. Educ., 2024, 101, 877–881 CrossRef CAS.
- G. S. Prakash, C. Panja, T. Mathew, V. Surampudi, N. A. Petasis and G. A. Olah, Org. Lett., 2004, 6, 2205–2207 CrossRef CAS.
- R. R. Yadav, R. A. Vishwakarma and S. B. Bharate, Tetrahedron Lett., 2012, 53, 5958–5960 CrossRef CAS.
- L. Bougdid, A. Heynderickx, S. Delbaere and C. Moustrou, Tetrahedron, 2007, 63, 8242–8249 CrossRef CAS.
- X. F. Wu, J. Schranck, H. Neumann and M. Beller, Chem. Commun., 2011, 47, 12462–12463 RSC.
- H. Yang, Y. Li, M. Jiang, J. Wang and H. Fu, Chem. –Eur. J., 2011, 17, 5652–5660 CrossRef CAS PubMed.
- S. Manna, S. Maity, S. Rana, S. Agasti and D. Maiti, Org. Lett., 2012, 14, 1736–1739 CrossRef CAS PubMed.
- S. Wang, C. C. Shu, T. Wang, J. Yu and G. B. Yan, Chin. Chem. Lett., 2012, 23, 643–646 CrossRef CAS.
- G. Yan, L. Zhang and J. Yu, Lett. Org. Chem., 2012, 9, 133–137 CrossRef CAS.
- A. C. Lindsay and J. Sperry, Synlett, 2013, 24, 461–464 CrossRef CAS.
- M. Jiang, H. Yang, Y. Li, Z. Jia and H. Fu, RSC Adv., 2013, 3, 25602–25604 RSC.
- G. S. Prakash, L. Gurung, P. C. Schmid, F. Wang, T. E. Thomas, C. Panja, T. Mathew and G. A. Olah, Tetrahedron Lett., 2014, 55, 1975–1978 CrossRef CAS.
- N. Chatterjee, D. Bhatt and A. Goswami, Org. Biomol. Chem., 2015, 13, 4828–4832 RSC.
- G. Shen, L. Zhao, W. Liu, X. Huang, H. Song and T. Zhang, Synth. Commun., 2017, 47, 10–14 CrossRef CAS.
- A. Mahanta, N. K. Gour, P. J. Sarma, R. K. Borah, P. K. Raul, R. C. Deka, A. J. Thakur and U. Bora, Appl. Organomet. Chem., 2019, 33, e4951 CrossRef.
- M. Zarei, E. Noroozizadeh, A. R. Moosavi-Zare and M. A. Zolfigol, J. Org. Chem., 2018, 83, 3645–3650 CrossRef CAS.
- J. I. Murray, D. B. Brown, M. V. Silva Elipe and S. Caille, Org. Process Res. Dev., 2022, 26, 657–660 CrossRef CAS.
- M. Al-Masum, N. Saleh and T. Islam, Tetrahedron Lett., 2013, 54, 1141–1144 CrossRef CAS.
- S. Saito and Y. Koizumi, Tetrahedron Lett., 2005, 46, 4715–4717 CrossRef CAS.
- P. LaBeaume, M. Placzek, M. Daniels, I. Kendrick, P. Ng, M. McNeel, R. Afroze, A. Alexander, R. Thomas, A. E. Kallmerten and G. B. Jones, Tetrahedron Lett., 2010, 51, 1906–1909 CrossRef CAS.
- S. U. Paik and M. G. Jung, Bull. Korean Chem. Soc., 2012, 33, 689–691 CrossRef CAS.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 12898–12899 CrossRef CAS.
- P. A. Joseph, S. Priyadarshini, M. L. Kantam and H. Maheswaran, Tetrahedron Lett., 2012, 53, 1511–1513 CrossRef.
- X. H. Li, L. G. Li, X. L. Mo and D. L. Mo, Synth. Commun., 2016, 46, 963–970 CrossRef CAS.
- M. Reitti, P. Villo and B. Olofsson, Angew. Chem., 2016, 128, 9074–9078 CrossRef.
- G. A. Lee, H. C. Lin, H. Y. Lee, C. H. Chen and H. Y. Huang, Asian J. Org. Chem., 2017, 6, 1733–1736 CrossRef CAS.
- H. Sepehrmansourie, M. Zarei, M. A. Zolfigol, S. Kalhor and H. Shi, Mol. Catal., 2022, 531, 112634 CrossRef CAS.
-
(a) J. L. Tu, Y. Zhu, P. Li and B. Huang, Org. Chem. Front., 2024, 11, 5278 RSC;
(b) J. L. Tu and B. Huang, Chem. Commun., 2024, 60, 11450 RSC.
- Z. Chen, S. Liu, Z. Xu, Z. Gan, R. Xu, Z. Xue and Y. Jin, J. Org. Chem., 2025, 90, 7458–7467 CrossRef.
- K. Zhang, A. Budinska, A. Passera and D. Katayev, Org. Lett., 2020, 22, 2714–2719 CrossRef CAS.
- S. Liu, Y. Lu, H. Wang, Z. Xue, Z. Xu, H. Wan, Q. Yin, T. Lv, S. X. Liu and Y. Jin, ACS Catal., 2025, 15, 3306–3313 CrossRef CAS.
- Y. Zheng, Z. Liu, Q. Huang and Y. Xie, Org. Lett., 2025, 27, 2997–3002 CrossRef CAS.
- C. Wu, Q. Bian, T. Ding, M. Tang, W. Zhang, Y. Xu, B. Liu, H. Xu, H. B. Li and H. Fu, ACS Catal., 2021, 11, 9561–9568 CrossRef CAS.
- D. Liang, X. Zhang, Y. Li, Y. Zhou, M. Ren, X. Zhang, Y. Li and N. Zhu, Sustainable Chem. Pharm., 2023, 33, 101077 CrossRef CAS.
- P. Cogolli, F. Maiolo, L. Testaferri, M. Tiecco and M. Tingoli, J. Heterocycl. Chem., 1979, 16, 1495–1496 CrossRef CAS.
- S. S. Mochalov, F. M. Abdel'razek, T. P. Surikova and Y. S. Shabarov, Chem. Heterocycl. Compd., 1980, 16, 339–344 CrossRef.
- N. K. Downer and Y. A. Jackson, Org. Biomol. Chem., 2004, 2, 3039–3043 RSC.
- N. Malecki, P. Carato, R. Houssin, P. Cotelle and J. P. Hénichart, Monatsh. Chem., 2005, 136, 1601–1606 CrossRef CAS.
- M. K. Mamarakhmonov, L. I. Belen kii, N. D. Chuvylkin, M. A. Ashirmatov, B. Z. Elmuradov, I. S. Ortikov and K. M. Shakhidoyatov, Russ. Chem. Bull., 2015, 64, 534–539 CrossRef CAS.
- E. Lukevics, L. Ignatovich, Y. Coldberg, F. Polyak, A. Gaukhman, S. Rozite and J. Popelis, J. Organomet. Chem., 1988, 348, 11–23 CrossRef CAS.
- P. L. Coe, A. M. Stuart and D. J. Moody, J. Fluor. Chem., 1998, 92, 27–32 CrossRef CAS.
- I. Mosiagin, A. J. Fernandes, A. Budinská, L. Hayriyan, K. E. Ylijoki and D. Katayev, Angew. Chem., Int. Ed., 2023, 62, e202310851 CrossRef CAS PubMed.
- C. K. De, F. Pesciaioli and B. List, Angew. Chem., 2013, 125 Search PubMed.
- P. Sutter and C. D. Weis, J. Heterocycl. Chem., 1986, 23, 29–32 CrossRef CAS.
- D. V. Joshi, J. R. Merchant and R. C. Shah, J. Org. Chem., 1956, 21, 1104–1109 CrossRef CAS.
- Y. Ohishi, T. Mukai, M. Nagahara, M. Yajima and N. Kajikawa, Chem. Pharm. Bull., 1989, 37, 2398–2405 CrossRef CAS PubMed.
- Q. Y. Zheng, C. F. Chen and Z. T. Huang, Tetrahedron, 1997, 53, 10345–10356 CrossRef CAS.
- W. Verboom, A. Durie, R. J. Egberink, Z. Asfari and D. N. Reinhoudt, J. Org. Chem., 1992, 57, 1313–1316 CrossRef CAS.
- O. Mogck, P. Parzuchowski, M. Nissinen, V. Böhmer, G. Rokicki and K. Rissanen, Tetrahedron, 1998, 54, 10053–10068 CrossRef CAS.
- Z. Qi-Yu, C. Chuan-Feng and H. Zhi-Tang, Chin. J. Chem., 2000, 18, 104–111 CrossRef.
- F. Vocanson, P. Seigle-Ferrand, R. Lamartine, A. Fort, A. W. Coleman, P. Shahgaldian, J. Mugnier and A. Zerroukhi, J. Mater. Chem., 2003, 13, 1596–1602 RSC.
- C. Danila, M. Bolte and V. Böhmer, Org. Biomol. Chem., 2005, 3, 172–184 RSC.
- S. Le Gac, X. Zeng, O. Reinaud and I. Jabin, J. Org. Chem., 2005, 70, 1204–1210 CrossRef CAS PubMed.
- O. Hudecek, J. Budka, V. Eigner and P. Lhoták, Tetrahedron, 2012, 68, 4187–4193 CrossRef CAS.
- M. Lejeune, J. F. Picron, A. Mattiuzzi, A. Lascaux, S. De Cesco, A. Brugnara, G. Thiabaud, U. Darbost, D. Coquiere, B. Colasson and O. Reinaud, J. Org. Chem., 2012, 77, 3838–3845 CrossRef CAS PubMed.
- A. Brugnara, L. Fusaro, M. Luhmer, T. Prangé, B. Colasson and O. Reinaud, Org. Biomol. Chem., 2014, 12, 2754–2760 RSC.
- T. Yamato, Y. Kinosaki and F. Zhang, Org. Prep. Proced. Int., 1999, 31, 167–172 CrossRef CAS.
- T. Yamato, H. Kamimura and T. Furukawa, J. Org. Chem., 1997, 62, 7560–7564 CrossRef.
- S. S. Mochalov, A. N. Fedotov, R. A. Gazzaeva, B. P. Archegov, E. V. Trofimova and N. S. Zefirov, Russ. J. Org. Chem., 2001, 37, 889–890 CrossRef CAS.
- T. Yamato, K. Tsuchihashi, N. Nakamura, M. Hirahara and H. Tsuzuki, Can. J. Chem., 2002, 80, 207–215 CrossRef CAS.
- S. Redon, Y. Li and O. Reinaud, J. Org. Chem., 2003, 68, 7004–7008 CrossRef CAS PubMed.
- P. Lhoták, J. Svoboda and I. Stibor, Tetrahedron, 2006, 62, 1253–1257 CrossRef.
- H. Xiao, H. Yin, L. Wang, C. Mei and X. Zhang, Monatsh. Chem., 2012, 143, 683–686 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Shadpour Mallakpour
*b,
Shadpour Mallakpour