
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc determination in aqueous samples using energy-dispersive X-ray fluorescence spectrometry after magnetic solid-phase microextraction using Fe3O4 nanoparticles
Guillermo Roth a,
Javier Silvaa,
Ricardo Facciob and
Mariela Pistón*a
a,
Javier Silvaa,
Ricardo Facciob and
Mariela Pistón*a
aGrupo de Análisis de Elementos Traza y Desarrollo de Estrategias Simples para Preparación de Muestras (GATPREM), Analytical Chemistry, DEC, Facultad de Química, Universidad de la República, Gral. Flores 2124, Montevideo, Uruguay. E-mail: mpiston@fq.edu.uy
bÁrea Física and Centro NanoMat, DETEMA, Facultad de Química, Universidad de la República, Montevideo, Uruguay
First published on 28th March 2025
Abstract
A novel analytical method for zinc (Zn) determination in aqueous samples was developed and validated using magnetic solid-phase microextraction (MSPME) combined with element detection via energy-dispersive X-ray fluorescence (EDXRF). Zn was extracted from aqueous samples using Fe3O4 nanoparticles impregnated with 1-(2-pyridylazo)-2-naphthol (PAN). Particle preparation involved two steps: synthesising magnetite(Fe3O4) particles and impregnating them with PAN. These impregnated nanoparticles were used to pre-concentrate and separate Zn before analytical quantification. The synthesised Fe3O4 particles were characterised as nanoparticles using several spectrometric techniques (X-ray powder diffraction (XRPD) and Raman spectroscopy). The efficiency of PAN impregnation was confirmed using Fourier Transform infrared spectroscopy (FT-IR). A single synthetic batch yielded sufficient material for 54 analyses. The validation showed limits of detection and quantification (LoD and LoQ, respectively) suitable for monitoring Zn levels in drinking water. Good alignment with a certified wastewater sample value was obtained with a mean recovery value of 96%. Moreover, a range of recoveries (106–109%) was obtained from spiked samples, indicating the trueness of the evaluation, and the precision was 8.0%. These results indicated that this analytical method was accurate and reliable for analysing these samples. In addition, EDXRF, a non-destructive technique, was applied to Zn quantification directly on the solid phase, making the proposed analytical method greener than classical atomic techniques such as flame atomic absorption spectroscopy (FAAS) or inductively coupled plasma atomic emission spectroscopy. This nanomaterial is postulated to be a solid-phase material applicable to MSPME procedures aimed at quantifying Zn in monitoring or remediation in environmental applications.
Introduction
Magnetic solid-phase microextraction (MSPME) is a separation and sample preparation technique that utilises a magnetic solid as the extractive phase, impregnated or functionalised with an organic compound (chelating agent) bonded to its surface. By dispersing the phase in a liquid sample, the analytes are adsorbed, and once the extraction is completed, the solid can be physically separated by applying an external magnetic field. These procedures offer several advantages compared with traditional solid-phase extraction methods, with one of the most significant being the requirement for only a small amount of sorbent.1 If the solid-phase synthesis achieves a high yield, it can also be remarkably convenient and cost-effective. Additionally, this kind of analytical method aligns with the principles of green analytical chemistry (GAC).2–4 Magnetic nanoparticles can be impregnated and thus transformed into an efficient sorbent for metal preconcentration. As a chelating agent, the organic compound 1-(2-pyridylazo)-2-naphthol (PAN) showed satisfactory results for preconcentrating and determining lead and cobalt when SiO2 nanoparticles were impregnated with this organic agent.5,6Combining MSPME with energy-dispersive X-ray fluorescence (EDXRF) has proven to be effective for determining trace elements in several matrices using graphene and CoFe2O4 nanoparticles.7–11 However, EDXRF has limitations in terms of the detectability of metals in liquid samples.8 These limitations can be mitigated by preconcentrating the analyte, which can be achieved by quantitatively extracting the analyte onto a solid phase from the original liquid sample, enabling EDXRF determinations to be performed directly on this material.
Zinc (Zn), selected as the target analyte, impacts the organoleptic properties of drinking water, as stated by the World Health Organisation (WHO),12 noting that Zn can impart an undesirable astringent taste to water. The WHO refers to tests indicating that 5% of the population can distinguish between Zn-free water and water containing Zn at a concentration of 4 mg L−1. In addition, water containing Zn at concentrations in the range of 3–5 mg L−1 tends to appear opalescent. Therefore, current recommendations state that drinking water should not exceed a Zn concentration of 5 mg L−1.12,13
Herein, a method for the quantitative determination of Zn in aqueous samples was developed and validated using MSPME combined with direct determination by EDXRF. Initially, Zn was extracted using Fe3O4 nanoparticles impregnated with PAN.
The sorbent was synthesised and characterised using spectrometric techniques. Fe3O4/PAN nanoparticles proved to be a promising sorbent for preconcentrating Zn, providing reliable results for its extraction (or remediation) from aqueous samples. This procedure could be applied to monitor the levels of Zn in drinking waters that may cause organoleptic issues. Furthermore, the synthesis of the extractive phase is detailed to ensure high reproducibility and yield. The resulting sorbent allows multiple analyses (54 in this study) using non-destructive techniques such as EDXRF. The implementation of this technique ensures the analytical procedure is more sustainable and greener than traditional approaches using atomic absorption spectroscopy that require flammable gases. To the best of our knowledge, this is a novel procedure with application in the environmental impact assessment of this metal in water and wastewater.
Experimental
Reagents
All chemicals employed in this study were of analytical grade or higher. For the synthesis of Fe3O4 particles, the following reagents were used: iron(III) chloride hexahydrate (FeCl3·6H2O, 97% Fluka, Switzerland), ammonium iron(II) sulphate hexahydrate (NH4)2Fe(SO4)2·6H2O, Mohr's salt (100.3% ICN Biomedical, USA), sodium hydroxide (NaOH, 98.6% Mallinckrodt, USA), sucrose (C12H22O11, [α]25D= +65.5 Baker, USA). For the particle impregnation step, PAN (99% Fluka, Switzerland) and sodium dodecyl sulphate (SDS, 98.5% Merck, Germany) were utilised.Zn calibration standards were prepared from a 1000 mg L−1 commercial standard solution used for atomic absorption spectroscopy (Fluka, Switzerland) using appropriate dilutions in ultrapure water (resistivity 18.2 MΩ cm at 25 °C), obtained from a purification system (Millipore, DirectQ3 UV, Germany).
A certified reference material (CRM) for wastewater, code MRC.INO.102 L002 (sourced from the Technological Laboratory of Uruguay, LATU) was employed to evaluate the trueness of the proposed analytical method.
Before performing the experimental work, the materials were immersed overnight in a 10% (v/v) nitric acid solution (Merck, Germany) and rinsed with ultrapure water.
Synthesis of Fe3O4 nanoparticles
The coprecipitation synthesis of magnetite (Fe3O4) nanoparticles described below yields a maximum of 29.5 g of Fe3O4 per batch.First, a solution containing 85.0 mmol of Fe(III) (Solution 1) was prepared. A mass of 22.975 g of FeCl3·6H2O was weighed into a beaker, followed by 50 mL of ultrapure water was added using a graduated cylinder. Separately, a solution containing 42.5 mmol of Fe(II) (Solution 2) was prepared by weighing 16.666 g of Mohr's salt (NH4)2Fe(SO4)2·6H2O, into a beaker and 50 mL of ultrapure water was added using a graduated cylinder. Both solutions were stirred individually with a glass rod until the solids had dissolved completely.
In addition, a 20.1 mmol L−1 sucrose solution (Solution 3) was prepared by weighing 0.342 g of sucrose into a beaker, followed by adding 50 mL of ultrapure water using a graduated cylinder. Finally, a 3 mol L−1 NaOH solution (Solution 4) was prepared by weighing 21.6 g of NaOH into an Erlenmeyer flask and adding 180 mL of ultrapure water using a graduated cylinder. Each solution was stirred separately with a glass rod until the solids were fully dissolved.
After preparing the solutions described above, Solutions 1 and 2 were combined in a 500 mL Erlenmeyer flask and mixed with continuous agitation using a magnetic stirrer. The prepared volumes of both solutions were used in their entirety. Care should be taken to avoid forming a pronounced vortex during this operation to minimise the incorporation of O2 and consequent oxidation of Fe(II). While stirring continuously, the entire prepared volume of Solution 3 was added. Then, Solution 4 was carefully dispensed dropwise using a peristaltic pump, with the discharge rate set to approximately one drop per second. The gradual addition of NaOH resulted in the precipitation of Fe3O4 as a black solid. The endpoint of the synthesis was determined when a pH of 13 was reached. After adding 100 mL of Solution 4, the pH was monitored. Stirring was stopped, and the solution was allowed to settle until a 5 mm layer of supernatant became visible to evaluate the pH. Then, drops of supernatant were collected with a glass rod, and the pH was determined using pH strips, continuing until pH 13 was reached. After this condition was achieved, stirring was continued for 30 min to ensure reaction completion.
After 30 min, a large neodymium magnet was placed underneath the flask, attracting the precipitated particles. Then, the supernatant was carefully decanted without removing the magnet. Afterwards, the magnet was removed, and the solid was resuspended in ultrapure water. This suspension was allowed to settle briefly, and the pH was measured. The washing procedure was iteratively repeated until the pH of the supernatant reached 7. Subsequently, the wet solid was transferred to large glass Petri dishes and placed in an oven (Heraeus RT360, Heraeus, Germany) to dry for 24 hours at 60 °C. After drying, the dishes were transferred to a muffle furnace (Atec HFA10, Atec, Uruguay) and the solid was calcined at 250 °C for 5 hours at a heating rate of 5 °C per min. The solids from all dishes were combined, and particle size reduction was conducted in a glass mortar. Finally, the synthesised magnetic particles were weighed and stored in a clean, dry glass container.
Impregnation of the Fe3O4 nanoparticles
The extractive phase for MSPME was prepared by impregnating an initial mass of 4.0 g of Fe3O4, providing a batch that allowed for 10 subsequent determinations. For larger masses, the procedure described below can be scaled proportionally. A 0.005 mol L−1 solution of PAN was prepared by weighing 0.025 g of PAN and adding 20 mL of ultrapure water. Separately, a 0.018 mol L−1 solution of SDS was prepared in a beaker by weighing 0.052 g of SDS and adding 10 mL of ultrapure water. These solutions were mixed, and 4.0 g of the previously synthesised Fe3O4 particles were added. Then, the mixture was placed in an ultrasonic bath (Cole-Parmer 8893, Cole-Parmer, USA) for 25 min and occasionally stirred with a glass rod. Once the impregnation step was completed, the particles were washed with ultrapure water. A neodymium magnet (Fig. 1) was placed underneath the beaker, and the particles were rinsed several times using a wash bottle. Afterwards, the particles were transferred to glass Petri dishes. The solid was dried overnight at room temperature using forced air convection, with the dishes placed in a custom-designed tunnel dryer. Upon drying, the solids from all the dishes were combined, the particle size was reduced in a glass mortar, and the particles were stored in a clean glass vial. Thus, the Fe3O4/PAN particles were ready for MSPME. The process of synthesis and impregnation is shown in Fig. 1.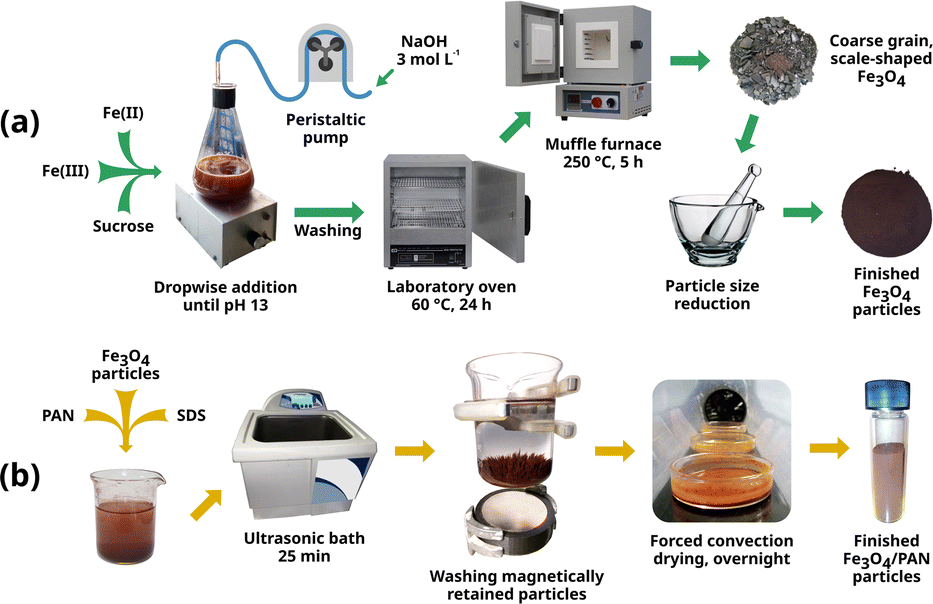 | ||
| Fig. 1 Summary of the process of synthesis (a) and impregnation (b) of Fe3O4 particles. | ||
Characterisation
Confocal Raman imaging was applied to non-impregnated and impregnated particles using a WITec instrument, the Alpha 300RA model (WITec, Germany). The Raman spectra were collected utilising a 532 nm excitation wavelength. Representative spectra were collected using 100 accumulations with an acquisition time of 0.5 seconds per spectra in selected area of 50 × 50 μm × μm.
Analytical determination
The optimised MSPME method comprises the following procedure: in a 15 mL conical tip tube, 0.4 g of Fe3O4/PAN particles (impregnated material) were weighed. The pH of the sample was adjusted to 5.9 using 0.1 mol L−1 NaOH or 0.1 mol L−1 HCl (depending on the pH of the original sample). Notably, the pH adjustment changes the volume of the solution, such that the volume of the sample aliquot and the final volume must be known for an accurate determination. The use of an acetic acid-acetate buffer should be avoided because it induces the release of PAN from the surfaces of the particles; thus, for pH adjustment, exactly 10 mL of the sample (standard or blank, with the previous pH adjustment) was added and shaken in a vortex or orbital shaker for 10 min. Afterwards, a neodymium magnet was placed adjacent to the outer wall of the conical tip tube to retain the particles, allowing the supernatant to develop a transparent appearance. Without removing the magnet from its position, the supernatant was removed (for example, using a micropipette). Fig. 2 displays a schematic of the MSPME procedure. | ||
| Fig. 2 Scheme of the MSPME process applied to standards, blanks, and samples. | ||
For the Zn quantification via EDXRF, a Shimadzu EDX7200 (Shimadzu, Japan) spectrometer with a rhodium target X-ray tube was employed. The instrumental operative conditions were air atmosphere, irradiation time of 100 s, tube voltage of 50 kV, and a collimator diameter of 3 mm. For quantification, the Kα analytical line was selected (Zn: 8.64 keV). The Fe3O4/PAN particles (0.1 g dried) with retained Zn after the extraction were placed in a Micro X-Cell (Shimadzu, 3577) with an outer diameter of 31.6 mm, 0.5 mL volume and a polypropylene film at the bottom.
The determinations were performed using the “Fundamental Parameters” approach to calibration in XRF.16 Before conducting the measurements, adjusting specific condition parameters in the fundamental parameter's method, included in the software instrument (PCEDX-Pro™), was necessary. Therefore, a working standard is required to determine the reference intensities of Zn using a known sample to calibrate and compensate for the background effects. This working standard consisted of a piece of a copper-based alloy containing Zn purchased from a local supplier. The selected material was a ternary alloy known as nickel–silver. To determine the exact Zn content of this alloy, the sample was segmented into three fragments massing 0.1 g each and individually dissolved using a wet digestion procedure at 100 °C on a hot plate and using 7.0 mL of a digestion mixture containing concentrated HCl 37%, HNO3 65%, and H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:24).
:3:24).
The solutions containing dissolved alloy were quantitatively transferred to volumetric flasks; their pH was adjusted and then diluted with ultrapure water. The analytical determinations for this purpose were conducted using a PerkinElmer AAnalyst 200 flame atomic absorption spectrometer (FAAS, PerkinElmer, USA), air–acetylene flame, equipped with a 10 cm burner and hollow cathode lamps (PerkinElmer, USA and Photron, Thailand) containing Cu, Ni and Zn, with primary wavelengths at 324.8 nm, 232.0 nm, and 213.9 nm, respectively. The theoretical composition of this material, according to the Unified Numbering System designation number C75720, should be Cu (60–65) % w/w, Ni (11–13) % w/w, and the remainder representing Zn.17 The experimental values were Cu 61.0% w/w, Ni 13.0% w/w and the remainder was Zn at 21.0% w/w, thus corresponding to the alloy C75720. Thus, this alloy was polished and used as a solid working standard to standardise the conditions for Zn determinations using the fundamental parameters method. Thus, a customized method was created, this does not evade the fact that a calibration curve must be performed. The instrumental measurements of the standards were performed to construct a calibration curve using the intensities (cps) vs. Zn concentration (mg L−1 in the original solution). The calibration curve was prepared using adequate aliquots from the Zn stock solution (1000 mg L−1) to obtain the following final concentrations: 0.5, 1.0, 1.5, 3.0, and 6.0 mg L−1; the solvent was ultrapure water with the pH adjusted to 5.9 using NaOH 0.1 mol L−1. In addition, blanks were prepared and measured. These standards, blanks, and samples were subjected to the whole MSPME procedure described above.
Optimisation and validation
To optimise the MSPME method, a fractional central composite experimental design was applied. The studied variables included sample pH, contact time of the nanoparticle concentration within the sample, and sample volume. Using a 33−1 fractional experimental design enabled a reduction in the number of required experiments from 27 in a 33 full factorial design to merely 9. Table 1 shows the conditions in each experiment.18| Parameters | Fe3O4 impregnated with PAN | Fe3O4 non-impregnated |
|---|---|---|
| a (Å) | 8.3160 (3) | 8.3482 (3) |
| Volume (Å3) | 575.12 (6) | 581.82 (5) |
| Crystalline size domain (nm) | 59.4 (5) | 45.9 (3) |
| wR2 | 7.38 | 6.485 |
| g.o.f | 2.63 | 2.41 |
| Nobs | 8701 | 8701 |
| Nvars | 23 | 23 |
The analytical methodology was validated following the recommendations of the Eurachem Guide, evaluating the figures of merit, detection and quantification limits (LoD and LoQ), linearity, precision, and trueness.19
This method was applied to determine Zn in the CRM for wastewater with satisfactory recoveries. Additionally, a spike recovery assay was performed using tap water.
Results and discussion
Characterisation of the non-impregnated and impregnated Fe3O4 nanoparticles
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, with refined lattice parameters as presented in Table 1, thus confirming their high-purity phase. The refined lattice parameters showed a slightly smaller value for the impregnated sample (a = 8.3160(3)) Å, V = 575.12(6) Å3 compared with the non-impregnated sample (a = 8.3482(3) Å, V = 581.82(5) Å3), possibly due to structural distortions or foreign ions incorporated during impregnation.
m space group, with refined lattice parameters as presented in Table 1, thus confirming their high-purity phase. The refined lattice parameters showed a slightly smaller value for the impregnated sample (a = 8.3160(3)) Å, V = 575.12(6) Å3 compared with the non-impregnated sample (a = 8.3482(3) Å, V = 581.82(5) Å3), possibly due to structural distortions or foreign ions incorporated during impregnation.
 | ||
| Fig. 3 XRPD patterns for impregnated and non-impregnated Fe3O4 nanoparticles according to the Rietveld refinements. | ||
The crystalline domain size was greater in the impregnated sample 59.4(5) nm compared with the non-impregnated sample 45.9(3) nm, suggesting enhanced crystal growth that was facilitated during the impregnation process. Despite these differences, the XRPD patterns for both samples were similar, indicating that impregnation did not significantly alter the phase purity or structural features. The quality of the Rietveld refinements was reliable, with reasonable statistical indicators (wR2 and g.o.f.) acquired for both samples; these profiles are presented in Fig. 4. These results demonstrate that the impregnation process influences the lattice parameters and crystalline domain size while preserving the structural integrity and purity of the Fe3O4 nanoparticles.
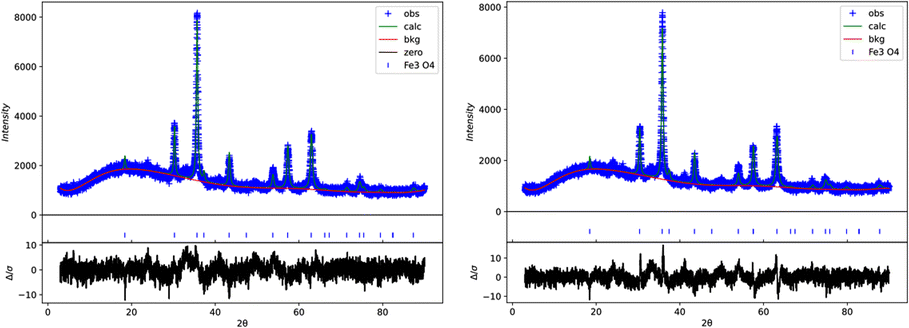 | ||
| Fig. 4 Rietveld refinement for impregnated (left) and non-impregnated (right) Fe3O4 nanoparticles according to the Rietveld refinements. | ||
Scanning Electron Microscopy (SEM) analysis was conducted to assess the morphology and size distribution of the nanoparticles. The SEM images (Fig. 5) revealed a significant size variation among the nanoparticles. It also indicates that while the particles exhibited some degree of agglomeration, individual nanoparticles were distinguishable within the observed range. Additionally, the SEM images confirmed an irregular morphology, consistent with nanoparticles synthesized under similar conditions.
 | ||
| Fig. 5 SEM image for impregnated (left) and non-impregnated (right) Fe3O4 nanoparticle. | ||
 | ||
| Fig. 6 Raman spectra (@532 nm) for impregnated and non-impregnated Fe3O4 nanoparticles. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations24 specifically, the band at 1130 cm−1 can be assigned to δ(C–H) in both rings, while the band at 1200 cm−1 corresponds to δ(C–H) (ring) and ν(C–C) (ring). The band at 1240 cm−1 is attributable to δ(C–H) (ring), ν(C–C) (ring), and δ(OH). Additionally, the band at 1300 cm−1 is associated with δ(C–C, C–N) (both rings), ν(C–N), and δ(C–H) (both rings). Finally, the band at 1355 cm−1 corresponds to νs(C–C) (ring), ν(NN, C–OH), and δ(C–H) (ring). The presence of these bands confirms that the functionalisation with PAN was successfully achieved.
N stretching vibrations24 specifically, the band at 1130 cm−1 can be assigned to δ(C–H) in both rings, while the band at 1200 cm−1 corresponds to δ(C–H) (ring) and ν(C–C) (ring). The band at 1240 cm−1 is attributable to δ(C–H) (ring), ν(C–C) (ring), and δ(OH). Additionally, the band at 1300 cm−1 is associated with δ(C–C, C–N) (both rings), ν(C–N), and δ(C–H) (both rings). Finally, the band at 1355 cm−1 corresponds to νs(C–C) (ring), ν(NN, C–OH), and δ(C–H) (ring). The presence of these bands confirms that the functionalisation with PAN was successfully achieved.
 | ||
| Fig. 7 FTIR spectra for impregnated and non-impregnated Fe3O4 nanoparticles. | ||
Analytical method: optimisation, validation, and application
The Zn content in the solid phase was measured directly using EDXRF, and that remaining in solution after the extraction (standard or CRM) was measured using FAAS. Additionally, Zn measurements were conducted using FAAS before and after the MSPME to confirm the efficiency of the procedure. For this purpose, a 2.5 mg L−1 Zn standard solution was measured before and after the extraction process, and the difference in concentration was considered to be an indirect indicator of the extraction efficiency. The experimental conditions are presented in Table 2.
| Levels | −1 | 0 | 1 |
| pH (sample) | 2.4 | 5.9 | 7.5 |
| Extraction time (min) | 5.0 | 10.0 | 15.5 |
| Sample volume (mL) | 5.0 | 10.0 | 20.0 |
In Fig. 8, the optimization results are presented as response surface plots which were generated using RStudio software. The use of surface response methods (RSM) allowed to visualize optimal conditions. A compromise between a practical MSPME process and 100% efficiency, the total migration of Zn from the aqueous solution to the solid sorbent phase, Fe3O4/PAN, was considered when selecting the optimal experimental conditions results obtained by the RSM were also compared with the remaining Zn concentration in the original solutions after the extraction. The optimal compromise between an extraction efficiency near 100% Zn migration onto the solid phase and the studied variables and practice were achieved at pH 5.9, contact time 10 min and sample volume 10 mL.
 | ||
| Fig. 8 Response surfaces obtained from the experimental design using RStudio software combined with the studied variables. (a) Variation in Zn% in the solid phase with contact time and pH, (b) variation in Zn% in the solid phase with contact time and sample volume, (c) variation in Zn% of Zn in the solid phase with sample volume and pH. | ||
The linearity was studied up to 6.0 mg L−1, and the calibration function obtained was: intensity (cps) = 0.0266 C + 0.45 (C in mg L−1 of Zn in the original solution before the MSPME process), R2 = 0.99. The trueness was evaluated using a wastewater CRM with a certified Zn value of 0.502 ± 0.025 mg L−1; the mean recovery value was 96% or 0.482 ± 0.038 mg L−1. Table 3 presents the figures of merit for the validation. In addition, spike-recovery assays were performed on real samples of tap water with recoveries (%R) in the range of 106–109% (samples were spiked at two levels, 1.0 mg L−1 and 2.0 mg L−1, respectively). The addition was needed since tap water samples usually contain low levels of Zn.
| Parameter | Result |
|---|---|
| a LoD: limit of detection, LoQ: limit of quantification.b Calibration curve (intensity of EDXRF vs. concentration of Zn in the original aqueous solution in mg L−1).c Using spiked tap water samples. | |
| LoD (mg L −1) | 0.45 |
| LoQ (mg L −1) | 1.49 |
| Trueness (%R, n = 6) | 96.0 |
| Linearityb | Up to 6.0 mg L−1 |
| Precision (%RSD, n = 4)c | 8.0 |
The CRM used for trueness evaluation is a wastewater sample containing other trace elements such as Fe 0.582 ± 0.032 mg L−1; Mn 0.880 ± 0.040 mg L−1; Cu 0.294 ± 0.012 mg L−1; Al 2.57 ± 0.18 mg L−1; Cr 0.541 ± 0.021 mg L−1; Ni 0.244 ± 0.009 mg L−1. Thus, this CRM, a complex aqueous matrix, is suitable not only to confirm the reliability of Zn determination but also to confirm that other inorganic elements, at concentrations expected in these types of samples, do not interfere with the assay.
The proposed method can produce sufficient material to perform 54 analyses. The obtained results were adequate for either Zn determination in aqueous samples or Zn removal to reduce the load of this metal in effluents or wastewater for environmental protection purposes.
The solid sorbent was characterised using complementary spectrometric techniques and indicated that the synthesis provided a high-quality nanomaterial, that remains free of significant modifications after the impregnation process. Thus, maintaining the advantages of these nanoparticles, such as high surface areas that function as an excellent sorbent in separating Zn. Moreover, this material could be applied to other divalent metals, such as Cu or Ni. In addition, EDXRF is a non-destructive technique that allows for the potential reuse of this material. For reuse, the sorbent must be exhaustively washed with diluted HCl; however, this procedure resulted in the release of PAN into the washing solution, as indicated by the strong colouration, meaning the impregnation process would need to be repeated. A more thorough approach was also tested, aimed at removing the entire impregnation layer and recovering only the magnetite particles. This was achieved by washing the particles repeatedly with 95% w/w ethanol until no coloration was observed in the supernatant. However, the procedure proved to be labor-intensive and generated significant waste, despite using small amounts of ethanol in each wash. Both acetone and 99.8% w/w isopropanol were found to be ineffective as washing solvents.
Conclusion
A novel analytical method using Fe3O4 nanoparticles impregnated with PAN as a solid-phase sorbent for MSPME was successfully developed and validated for Zn preconcentration and subsequent direct determination using EDXRF. A characterisation of the material was performed using XRPD and Raman spectroscopy, resulting in a suitable magnetic nanomaterial for the proposed objectives. In addition, FT-IR spectroscopy showed that the developed impregnation protocol was effective. A single batch of material produced using this impregnation procedure yielded sufficient material for 54 analyses. Trueness was 96.0% for the CRM and 106–109% for spike-recovery assays, with an RSD better than 8.0%, these figures of merit indicated that this analytical method is accurate, and the determination by EDXRF was simple and aligned well with GAC principles using only 0.4 g of solid phase for each determination.This procedure can be considered a potential approach for Zn removal from aqueous samples, thus having application potential in the remediation of wastewater and monitoring of drinking water.
Data availability
The data supporting this article have been included as part of the manuscript.Author contributions
Guillermo Roth: data curation, formal analysis, methodology, validation, investigation, writing–review & editing. Javier Silva: data curation, formal analysis, investigation, methodology, validation, writing–review & editing. Ricardo Faccio: data curation, formal analysis, methodology, investigation, supervision, software, writing–review & editing. Mariela Pistón: conceptualisation, funding acquisition, investigation, methodology, project administration, supervision, writing–original draft, writing–review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Comisión Sectorial de Investigación Científica (CSIC) Project 2311 and PEDECIBA-Química.References
- G. Islas, I. Ibarra, P. Hernandez, J. Miranda and A. Cepeda, Int. J. Anal. Chem., 2017, 1, 8215271 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- L. H. Keith, L. U. Gron and J. L. Young, Chem. Rev., 2007, 107, 2695–2708 CAS.
- J. Silva, V. Bühl, F. Iaquinta and M. Pistón, Heliyon, 2023, 9, e20967 CAS.
- A. Kaur and U. Gupta, Chin. J. Chem., 2009, 27, 1833–1838 CAS.
- M. Khan, E. Yilmaz and M. Soylak, J. Mol. Liq., 2016, 224, 639–647 CrossRef CAS.
- M. Khajeh and E. Sanchooli, J. Food Compos. Anal., 2010, 23, 677–680 CrossRef CAS.
- E. Marguí, R. Van Grieken, C. Fontàs, M. Hidalgo and I. Queralt, Appl. Spectrosc. Rev., 2010, 45, 179–205 CrossRef.
- K. Kocot, R. Leardi, B. Walczac and R. Sitko, Talanta, 2015, 134, 360–365 CrossRef CAS PubMed.
- K. Kocot and R. Sitko, Spectrochim. Acta, Part B, 2014, 95, 7–13 CrossRef.
- L. A. Meira, J. S. Almeida, F. de S. Dias, P. P. Pedra, A. L. Costa Pereira and L. S. G. Teixeira, Microchem. J., 2018, 142, 144–151 CrossRef CAS.
- World Health Organization, Guidelines for Drinking-Water Quality, 2nd edn, vol. 2, World Health Organization, Geneva, 1996 Search PubMed.
- U.S. Environmental Protection Agency, https://www.epa.gov/sdwa/drinking-water-regulations-and-contaminants, (accessed 27th January 2025).
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- V. Thomsen, Spectroscopy, 2007, 22, 46–50 CAS.
- Unified Numbering System for Copper + Copper Alloys, https://unscopperalloys.org/wrought/nickel-silvers.php, (accessed 27th January 2025).
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, 6th edn, Ashford Colour Press Ltd., UK, Gosport, 2010 Search PubMed.
- Eurachem Guide, The fitness for purpose of analytical methods – a laboratory guide to method validation and related topics, https://www.eurachem.org, accessed 27th January 2025.
- Á. de Jesús Ruíz-Baltazar, S. Y. Reyes-López and R. Pérez, Results Phys., 2017, 7, 1828–1832 CrossRef.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2804–2812 CrossRef CAS.
- R. D. Waldron, Phys. Rev., 1955, 99, 1727–1735 CAS.
- A. K. Bordbar, A. A. Rastegari, R. Amiri, E. Ranjbakhsh, M. Abbasi and A. R. Khosropour, Biotechnol. Res. Int., 2014, 2014, 1–6 Search PubMed.
- L. Szabó, H. Krisztian, N. E. Mircescu, A. Fălămaş, L. F. Leopold, N. Leopold, C. Buzumurgă and V. Chiş, Spectrochim. Acta, Part A, 2012, 93, 266–273 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |