
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkoxy-modified terpyridine complexes as efficient photocatalysts for diclofenac potassium degradation in pharmaceutical wastewater†
Ehsan Ullah Mughal *a,
Nafeesa Naeema,
Farwa Zainaba,
Ayesha Javaidb,
Muhammad Imran*b,
Sujeet Kumar Pandeyc,
Amina Sadiqd,
Ayza Jabeena,
Hanan A. Ogalye,
Fatimah A. M. Al-Zahranie and
Abdullah Yahya Abdullah Alzahranif
*a,
Nafeesa Naeema,
Farwa Zainaba,
Ayesha Javaidb,
Muhammad Imran*b,
Sujeet Kumar Pandeyc,
Amina Sadiqd,
Ayza Jabeena,
Hanan A. Ogalye,
Fatimah A. M. Al-Zahranie and
Abdullah Yahya Abdullah Alzahranif
aDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
bSchool of Chemistry, University of the Punjab, Lahore, 54000, Pakistan. E-mail: imran.hons@pu.edu.pk
cDepartment of Chemical and Biochemical Engineering, Rajiv Gandhi Institute of Petroleum Technology, Jais, Amethi 229304, India
dDepartment of Chemistry, Govt. College Women University, Sialkot-51300, Pakistan
eChemistry Department, College of Science, King Khalid University, Abha 61421, Saudi Arabia
fDepartment of Chemistry, Faculty of Science and Arts, King Khalid University, Mohail Assir, Saudi Arabia
First published on 4th April 2025
Abstract
This study explores the synthesis and application of terpyridine-based metal complexes (C1–C6) for the degradation of diclofenac potassium (DCF), a widely used pharmaceutical pollutant. Terpyridine (TPY) ligands and their metal complexes were synthesized and characterized using FTIR, UV-Vis, 1H NMR, 13C NMR spectroscopy, and mass spectrometry. The novelty of this study lies in the fact that, for the first time, such structures were investigated/explored as potential photocatalysts for the degradation of DCF. The study demonstrates that the target complex compounds exhibit remarkable catalytic efficiency, facilitating the effective degradation of the target pollutant, DCF. The catalytic efficiency of the complexes (C1–C6) in the degradation process was investigated under optimized conditions, including pH and catalyst concentration. A detailed mechanistic study was conducted to elucidate the pathways involved in diclofenac degradation. Kinetic parameters were determined, demonstrating the reaction followed pseudo first-order kinetics. Furthermore, the recyclability of the TPY complexes was assessed over multiple degradation cycles, confirming their stability and reusability. Moreover, computational chemistry techniques utilizing density functional theory (DFT) efficiently explain the electrical properties of the target molecules. The molecular electrostatic potential (MEP) is a theoretical model widely utilized in catalyst chemistry to examine potential sites for nucleophilic and electrophilic attacks on photocatalysts and pollutants using a reactivity map. The density of states (DOS) indicated that C5 exhibited enhanced photoactivity owing to the more prominent localization and delocalization of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The adsorption route of DCF on C2, C4, and C5 was accurately predicted using molecular electrostatic potential (MEP) descriptors. This study highlights the potential of TPY-based catalysts as efficient and sustainable materials for the remediation of pharmaceutical contaminants in water systems. The enhanced photocatalytic performance is attributed to the optimized electronic structure and the strategic incorporation of alkoxy-functionalized TPY scaffolds. Compared to previous studies on methylene blue (MB) degradation, the newly synthesized complexes exhibit significantly higher efficiency, particularly in the degradation of DCF, demonstrating superior reaction kinetics and recyclability. These findings underscore their promise as next-generation photocatalysts for environmental applications.
1. Introduction
Diclofenac potassium (DCF), a widely used non-steroidal anti-inflammatory drug (NSAID), is commonly prescribed for the treatment of pain, inflammation, and fever.1 Due to its high solubility, rapid absorption, and therapeutic effectiveness, DCF has become an essential pharmaceutical for human and veterinary medicine.2 However, the frequent and extensive use of DCF has led to its persistent release into aquatic environments through wastewater treatment plants, industrial discharges, and improper disposal of pharmaceuticals.3–5As an emerging pollutant, DCF is categorized under pharmaceutical contaminants that pose a serious threat to environmental health.6 Conventional wastewater treatment systems are often insufficient for the complete removal of such organic micropollutants, resulting in their accumulation in water bodies.7,8 Residues of DCF have been detected in surface water, groundwater, and even drinking water at trace levels, raising significant environmental and health concerns.9
The presence of DCF in aquatic systems is harmful to both aquatic life and human health.10 Ecotoxicological studies have shown that even at low concentrations, DCF disrupts aquatic ecosystems by causing oxidative stress, endocrine disruption, and cellular damage in aquatic organisms such as fish and invertebrates.11,12 Long-term exposure can lead to developmental abnormalities, reduced reproduction rates, and mortality in aquatic species.13 Furthermore, its persistence in the environment contributes to bioaccumulation and biomagnification across the food chain, ultimately posing a risk to human health when contaminated water is consumed.14,15
To address this environmental challenge, the development of efficient and sustainable degradation methods is imperative. Among the strategies explored, photodegradation has emerged as a promising approach for the breakdown of pharmaceutical pollutants.16 Photodegradation processes, facilitated by photocatalysts, leverage light energy to degrade complex organic molecules into less harmful byproducts.17 Various materials have been explored for the degradation of diclofenac in recent years, including metal oxides, photocatalysts, carbon-based materials, and enzymatic systems.18–21 These approaches have demonstrated varying degrees of success in breaking down the compound, with challenges such as incomplete degradation, formation of toxic by-products, and limited recyclability often reported.22,23 Several materials have been studied for DCF degradation, each with distinct benefits and challenges. Titanium dioxide (TiO2) photocatalysts are highly efficient under ultra violet (UV) light but exhibit limited activity in visible light and are prone to deactivation.24 Carbon-based materials, such as graphene oxide and biochar, offer high adsorption capacity but are hindered by charge recombination and slow electron transfer.25,26 Metal–organic frameworks (MOFs) show promising catalytic performance but face issues with structural instability in aqueous conditions.27 Enzymatic methods using laccases and peroxidases provide eco-friendly alternatives but are constrained by high costs and enzyme instability.28 These limitations emphasize the need for more durable and efficient catalytic systems for diclofenac removal from wastewater.29,30
Recent studies have highlighted the potential of coordination complexes as effective catalysts for pharmaceutical degradation.31,32 Among these, TPY-based complexes have gained attention for their ability to facilitate redox reactions, promote selective degradation pathways, and exhibit remarkable recyclability.33 In our earlier work, we successfully demonstrated the degradation of methylene blue using TPY metal complexes, revealing their robust catalytic efficiency and stability under diverse conditions.34,35 In this context, the use of metal complexes has gained significant attention due to their unique electronic structures, stability, and ability to promote efficient photodegradation reactions.
TPY-based metal complexes (Fig. 1) have proven to be versatile compounds in catalysis and photodegradation studies.36,37 TPYs, as polydentate ligands, exhibit excellent coordination ability with transition metals, forming stable complexes with tailored electronic properties.38 These complexes often act as effective photosensitizers and photocatalysts under light irradiation, enabling the activation of molecular oxygen or generation of reactive oxygen species (ROS), which are crucial for the degradation of organic pollutants.39,40 The tunability of TPY ligands and their corresponding metal complexes allows for enhanced photophysical properties, which can be optimized for specific environmental applications.41,42
 | ||
| Fig. 1 General structure of TPY-based metal complex. | ||
Recent studies have highlighted the potential of TPY complexes in addressing environmental pollution through photodegradation mechanisms.34,35 Their ability to degrade organic pollutants, such as dyes, pesticides, and pharmaceutical residues, has demonstrated significant advancements in pollutant removal efficiency, recyclability, and kinetics.43 However, comprehensive studies focusing on the degradation of DCF using TPY-based systems remain limited, necessitating further investigation into their synthesis, mechanism, and practical applicability.
Inspired by these findings, the current study extends the application of these complexes to the degradation of DCF, aiming to establish a sustainable and effective method for its removal from water systems. This work contributes to advancing the field by exploring the catalytic potential of TPY complexes for addressing persistent pharmaceutical pollutants. This study aims to explore the application of TPY-based metal complexes for the photodegradation of DCF. The work focuses on the synthesis and characterization of the complexes, their photodegradation mechanisms, kinetics, and recyclability under environmentally relevant conditions. By addressing these aspects, the present study contributes to the development of efficient and sustainable approaches for the removal of pharmaceutical pollutants, offering a potential solution for mitigating environmental contamination caused by DCF. Furthermore, we have correlated our experimental photocatalytic degradation results with density functional theory (DFT) calculations. This computational analysis offered significant insights into the reaction mechanisms and provided an excellent agreement with the experimental data, reinforcing the reliability and efficiency of the proposed catalytic system. This integrative approach not only deepens the understanding of the degradation process but also demonstrates the potential of TPY complexes for broader environmental applications.
2. Materials and methods
2.1. Materials
All chemicals used in this study were purchased from established suppliers, including Merck and Sigma-Aldrich, and were utilized as received without any additional purification. Melting points were measured on an Electrothermal apparatus, and the values reported are uncorrected. Infrared (IR) spectra were recorded using a Bio-Rad spectrophotometer, and the results are expressed in wavenumber (ῡ) units. UV-Vis spectra were obtained using a Jasco V-660 UV-Vis spectrophotometer with quartz cells.2.2. General procedure for the synthesis of ligands (L1–L4) and complexes (C1–C6)
The synthesis of TPY-based ligands and their corresponding metal complexes has been reported in our previous publication.442.3. Photocatalytic activity
The photocatalytic experiments were carried out for the removal of DCF using as-synthesized complexes as photocatalysts.16 For a standard catalytic run, 10 mg of each catalyst was added to the 100 mL of a DCF solution (10 mg L−1). The resulting suspension was placed in the dark for approximately half an hour under continuous magnetic stirring. A small aliquot of this suspension was withdrawn using a syringe fitter to remove any catalyst particles, and the absorbance was measured with a UV-Vis spectrophotometer at the maximum wavelength of 276 nm. According to Beer–Lambert law, this absorbance is proportional to the concentration and was considered the initial concentration (C0) before light exposure. Subsequently, the suspension was irradiated using a 300 W xenon lamp as the light source. The aliquots were taken at 15 min intervals, their absorbance was recorded, and termed as DCF concentration Ct at certain time interval (t). In this way, the obtained data was used to calculate the photocatalytic efficiency using following equation (eqn (1)).
 | (1) |
2.4. Computational study
Computational chemistry methods employing DFT effectively elucidate the electrical characteristics of chemical compounds.48 These techniques provide a deeper comprehension of the interactions between compounds and pollutants, hence enhancing the knowledge of the degradation mechanism and contributing to the creation of effective compounds. The electronic aspects of the compounds were computed using the Density of States (DOS) to examine the effects of compounds. DOS is an excellent tool for visually assessing the frontier molecular orbitals.49
3. Results and discussion
3.1. Chemistry
The TPY ligands and their metal complexes were synthesized according to established literature methods44 (as outlined in Scheme 1 and Table 1). The characterization of ligands L1–L4 and complexes C1–C6 was performed using FTIR, UV-Vis, 1H NMR, 13C NMR spectroscopy, and mass spectrometry. | ||
| Scheme 1 Synthesis of TPY-based metal complexes (C1–C6). | ||
| Compound no. | Chemical structure |
|---|---|
| L1 |  |
| L2 | 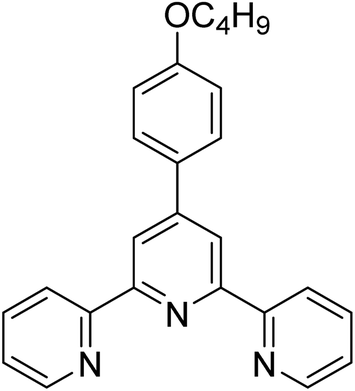 |
| L3 |  |
| L4 |  |
| C1 | 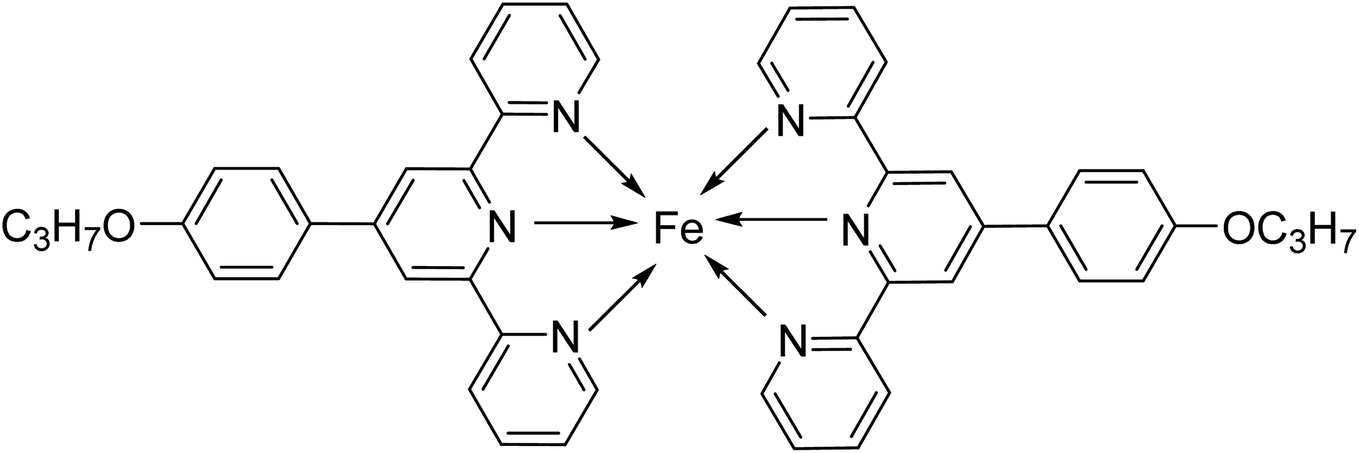 |
| C2 |  |
| C3 |  |
| C4 |  |
| C5 | 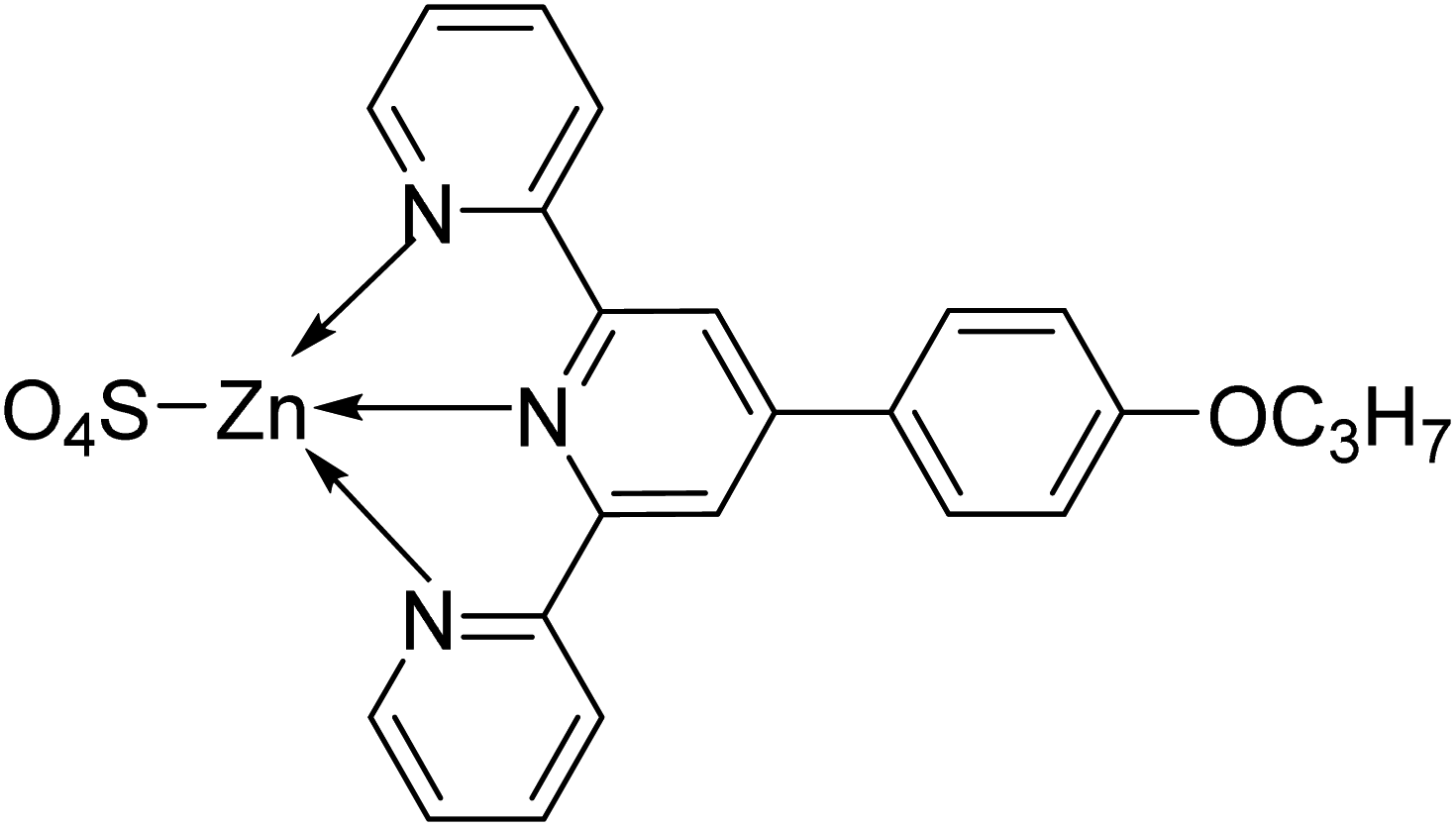 |
| C6 |  |
The spectral data of already reported compound L4 is reported in the literature.44 The spectroscopic data of all the newly synthesized ligands and complexes are given below.
 ), 1.82–1.75 (m, 2H,
), 1.82–1.75 (m, 2H,  ), 1.03 (t, J = 10.0 Hz, 3H,
), 1.03 (t, J = 10.0 Hz, 3H,  ); 13C NMR (126 MHz, DMSO-d6): δ 160.4, 156.0, 155.5, 149.8, 149.5, 138.0, 129.8, 128.6, 125.0, 121.4, 117.7, 115.7, 69.6, 22.5, 10.8; accurate mass (ESI) of [M + H]+: calculated for C24H22N3O 368.1762; found 368.1753.
); 13C NMR (126 MHz, DMSO-d6): δ 160.4, 156.0, 155.5, 149.8, 149.5, 138.0, 129.8, 128.6, 125.0, 121.4, 117.7, 115.7, 69.6, 22.5, 10.8; accurate mass (ESI) of [M + H]+: calculated for C24H22N3O 368.1762; found 368.1753. ), 1.83–1.76 (m, 2H,
), 1.83–1.76 (m, 2H, 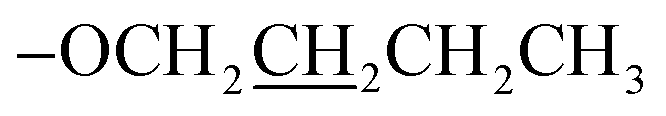 ), 1.60–1.56 (m, 2H,
), 1.60–1.56 (m, 2H,  ), 1.02 (t, J = 10.0 Hz, 3H,
), 1.02 (t, J = 10.0 Hz, 3H,  ); 13C NMR (151 MHz, DMSO-d6): δ 160.4, 156.1, 155.4, 149.8, 149.4, 138.1, 129.7, 128.5, 125.0, 121.4, 117.7, 115.6, 69.7, 31.1, 22.4, 10.8; accurate mass (ESI) of [M + H]+: calculated for C25H24N3O 382.1919; found 380.1906.
); 13C NMR (151 MHz, DMSO-d6): δ 160.4, 156.1, 155.4, 149.8, 149.4, 138.1, 129.7, 128.5, 125.0, 121.4, 117.7, 115.6, 69.7, 31.1, 22.4, 10.8; accurate mass (ESI) of [M + H]+: calculated for C25H24N3O 382.1919; found 380.1906. ), 1.94–1.87 (m, 2H,
), 1.94–1.87 (m, 2H, 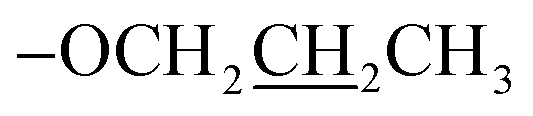 ), 1.15 (t, J = 12.0 Hz, 3H,
), 1.15 (t, J = 12.0 Hz, 3H,  ); 13C NMR (151 MHz, acetone-d6): δ 161.7, 160.5, 158.5, 153.3, 150.1, 138.8, 129.3, 128.3, 128.2, 127.5, 124.0, 120.6, 115.53, 115.51, 69.6, 22.3, 9.9; accurate mass (ESI) of [M + H]+: calculated for C48H43FeN6O2 791.2797; found 791.2780.
); 13C NMR (151 MHz, acetone-d6): δ 161.7, 160.5, 158.5, 153.3, 150.1, 138.8, 129.3, 128.3, 128.2, 127.5, 124.0, 120.6, 115.53, 115.51, 69.6, 22.3, 9.9; accurate mass (ESI) of [M + H]+: calculated for C48H43FeN6O2 791.2797; found 791.2780.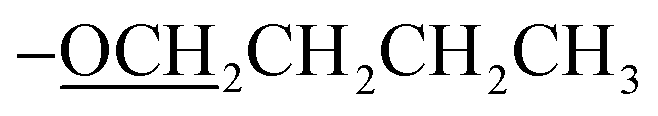 ), 1.89–1.84 (m, 2H,
), 1.89–1.84 (m, 2H, 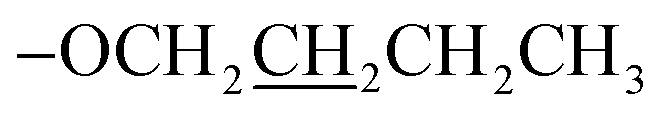 ), 1.61–1.55 (m, 2H,
), 1.61–1.55 (m, 2H,  ), 1.05 (t, J = 6.0 Hz, 2H,
), 1.05 (t, J = 6.0 Hz, 2H, 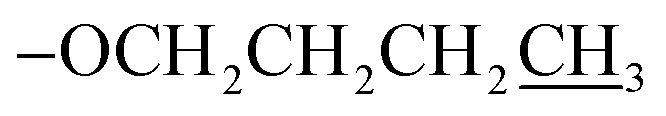 ); 13C NMR (151 MHz, acetone-d6): δ 162.1, 156.0, 150.0, 148.4, 148.1, 141.3, 129.6, 127.7, 127.6, 123.3, 120.3, 115.4, 68.0, 31.1, 19.0, 13.2; accurate mass (ESI) of [M + H]+: calculated for C25H24N3O5SZn 542.0728; found 542.0720.
); 13C NMR (151 MHz, acetone-d6): δ 162.1, 156.0, 150.0, 148.4, 148.1, 141.3, 129.6, 127.7, 127.6, 123.3, 120.3, 115.4, 68.0, 31.1, 19.0, 13.2; accurate mass (ESI) of [M + H]+: calculated for C25H24N3O5SZn 542.0728; found 542.0720. ), 1.88–1.81 (m, 2H,
), 1.88–1.81 (m, 2H,  ), 1.08 (t, J = 10.0 Hz, 3H,
), 1.08 (t, J = 10.0 Hz, 3H,  ); 13C NMR (126 MHz, DEPT, DMSO-d6): δ 148.2, 141.7, 135.3, 130.3, 130.2, 128.0, 124.0, 120.4, 115.7, 69.8, 22.5, 11.0; accurate mass (ESI) of [M + H]+: calculated for C24H22N3O5SZn 528.0572; found 528.0554.
); 13C NMR (126 MHz, DEPT, DMSO-d6): δ 148.2, 141.7, 135.3, 130.3, 130.2, 128.0, 124.0, 120.4, 115.7, 69.8, 22.5, 11.0; accurate mass (ESI) of [M + H]+: calculated for C24H22N3O5SZn 528.0572; found 528.0554.3.2. Photocatalytic activity
The photocatalytic capability of synthesized complexes was evaluated to catalyze the removal of DCF in aqueous media (Fig. 2). Initially, the DCF degradation without any catalyst and utilizing light irradiation alone was carried out as a blank experiment. It was noticed that degradation (2.77%) of DCF was negligible in the absence of the catalyst revealing that photolysis is insufficient for the DCF removal. Similarly, additional control experiments were employed under dark conditions containing synthesized complexes only. No considerable change in degradation efficiency was observed in these experiments, indicating that the complexes exhibited minimal catalytic activity and only slight adsorptive behavior. This emphasizes the necessity of both light and the catalyst working together to achieve effective removal of DCF. Upon light irradiation, the photocatalytic efficiency boosted to a greater extent due to the synergistic role of light and the catalysts, facilitating the degradation of DCF. Fig. 2(A) shows that there is an abrupt enhancement in the degradation efficiency during the initial 15 to 30 min of irradiation, ascribed to the abundance of available catalytic sites at the start of the reaction. As reaction proceeds further, there is only a nominal increase in degradation efficiency and ultimately became almost constant between 75 to 90 min of light exposure. The occupation of maximum catalytic sites with DCF molecules at this stage of reaction were responsible for this behavior. It can be explained by the saturation of catalytic sites with DCF molecules, reducing the availability of free sites for further degradation. Additionally, the accumulation of reaction intermediates or byproducts on the catalyst surface could hinder the accessibility of fresh DCF molecules to the active sites.50 When photocatalytic activity of synthesized complexes was compared among themselves, it was found that photocatalytic process is structural dependent where nature of central metal, type of ligand, and geometry attained by the metal complex play a crucial role. The observed degradation efficiencies for synthesized complexes were in the order: C5 (84.3%) > C2 (81.5%) > C4 (77.2%) > C6 (72.6%) > C1 (68.5%) > C3 (64.1%) (Fig. 2(B)). The highest degradation efficiency of C5 is due to the small steric hindrance of propoxy group attached to the phenyl ring, which facilitates easier access of substrate molecules towards zinc metal centre. Moreover, the propoxy group, being a strong electron-donating group, stabilizes the Zn–ligand bond and enhances the catalytic capability of the zinc center. In case of C2, the photocatalytic efficiency slightly decreased because the butoxy group introduces greater steric hindrance compared to the propoxy group. The longer chain length of the butoxy group also weakens the Zn–ligand interaction, affecting charge transfer tendency of the complex. The presence of benzyloxy group in C4 further reduced the degradation efficiency attributed to significant steric hindrance caused by the bulky benzene ring.37 This bulkiness restricts substrate access to the Zn center, thus reducing catalytic efficiency. Although the benzyloxy group is an excellent electron-donor, it is less effective than alkoxy groups because of extensive delocalization over the aromatic ring. This phenomenon leads to reduced activation of the catalytic center. The superior performance of the zinc complexes is also attributed to their coordination number of 4, which allows maximum interaction with drug molecules as the zinc centre retains vacant sites for pollutant interaction.35 Moreover, the stable d10 configuration of Zn2+ and its resulting redox inertness contribute to the stability of the complex51 which may help to maintain an active substrate–host interaction for catalytic activity. This stability could also refrain the metal complex from participating in side-reactions or decompositions, allowing catalytic action only. | ||
| Fig. 2 (A) Photocatalytic efficiency of a synthesized complexes for the removal of DCF as a function of light irradiation time, (B) comparative photocatalytic efficiency, and (C) reaction kinetics of synthesized complexes. | ||
Among the remaining complexes, the Cu complex (C6) demonstrated better performance than the Fe complexes (C1 and C3). The lower catalytic activity of Cu and Fe complexes compared to Zn complexes is primarily due to their coordination number of 6, forming an octahedral geometry where all sites are fully occupied by ligands.36,52 This structure limits the exposure of the metal center to pollutant molecules, thereby reducing charge transfer between metal and ligand. Although both copper and iron complexes have similar coordination number, the ligand environment significantly influences photocatalytic performance. In C6, the ligand has dimethoxy groups that are relatively small, with less steric hindrance compared to the bulky ligands in C1 and C3. Furthermore, the dimethoxy groups exhibit strong electron-donating capabilities due to their inductive and resonance effects, facilitating efficient electron transfer from the ligand to the metal center. In case of C1 and C3, steric effects of the ligands also contribute to the diminished catalytic activity of Fe complexes. Despite the similar co-ordination number of Fe and Cu, the slightly higher catalytic efficiency of Cu complexes could be attributed to its higher intrinsic Lewis acidity53 which could facilitate effective adsorption of substrate molecules onto the metal complex surface, initiating catalytic action.54 Moreover, Lewis acidity is also associated with the separation of photoinduced electron–hole pairs, suppressing their recombination rate.55
3.3. DCF photodegradation kinetics
Kinetic study of the photocatalytic process is essential for understanding the pathway by which the substrate interacts with the catalyst and it helps to determine the reaction rate constant. The Langmuir–Hinshelwood kinetic model, modified to the pseudo first-order kinetics, is commonly applied to describe the degradation of organic contaminants.56 The experimental data of photocatalytic reaction fitted well to the pseudo first-order kinetic model equation (eqn (2)) followed by the determination of rate constants from the slope of the plot between time and ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C0/Ct (Fig. 2(C)).
C0/Ct (Fig. 2(C)).
 | (2) |
The obtained values of k for C1, C2, C3, C4, C5, and C6 were 0.00445 min−1, 0.01716 min−1, 0.00343 min−1, 0.01907 min−1, 0.02355 min−1, and 0.01583 min−1, respectively. The highest rate constants of Zn complexes (C2, C4, C5) among all the synthesized complexes were in agreement with their photocatalytic efficiencies indicating fast reaction kinetics of DCF degradation in the presence of these complexes.
In photocatalytic process, electric energy consumption helps to estimate operating costs and can be evaluated using “electric energy per order” (EEO) for a pseudo first-order kinetic reaction. The EEO can be calculated from the following equation (eqn (3)).
 | (3) |
3.4. Operational parameters affecting photodegradation process
It has been known that operating conditions of pollutant concentration, catalyst dosage, and solution pH influence the photocatalytic efficiency, therefore, these parameters were studied in a specific range to determine optimal conditions. The photocatalyst (C5) with highest degradation efficiency was employed for further optimization studies. | ||
| Fig. 3 Effect of operational parameters on the photocatalytic degradation of DCF under visible light irradiation using C5 as photocatalyst: (A) DCF initial concentration, (B) catalyst dosage, (C) solution pH and (D) plot of change in pH vs. initial pH for the determination of pHpzc of C5 (intercept = pHpzc). | ||
63 whereas pHpzc of the catalyst was experimentally determined to be 5.8 (Fig. 3(D)). In fact, these two important considerations (pHpzc of the catalyst and pKa of the DCF) helped to select the pH range to be studied and the pH value at which maximum degradation efficiency was obtained was considered as optimum pH. Initially, in highly acidic conditions (pH = 3), where pKa > pH < pHpzc, both the drug and the catalyst are positively charged. This results in electrostatic repulsion between the two surfaces, thereby limiting the adsorption of the drug onto the catalyst surface, a prerequisite for catalytic action. The mild acidic (pH = 5) conditions favored this adsorption because at this pH DCF begins to deprotonate and acquires a negative charge, which attracts the positively charged catalyst surface. However, as the pH increases further (pH = 7), the catalyst surface also becomes negatively charged since this pH is higher than the pHpzc. Consequently, electrostatic repulsion between the negatively charged DCF and the catalyst surface starts to dominate, reducing the degradation efficiency. At even higher pH values (pH = 9), this repulsion intensifies because the negative charge on DCF increases due to its complete deprotonation, further limiting its adsorption onto the catalyst surface.64 The study of pH parameter highlights its critical role in determining the surface charge characteristics of both the drug and the catalyst, emphasizing the importance of optimizing pH conditions to achieve maximum photocatalytic performance.3.5. Photocatalyst reusability
Reusability of a photocatalyst without significant loss in catalytic efficiency ensures sustainability, cost-effectiveness, and practical applicability of the material. The stability of C5 was assessed by reusing the immobilized catalyst under optimal conditions in subsequent runs following the initial standard run. The experimental results demonstrated that the photocatalytic performance retains satisfactory reusability for up to four cycles (Fig. 4(A)). The observed degradation efficiencies were 90.2%, 89.7%, 88.0%, and 86.5% at 1st, 2nd, 3rd, and 4th catalytic runs, respectively. The slight reduced activity observed during the 4th cycle could potentially be attributed to the accumulation of intermediate compounds adsorbed on the surface of the photocatalyst.65 Overall, minor catalytic loss (3.7%) over repeated runs suggests the scalable operational efficiency of C5.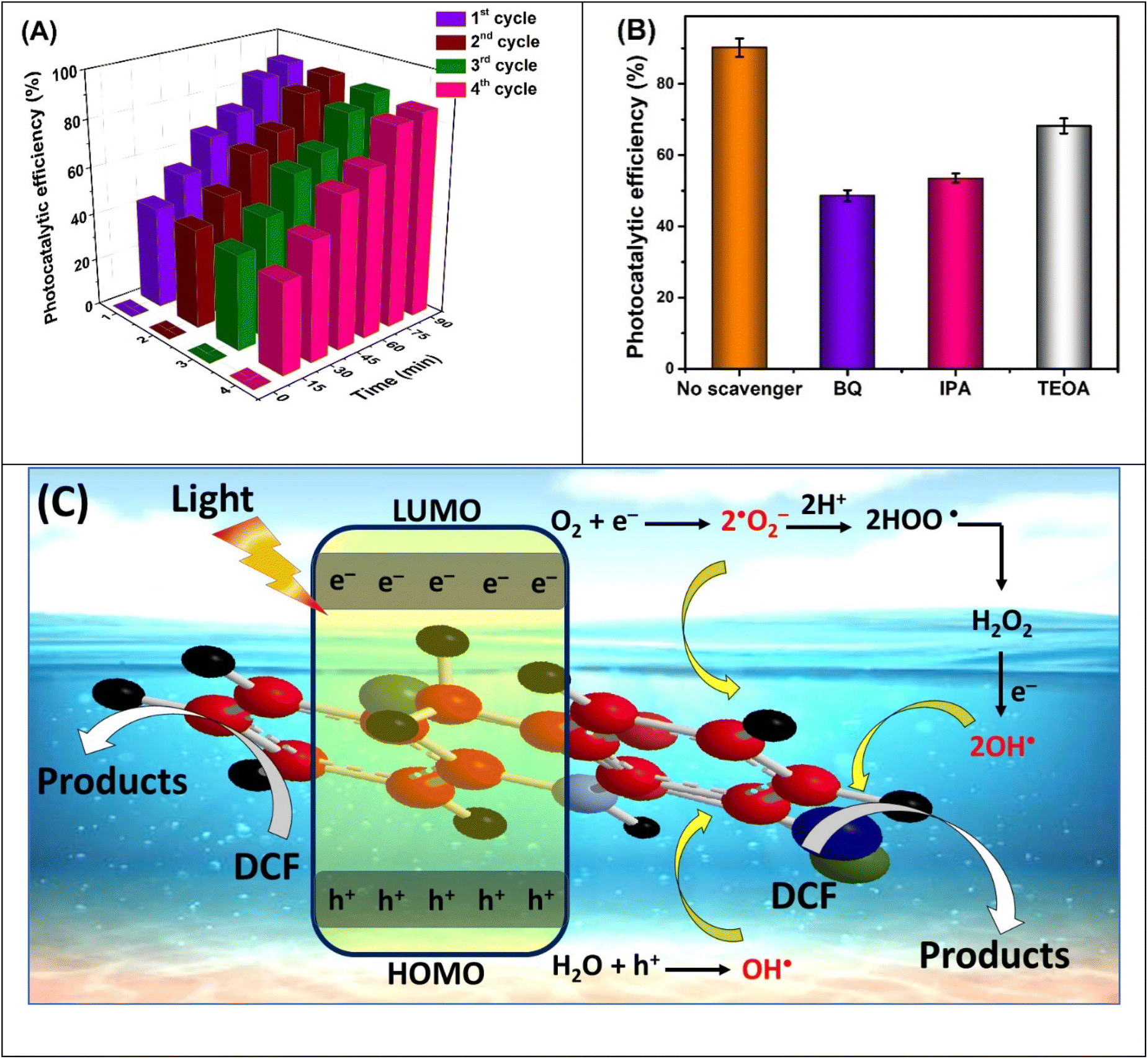 | ||
| Fig. 4 (A) Reusability, (B) photocatalytic experiments in the presence of scavengers, and (C) plausible DCF degradation mechanism employing C5 as photocatalyst under optimized conditions. | ||
3.6. Trapping experiments and reaction mechanism
The reactive oxygen species (ROS) generated at the catalyst surface upon light irradiation play a crucial role in the degradation process. To determine the contribution of these species, trapping experiments were conducted, where photocatalytic activity was evaluated in the presence of scavenging agents. A scavenging agent has a tendency to quench specific reactive species; therefore, when added to a photocatalytic reaction, it inhibits the activity due to the reduced availability of reactive species. The extent of reduction in photocatalytic efficiency in the presence of a specific scavenger correlates with the dominant role of that species in driving the degradation process. In this study, various scavenging agents were employed to validate the role of ROS (Fig. 4(B)). For instance, benzoquinone (BQ), a quencher for ˙O−2, was used to confirm its presence in the degradation mechanism. The significant suppression of DCF removal efficiency (48.6%) in the presence of BQ indicates that ˙O−2 was actively generated throughout the process, and played a key role in the degradation activity.66 Similarly, 2-propanol (iso-propanol, IPA) was employed as a scavenger for hydroxyl radicals (OH˙). The degradation of DCF was significantly inhibited (53.5%) in the presence of 2-propanol indicating that OH˙ was also essential for DCF removal processes. Additionally, photoinduced holes (h+) are generated simultaneously during the reaction. To investigate their contribution, triethanolamine (TEOA) was used as a scavenger in the aqueous solution.67 A slight reduction in the degradation efficiency of DCF (68.2%) was observed, suggesting a limited contribution of holes to the photodegradation process. Based on these findings, a probable reaction mechanism is proposed to explain the involvement of these photoinduced species (Fig. 4(C)). The electrons in the highest occupied molecular orbital (HOMO) of C5 are excited to lowest unoccupied molecular orbital (LUMO) upon absorbing energy from the incident photons. As a result, LUMO becomes electron-rich, whereas positive vacancies known as holes are left behind in the HOMO and it becomes electron-deficient.68 The electrons present in LUMO reduce adsorbed oxygen molecules into superoxide anions which further react with nearby water molecules converting them into hydroperoxyl (OOH˙) radicals, eventually converting into hydroxyl radicals (OH˙).69 In parallel, some hydroxyl radicals are directly generated through the oxidation of water molecules by the holes in the HOMO. These reactive species (˙O−2 and OH˙), being highly unstable and short-lived, attack the DCF molecules, breaking them down into simpler compounds through a series of steps.3.7. Performance comparison of C5 with other photocatalysts
Table 2 provides a comparative analysis of the photodegradation efficiency of C5 for DCF removal, benchmarked against other materials previously reported for the same application. A comprehensive literature scan indicates a notable gap in the use of coordination polymers and metal complexes specifically for DCF removal. To address this, the comparison incorporates a diverse range of materials, reported for similar studies. The results highlight the superior performance of our fabricated C5 complex. Notably, C5 achieves exceptional photocatalytic efficiency for DCF removal under visible light irradiation in the shortest time span, underscoring its potential as a highly effective and practical material for wastewater remediation applications.| Photocatalyst | DCF concentration | Catalyst loading | Source of light | Degradation efficiency (%) | Rate constant (min−1) | Reference |
|---|---|---|---|---|---|---|
| a CBS: Chitosan Boc-serine. | ||||||
| g-C3N4/NH2-MIL-125 | 10 mg L−1 | 250 mg L−1 | UV LED | 100% in 2 h | 0.0282 | 70 |
| MIL-88 (Fe)/Ag3PO4/GCN | 5 mg L−1 | 0.5 g | 300 W xenon lamp | >90% in 120 min | 0.076 | 71 |
| ZnO-CBS-Ni complex | 50 mg L−1 | 50 mg L−1 | Visible light | 93% in 180 min | — | 72 |
| O-g-C3N4/ZnO/TiO2@halloysite nanotubes | 10 mg L−1 | 1.0 g L−1 | 15 W UVA lamp | 100% in 50 min | 0.060 | 73 |
| Cu2ZnSnS4 | 10 mg L−1 | 1.0 g L−1 | 300 W xenon lamp | 90% in 120 min | — | 74 |
| C5 | 10 mg L−1 | 20 mg | 300 W xenon lamp | 90.2% in 90 min | 0.02355 | This work |
3.8. Comparative performance evaluation of complexes based on the same TPY scaffold for methylene blue dye and diclofenac potassium degradation
The synthesized terpyridine-based metal complexes (C1–C6) were employed as heterogeneous photocatalysts for the degradation of DCF, a pharmaceutical pollutant. The photocatalytic efficiency of the complexes followed the order: C5 (84.3%) > C2 (81.5%) > C4 (77.2%) > C6 (72.6%) > C1 (68.5%) > C3 (64.1%). Among these, C5 demonstrated the highest degradation efficiency, effectively removing 10 mg L−1 of DCF with a remarkable 90.2% degradation rate within 90 minutes under light irradiation, utilizing only 20 mg of catalyst at pH 5. The degradation kinetics conformed to the pseudo-first-order model, with the highest rate constant observed for C5 (0.02355 per min) (Table 3).| Photocatalyst | DCF concentration | MB dye concentration | Catalyst loading | Source of light | Degradation efficiency (%) | Rate constant (min−1) | Reference |
|---|---|---|---|---|---|---|---|
| Complex 1 [Zn(4′-(4-methylphenyl)-2,2′:6′,2′′-terpyridine)(SO4)2−] | — | 10 mg L−1 | 20 mg | 18 W UV lamp | 67.31% in 40 min | 0.0099 | 35 |
| Complex 7 [Zn(N,N-diphenyl-4-(2,2′:6′,2′′-terpyridin-4′-yl)aniline)(SO4)2−] | — | 10 mg L−1 | 20 mg | 18 W UV lamp | 65.91% in 40 min | 0.0110 | 35 |
| Complex 5 [Zn (3,4-dimethoxyphenyl-2,2′:6′,2′′-terpyridine)(SO4)2−] | — | 15 mg L−1 | 10 mg | UV lamp | 79.84 in 120 min | 0.009 | 34 |
| Complex C5 [Zn(4′-(4-propoxyphenyl)-2,2′:6′,2′′-terpyridine)SO42−] | 10 mg L−1 | — | 20 mg | 300 W xenon lamp | 90.2% in 90 min | 0.02355 | This work |
A comparative analysis with our previously published studies on methylene blue (MB) degradation highlights the superior efficiency of the newly synthesized complexes. In our earlier work, Fe(II), Co(II), and Zn(II) terpyridine complexes exhibited significant photocatalytic potential against MB degradation. Zinc-based complexes, particularly Complex 1 (67.31%) and Complex 7 (65.91%), showed the highest degradation efficiencies under optimized conditions (10 mg L−1 dye concentration, 20 mg catalyst dose, pH 4, 40 min irradiation time). The rate constants for these complexes were 0.009 min−1 and 0.011 min−1, respectively, both following pseudo-first-order kinetics. Reusability tests revealed minimal loss of activity (8.62% and 7.01% over four cycles), indicating their sustainability in wastewater treatment applications35 (Table 3).
Similarly, in another study from our group, the series C1–C6 exhibited varying degradation efficiencies against MB, with C5 achieving the highest efficiency (79.84%) and a rate constant of 0.009 min−1. Zn-based complexes consistently outperformed their Fe-based counterparts despite having the same ligands, suggesting that the metal center significantly influences catalytic activity. The excellent recyclability of these catalysts further emphasized their practicality for long-term use in environmental remediation34 (Table 3).
Compared to our previous research (Table 3),34,35 the present study demonstrates significantly enhanced photocatalytic efficiency, particularly in the degradation of DCF. Complex C5 not only outperformed its counterparts in degrading DCF (90.2% efficiency with a rate constant of 0.02355 min−1) but also exhibited a remarkable improvement over the best-performing complexes from the MB studies. The increased efficiency can be attributed to the optimized electronic structure, which facilitates better charge transfer, enhanced generation of reactive oxygen species, and improved stability during repeated use. The incorporation of alkoxy (–OR) substituents on the TPY scaffold plays a crucial role in modulating the electronic properties and overall photocatalytic efficiency of metal complexes. Alkoxy groups are known to be electron-donating, which significantly influences the electronic structure, charge distribution, and photophysical behavior of the TPY ligand. The findings reinforce the versatility of terpyridine-based metal complexes as effective and sustainable photocatalysts, offering substantial advancements in wastewater treatment, particularly for pharmaceutical pollutants. The synergistic effect of electronic tuning and structural modifications conferred by alkoxy groups underscores their importance in designing highly efficient TPY-based photocatalysts. These findings provide valuable insights for the rational development of next-generation metal complexes with superior performance in environmental remediation and wastewater treatment.
Henceforth, the newly synthesized complexes, especially C5, demonstrate superior photocatalytic performance compared to previously studied systems. Their remarkable degradation efficiency, favorable reaction kinetics, and excellent recyclability highlight their potential for real-world applications in environmental remediation. This study underscores the importance of electronic structure optimization in designing next-generation photocatalysts for the effective removal of persistent organic pollutants from aqueous systems.
4. Structure–property relationship
The photocatalytic activity of the synthesized metal complexes demonstrated a strong correlation with their structural characteristics, including the nature of the central metal, ligand type, and overall geometry (Fig. 5). The degradation efficiency of diclofenac (DCF) varied significantly among the complexes, following the order: C5 (84.3%) > C2 (81.5%) > C4 (77.2%) > C6 (72.6%) > C1 (68.5%) > C3 (64.1%). | ||
| Fig. 5 Structure–property relationship of the target terpyridine-based complexes. | ||
The superior photocatalytic performance of C5 can be attributed to the propoxy (–OPr) substituent, which provides minimal steric hindrance and facilitates greater substrate accessibility to the zinc center. Additionally, the electron-donating nature of the propoxy group strengthens the Zn–ligand bond, enhancing charge transfer efficiency. In contrast, C2, featuring a butoxy (–OBu) group, exhibited a slight reduction in efficiency due to the increased steric bulk, which weakens the Zn–ligand interaction and reduces electron transfer capacity. Similarly, the presence of a benzyloxy (–OCH2Ph) substituent in C4 further diminished degradation efficiency, primarily due to the significant steric hindrance imposed by the bulky benzene ring. While benzyloxy groups possess electron-donating properties, their extensive delocalization reduces the electron density available for efficient catalytic activation.
Among the remaining complexes, C6 (Cu complex) outperformed the Fe-based counterparts (C1 and C3) due to the presence of dimethoxy (–OMe) substituents. These groups, being relatively small, introduce less steric hindrance while enhancing electron transfer through both inductive and resonance effects. The reduced catalytic activity of Fe complexes is attributed to their increased steric hindrance and lower ligand-field stabilization, which limits effective interaction with the substrate.
The observed trend in photocatalytic efficiency can be further explained by the coordination number and geometry of the metal centers. Zinc complexes (C2, C4, C5) adopt a tetrahedral coordination, providing vacant sites that enhance pollutant adsorption and electron transfer. In contrast, Fe and Cu complexes (C1, C3, C6) exhibit octahedral coordination, where ligand saturation restricts substrate interaction, thereby lowering catalytic activity. This structural constraint inhibits efficient charge transfer from the metal center to the substrate, ultimately reducing photocatalytic performance.
Overall, the structure–property relationship highlights those steric effects, electronic properties, and coordination geometry play crucial roles in dictating the catalytic activity of these metal complexes. Zinc-based complexes, particularly those with small electron-donating substituents, exhibited superior photocatalytic efficiency, emphasizing the importance of rational ligand design in optimizing photocatalyst performance.
5. DFT studies
Fig. 6 and 7 illustrate the optimized structures of C2, C4, C5, and DCF, as well as the DOS for C2, C4, and C5, respectively. Fig. 6 demonstrates that there is no major alteration in the HOMO–LUMO gap.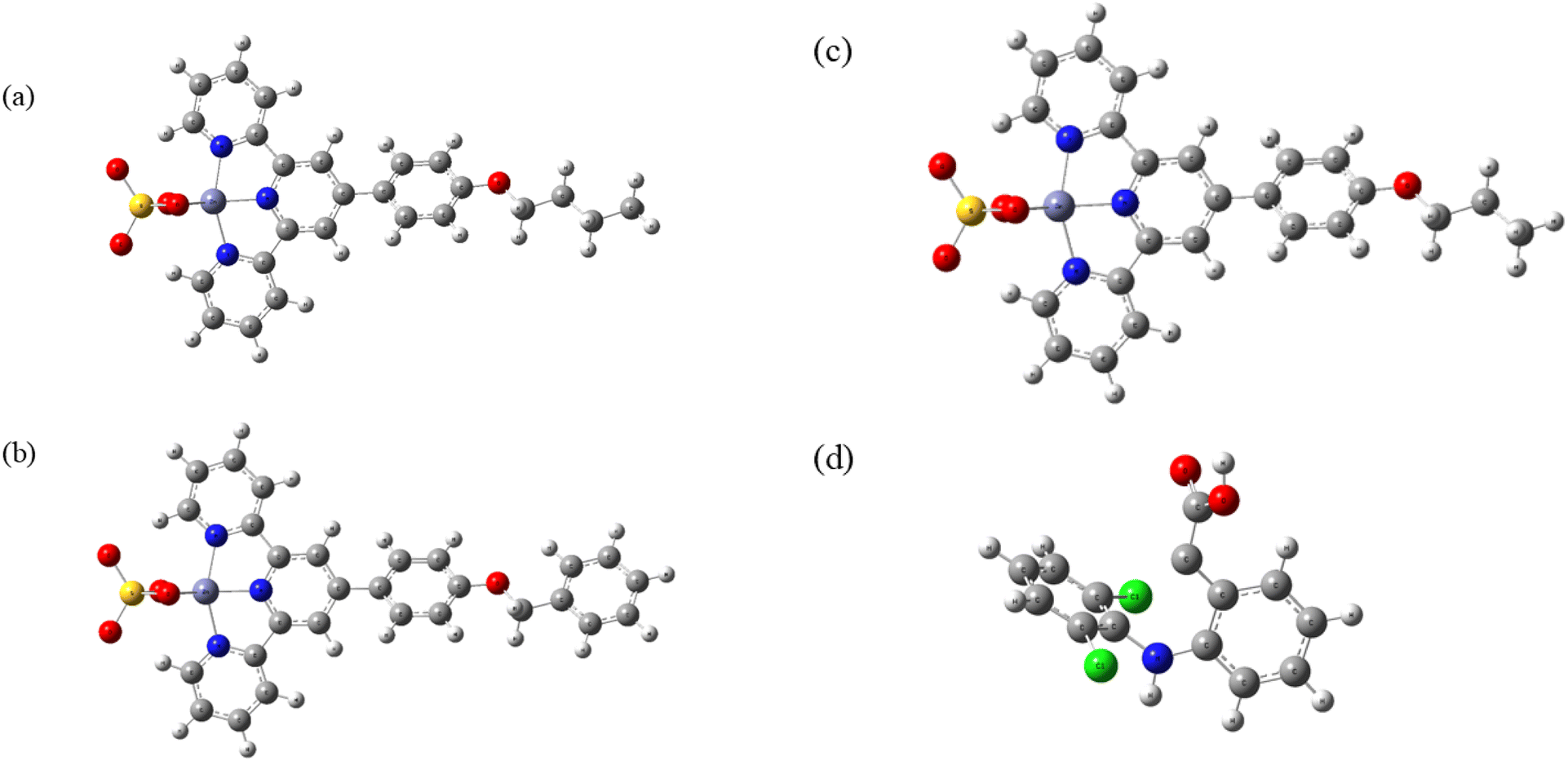 | ||
| Fig. 6 The optimized structure of (a) C2, (b) C4, (c) C5 and (d) DCF. | ||
 | ||
| Fig. 7 The DOS and HOMO, LUMO of the C2, C4 and C5. | ||
Theoretical model widely utilized is MEP in catalyst chemistry to investigate possible areas for nucleophilic and electrophilic attacks on photocatalysts and pollutants using map of reactivity. Potential on the surface has been categorized into five distinct colors, in this manner: blue < green < yellow < orange < red. Fig. 8 illustrates that the deep red areas close to the atoms denote the locations of the highest chance for an electrophilic attack. The blue zone at the atoms indicates the probable sites for nucleophilic attack in the interaction between the photocatalysts and the pollutant. To explain the reaction process among the photocatalysts (C2, C4, C5) and DCF, descriptors were assessed in the subsequent section.
 | ||
| Fig. 8 The MEP of the (a) C2, (b) C4, (c) C5 and (d) DCF. | ||
The reactivity descriptors of C2, C4, C5, and DCF were computed and analyzed to investigate the correlation among structure, stability, and chemical reactivity. The difference between HOMO–LUMO is a reliable predictor of the noncovalent interaction between photocatalysts and reactants.75 The energy difference between HOMO–LUMO is a crucial characteristic for comparing the reactivity of C2, C4, and C5 with respect to DCF. A smaller gap corresponds to increased reactivity among the photocatalysts.
According to the difference between HOMO–LUMO presented in Table 4, the photocatalysts reactivity and DCF may be ordered as follows: DCF (2.3459 eV) < C2 = C5 (2.5274 eV) ≈ C4 (2.5130 eV). According to the values, during the photodegradation of DCF utilizing C2, C4, and C5 as photocatalysts. C2, C4, and C5 function as nucleophiles, while DCF acts as an electrophile.76
| Name | HOMO (eV) | LUMO (eV) | η (eV) | μ (eV) | σ (eV−1) | χ (eV) | LUMO–HOMO (eV) | ΔN (eV) |
|---|---|---|---|---|---|---|---|---|
| C2 | −5.2357 | −2.7083 | 1.2637 | −3.9720 | 0.7913 | 3.972 | 2.5274 | −12.485 |
| C4 | −5.2722 | −2.7592 | 1.2563 | −4.0157 | 0.7959 | 4.0157 | 2.5130 | −12.839 |
| C5 | −5.2390 | −2.7116 | 1.2637 | −3.9753 | 0.7913 | 3.9753 | 2.5274 | −12.505 |
| DCF | −5.0365 | −2.6906 | 1.1729 | −3.8635 | 0.8526 | 3.8635 | 2.3459 | −12.726 |
Some important reactivity parameters such as chemical hardness(η), chemical potential (μ), electronegativity (χ), and softness (σ). The compounds' reactivity and chemical stability were assessed using absolute hardness (η) and softness (σ). A lower value of η correlates with increased reactivity among the chemicals. In this investigation, C2, C4, and C5 exhibit almost equivalent values (1.255 eV). The results corresponded with the computed difference between HOMO–LUMO, indicating that photocatalysts with a smaller difference between HOMO–LUMO exhibit the greatest η, suggesting that the compound has the tendency to donate electrons to an acceptor during the process. The highest charge transfers (ΔNmax) for DCF (−12.726 eV), C2 (−12.485 eV), C4 (−12.839 eV), and C5 (−12.505 eV) were computed using eqn (4).
According to the data observed from eqn (5), DCF experiences electrophilic nucleophilic and nucleophilic attacks when interacting with C2, C4, and C5, respectively. The electrophilic charge transfer (ECT) was calculated using eqn (5). The ECT values for the DCF–C2 (0.241 eV) and DCF–C5 (0.221 eV) complexes are positive, whereas the value for the DCF–C4 complex (−0.113 eV) is negative. According to this study, C2, C4 and C5 engage in nucleophilic and electrophilic substitution by either giving or accepting an electron from DCF, therefore establishing a non-covalent contact.75,76
 | (4) |
 | (5) |
The HOMO and LUMO indicate the molecular chemical stability, with red indicating negative charge and green signifying positive charges associated with the compound. The HOMO and LUMO denote the electron donor and electron acceptor, respectively. Fig. 7 indicates that the HOMO and LUMO electron densities of all the catalysts exhibit small variations.
The results for the HOMO of the compound studied are shown in Fig. 9, and the LUMO of the compound studied is shown in Fig. 10. The main contributors to degradation were hydroxyl radicals (OH˙) and superoxide radical anion (˙O−2). The HOMO and LUMO positioning are responsible for OH˙ and (˙O−2) respectively.77 The HOMO position is more negative, representing more OH˙ contributors, and if LUMO is more positive, it represents more (˙O−2) contributors. From Fig. 8 and 9 we can say that C5 has more OH˙ contributors than C2 and more (˙O−2) contributors than C4. From here, we can say that both contributors make C5 the most efficient compound for degradation.
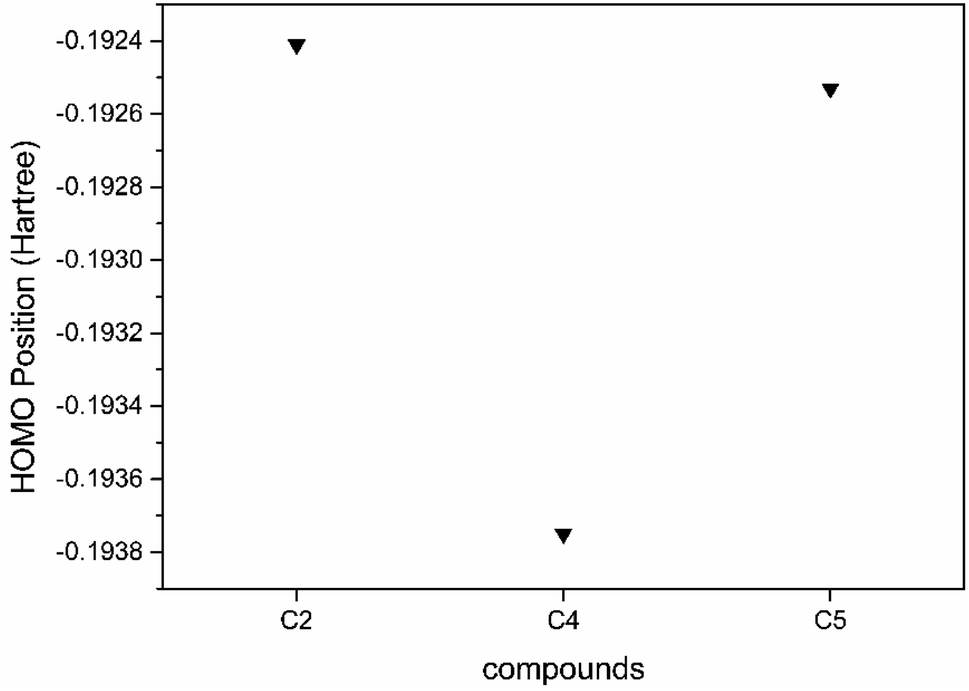 | ||
| Fig. 9 HOMO positioning of C2, C4, and C5 compounds. | ||
 | ||
| Fig. 10 LUMO positioning of C2, C4, and C5 compounds. | ||
Fig. 11 illustrates the formation energy of various compounds, revealing that C5 has a higher energy than C2 and C4, which could potentially account for its high degradation efficiency. High energy means less stability, which increases the tendency of a compound to interact with a pollutant for degradation. The HOMO–LUMO gaps of C2, C4, and C5 were found to be similar at 2.52 eV with a difference of ±0.01 eV. These minor differences in the energy gaps can be attributed to the synthesis methods used.
 | ||
| Fig. 11 Formation energies of C2, C4, and C5 compounds. | ||
6. Conclusions
In summary, we have successfully accomplished the synthesis of the target complexes (C1–C6) with high yields, and their structural features were thoroughly characterized using various spectroscopic techniques (UV, IR, NMR and mass spectrometry). The synthesized complexes were employed in heterogeneous photocatalysis to degrade a water pollutant DCF. The findings of this study demonstrate a substantial advancement in the photocatalytic application of TPY-based metal complexes, particularly in the degradation of pharmaceutical pollutants such as diclofenac potassium, surpassing previous studies on methylene blue degradation by exhibiting higher efficiency, superior charge transfer characteristics, and excellent recyclability, thus establishing these novel alkoxy-functionalized TPY complexes as promising candidates for sustainable wastewater treatment technologies. The photocatalytic efficiencies of the complexes were in the order: C5 (84.3%) > C2 (81.5%) > C4 (77.2%) > C6 (72.6%) > C1 (68.5%) > C3 (64.1%). Among different complexes of transition metal with a TPY backbone, C5 effectively removed 10 mg L−1 of DCF, achieving a degradation efficiency of 90.2% within 90 min of light irradiation employing only 20 mg of the catalyst at pH 5. The reaction kinetics was in accordance with the pseudo-first-order kinetics with the highest rate constant observed for C5 at 0.02355 min−1 and the sustainable photocatalytic performance of C5 up to four repeated cycles indicated its economic feasibility. The photocatalytic experiments carried out in the presence of scavengers revealed that the prime reactive species involved in the degradation process were ˙O−2 and OH˙. Furthermore, the correlation between DFT results and experimental findings provides insight into the photocatalytic efficiencies of the studied complexes. The HOMO–LUMO gap analysis indicates that C2, C4, and C5 exhibit similar reactivity (∼2.52 eV). Additionally, the DOS study revealed that C5 exhibits an improved photoactivity value owing to the enhanced localization and delocalization of the HOMO and LUMO in C5 compared to C2 and C4. The position of the HOMO and LUMO is responsible for OH˙ and (˙O2−), respectively, which makes C5 a suitable option for photoactivity. According to the DFT calculation, the nucleophilic and electrophilic attacks occur during the formation of the C2–DCF, C5–DCF, and C4–DCF complexes, respectively. DFT results align with experimental findings, reinforcing the role of electronic structure in photocatalytic performance and highlighting computational methods as valuable tools for screening effective photocatalysts for environmental remediation. In conclusion, the C5 is an efficient candidate for promising applications of rapid degradation DCF in pharmaceutical wastewater. However, the practical utility of this catalyst requires upscaled synthesis and photocatalytic studies employing real wastewater to assess its industrial feasibility. The catalyst should also be evaluated against other emerging contaminants, such as pesticides, microplastics, and various personal care products, to further broaden its environmental impact. Moreover, assessing the potential toxicity associated with this catalyst is crucial to ensure environmental regulatory standards. These future directions could significantly strengthen the applicability of TPY-based metal complexes for sustainable wastewater treatment.Data availability
All the data is provided in the manuscript and ESI file.†Author contributions
Ehsan Ullah Mughal: Main idea, supervision, methodology, final writing the manuscript. Nafeesa Naeem: Data analysis and collection, reviewing, editing, first-draft preparation. Farwa Zainab: Experimental work performance. Ayesha Javaid: Performed photodegradation studies, first-draft preparation. Muhammad Imran: Methodology, performed photodegradation studies. Sujeet Kumar Pandey: Computational studies. Amina Sadiq: Formal analysis. Ayza Jabeen: Formal analysis. Hanan A. Ogaly: Formal analysis. Fatimah AM Al-Zahrani: Formal analysis. Abdullah Yahya Abdullah Alzahrani: Formal analysis. All authors gave final approval for publication.Conflicts of interest
No conflict of interest to declare.Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Research Project under grant number RGP2/152/45.References
- K. P. Garnock-Jones, CNS Drugs, 2014, 28, 761–768 CAS.
- B. Chuasuwan, V. Binjesoh, J. Polli, H. Zhang, G. Amidon, H. Junginger, K. Midha, V. Shah, S. Stavchansky and J. Dressman, J. Pharm. Sci., 2009, 98, 1206–1219 CrossRef CAS PubMed.
- A. Saylan, C. A. Turel, I. Turel, A. Cetinkaya, I. E. Torun and H. Celik, Exp. Biomed. Res., 2023, 6, 77–87 CAS.
- H. Mabrouki and D. Akretche, Desalin. Water Treat., 2016, 57, 6033–6043 CAS.
- S. Chime, A. Attama, F. Kenechukwu, E. Umeyor and G. Onunkwo, Br. J. Pharm. Res., 2013, 3, 90 Search PubMed.
- I. Alessandretti, C. V. T. Rigueto, M. T. Nazari, M. Rosseto and A. Dettmer, J. Environ. Chem. Eng., 2021, 9, 106743 CAS.
- A. C. F. P. Fuhr, C. F. de Azevedo, N. Ahmad, S. Mohandoss, G. L. Dotto and F. M. Machado, J. Mol. Liq., 2024, 407, 125191 Search PubMed.
- R. Wang, Z. Wang, H. Yuan, C. Li and N. Zhu, J. Hazard. Mater., 2024, 479, 135603 CAS.
- L. Lonappan, S. K. Brar, R. K. Das, M. Verma and R. Y. Surampalli, Environ. Int., 2016, 96, 127–138 CrossRef CAS PubMed.
- K. Onwuka, C. F. Aaron, C. I. Nosiri, O. C. Atasie, C. Aguwamba and N. I. Uzoamaka, JCNB, 2024, 5, 37–55 CrossRef.
- J. M. Angosto, M. J. Roca and J. A. Fernández-López, Water, 2020, 12, 3567 CAS.
- F.-P. Huang, W.-J. Qin, X.-Y. Pan, K. Yang, K. Wang and Q.-H. Teng, J. Org. Chem., 2024, 89, 4395–4405 CrossRef CAS PubMed.
- N. S. dos Santos, L. F. Marquiza, C. S. C. Calheiros, P. S. Cavalheri, B. S. Machado, G. H. Cavazzana and F. J. C. M. Filho, Water, 2021, 13, 1043 CrossRef CAS.
- C. F. de Azevedo, N. F. de Souza, F. B. Cardoso, A. C. F. P. Fuhr, E. C. Lima, A. G. Osório and F. Machado Machado, Environ. Sci. Pollut. Res., 2024, 31, 48650–48662 CrossRef CAS PubMed.
- X. Yang, J. Yang, T. Zhao, W. Qian, Y. Wang, A. Holmen, W. Jiang, D. Chen and H. Ben, Chem. Eng. J., 2022, 445, 136655 CrossRef CAS.
- A. Javaid, M. Imran, F. Kanwal and S. Latif, Mater. Sci. Semicond. Process., 2024, 177, 108350 CrossRef CAS.
- N. Zhang, J. Li, G. Liu, X. Chen and K. Jiang, Water Sci. Technol., 2017, 75, 2163–2170 CrossRef CAS PubMed.
- S. K. Srivastava, RSC Appl. Interfaces, 2024, 1(3), 340–429 RSC.
- K. Prakruthi, M. P. Ujwal, S. R. Yashas, B. Mahesh, N. Kumara Swamy and H. P. Shivaraju, Environ. Sci. Pollut. Res., 2022, 29, 4930–4957 CrossRef CAS PubMed.
- A. Javaid, M. Imran, F. Kanwal, S. Latif, S. F. Adil, M. R. Shaik and M. Khan, Molecules, 2023, 28, 7979 CrossRef CAS PubMed.
- G.-R. Wu, J.-K. Xu, L.-J. Sun, Y.-F. Li, S.-Q. Gao and Y.-W. Lin, J. Environ. Chem. Eng., 2023, 11, 111471 CrossRef CAS.
- A. Intisar, A. Ramzan, S. Hafeez, N. Hussain, M. Irfan, N. Shakeel, K. A. Gill, A. Iqbal, M. Janczarek and T. Jesionowski, Chemosphere, 2023, 336, 139203 CrossRef CAS PubMed.
- G. Amariei, K. Boltes, R. Rosal and P. Leton, PLoS One, 2020, 15, e0227267 CrossRef CAS PubMed.
- I. Tbessi, M. Benito, J. Llorca, E. Molins, S. Sayadi and W. Najjar, J. Alloys Compd., 2019, 779, 314–325 CrossRef CAS.
- A. B. Ganganboina, M. D. Nguyen, T. H. L. Nguyen, E. P. Kuncoro and R.-A. Doong, Chem. Eng. J., 2021, 425, 131520 CrossRef.
- W. Liu, Y. Li, F. Liu, W. Jiang, D. Zhang and J. Liang, Water Res., 2019, 151, 8–19 CrossRef CAS PubMed.
- S. Li, J. Cui, X. Wu, X. Zhang, Q. Hu and X. Hou, J. Hazard. Mater., 2019, 373, 408–416 CrossRef CAS PubMed.
- A. Tufail, S. Alharbi, J. Alrifai, A. Ansari, W. E. Price and F. I. Hai, Process Saf. Environ. Prot., 2021, 145, 110–119 CrossRef CAS.
- F. T. Geldasa, M. A. Kebede, M. W. Shura and F. G. Hone, RSC Adv., 2023, 13, 18404–18442 CAS.
- X. Liu, F. Li, Y. Liu, P. Li, L. Chen, B. Li, T. Qian and W. Liu, J. Environ. Chem. Eng., 2022, 10, 107545 CrossRef CAS.
- T. Velempini, E. Prabakaran and K. Pillay, Mater. Today Chem., 2021, 19, 100380 CrossRef CAS.
- V. Calisto, M. R. M. Domingues and V. I. Esteves, Water Res., 2011, 45, 6097–6106 CrossRef CAS PubMed.
- S. Kothawade and P. Shende, Coord. Chem. Rev., 2024, 510, 215851 CrossRef CAS.
- E. U. Mughal, S. F. Kainat, N. Naeem, M. Imran, A. Javaid, A. Sadiq, A. Y. A. Alzahrani, S. B. Moussa and S. A. Ahmed, Dyes Pigm., 2024, 112254 CrossRef CAS.
- E. U. Mughal, A. Javaid, M. Imran, M. A. Abourehab, E. B. Elkaeed, N. Naeem, A. Y. A. Alzahrani, A. Sadiq and S. F. Kainat, Inorg. Chim. Acta, 2023, 546, 121329 CrossRef CAS.
- C. Wei, Y. He, X. Shi and Z. Song, Coord. Chem. Rev., 2019, 385, 1–19 CrossRef CAS PubMed.
- A. Winter and U. S. Schubert, ChemCatChem, 2020, 12, 2890–2941 CrossRef CAS.
- S. F. Kainat, M. B. Hawsawi, E. U. Mughal, N. Naeem, A. M. Almohyawi, H. M. Altass, E. M. Hussein, A. Sadiq, Z. Moussa and A. S. Abd-El-Aziz, RSC Adv., 2024, 14, 21464–21537 RSC.
- Y. Wang, T. Liu, L. Chen and D. Chao, Inorg. Chem., 2021, 60, 5590–5597 CrossRef CAS PubMed.
- D. Saccone, C. Magistris, N. Barbero, P. Quagliotto, C. Barolo and G. Viscardi, Materials, 2016, 9, 137 CrossRef PubMed.
- K. Liu, L. Du and T. Wang, Inorg. Chem., 2024, 63, 4614–4627 CrossRef CAS PubMed.
- E. U. Mughal, R. J. Obaid, A. Sadiq, M. A. Alsharif, N. Naeem, S. Kausar, A. A. Altaf, R. S. Jassas, S. Ahmed and R. I. Alsantali, Dyes Pigm., 2022, 201, 110248 CrossRef CAS.
- X. Sun, H. D. Cole, G. Shi, V. Oas, A. Talgatov, C. G. Cameron, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2024, 63, 21323–21335 CrossRef CAS PubMed.
- E. U. Mughal, M. Mirzaei, A. Sadiq, S. Fatima, A. Naseem, N. Naeem, N. Fatima, S. Kausar, A. A. Altaf and M. N. Zafar, R. Soc. Open Sci., 2020, 7, 201208 CrossRef CAS PubMed.
- V. Barone, J. Bloino and M. Biczysko, Vibrationally-resolved electronic spectra in GAUSSIAN 09. Revision a, 2(1), 2009, pp. 1–20 Search PubMed.
- M. Frisch and F. Clemente, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino and G. Zhe, Gaussian, 9 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView 6.0.16, Semichem Inc., Shawnee Mission, KS, USA, 2016, pp. 143–150 Search PubMed.
- N. Chakinala, P. Ranjan, A. G. Chakinala and P. R. Gogate, Catal. Commun., 2023, 174, 106589 CrossRef CAS.
- G. Wu, Y. Liu, G. Liu, R. Hu and G. Gao, J. Mol. Liq., 2021, 328, 115411 CrossRef CAS.
- A. Akbari, Z. Sabouri, H. A. Hosseini, A. Hashemzadeh, M. Khatami and M. Darroudi, Inorg. Chem. Commun., 2020, 115, 107867 CrossRef CAS.
- C. Paraschiv, A. Cucos, S. Shova, A. M. Madalan, C. Maxim, D. Visinescu, B. Cojocaru, V. I. Parvulescu and M. Andruh, Cryst. Growth Des., 2015, 15, 799–811 CrossRef CAS.
- S. Ranjan Jena, T. Mandal and J. Choudhury, Chem. Rec., 2022, 22, e202200165 CrossRef CAS PubMed.
- M. Hernick and C. Fierke, Comprehensive Natural Products II, 2010, vol. 8, pp. 547–581 Search PubMed.
- S. Wang, Z. Liu, Y. Zhao, W. Song, Z. Sun and J. Ma, J. Environ. Chem. Eng., 2024, 12, 113087 CrossRef CAS.
- Z. Xiao, W. Yang, J. Wang, S. Cao, J. Yang and M. Zhu, Chem. Eng. J., 2025, 504, 159022 CrossRef CAS.
- F. Han, X. Ye, Q. Chen, H. Long and Y. Rao, Sep. Purif. Technol., 2020, 232, 115967 CrossRef CAS.
- M. A. Behnajady, H. Eskandarloo, N. Modirshahla and M. Shokri, Dig. J. Nanomater. Biostructures, 2011, 6, 1887–1895 Search PubMed.
- A. Javaid, M. Imran, M. P. Rayaroth, X. Sun, C. Wang, G. Boczkaj and M. Momotko, Curr. Opin. Chem. Eng., 2024, 46, 101054 CrossRef.
- P. John, K. Johari, N. Gnanasundaram, A. Appusamy and M. Thanabalan, Environ. Technol. Innovation, 2021, 22, 101412 CrossRef CAS.
- K. V. Plakas, V. C. Sarasidis, S. I. Patsios, D. A. Lambropoulou and A. J. Karabelas, Chem. Eng. J., 2016, 304, 335–343 CrossRef CAS.
- N. Ahmadpour, M. H. Sayadi, S. Sobhani and M. Hajiani, J. Environ. Manage., 2020, 271, 110964 CrossRef CAS PubMed.
- L. Rueda-Salaya, A. Hernández-Ramírez, L. Hinojosa-Reyes, J. Guzmán-Mar, M. Villanueva-Rodríguez and E. Sánchez-Cervantes, J. Photochem. Photobiol., A, 2020, 391, 112364 CrossRef CAS.
- L. Gao, B. Zhou, F. Wang, R. Yuan, H. Chen and X. Han, Environ. Sci. Pollut. Res., 2020, 27, 2044–2053 CrossRef CAS PubMed.
- N. Aghababaei, M. Abdouss, H. Hosseini-Monfared and F. Ghanbari, J. Environ. Chem. Eng., 2023, 11, 110477 CrossRef CAS.
- Z. Wu, J. Liu, J. Shi and H. Deng, Environ. Res., 2024, 249, 118361 CrossRef CAS PubMed.
- A. Javaid, M. Imran, F. Kanwal, S. Latif, M. Khan, M. Kuniyil, M. R. Shaik, M. R. H. Siddiqui and S. F. Adil, Ceram. Int., 2024, 50, 41915–41930 CrossRef CAS.
- A. Javaid, M. Imran, F. Kanwal, S. Latif and M. N. Khan, J. Water Process Eng., 2025, 69, 106665 CrossRef.
- S.-Q. Li, M. Ray, A.-J. Zhu, X. Zhang, A. K. Pradhan, A. Mohanty, M. Muddassir and J.-C. Jin, Polyhedron, 2024, 262, 117166 CrossRef CAS.
- Y. Wen, M. Feng, P. Zhang, H.-C. Zhou, V. K. Sharma and X. Ma, ACS ES&T Eng., 2021, 1, 804–826 Search PubMed.
- V. Muelas-Ramos, M. Sampaio, C. Silva, J. Bedia, J. Rodriguez, J. Faria and C. Belver, J. Hazard. Mater., 2021, 416, 126199 CrossRef CAS PubMed.
- K. Hemkumar, P. Ananthi and A. Pius, Mater. Sci. Eng., B, 2023, 292, 116453 CrossRef CAS.
- D. Pourkodee, D. R. Devee and E. Sailatha, Inorg. Chem. Commun., 2024, 113585 Search PubMed.
- N. Aghababaei, M. Abdouss, H. Hosseini-Monfared and F. Ghanbari, J. Water Process Eng., 2023, 53, 103702 CrossRef.
- G. Tseberlidis, V. Trifiletti, A. H. Husien, A. L'Altrella, S. Binetti and F. Gosetti, Appl. Sci., 2024, 14, 9923 CrossRef CAS.
- J. Sharma, I. Mishra and V. Kumar, J. Environ. Manage., 2016, 166, 12–22 Search PubMed.
- F. B. Shittu, A. Iqbal, M. N. Ahmad, M. R. Yusop, M. N. M. Ibrahim, S. Sabar, L. D. Wilson and D. H. Y. Yanto, RSC Adv., 2022, 12, 10409–10423 RSC.
- S. Kaya and C. Kaya, Comput. Theor. Chem., 2015, 1060, 66–70 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01117e |
| This journal is © The Royal Society of Chemistry 2025 |