
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient oxidation of methane into methanol over Ni-promoted Cu/ZSM-5†
Zilong Shena,
Wenzhi Li *a,
Jingting Jina,
Zhiheng Lub,
Liqun Wanga,
Yihang Jianga and
Liang Yuanb
*a,
Jingting Jina,
Zhiheng Lub,
Liqun Wanga,
Yihang Jianga and
Liang Yuanb
aLaboratory of Clean Low-Carbon Energy, Department of Thermal Science and Energy Engineering, University of Science and Technology of China, Hefei 230023, P. R. China. E-mail: liwenzhi@ustc.edu.cn
bNational & Local Joint Engineering Research Center of Precision Coal Mining, Anhui University of Science and Technology, Huainan 232001, P. R. China
First published on 17th March 2025
Abstract
Direct functionalization of methane in natural gas is of paramount importance but faces tremendous challenges. We reported a nickel-modified copper zeolite catalyst for the selective oxidation of methane into methanol. Using H2O2 as an oxidant in the liquid phase at 80 °C, Cu1Ni0.75/ZSM-5 catalyst presented a relatively high methanol yield of 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 162 μmol gcat−1 h−1 (with a methanol selectivity of ∼74%). Combining series of designed experiments and thorough characterization analysis, including electron microscopy, X-ray photoelectric spectroscopy, Fourier transform infrared reflection as well as in situ diffuse reflectance infrared Fourier transform spectroscopy, abundant CuI active sites were found on Ni-Promoted Cu/ZSM-5, differing from the dominating CuII active sites over Cu/ZSM-5. CuI active sites had an excellent ability to promote CH4 adsorption, CH4 activation and CH3OH generation compared to CuII active sites. This work elucidates a constellation of insightful and potent perspectives for further improvement of metal-zeolite catalysts for the direct oxidation of methane to methanol.
162 μmol gcat−1 h−1 (with a methanol selectivity of ∼74%). Combining series of designed experiments and thorough characterization analysis, including electron microscopy, X-ray photoelectric spectroscopy, Fourier transform infrared reflection as well as in situ diffuse reflectance infrared Fourier transform spectroscopy, abundant CuI active sites were found on Ni-Promoted Cu/ZSM-5, differing from the dominating CuII active sites over Cu/ZSM-5. CuI active sites had an excellent ability to promote CH4 adsorption, CH4 activation and CH3OH generation compared to CuII active sites. This work elucidates a constellation of insightful and potent perspectives for further improvement of metal-zeolite catalysts for the direct oxidation of methane to methanol.
1. Introduction
Methane is the major constituent of coalbed methane, natural gas, combustible ice, etc., but has drawbacks of high storage cost and low calorific value as a fuel.1,2 The highly efficient conversion of methane into commodity chemicals and liquid fuels,3–5 especially its conversion into methanol (a clean liquid fuel suitable for transportation and storage),6–8 has drawn extensive attention. The current industrial route is a two-step process, and depends on step-wise catalytic oxidation to synthesis gas, a mixture of H2 and CO, which can react to form methanol (CH4 + H2O → CO + 3H2, CO + 2H2 → CH3OH) under high temperature and pressure.9,10 This process, however, is quite complicated and energy-intensive,11 thus, the direct oxidation of methane to methanol under mild reaction conditions is urgently needed. Owing to the high carbon-hydrogen (C–H) bond energy (438.8 kJ mol−1) in the non-polarized methane, direct oxidation of methane to methanol remains a major challenge.12–14 A large number of noble metal catalysts (e.g., AuPd@ZSM-5-C16,15 AuPd nanocatalyst,16 IrCuPd-ZSM-5,17 GR-Rh/Ni (111) catalyst18 and so on) have been proposed due to their excellent ability in methane activation. Moreover, a portion of researchers chose to design catalysts with unique active sites (i.e., Cu/CN,19 metal–organic framework (MOF),20,21 PdxAuy nanosheets22) to achieve efficient direct conversion of methane to methanol. Yet, these catalysts are either expensive to prepare or difficult to synthesize, which makes them unreliable for large-scale production.23,24 Inspired by the biocatalytic enzyme systems in nature that can achieve direct methane to methanol conversion under mild conditions, metal-containing zeolites with the enzyme-like active Cu-oxo active sites have received great attention.25–29In recent years, direct conversion of methane into methanol over the copper-exchanged zeolites such as Cu-MOR,30 Cu-CHA,11 Cu-ZSM-5,31 using O2,11,30 N2O32 or H2O2 (ref. 31) as oxidant, has garnered significant interest. Sushkevich et al. achieved 97% methanol selectivity on Cu-MOR using water steam as the oxidant at 473–673 K.30 Zhang et al. reported that Cu–OH single sites confined within the 6-membered ring (6 MR) voids of SSZ-13 zeolite exhibited high efficiency in continuous methane to methanol conversion using water as an oxidant at 400 °C. In addition, the bare Cu(II) single atom sites in 6 MR exhibited activity in methane C–H activation, while their stable four coordination structure hindered their reactivity at lower temperatures.33 The direct conversion of methane in gas-phase systems mostly occurred under high-temperature conditions, which contradicts the intention to reduce energy consumption. Therefore, many researchers delved their interest into the low-temperature liquid-phase reaction systems, using H2O2 as an oxidant. Jin et al. used copper oxysalt as the precursor to prepare Cu/ZSM-5 by calcination method, on which the methanol yield exhibited a special M-shaped curve with the highest yield of 15975.73 μmol gcat−1 h−1.31 Nevertheless, single metal catalysts may not be able to effectively distinguish methane from generated intermediates, resulting in increased side reactions and decreased methanol selectivity. In the process of methane conversion, it is necessary to construct more refined active sites to improve the overall efficiency.
It was found that bimetallic catalysts have superior ability to promote CH4 activation and CH3OH generation compared to monometallic catalysts. For example, Yu et al. reported a bimetallic catalyst (Cu–Fe(2/0.1)/ZSM-5), exhibiting a superior methanol yield of 431 molMeOH molFe−1 h−1, which is at least one order of magnitude greater than that of any catalysts previously reported.34 Wang et al. elaborated that Pd and Cu synergistically disaggregated methane, which facilitated the formation of important intermediate (metal–CH3) with a relatively higher CH3OH yield of ∼31800 μmol gcat−1 h−1.35 Yu et al. designed and constructed atomically dispersed Ag and Cu dual single atoms anchored on ZSM-5 through an improved co-adsorption strategy as an efficient catalyst to promote methane conversion. This work demonstrated that the synergistic effect between Ag and Cu single atoms anchored within the zeolite channels promoted the formation of highly active surface hydroxyl species to activate C–H bonds, and correspondingly enhanced the yield, selectivity, and stability.36 Xu et al. found that regulating Au coverage on PdxAuy nanosheets could control the energy barrier of the triggering step to obtain an excellent CH3OH production rate of 147.8 mmol gPd−1 h−1, with a CH3OH selectivity of 98% at 70 °C.22 However, the effect of bimetallic synergy on the direct conversion of methane into methanol is still a noteworthy issue. Moreover, most existing researches used noble metals to modify copper exchange zeolites, which were costly, thus, we plan to find a suitable non-noble metal modification pathway to promote Cu-zeolites’ reactivity.
Herein, we noticed that the addition of Ni during the preparation process of Cu/ZSM-5 catalysts greatly increased the yield of methanol, while maintaining methanol selectivity, thus, we devised a series of Ni-Promoted Cu/ZSM-5 catalysts using ammonia evaporation method and were applied for direct oxidation of methane to methanol (DOMM) in the liquid phase using H2O2 as an oxidant at 80 °C. We found that Ni improved the CH3OH yield of Cu/ZSM-5 catalysts. Therefore, a series of experiments and characterization methods (such as electron microscopy, spectroscopic techniques to in situ or ex situ, Electron Spin Resonance) were used to explain this phenomenon, from which we explained the reason why Ni promoted Cu/ZSM-5 catalysts: Ni promoted the conversion of CuII sites to CuI sites, while CuI sites had excellent ability to promote CH4 adsorption, CH4 activation and CH3OH generation compared to CuII sites, and then discovered a possible free radicals pathway for the high-yield conversion of CH4 to C1 products (CH3OH and HCOOH).
2. Materials and methods
2.1. Catalyst preparation
The pretreatment of zeolite (HZSM-5):H-ZSM-5 was calcined at 500 °C with a heating ramp of 2 °C min−1 for 4 h in air.Ammonia evaporation method: First, 1 mL of NH3·H2O was added to 20 mL of deionized water dispersed with 1g of HZSM-5 support and stirred for 10 min. Once 5 mL of the required concentration of Cu(NO3)2·3H2O and Ni(NO3)2·6H2O solution was added, then sealed the suspension with Parafilm and stirred for 1 hour. Next, removed the Parafilm and evaporated the solvent at 90 °C for 1 hour. After obtaining the catalyst precursor through vacuum filtration, it was dried in an oven at 110 °C for 8 h. Finally, the sample was calcined at 500 °C with a heating ramp of 2 °C min−1 for 4 h in air. The obtained catalyst was denoted as CuxNiy/ZSM-5 (x/y refers to the ratio of weight percentage of Cu/Ni). Cu1/ZSM-5 refers to the sample with 1 wt% of Cu.
2.2. Catalyst testing
The methane oxidation was tested in a 100 mL stainless steel reactor. The vessel was loaded with an aqueous solution of H2O2 (40 mL, 0.5 M) and 10 mg of catalyst, purged three times with methane, and then charged with methane to 30 bar. The reaction mixture was heated to 80 °C while being stirred at a speed of 900 rpm and maintained under these conditions for a duration of 30 minutes. Upon completion of the reaction, the vessel was rapidly cooled using an ice-bath.The gaseous phase products were further analyzed by a gas chromatograph (GC 5190) equipped with a thermal conductivity detector (TCD). The liquid products were quantified by 1H-NMR on a 600 MHz JNM-ECZ600R/S1 Superconducting Fourier Nuclear Magnetic Resonance Spectrometer. Typically, 0.7 mL of the sample solution and 0.3 mL of D2O were combined in an NMR tube. A solvent suppression program was employed to minimize the signal originating from the solvent. A representative example of 1H NMR spectrum obtained from products of methane oxidation reaction is shown in Fig. S1.†
The CH3OH yield, HCOOH yield, C1 yield and CH3OH selectivity were calculated through the following equations:
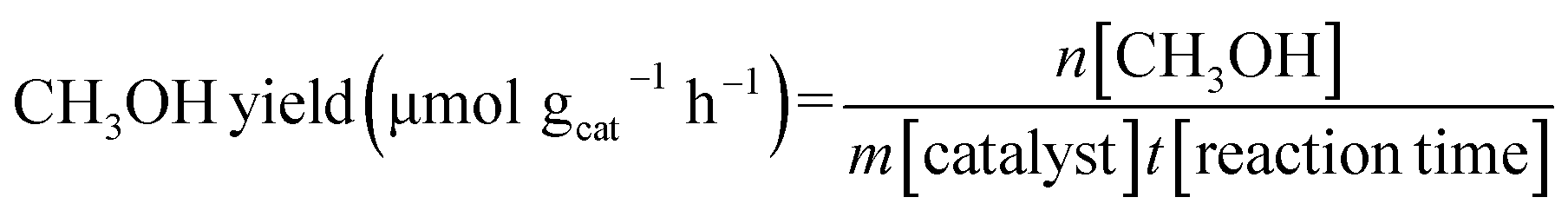



2.3. Catalyst characterization
The Transmission Electron Microscope (TEM) and High-Angle Annular Dark Field Scanning Transmission Electron Microscope (HAADF-STEM) mapping images were characterized by two TEM instruments (JEM-2100F FETEM and FEI Talos F200S), which showed the morphology and surface crystal structure of the catalysts prepared in this article. The X-ray Diffraction (XRD) patterns were performed on a Rigaku SmartLab (9) Multifunctional Rotating-anode X-ray Diffractometer (Cu Kα) in the range of 5–60° with a step size of 0.02°, operated at 40 kV and 40 mA. The X-ray Photoelectron Spectroscopy (XPS) was performed using a Thermo Scientific ESCALAB 250Xi X-ray photoelectron spectrometer (Al Kα 150 W, hν = 1486.6 eV). The binding energies were calibrated using C1s peak of contaminant carbon (BE = 284.8 eV) as an internal standard. The loading amounts of Cu and Ni were determined by the Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES) instrument. Before analysis, a proper amount of powder sample was treated with nitric acid to obtain a clear solution. The surface area and pore-size distribution of catalysts were tested through nitrogen adsorption and desorption method on a Micromeritics ASAP 2460 instrument using the BET equation and the BJH method, respectively. Additionally, adsorption/desorption isotherms for CH4 in the catalysts were also measured. Fourier Transform Infrared Reflection (FT-IR) and in situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) were analyzed on the NICOLET iS50 FT-IR spectrometer equipped with a MCT detector. High-performance Liquid Chromatography (HPLC) was used to test the yield of formic acid. 5,5-dimethyl-1-pyrroline N-oxide (DMPO) spin-trapping Electron Paramagnetic Resonance (EPR) experiments were conducted on a JES-FA 200 (JEOL) Electron Paramagnetic Resonance Spectrometer at room temperature. The samples (1 mL) after methane oxidation at 80 °C for 5 min were quickly collected and 1 mL DMPO solution (10 mg mL−1) was added into the samples, and then filtered and frozen by liquid nitrogen. These samples were thawed subsequently for the EPR measurements.3. Results and discussions
3.1. Catalytic performance
The CH3OH yield, CH3OH selectivity and CH4 conversion rate over Cu1/ZSM-5 and Cu1Ni0.75/ZSM-5 under different reaction conditions were tested. Methanol, formic acid, unreacted methane, hydrogen and oxygen produced by the decomposition of hydrogen peroxide could be detected through 1H NMR and gas chromatograph equipped with a thermal conductivity detector.The CuxNiy/ZSM-5 catalysts which were prepared by ammonia evaporation method exhibited excellent catalytic performance for DOMM under the standard reaction conditions (x/y referred to the ratio of weight percentage of Cu/Ni and the actual loading amounts were shown in Table S1†). Only liquid phase products (CH3OH and HCOOH) were detected after the DOMM reaction, and no gaseous phase products like CO and CO2 were detected, indicating this catalyst have by-passed the unwanted over-oxidation (Fig. S2†). A volcano-like Ni loading-catalytic performance relationship was exhibited where the maximum catalytic activity appeared at Cu1Ni0.75/ZSM-5 with a CH3OH yield of 82162 μmol gcat−1 h−1 and selectivity of 74% (Fig. 1A).
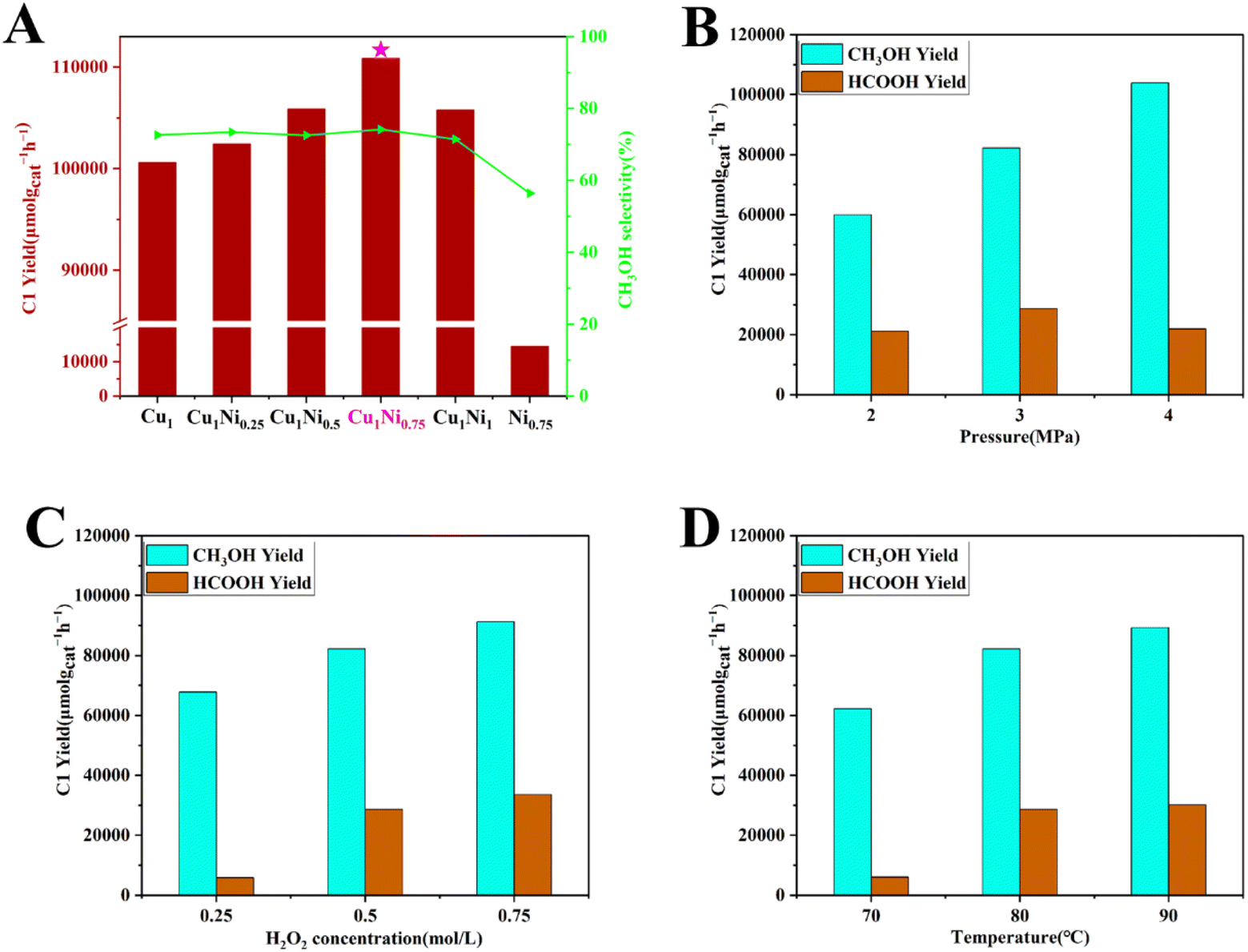 | ||
| Fig. 1 Effect of (A) different Ni loading amounts on C1 yield and CH3OH selectivity (CuxNiy refer to CuxNiy/ZSM-5 catalysts). Effect of (B) different CH4 pressure, (C) H2O2 concentration, (D) reaction temperature on CH3OH and HCOOH yield. Standard reaction conditions for this work unless otherwise stated were 38 mL H2O, 2 mL 30% H2O2, feed CH4 at 3.0 MPa, react at 80 °C for 30 min, and 10 mg CuxNiy/ZSM-5 catalysts. | ||
In the control experiment without methane or H2O2, there were no products. When no catalyst was added, there were still a trace amount of CH3OH, which could resulted from the ˙OH radicals generated by H2O2 spontaneous decomposition (Fig. S3†). For DOMM reaction over the pure HZSM-5 zeolite support, the CH3OH yield was not significantly different from the situation without catalyst, which indicated that pure HZSM-5 zeolite support had merely catalytic effect on the DOMM reaction (Fig. S4†). For DOMM over Cu1/ZSM-5 at 80 °C, the yield of CH3OH was 73050 μmol gcat−1 h−1 (Fig. 1A), which was much higher than that of pure HZSM-5 zeolite support. Obviously, the load of Cu greatly promoted the DOMM reaction, which proved that Cu was the main active site of the reaction. However, when the Ni-Promoted Cu/ZSM-5 catalysts were used in the DOMM reaction, the yields of CH3OH were further improved by 12.5% compared to the Cu/ZSM-5 catalysts, which showed that the addition of Ni improved the activity of Cu species over Cu1Ni0.75/ZSM-5 catalyst. Furthermore, in order to eliminate the possibility that the observed activity may only come from a combination of Ni and Cu active sites, and considering the quantities of Cu, Ni and ZSM-5 in both reaction systems should be equal, we used physically mixed Cu1/ZSM-5 and Ni0.75/ZSM-5 (10 mg each, reaction 6) and physically mixed Cu1Ni0.75/ZSM-5 and ZSM-5(10 mg each, reaction 7) as catalysts for the DOMM reaction to make a comparison (Table S2†). The yield of CH3OH over the physically mixed Cu1Ni0.75/ZSM-5 and ZSM-5 (10 mg each) for DOMM was 857.22 μmol h−1, which was 11.39% higher than that of the physically mixed Cu1/ZSM-5 and Ni0.75/ZSM-5. The above results distinctly suggested that there was a remarkable synergistic effect between Cu and Ni, which enhanced the catalytic activity.
To improve the reaction conditions for the catalytic conversion of methane using Cu1Ni0.75/ZSM-5 catalyst, a series of reaction conditions (such as CH4 pressure, H2O2 concentration, and reaction temperature) were studied. The effect of the CH4 pressure on the DOMM was firstly studied. With the increase of the CH4 pressure, the yield of all products increased, specifically the CH3OH selectivity also increased (Fig. 1B and Table S3†). Next, the H2O2 concentration was studied. Similar to CH4 pressure, the H2O2 concentration was positively correlated with C1 yield. Even so, the CH3OH selectivity possessed the opposite trend (Fig. 1C and Table S3†), evidencing that the ˙OH radicals generated by H2O2 decomposition are crucial in the DOMM reaction as the increase in H2O2 will lead to excessive oxidation of CH4 to HCOOH. Another important reaction condition is temperature, which would improve DOMM reaction (Fig. 1D).
In addition, the stability of catalysts was tested through cyclic experiments under the standard reaction condition (Fig. S5†). CH4 was converted into CH3OH at a rate ranging from 74554 to 82600 μmol gcat−1 h−1 with a CH3OH selectivity kept at 74–79%. Our best catalyst with Cu/Ni loadings at 1/0.75 wt% had a more excellent CH3OH yield compared to other recently reported catalysts, which indicated that Ni-Promoted Cu/ZSM-5 catalyst showed promising application prospects in methane conversion under mild conditions (Fig. S6 and Table S4†). Above all, the Cu1Ni0.75/ZSM-5 catalyst could be considered as a stable and efficient catalyst for the DOMM reaction.
3.2. Structure characterization of catalysts
The activity of catalysts for methane conversion is closely related to their structure,37 thus, a complete structure characterization of the CuxNiy/ZSM-5 catalysts was in progress. First, XRD patterns of the different catalysts were shown (Fig. 2A). After the loading of metal (Cu and/or Ni), no metal peaks were detected (Cu2O:JCPD Card 65-3288, CuO:JCPD Card 45–0937, Ni:JCPD Card 04-0850, NiO:JCPD Card 44-1159; the detailed comparison results were shown in the Fig. S7†).In addition, we could observe the uniform distribution of Cu and Ni elements through the EDS mapping (Fig. 2B), similarly, the ICP-OES results confirmed the existence of Cu and Ni elements, which explained the high dispersibility of Cu and Ni in zeolite support. The TEM images were displayed in Fig. S8,† which indicated that CuNi existed on the surface of ZSM-5 zeolite in the form of nanoparticles. Next, the structure of CuxNiy/ZSM-5 catalysts were further studied by FT-IR (Fig. 2C). The 550 cm−1 vibration band was assigned to the unique vibration way of double pentadecasil ring, a typical characteristics of ZSM-5 zeolite, which showed that the loading of metal did not damage the structure of the ZSM-5 zeolite.38 The 462 cm−1 signal is attributed to the bending vibration of Si(Al)–O bonds, the peaks at 790 cm−1,1080 cm−1 and 1230 cm−1 were assigned to the asymmetric stretching vibrational of Si–O–Si bonds.39 After the loading of metal (Cu or CuNi), the 1080 cm−1 and 1230 cm−1 bands become sharper, which marks the substitution of metal atoms in the ZSM-5 zeolite framework to form Si–O–Cu bonds.40 According to previous reports, this was attributed to the alkalinity of the ammonia evaporation method led to partial desilication, and then copper atoms replaced the position of silicon atoms to form Si–O–Cu bonds in the zeolite framework.41–43 | ||
| Fig. 2 (A) XRD patterns of ZSM-5, Cu1/ZSM-5, Cu1Ni0.75/ZSM-5, and Ni0.75/ZSM-5 catalysts. (B) STEM-EDS elemental mappings of Cu1Ni0.75/ZSM-5 catalyst. (C) The FT-IR spectrum of CuxNiy/ZSM-5 catalysts in the range of 400–2500 cm−1. | ||
The EDS mapping images show the distribution of Cu and Ni elements to be uniformed (Fig. 2B). To further study the elemental coordination information of Cu and Ni, XPS analysis was used to analyze chemical states (Fig. 3A). The Cu 2p signal of the Cu1Niy/ZSM-5 catalysts can be fitted into two peaks located at 932.6 and 933.8 eV, respectively for CuI and CuII species.44,45 In addition, the low intensity of CuII satellite peak located at 943.0 eV may be due to the high dispersibility of CuII in the zeolite,46,47 which has been previously demonstrated by XRD, ICP-OES and EDS mapping. When only Cu was loaded, CuII sites are predominant on the catalyst, with only a small portion of CuI sites present. As the loading of Ni increases, the CuI/CuII ratio rapidly increased from 0.19 to 0.86 (Table 1), correspondingly, the NiII/Ni0 ratio increased from 0.31 to 1.25 (Table S5†), which means that the ratio of CuI and NiII increased.48 Combined with the limited reactivity over Ni/ZSM-5 sample, it could be assumed that CuI plays the dominating role in DOMM reaction. Moreover, the variation of binding energy for Cu 2p and Ni 2p indicates electron transfer from Ni to Cu(Fig. S10†). The electronic perturbation of Cu by Ni is known as ligand effect, these currently electron-rich Cu sites could form strong polar interactions with carbon atoms in methane molecules, which helps to break the symmetry and stability of C–H bonds, ergo boosting CH4 cleavage to *CH3. And the increase of *CH3 could be confirmed through in situ CH4-DRIFTS spectra described below (Fig. 4A).
 | ||
| Fig. 3 (A) Cu 2p XPS spectra of different Cu1Niy/ZSM-5 catalysts. (B) In situ NO-DRIFTS spectra of Cu1/ZSM-5 and Cu1Ni0.75/ZSM-5 catalysts at room temperature. (C) N2 adsorption–desorption isotherms of the ZSM-5, Cu1/ZSM-5 and Cu1Ni0.75/ZSM-5 catalysts at 77 K. (D) Adsorption–desorption isotherms for CH4 in ZSM-5, Cu1/ZSM-5 and Cu1Ni0.75/ZSM-5 catalysts at 283 K. | ||
| Entry | Sample | Binding energy | CuI/CuII ratio | |
|---|---|---|---|---|
| CuI | CuII | |||
| 1 | Cu1/ZSM-5 | 932.6 | 933.8 | 0.19 |
| 2 | Cu1Ni0.25/ZSM-5 | 932.6 | 933.8 | 0.27 |
| 3 | Cu1Ni0. 5/ZSM-5 | 932.6 | 933.8 | 0.53 |
| 4 | Cu1Ni0.75/ZSM-5 | 932.6 | 933.8 | 0.70 |
| 5 | Cu1Ni1/ZSM-5 | 932.6 | 933.8 | 0.86 |
 | ||
| Fig. 4 (A) In situ DRIFTS spectra of CH4 adsorption at 80 °C for Cu1/ZSM-5 and Cu1Ni0.75/ZSM-5 catalysts. (B) EPR free radical capture experiments using DMPO as the radical trapping agent over Cu1/ZSM-5 and Cu1Ni0.75/ZSM-5 catalysts. (C) C1 yield during direct oxidation of methane to methanol using ascorbic acid as free radical scavenger. | ||
In situ NO-DRIFTS spectra was used to further distinguish CuI and CuII sites (Fig. 3B). The signal at 1800 cm−1 was identified as the absorption peak of CuI(NO). The signals at 1715 cm−1 and 1830 cm−1 were attributed to the absorption peaks of CuI(NO)2, correspondingly, the absorption peaks at 1906 cm−1, 1942 cm−1 and 1991 cm−1 related to CuII(NO),30,49,50 which indicated the coexistence of CuI and CuII sites in the CuxNiy/ZSM-5 catalysts. Meanwhile, the XRD, FT-IR, XPS and In situ NO-DRIFTS comparison of the recycled Cu1Ni0.75/ZSM-5 was carried out (Fig. S11†). The CuI/CuII ratio of recycled Cu1Ni0.75/ZSM-5 was 0.69, which was close to fresh Cu1Ni0.75/ZSM-5 (0.70). These characterizations of Cu1Ni0.75/ZSM-5 after reaction remain almost the same as the fresh samples, confirming the stability of catalyst structure.
N2/CH4 isotherm adsorption–desorption experiments results were showed in Fig. 3C, D and Table S6.† Compared with ZSM-5, the BET surface area remained almost unchanged and the microporous structure was slightly lost, but, the adsorption of CH4 by Cu1/ZSM-5 was significantly improved, which was clearly attributed to the presence of Cu sites. When Ni are further introduced, the BET specific surface area and total pore volume of the Cu1Ni0.75/ZSM-5 catalyst were slightly reduced, which may be due to the loading of Ni caused changes in the physical structure of ZSM-5 carrier, as the length of Ni–O bonds (∼2.0 Å) does not match that of Si–O bonds (∼1.6 Å). Therefore, this substitution of Ni for Si would present a minor variation of the shape and size of ZSM-5 channels. This could explain the decrease in activity when the loading of Ni exceeds 0.75 wt%, which is that the excessive Ni species blocked the micropores, thereby hindering the transfer of reactants. In addition, there was no significant change in the adsorption of CH4 by Cu1Ni0.75/ZSM-5 compared to Cu1/ZSM-5.
3.3. Reaction mechanism
The coexistence of CuI and CuII sites in the CuxNiy/ZSM-5 catalysts has been confirmed, but their effect on the DOMM reaction has not been determined yet. Therefore, in situ CH4-DRIFTS spectra were used to detect the function of the CuII and CuI sites (Fig. 4A). Several strong absorption peaks attributed to *CH4 (3016 cm−1 and 1305 cm−1)51–53 and formed M–CH3 (1230 cm−1, M:Cu or Ni) 33 can be detected on Cu1Ni0.75/ZSM-5 catalyst with abundant CuI sites. While the Cu1/ZSM-5 catalyst with more CuII sites also had the absorption peaks of at 3016 cm−1 and 1305 cm−1, but the absorption peaks at Cu1/ZSM-5 catalyst were significantly lower than that at Cu1Ni0.75/ZSM-5 catalyst, which showed CuI sites are apparently conducive to methane adsorption (Fig. S12†). In addition, the absorption peak of M–CH3 at 1230 cm−1 was nearly invisible compared to that at Cu1Ni0.75/ZSM-5 catalyst, again testified the constrained CH4 activation ability of pure Cu sample with limited CuI (Fig. S13†). We believe that the reason for the above results is: the Cu1Ni0.75/ZSM-5 catalyst with the majority of CuI sites has a stronger ability to promote the breaking of the first C–H bonds of CH4. The *CH3 adsorbed on metal sites could react with ˙OH in the liquid phase to generate CH3OH, thus, explaining the high yield of CH3OH in Cu1Ni0.75/ZSM-5 for its metal sites were more valid to form *CH3. In addition, no signals for OCH3* species (2836 cm−1 and 1047 cm−1) were found,54–56 which showed *CH3 did not tend to adsorb on O sites, but rather tended to adsorb on metal sites (Fig. S14†). Combining N2/CH4 isotherm adsorption–desorption experiments results and in situ CH4-DRIFTS spectra, it was speculated that CH4 was adsorbed on Cu sites, and CuI sites had better ability to promote CH4 adsorption, CH4 activation and CH3OH generation compared to CuII sites. Therefore, we found that the CuI sites plays a role in activating CH4, and
at 3016 cm−1 and 1305 cm−1, but the absorption peaks at Cu1/ZSM-5 catalyst were significantly lower than that at Cu1Ni0.75/ZSM-5 catalyst, which showed CuI sites are apparently conducive to methane adsorption (Fig. S12†). In addition, the absorption peak of M–CH3 at 1230 cm−1 was nearly invisible compared to that at Cu1Ni0.75/ZSM-5 catalyst, again testified the constrained CH4 activation ability of pure Cu sample with limited CuI (Fig. S13†). We believe that the reason for the above results is: the Cu1Ni0.75/ZSM-5 catalyst with the majority of CuI sites has a stronger ability to promote the breaking of the first C–H bonds of CH4. The *CH3 adsorbed on metal sites could react with ˙OH in the liquid phase to generate CH3OH, thus, explaining the high yield of CH3OH in Cu1Ni0.75/ZSM-5 for its metal sites were more valid to form *CH3. In addition, no signals for OCH3* species (2836 cm−1 and 1047 cm−1) were found,54–56 which showed *CH3 did not tend to adsorb on O sites, but rather tended to adsorb on metal sites (Fig. S14†). Combining N2/CH4 isotherm adsorption–desorption experiments results and in situ CH4-DRIFTS spectra, it was speculated that CH4 was adsorbed on Cu sites, and CuI sites had better ability to promote CH4 adsorption, CH4 activation and CH3OH generation compared to CuII sites. Therefore, we found that the CuI sites plays a role in activating CH4, and 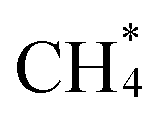 adsorbs onto them at the beginning of the DOMM reaction and generating initial *CH3. Subsequently, *CH3 was converted to ˙CH3 under the induction of a free radical rich aqueous solution, and then combined with ˙OH to generate CH3OH.
adsorbs onto them at the beginning of the DOMM reaction and generating initial *CH3. Subsequently, *CH3 was converted to ˙CH3 under the induction of a free radical rich aqueous solution, and then combined with ˙OH to generate CH3OH.
Usually, the mechanisms of the DOMM reaction can be divided into free radical mechanism57,58 and non-free mechanism59,60 according to the different intermediate transition species. In the control experiment without H2O2, there were no products (Fig. S3†). Moreover, the activity of DOMM reaction was significantly limited when there was no catalyst or only pure ZSM-5 zeolite. Next, the reaction activities were greatly enhanced when ZSM-5 support was loaded with Cu or CuNi, thus, the DMPO spin-trapping Electron Paramagnetic Resonance experiments were used to give in-depth insight into the behaviors of free radicals during the reaction process (Fig. 4B). The presence of ˙CH3 and ˙OH could be clearly observed through free radical capture experiments using DMPO, and the concentrations of ˙CH3 and ˙OH in the DOMM reaction catalyzed by Cu1Ni0.75/ZSM-5 were significantly higher than those catalyzed by Cu1/ZSM-5, which explained Cu1Ni0.75/ZSM-5 catalyst had excellent ability to promote the breaking of C–H bonds in CH4 to form *CH3 and the dissociation of H2O2 to produce ˙OH, and then the ˙OH radicals generated by H2O2 abstracted the metal adsorbed *CH3 to ˙CH3. In addition, a small amount of ˙OH radicals was detected over Cu1/ZSM-5, indicating that the contribution of Cu species to the generation of ˙OH radicals was limited, while a large amount of ˙OH was formed over Cu1Ni0.75/ZSM-5, indicating that the combination of Cu and Ni species had better ability to promote the decomposition of H2O2 to produce ˙OH radicals compared to single Cu species. This phenomenon was known as the Fenton-like reaction, which explained the high ˙OH concentration over Cu1Ni0.75/ZSM-5.61
In order to explore the source of another important C1 product HCOOH, subsequent oxidation experiments were conducted to determine the order of its production with CH3OH, specifically, 40 mL aqueous solution containing 0.01 M CH3OH and 0.5 M H2O2 and 30 bar N2 was used. The products (0.009 M CH3OH and 0.001 M HCOOH), indicated that HCOOH was produced by CH3OH peroxidation, i.e. CH4 to HCOOH is a two-step reaction (CH4 → CH3OH → HCOOH) (Fig. S15†).
The pathway of DOMM reaction under the influence of various free radicals was studied by combining the volcano-shaped curve of methanol yield and characterization method. Firstly, *CH4 adsorbed onto metal sites at the beginning of the DOMM reaction and generated initial *CH3. Subsequently, *CH3 was converted to ˙CH3 under the induction of a free radical rich aqueous solution. Then, we found that the yield of CH3OH significantly decreased when ˙OH was quenched, which confirmed the hypothesis that CH3OH was composed of ˙OH capturing ˙CH3. Finally, we believed that a small portion of CH3OH was further oxidized to form the byproduct HCOOH under the influence of ˙OH based on the control experiment using CH3OH as the sole carbon source and corresponding ˙OH quenching experiment as described above (Fig. S15†).
A comprehensive and feasible reaction pathway for direct oxidation of methane to methanol over Ni-promoted Cu/ZSM-5 using H2O2 as oxidant had been proposed according to a series of controlled experiments, EPR free radical capture experiments, in situ CH4-DRIFTS spectra and free radical quenching experiments. In the liquid-phase system containing Ni-Promoted Cu/ZSM-5 and H2O2, CuI promoted the adsorption of methane and efficiently polarized the first C–H bond of methane to form *CH3. Subsequently, *CH3 was converted to ˙CH3 under the induction of a free radical rich aqueous solution, and then combined with ˙OH radicals generated by H2O2 to generate the target product (CH3OH) of the DOMM reaction, but partial CH3OH would be attacked by ˙OH and undergo excessive oxidation to generate HCOOH.
4 Conclusion
In conclusion, we have prepared a series of CuxNiy/ZSM-5 catalysts with different Ni loadings through the ammonia evaporation method for the direct oxidation of methane to methanol using H2O2 as the oxidant. A volcano-type catalytic performance was exhibited with the increase of Ni loading amounts, where the maximum catalytic activity appeared at Cu1Ni0.75/ZSM-5 with a CH3OH yield of 82162 μmol gcat−1 h−1 and CH3OH selectivity of 74%, which was obviously superior than monometallic Cu1/ZSM-5 catalyst. We had explained the reason why the addition of Ni effectively promoted the catalytic activity of Cu/ZSM-5 for methane conversion through a combination of various experiments (especially the control experiments and ˙OH partial quenching experiment) and characterization methods: on the one hand, Ni promoted the conversion of CuII to CuI, these currently electron-rich Cu sites (CuI sites) could form strong polar interactions with carbon atoms in methane molecules, which helped to break the symmetry and stability of C–H bonds, ergo boosting CH4 cleavage to *CH3; on the other hand, the combination of Cu and Ni species, instead of single Cu species, promoted the decomposition of H2O2 to produce more ˙OH radicals, thus, the methanol yield increased accordingly. Correspondingly, a small amount of methanol would undergo excessive oxidation to form formic acid under the action of ˙OH radicals. This work provides useful and valuable opinion into the direct oxidation of methane to methanol over the metal-zeolite catalysts in the H2O2 liquid phase system, and opens up whole new vistas for the design of highly efficient metal-zeolite catalysts for direct oxidation of methane to methanol.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
W. L. and Z. S. conceived and supervised the research. Z. S. designed and performed the experiments and data analysis. Z. S. and J. J. wrote the paper. All authors discussed the results and commented on the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Anhui Natural Science Foundation (2408085ME143), Key Research and Development Projects in Anhui Province (2022107020013) and the Institute of Energy, the Hefei Comprehensive National Science Center under Grant No. 21KZS219.References
- L. He, Y. Fan, J. Bellettre, J. Yue and L. Luo, Renewable Sustainable Energy Rev., 2020, 119, 109589 CrossRef CAS.
- D. Allen, Acc. Chem. Res., 2016, 49, 1344–1350 CrossRef CAS PubMed.
- Y. Xu, X. Li, J. Gao, J. Wang, G. Ma, X. Wen, Y. Yang, Y. Li and M. Ding, Science, 2021, 371, 610–613 CrossRef CAS PubMed.
- A. K. M. K. Aurnob, K. Ding, D. R. Kauffman and J. J. Spivey, Appl. Catal., B, 2023, 322, 122107 CrossRef CAS.
- T. N. Le, T. B. N. Le, P. T. Nguyen, T. T. Nguyen, Q. N. Tran, T. T. Nguyen, Y. Kawazoe, T. B. Phan and D. M. Nguyen, RSC Adv., 2023, 13, 15926–15933 RSC.
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- B. An, Z. Li, Z. Wang, X. Zeng, X. Han, Y. Cheng, A. M. Sheveleva, Z. Zhang, F. Tuna, E. J. L. McInnes, M. D. Frogley, A. J. Ramirez-Cuesta, L. S. Natrajan, C. Wang, W. Lin, S. Yang and M. Schröder, Nat. Mater., 2022, 21, 932–938 CrossRef CAS PubMed.
- M. H. Mahyuddin, Y. Shiota, A. Staykov and K. Yoshizawa, Acc. Chem. Res., 2018, 51, 2382–2390 CrossRef CAS PubMed.
- K. Aasberg-Petersen, I. Dybkjær, C. V. Ovesen, N. C. Schjødt, J. Sehested and S. G. Thomsen, J. Nat. Gas Sci. Eng., 2011, 3, 423–459 CrossRef CAS.
- C. Palmer, D. C. Upham, S. Smart, M. J. Gordon, H. Metiu and E. W. McFarland, Nat. Catal., 2020, 3, 83–89 CrossRef CAS.
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dinca and Y. Roman-Leshkov, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS PubMed.
- L. Luo, J. Luo, H. Li, F. Ren, Y. Zhang, A. Liu, W. Li and J. Zeng, Nat. Commun., 2021, 12, 1218 CrossRef CAS PubMed.
- Y. Fan, W. Zhou, X. Qiu, H. Li, Y. Jiang, Z. Sun, D. Han, L. Niu and Z. Tang, Nat. Sustain., 2021, 4, 509–515 CrossRef.
- C. Feng, S. Zuo, M. Hu, Y. Ren, L. Xia, J. Luo, C. Zou, S. Wang, Y. Zhu, M. Rueping, Y. Han and H. Zhang, Nat. Commun., 2024, 15, 9088 CrossRef CAS PubMed.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F. S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- R. Serra-Maia, F. M. Michel, T. A. Douglas, Y. Kang and E. A. Stach, ACS Catal., 2021, 11, 2837–2845 CrossRef CAS.
- M. Li, J. Shan, G. Giannakakis, M. Ouyang, S. Cao, S. Lee, L. F. Allard and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2021, 292, 120124 CrossRef CAS.
- Y. Xi and A. Heyden, ACS Catal., 2019, 9, 6073–6079 CrossRef CAS.
- J. Li, P. Yan, K. Li, J. You, H. Wang, W. Cui, W. Cen, Y. Chu and F. Dong, J. Mater. Chem. A, 2019, 7, 17014–17021 RSC.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- M. C. Simons, S. D. Prinslow, M. Babucci, A. S. Hoffman, J. Hong, J. G. Vitillo, S. R. Bare, B. C. Gates, C. C. Lu, L. Gagliardi and A. Bhan, J. Am. Chem. Soc., 2021, 143, 12165–12174 CrossRef CAS PubMed.
- Y. Xu, D. Wu, Q. Zhang, P. Rao, P. Deng, M. Tang, J. Li, Y. Hua, C. Wang, S. Zhong, C. Jia, Z. Liu, Y. Shen, L. Gu, X. Tian and Q. Liu, Nat. Commun., 2024, 15, 564 CrossRef CAS PubMed.
- A. Li, T. Wang, C. Li, Z. Huang, Z. Luo and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 3804–3808 CrossRef CAS PubMed.
- B. Wang, F. Yang, Y. Dong, Y. Cao, J. Wang, B. Yang, Y. Wei, W. Wan, J. Chen and H. Jing, Chem. Eng. J., 2020, 396, 125255 CrossRef CAS.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- X. Liu, C. Wang, J. Zhou, C. Liu, Z. Liu, J. Shi, Y. Wang, J. Teng and Z. Xie, Chem. Soc. Rev., 2022, 51, 8174–8200 RSC.
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Acc. Chem. Res., 2021, 54, 2894–2904 CrossRef CAS PubMed.
- H. M. Rhoda, A. J. Heyer, B. E. R. Snyder, D. Plessers, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2022, 122, 12207–12243 CrossRef CAS PubMed.
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523 CrossRef CAS PubMed.
- J. Jin, W. Li, L. Zhang, L. Zhu, L. Wang and Z. Zhou, J. Colloid Interface Sci., 2023, 645, 964–973 CrossRef CAS PubMed.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- H. Zhang, P. Han, D. Wu, C. Du, J. Zhao, K. H. L. Zhang, J. Lin, S. Wan, J. Huang, S. Wang, H. Xiong and Y. Wang, Nat. Commun., 2023, 14, 7705 CrossRef CAS PubMed.
- T. Yu, Z. Li, L. Lin, S. Chu, Y. Su, W. Song, A. Wang, B. M. Weckhuysen and W. Luo, ACS Catal., 2021, 11, 6684–6691 CrossRef CAS.
- L. Wang, J. Jin, W. Li, C. Li, L. Zhu, Z. Zhou, L. Zhang, X. Zhang and L. Yuan, Energy Environ. Sci., 2024, 17, 9122–9133 RSC.
- B. Yu, L. Cheng, S. Dai, Y. Jiang, B. Yang, H. Li, Y. Zhao, J. Xu, Y. Zhang, C. Pan, X. M. Cao, Y. Zhu and Y. Lou, Adv. Sci., 2023, 10, 2302143 CrossRef CAS PubMed.
- S. Bai, Y. Xu, P. Wang, Q. Shao and X. Huang, ACS Catal., 2019, 9, 6938–6944 CrossRef CAS.
- H. Liu, H. Liu, J. Hu, W. Zhong, Z. Hu and H. Wang, J. Anal. Appl. Pyrolysis, 2024, 181, 106608 CrossRef CAS.
- E. Yuan, K. Zhang, G. Lu, Z. Mo and Z. Tang, J. Ind. Eng. Chem., 2016, 42, 142–148 CrossRef CAS.
- S. K. Jesudoss, J. Judith Vijaya, K. Kaviyarasu, P. Iyyappa Rajan, S. Narayanan and L. John Kennedy, J. Photochem. Photobiol., B, 2018, 186, 178–188 CrossRef CAS PubMed.
- Y. Meng, H. C. Genuino, C. Kuo, H. Huang, S. Chen, L. Zhang, A. Rossi and S. L. Suib, J. Am. Chem. Soc., 2013, 135, 8594–8605 CrossRef CAS PubMed.
- J. Gong, H. Yue, Y. Zhao, S. Zhao, L. Zhao, J. Lv, S. Wang and X. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- J. Pang, M. Zheng, C. Wang, X. Yang, H. Liu, X. Liu, J. Sun, Y. Wang and T. Zhang, ACS Catal., 2020, 10, 13624–13629 CrossRef CAS.
- L. Artiglia, V. L. Sushkevich, D. Palagin, A. J. Knorpp, K. Roy and J. A. van Bokhoven, ACS Catal., 2019, 9, 6728–6737 CrossRef CAS.
- H. Xue, X. Guo, T. Meng, Q. Guo, D. Mao and S. Wang, ACS Catal., 2021, 11, 7702–7718 CrossRef CAS.
- E. P. Hessou, M. Badawi, L. Valentin, G. Atohoun, S. Dzwigaj, M. Calatayud and F. Tielens, Top. Catal., 2022, 65, 848–858 CrossRef CAS.
- A. Corma, A. Palomares and F. Marquez, J. Catal., 1997, 170, 132–139 CrossRef CAS.
- Y. Chen, B. Qiu, Y. Liu and Y. Zhang, Appl. Catal., B, 2020, 269, 118801 CrossRef CAS.
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741 RSC.
- Z. Zhou, M. Miao, W. Li, J. Jin, L. Wang, L. Zhang and L. Yuan, Mol. Catal., 2024, 562, 114237 CrossRef CAS.
- J. Ding, Z. Teng, X. Su, K. Kato, Y. Liu, T. Xiao, W. Liu, L. Liu, Q. Zhang, X. Ren, J. Zhang, Z. Chen, O. Teruhisa, A. Yamakata, H. Yang, Y. Huang, B. Liu and Y. Zhai, Chem, 2023, 9, 1017–1035 CAS.
- X. Sun, X. Chen, C. Fu, Q. Yu, X. Zheng, F. Fang, Y. Liu, J. Zhu, W. Zhang and W. Huang, Nat. Commun., 2022, 13, 6677 CrossRef CAS PubMed.
- C. Wang, Y. Xu, L. Xiong, X. Li, E. Chen, T. J. Miao, T. Zhang, Y. Lan and J. Tang, Nat. Commun., 2024, 15, 7535 CrossRef CAS PubMed.
- C. Zhang, L. Wang, Y. Chen, X. He, Y. Song, O. M. Gazit and Z. Zhong, Chem. Eng. J., 2023, 475, 146102 CrossRef CAS.
- X. Xiao, G. Wang, X. Cai, H. Liu, F. Si, H. Wang, T. Hou and Y. Li, Chem. Eng. J., 2024, 498, 155404 CrossRef CAS.
- J. D. Jiménez, P. G. Lustemberg, M. Danielis, E. Fernández-Villanueva, S. Hwang, I. Waluyo, A. Hunt, D. Wierzbicki, J. Zhang, L. Qi, A. Trovarelli, J. A. Rodriguez, S. Colussi, M. V. Ganduglia-Pirovano and S. D. Senanayake, J. Am. Chem. Soc., 2024, 146, 25986–25999 CrossRef PubMed.
- X. Song, C. Basheer and R. N. Zare, J. Am. Chem. Soc., 2023, 145, 27198–27204 CrossRef CAS PubMed.
- Q. Zhou, X. Tan, X. Wang, Q. Zhang, C. Qi, H. Yang, Z. He, T. Xing, M. Wang, M. Wu and W. Wu, ACS Catal., 2024, 14, 955–964 CrossRef CAS.
- H. Lee, C. Kwon, C. Keum, H. Kim, H. Lee, B. Han and S. Lee, Chem. Eng. J., 2022, 450, 138472 CrossRef CAS.
- Z. Fang, M. Huang, B. Liu, F. Jiang, Y. Xu and X. Liu, J. Catal., 2022, 405, 1–14 CrossRef CAS.
- M. Shen, Z. Huang, L. Qiu, Z. Chen, X. Xiao, X. Mo and L. Cui, J. Cleaner Prod., 2020, 268, 122174 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01115a |
| This journal is © The Royal Society of Chemistry 2025 |