
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExercise in 1-aryl-3-CF3-1H-pyrazoles: regioselective synthesis of 4-/5-iodides and cross-coupling reactions†
Kamil Świątek,
Greta Utecht-Jarzyńska and
Marcin Jasiński *
*
University of Lodz, Faculty of Chemistry, Department of Organic and Applied Chemistry, Tamka 12, 91-403 Łódź, Poland. E-mail: mjasinski@uni.lodz.pl
First published on 25th March 2025
Abstract
A series of 1-aryl-3-CF3-1H-pyrazoles was prepared and examined using iodination reactions. Treatment with n-BuLi followed by trapping of the corresponding lithium pyrazolide with elemental iodine produced 5-iodo derivatives exclusively, while CAN-mediated iodination with I2 afforded isomeric 4-iodides in a highly regioselective manner. The title iodides were demonstrated to be convenient building blocks for the preparation of more complex 3-trifluoromethylated pyrazoles through the model Suzuki–Miyaura and Sonogashira reactions.
Introduction
During the last decades, fluoromethylated pyrazoles have been recognized as privileged structural motifs for agrochemicals, pharmaceuticals and organic materials; therefore, they are highly attractive targets for organic synthesis.1 Among the variety of protocols for the preparation of fluoromethylated pyrazoles reported thus far,1,2 (3 + 2)-cycloaddition reactions of CHF2- and CF3-functionalized nitrile imines, formally derived from di- and tri-fluoroacetaldehyde, respectively, are highly efficient methods.3 For example, electron-rich dipolarophiles such as enol ethers and arynes4a,b as well as electron-deficient olefins, e.g. nitro- and cyano-alkenes,4c–f enones,4g,h and maleimides4i are suitable agents to trap in situ generated nitrile imines 1, leading to functionalized pyrazoles and the related fused systems. The approach presented in Scheme 1a (route A) is documented in the literature and offers straightforward access to target materials 2 of various substitution patterns; however, it requires access to a library of properly functionalized reactive dipolarophiles. Moreover, there are limitations to obtain regioselectivity for the reactions of 1 with non-activated ethylenes and some acetylene derivatives.5 An alternative route towards C(4)–/C(5)-functionalized 3-CF3-pyrazole 2 with nitrile imine 1 as a key substrate via post-cyclizative functionalization of the pyrazole ring in 3 (Scheme 1, route B) has not been thoroughly explored.6 Hence, the development of general protocols for the selective introduction of iodine at C(4) and C(5) in 1-aryl-3-CF3-pyrazole 3 is of interest considering the well-known utility of (hetero)aryl iodides in the synthesis of more complex systems available through cross-coupling reactions.7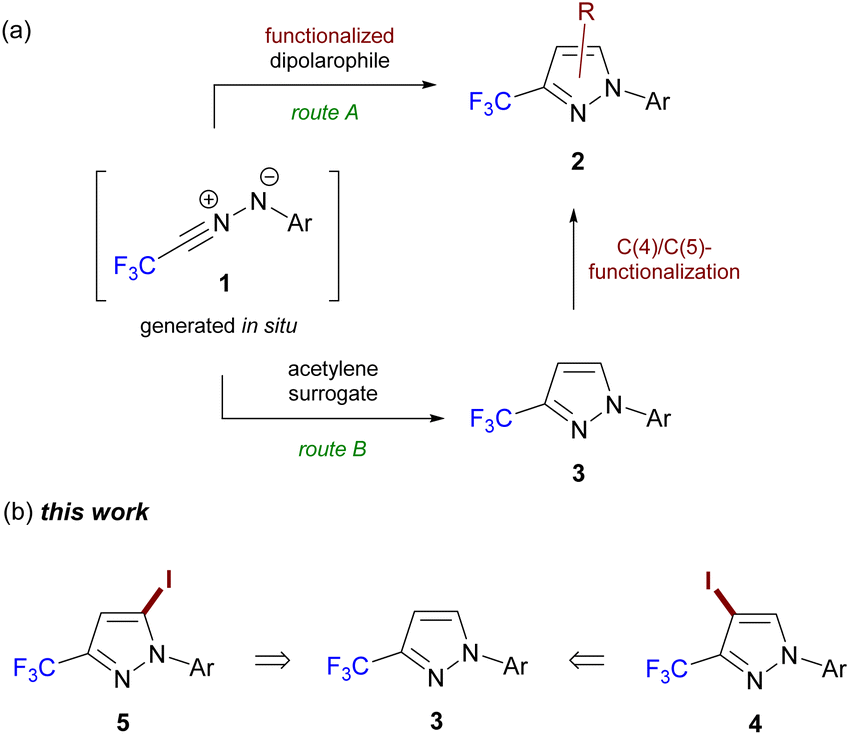 | ||
| Scheme 1 (a) General routes towards C(4)–/C(5)-substituted pyrazoles 2 with CF3-nitrile imine 1 as a key building block and (b) structures of iodides 4 and 5 reported herein. | ||
In continuation of our study on fluorinated azoles,8 herein, we reported on the efficient functionalization of 1-aryl-3-CF3-pyrazole 3 to produce 4-iodo and 5-iodo-analogues 4 and 5 in a highly selective fashion (Scheme 1b). Exemplary Suzuki–Miyaura and Sonogashira coupling reactions of model C(4)–/C(5)-iodides are also presented.
Results and discussion
The key intermediate 3 is easily obtained by trapping in situ generated nitrile imine 1 with suitable acetylene equivalents. For example, we previously disclosed a one-pot protocol based on (3 + 3)-cycloaddition of 1,3-dipole 1 with mercaptoacetaldehyde, followed by spontaneous Eschenmoser ring-contraction of the first formed 1,3,4-thiadiazine.9a More recently, the Hu group disclosed an alternative method with (2-bromoethyl)-diphenylsulfonium triflate applied as an acetylene surrogate.9b As shown in Scheme 2, we commenced our study with 1-aryl-3-CF3-pyrazole 3 prepared following the former general protocol. Starting with dimeric mercaptoacetaldehyde (1,4-dithiane-2,5-diol, 6) and hydrazonoyl bromides 7a–7h as nitrile imine precursors, a series of eight compounds 3a–3h bearing selected para-substituted phenyl substituents at N(1) (X = Me, iPr, H, Cl, CF3, CN, SO2NH2 and OMe) was prepared in 60–92% yield.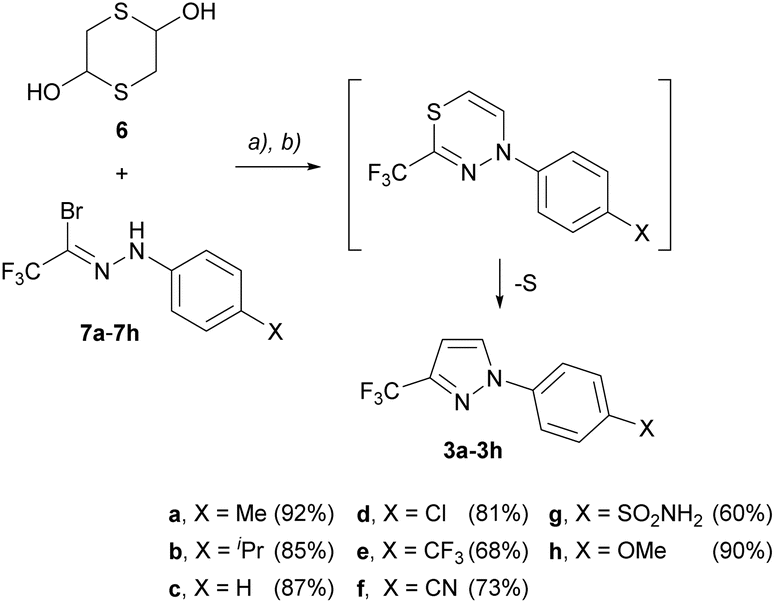 | ||
| Scheme 2 One-pot synthesis of 1-aryl-3-CF3-pyrazoles 3a–3h starting with dimeric mercaptoacetaldehyde 6 and nitrile imine precursors 7a–7h: (a) Et3N, DCM, 4 h, rt; (b) pTsCl, 16 h. | ||
The general methods for direct iodination of structurally diverse pyrazoles usually comprise the use of elemental iodine under oxidative conditions.10 Also, the applications of common inorganic salts such as NaI and KI–KIO3, as well as organic iodocompounds including NIS, Me4NI, 1,3-diiodo-5,5-dimethylhydantoin (DIH), and aryliodine(III) diacetates as iodine sources are known.11 More recently, Peglow and Nascimento reported an elegant one-pot protocol using potassium iodate (KIO3) and diphenyl diselenide (PhSe)2 as catalysts for the selective iodination of in situ generated pyrazoles under acidic conditions.11g Despite remarkable progress in iodination reactions of classical N-alkyl- and N-aryl-pyrazoles, synthetic methods towards fluoromethylated iodopyrazole derivatives are only reported to a limited extent.12
In the first part of this study, we followed the work by Rodríguez-Franco on the regioselective iodination of pyrazoles using I2 in the presence of ceric ammonium nitrate (CAN) as mild oxidant.10b The first experiment with model compound 3a (1.0 mmol) was conducted using a slight excess of I2 (0.6 mmol) and CAN (0.6 mmol) in MeCN. Trace conversion of 3a was observed after 24 h at room temperature, while increasing the temperature (reflux) and using an excess of iodinating agents (iodine: 1.3 equiv.; CAN: 1.1 equiv.) enabled complete consumption of the starting pyrazole in a reasonable reaction time (overnight) (Scheme 3).‡ Analysis of the crude mixture by 1H NMR spectroscopy indicated the formation of a single product, and the presence of the diagnostic singlet absorption at δ = 7.97 attributed to C(5)–H suggested the exclusive introduction of iodine at C(4) of the pyrazole ring. After standard aqueous workup followed by simple filtration through a short chromatography column (FCC), the expected product 4a was isolated in 81% yield. 13C and 19F NMR analyses of the obtained material supplemented by MS measurements and combustion analysis confirmed the structure of 4a as the desired 4-iodo-1-tolyl-3-trifluoromethylpyrazole and its analytical purity. In 13C NMR spectra, the expected remarkable high-field shift (Δ ≈ 50) is noteworthy for the signal attributed to C(4) from δ = 105.9 (q, 3JC–F ≈ 2.2 Hz) in 3a to δ = 55.7 (q, 3JC–F ≈ 1.6 Hz) in 4a.
 | ||
Scheme 3 Synthesis of 4- and 5-iodopyrazoles 4 and 5: (a) I2 (1.3 equiv.), CAN (1.1 equiv.), MeCN, reflux, 16 h; (b) NIS (1.5 equiv.), AcOH/TFA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 85 °C, 16 h; (c) n-BuLi (1.3 equiv.), −78 °C, THF, 10 min, then I2 (1.4 equiv.), −78 °C to rt, 4 h; (d) n-BuLi (3.0 equiv.), THF/TMEDA, −78 °C, 10 min, then I2 (3.2 equiv.), −78 °C to rt, 4 h. :1), 85 °C, 16 h; (c) n-BuLi (1.3 equiv.), −78 °C, THF, 10 min, then I2 (1.4 equiv.), −78 °C to rt, 4 h; (d) n-BuLi (3.0 equiv.), THF/TMEDA, −78 °C, 10 min, then I2 (3.2 equiv.), −78 °C to rt, 4 h. | ||
With the established reaction conditions, the iPr-functionalized derivative 3b was examined to afford the expected 4b as the sole material, which was isolated by FCC in 75% yield (Scheme 3). In this case, there was no competitive iodination of the benzylic-like position in the iPr group or the N-aromatic substituent. Notably, the reaction proceeded smoothly even in the case of substrates bearing electron-deficient N-aryl substituents (3d–3f), although the introduction of strong electron-withdrawing groups X such as CF3 and CN in 3e and 3f, respectively, decreased the reaction rate according to TLC. In addition, a competition experiment was conducted wherein a mixture of equimolar amounts of 3a (0.1 mmol) and 3f (0.1 mmol) was treated with a deficient amount of iodine source (I2/CAN; 0.05 mmol each) to produce a mixture of products 4a and 4f in a ca. 10:1 ratio after overnight heating (Scheme 4).
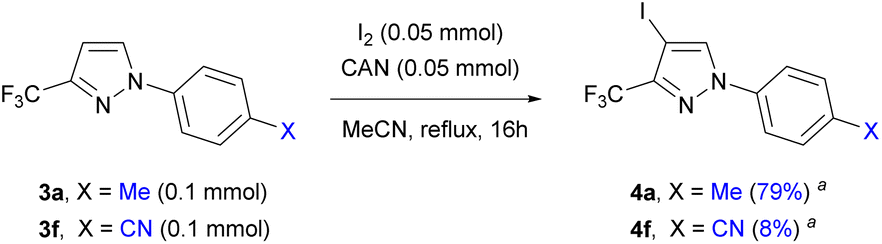 | ||
| Scheme 4 Competition experiment between 3a and 3f; a conversion of individual starting materials based on 1H NMR of the reaction mixture. | ||
In contrast, the attempted iodination of sulfonamide-functionalized pyrazole 3g under the applied conditions led to a complex mixture in which no desired 4-iodopyrazole 4g was detected. On the other hand, treatment of 3h (X = OMe) with CAN/I2 resulted in simultaneous iodination of the p-anisyl group to afford pyrazole 8 (30% yield), a minor component of the mixture, along with nitro analogue 9 (39% yield), isolated as the major product. The experiment revealed lesser-known nitrating activity of ceric ammonium nitrate in MeCN solutions, which is in accordance to previous reports on CAN-mediated ortho-selective nitrations of some electron-rich arenes.13 Hence, the reaction was conducted under acidic conditions using a 1:1 mixture of glacial acetic acid and TFA as reaction media and employing N-iodosuccinimide (NIS) as a source of iodide to suppress the nucleophilicity of functional groups in 3g and 3h.12b In this case, the desired sulphonamide-functionalized product 4g was obtained in 71% yield. However, in the case of 3h, the expected product 4h (36% yield) was obtained along with small amounts of another monoiodinated product, 1-(3-iodo-4-methoxyphenyl)-3-trifluoro-methylpyrazole (10, 10%), formed via alternative attack of electrophilic iodine onto the methoxyphenyl substituent.
In the synthesis of a second series of iodopyrazoles of type 5, we benefited from the remarkable acidity of the C(5)–H in starting 1-aryl-3-CF3-pyrazoles 3.9a As shown in Scheme 3, treatment of 3a with a slight excess of n-BuLi at −78 °C in dry THF smoothly generated the corresponding lithium 5-pyrazolide,14 which was trapped by elemental iodine to produce the target 5-iodo derivative 5a (86% yield) after standard aqueous workup. Following the devised protocol,‡ a set of isomeric iodides 5b–5h was obtained in 65–89% yield. Notably, threefold excess n-BuLi in the presence of TMEDA as the co-solvent enabled the efficient iodination of sulphonamide 3g (65% yield).9a
The spectroscopic properties of iodides 4 and 5 also deserve a brief comment. Increasing the electron-withdrawing character of substituent X in starting pyrazoles 3 increases shielding of 19F nuclei (Fig. 1). Analysis of chemical shifts demonstrate that they non-linearly correlate with the Hammett σp parameter.15 The introduction of iodine at C(4) and C(5) of the pyrazole ring (in 4 and 5, respectively) results in further up-field shift, although slightly stronger shielding is caused by the latter functionalization. The observed trends are according to so-called “normal” fluorine chemical shifts that are typical for trifluoromethylated benzene derivatives, which supplements the understanding of normal and reverse fluorine NMR chemical shifts.16
 | ||
| Fig. 1 Correlation of 19F chemical shifts for a series of pyrazoles 3–5. | ||
Next, compounds 4a and 5a were examined in model cross-coupling reactions to demonstrate practical utility of the newly synthetized iodides in the synthesis of more complex pyrazole analogues (Scheme 5). The expected 4-phenylpyrazole 11 was obtained in 56% yield following the treatment of 4a with phenylboronic acid under classical Suzuki–Miyaura reaction conditions using Pd(PPh3)4 as a catalyst. A similar result was observed for 5a to afford 5-phenylated derivative 12 (62%).
 | ||
| Scheme 5 Selected cross-coupling reactions of isomeric iodides 4a and 5a: (a) phenylboronic acid (1.3 equiv.), Pd(PPh3)4 (30 mol%), K2CO3, THF/H2O, reflux, 2 d; (b) phenylacetylene (1.25 equiv.), Pd(PPh3)4 (10 mol%), CuI (10 mol%), THF/Et3N, 80 °C, 24 h. | ||
Another pair of isomeric products 13 and 14 functionalized with the phenylethynyl group was prepared via Sonogashira coupling in >90% yield.
Conclusions
1-Aryl-3-CF3-pyrazoles were suitable building blocks for highly selective iodination to provide access to C(4)- and C(5)-iodides. The CAN-induced reaction with elemental iodine provided products functionalized at C(4) of the pyrazole ring, while the isomeric 5-iodo products were obtained by trapping of the respective in situ generated lithium pyrazolide with I2. The title products are valuable reagents for the synthesis of more complex trifluoromethylated pyrazoles as exemplified by the synthesis of phenyl- and phenylethynyl-functionalized derivatives via Suzuki–Miyaura and Sonogashira cross-coupling reactions, respectively.Data availability
The data supporting this article have been included as part of the ESI.† In addition, raw NMR data (FID) as well as pdf. copies of the IR, MS and EA analyses conducted for all new compounds are available from the corresponding author.Author contributions
MJ and GUJ designed experiments; KŚ and GUJ synthetized and characterized the studied materials; MJ drafted the manuscript; all authors contributed to the final version of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KŚ and MJ thank the University of Lodz for financial support in the framework of the IDUB grants (KŚ, #8/DGB/2022, and MJ, #14/IGB/2024).Notes and references
- (a) Z. Moussa, M. Ramanathan, S. M. Alharmoozi, S. A. S. Alkaabi, S. H. M. Al Aryani, S. A. Ahmed and H. T. Al-Masri, Heliyon, 2024, 10, e38894 CrossRef CAS PubMed; (b) S. P. Chandrasekharan, A. Dhami, S. Kumar and K. Mohanan, Org. Biomol. Chem., 2022, 20, 8787 RSC; (c) S. Fustero, A. Simón-Fuentes, O. Delgado and R. Román, Fluorinated Pyrazoles and Indazoles in Fluorine in Heterocyclic Chemistry, ed. V. Nenajdenko, Springer, Cham, Switzerland, 2014, vol. 1, pp. 279 Search PubMed.
- P. K. Mykhailiuk, Chem. Rev., 2021, 121, 1670 CrossRef CAS PubMed.
- (a) N. R. Yamaletdinova and R. R. Gataullin, Helv. Chim. Acta, 2024, 107, e202400058 CrossRef CAS; (b) Z. Dongxu, Beilstein J. Org. Chem., 2023, 19, 1741 CrossRef PubMed; (c) C. Jamieson and K. Livingstone, The Nitrile Imine 1,3-dipole. Properties, Reactivity and Applications, Springer, Cham, Switzerland, 2020 CrossRef.
- (a) G. Utecht, A. Fruziński and M. Jasiński, Org. Biomol. Chem., 2018, 16, 1252 RSC; (b) A. Kowalczyk, G. Utecht-Jarzyńska, G. Mlostoń and M. Jasiński, J. Fluorine Chem., 2021, 241, 109691 CrossRef CAS; (c) Y.-C. Tian, J.-K. Li, F.-G. Zhang and J.-A. Ma, Adv. Synth. Catal., 2021, 363, 2093 CrossRef CAS; (d) K.-H. Wang, H. Liu, X. Liu, C. Bian, J. Wang, Y. Su, D. Huang and Y. Hu, Asian J. Org. Chem., 2022, 11, e202200103 CrossRef CAS; (e) Y. Zhou, C.-F. Gao, H. Ma, J. Nie, J.-A. Ma and F.-G. Zhang, Chem.–Asian J., 2022, e202200436 CAS; (f) N. Zhang, H. Ma, C. W. Cheung, F.-G. Zhang, M. Jasiński, J.-A. Ma and J. Nie, Org. Biomol. Chem., 2023, 21, 5040 RSC; (g) A. Kowalczyk, G. Utecht-Jarzyńska, G. Mlostoń and M. Jasiński, Org. Lett., 2022, 24, 2499 CrossRef CAS PubMed; (h) Y. Feng, Y. Ren, D. Tang, K.-H. Wang, J. Wang, D. Huang, X. Lv and Y. Hu, Org. Biomol. Chem., 2024, 22, 2797 RSC; (i) G. Utecht-Jarzyńska, S. Jarzyński and M. Jasiński, RSC Mechanochem., 2025, 2, 79 RSC.
- (a) K. Tanaka, S. Maeno and K. Mitsuhashi, Chem. Lett., 1982, 11, 543 CrossRef; (b) K. Tanaka, S. Maeno and K. Mitsuhashi, J. Heterocycl. Chem., 1985, 22, 565 CrossRef CAS; (c) G. Utecht, G. Mlostoń and M. Jasiński, Synlett, 2018, 29, 1753 CrossRef CAS.
- (a) K. Yagi, T. Ogura, A. Numata, S. Ishii and K. Arai, Heterocycles, 1997, 45, 1463 CrossRef CAS; (b) S. M. Gaulier, R. McKay and N. A. Swain, Tetrahedron Lett., 2011, 52, 6000 CrossRef CAS; (c) Q. Huang, G. Tran, D. G. Pardo, T. Tsuchiya, S. Hillebrand, J.-P. Vors and J. Cossy, Tetrahedron, 2015, 71, 7250 CrossRef CAS; (d) A. Ziadi, N. Uchida, H. Kato, R. Hisamatsu, A. Sato, S. Hagihara, K. Itami and K. U. Torii, Chem. Commun., 2017, 53, 9632 RSC; (e) M. A. Tairov, et al., Org. Process Res. Dev., 2020, 24, 2619 CrossRef CAS.
- (a) C. Liu, Y. Gu, J. Wu, Y. Gu and Q. Sen, Chem. Sci., 2017, 8, 4848 RSC; (b) R.-Y. Zhu, L.-Y. Liu, H. S. Park, Y. Wu, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 16080 CrossRef CAS PubMed; (c) J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Chem. Sci., 2017, 8, 40 RSC; (d) M. L. Czyz, et al., Org. Biomol. Chem., 2018, 16, 1543 RSC; (e) Gajanan, K. Rathod and R. Jain, ChemistrySelect, 2022, 7, e202200773 CrossRef CAS; (f) J. DeLano, et al., Chem. Sci., 2021, 12, 7758 RSC.
- (a) W. K. Poper, J.-A. Ma and M. Jasiński, J. Org. Chem., 2024, 89, 15331 CrossRef CAS PubMed; (b) G. Mlostoń, M. Kowalczyk, M. Celeda, M. Jasiński, M. Denel-Bobrowska and A. B. Olejniczak, Molecules, 2022, 27, 3524 CrossRef PubMed.
- (a) K. Świątek, G. Utecht-Jarzyńska, M. Palusiak, J.-A. Ma and M. Jasiński, Org. Lett., 2023, 25, 4462 Search PubMed; (b) W.-J. Luo, X. Liang, M. Chen, K.-H. Wang, D. Huang, J. Wang, D.-P. Chen and Y. Hu, J. Org. Chem., 2024, 89, 10066 CrossRef CAS PubMed.
- (a) J. D. Vaughan, D. G. Lambert and V. L. Vaughan, J. Am. Chem. Soc., 1964, 86, 2857 CrossRef CAS; (b) M. I. Rodríguez-Franco, I. Dorronsoro, A. I. Hernández-Higueras and G. Antequera, Tetrahedron Lett., 2001, 42, 863 CrossRef; (c) M. M. Kim, R. T. Ruck, D. Zhao and M. A. Huffman, Tetrahedron Lett., 2008, 49, 4026 CrossRef CAS; (d) M. Gorjizadeh, M. Afshari and M. Naseh, Russ. J. Gen. Chem., 2016, 86, 1931 CrossRef CAS ; see also: ; (e) C. M. P. Pereira, F. H. Quina, F. A. N. Silva, D. J. Emmerich and A. Machulek Jr, Mini-Rev. Org. Chem., 2008, 5, 331 CrossRef CAS.
- (a) S. Guillou, F. J. Bonhomme, M. S. Ermolenko and Y. L. Janin, Tetrahedron, 2011, 67, 8451 CrossRef CAS; (b) B. V. Lyalin and V. A. Petrosyan, Russ. Chem. Bull. Int. Ed., 2013, 62, 1044 CrossRef CAS; (c) N. Lu, L. Huang, L. Xie and J. Cheng, Eur. J. Org Chem., 2018, 2018, 3437 CrossRef CAS; (d) K. Iida, S. Ishida, T. Watanabe and T. Arai, J. Org. Chem., 2019, 84, 7411 CrossRef CAS PubMed; (e) B. C. O. Rocha, G. P. Perecim and C. Raminelli, SynOpen, 2019, 3, 142 CrossRef CAS; (f) O. B. Bondarenko, G. L. Karetnikov, A. I. Komarov, A. I. Pavlov and S. N. Nikolaeva, J. Org. Chem., 2021, 86, 322 CrossRef CAS PubMed; (g) T. J. Peglow, J. P. S. S. C. Thomaz, P. C. Nobre, C. Scimmi, C. Santi and V. Nascimento, Chem.–Asian J., 2024, 19, e202400749 CrossRef CAS PubMed.
- (a) M. Schlosser, J.-N. Volle, F. Leroux and K. Schenk, Eur. J. Org Chem., 2002, 2913 CrossRef CAS; (b) C. Wiethan, W. C. Rosa, H. G. Bonacorso and M. Stradiotto, Org. Biomol. Chem., 2016, 14, 2352 RSC; (c) V. M. Muzalevskiy and V. G. Nenajdenko, Org. Biomol. Chem., 2018, 16, 7935 RSC; (d) A. F. Junges, E. P. Pittaluga, N. Zanatta, M. A. P. Martins and H. G. Bonaorso, Tetrahedron Lett., 2019, 60, 1385 CrossRef CAS; (e) A. Patnaik, et al., Bioorg. Med. Chem., 2020, 28, 115548 CrossRef CAS PubMed; (f) A. Boelke, T. J. Kuczmera, L. D. Caspers, E. Lork and B. J. Nachtsheim, Org. Lett., 2020, 22, 7261 CrossRef CAS PubMed.
- (a) S. Dinçtürk and J. H. Ridd, J. Chem. Soc., Perkin Trans. 2, 1982, 965 RSC; (b) M. Chakrabarty and A. Batabyal, Synth. Commun., 1994, 24, 1 CrossRef CAS; (c) M. A. Ponce and R. Erra-Balsells, J. Heterocycl. Chem., 2001, 38, 1071 CrossRef CAS; (d) K. Deleersnyder, S. Schaltin, J. Fransaer, K. Binnemans and T. N. Parac-Vogt, Tetrahedron Lett., 2009, 50, 4582 CrossRef CAS; (e) S. Wang, et al., Org. Chem. Front., 2021, 8, 5771 RSC; (f) Z. Iqubal, A. Joshi and S. R. De, Eur. J. Org Chem., 2022, 31, e202200746 CrossRef.
- J. Oslob, D. Aubele, J. Kim, R. McDowell, Y. Song, A. Sran, M. Zhong and MyoKardia, Inc., WO Pat., 2016/118774 A1, 2016 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- J. N. Dahanayake, C. Kasireddy, J. P. Karnes, R. Verma, R. M. Steinert, D. Hildebrandt, O. A. Hull, J. M. Ellin and K. R. Mitchell-Koch, Progress in our understanding of 19F chemical shifts, in Annual Reports on NMR Spectroscopy, Elsevier, 2018, vol. 5, p. 281 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01103e |
| ‡ General procedure A for the synthesis of 4-iodopyrazoles 4a–4f: A solution of 1-aryl-3-trifluoromethylpyrazole 3 (1.0 mmol), CAN (603 mg, 1.1 mmol), and elemental iodine (330 mg, 1.3 mmol) in MeCN (6 mL) was refluxed overnight. After the solvent was removed in vacuo, the residue was dissolved in DCM (15 mL), the mixture was washed with sat. aq. Na2S2O3 (5 mL), then with water (10 mL), and the organic layer was dried over Na2SO4. The solvent was evaporated and the crude product 4 was purified by filtration through a short silica gel pad (FCC) or by standard column chromatography (CC). 4-Iodo-1-(p-tolyl)-3-trifluoromethyl-1H-pyrazole (4a): FCC (SiO2, hexane/DCM 3:2); yellow oil, 285 mg (81%). 1H NMR (600 MHz, CDCl3) δ 2.42 (s, 3H), 7.29–7.31 (m, 2H), 7.53–7.55 (m, 2H), 7.97 (s, 1H, 5-H). 13C{1H} NMR (151 MHz, CDCl3) δ 21.1, 55.9 (q, 3JC–F ≈ 1.6 Hz), 119.7, 121.0 (q, 1JC–F = 270.2 Hz), 130.3, 134.4, 136.7, 138.5, 144.9 (q, 2JC–F = 37.2 Hz). 19F NMR (565 MHz, CDCl3): δ −61.72 (s, CF3). IR (neat) ν 1521, 1470, 1394, 1230, 1163, 1129, 1066, 992, 813 cm−1. ESI-MS (m/z): 353.1 (100, [M + H]+). Anal. calcd. for C11H8F3IN2 (352.1): C 37.52, H 2.29, N 7.96; found: C 37.45, H 2.33, N 7.85. General Procedure B for the synthesis of 4-iodopyrazoles 4g and 4h: To a solution of pyrazole 3 (1.0 mmol) in glacial acetic acid (1 mL) was added a solution of NIS (338 mg, 1.5 mmol) in TFA (1 mL) and the resulting mixture was heated overnight at 80 °C. The solution was cooled to room temperature, diluted with DCM (60 mL), washed with sat. aq. Na2S2O3 (2 × 5 mL), and then with sat. aq. NaHCO3 (3 × 5 mL). The organic layer was separated, dried over Na2SO4, and solvents were removed in vacuo. The products were purified by column chromatography. 4-(4-Iodo-3-trifluoromethyl-pyrazol-1-yl)-benzenosulfonamide (4g): CC (SiO2, hexane/EtOAc 2:3); light yellow solid, 296 mg (71%); mp 166–168 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.48 (sbr, 2H, NH2), 7.97–7.99 (m, 2H), 8.05–8.08 (m, 2H), 9.05 (s, 1H, 5-H). 13C{1H} NMR (151 MHz, DMSO-d6) δ 59.7 (br), 119.4, 120.9 (q, 1JC–F = 270.0 Hz), 127.4, 136.7, 140.2, 143.2, 144.1 (q, 2JC–F = 36.4 Hz). 19F NMR (565 MHz, DMSO-d6): δ −60.72 (s, CF3). IR (neat) ν 3340, 3247, 1592, 1477, 1387, 1331, 1234, 1156, 1118, 1096, 992, 910, 835 cm−1. HRMS ((−)-ESI-TOF) m/z: [M–H]− calcd. for C10H7F3IN3O2S 415.9177, found 415.9178. General Procedure C for the synthesis of 5-iodopyrazoles 5a–5h: To a solution of 1-aryl-3-CF3-pyrazole 3 (1.0 mmol) in dry THF (5 mL) was added n-BuLi (2.5 M in hexane, 0.52 mL, 1.3 mmol) dropwise under vigorous stirring at −78 °C. After 10 min, a solution of iodine (356 mg, 1.4 mmol) in dry THF (3 mL) was added and the mixture was allowed to reach room temperature within 4 h. The resulting was diluted with DCM (30 mL), washed with sat. aq. Na2S2O3 (10 mL), then with water (5 mL). The organic layer was dried over Na2SO4 and the solvents were removed under reduced pressure. Crude product was isolated by filtration through a short silica gel pad (FCC). 5-Iodo-1-(p-tolyl)-3-trifluoromethyl-1H-pyrazole (5a): FCC (SiO2, hexane/DCM 1:1); light orange solid, 303 mg (86%). 1H NMR (600 MHz, CDCl3) δ 2.44 (s, 3H), 6.86 (s, 1H, 4-H), 7.30–7.31 (m, 2H), 7.38–7.40 (m, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 21.3, 83.0, 115.1 (q, 3JC–F ≈ 2.1 Hz), 120.8 (q, 1JC–F = 269.3 Hz), 126.3, 129.7, 137.1, 139.9, 145.4 (q, 2JC–F = 38.8 Hz). 19F NMR (565 MHz, CDCl3): δ −62.39 (s, CF3). IR (neat) ν 1506, 1457, 1368, 1230, 1167, 1118, 1085, 980, 965, 824, 801 cm−1. ESI-MS (m/z): 353.1 (100, [M + H]+). Anal. calcd. for C11H8F3IN2 (352.1): C 37.52, H 2.29, N 7.96; found: C 37.62, H 2.31, N 7.77. |
| This journal is © The Royal Society of Chemistry 2025 |