
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of pesticide residue using an Fe3O4/GO/Ag nanocomposite as a SERS substrate and mechanism investigation†
Rui Wu *,
Xi Song,
Yulin Wang and
Qiang Zhang
*,
Xi Song,
Yulin Wang and
Qiang Zhang
Shaanxi Province Key Laboratory of Catalytic, School of Chemical and Environmental Science, Shaanxi University of Technology, Hanzhong 723001, P. R. China. E-mail: hftffc@163.com; wurui@snut.edu.cn
First published on 3rd April 2025
Abstract
Considering that pesticide residues cause significant harm to health and the environment, it is crucial and urgent to fabricate surface-enhanced Raman scattering (SERS) substrates with high sensitivity to detect pesticide residues. In this study, a three-layer structure of multifunctional Fe3O4/GO/Ag nanoparticles (NPs) as an active substrate for SERS was fabricated and comprehensively characterized through X-ray diffraction (XRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), energy dispersive spectroscopy (EDS), elemental mapping, and X-ray photoelectron spectroscopy (XPS). The active substrate was used to detect thiram pesticide through SERS. Combined with the synergistic effect, Ag NPs and graphene oxide (GO) possessed high-density and ordered hot spots to enhance the electromagnetic field; therefore, signal amplification and fascinating sensitivity were achieved. The lowest concentration of thiram for clear presentation of the peak was 10−12 mol L−1. There was a satisfactory linear relationship, ranging from 10−10 to 10−4 mol L−1, with a low limit of detection, high sensitivity, satisfactory enhancement factor (EF), stability, and repeatability. In addition, Fe3O4/GO/Ag was successfully applied to identify pesticide residues in a practical sample. Furthermore, the enhancement mechanism, including electromagnetic enhancement and chemical reinforcement, is discussed in detail. Fe3O4/GO/Ag is a potential active substrate for SERS and can be applied for rapid detection in the food industry, environmental fields and biomedicine with fascinating sensitivity.
1. Introduction
In modern agricultural production, an increasing number of pesticides are being used to control or eliminate pests to improve the quality and yield of crops. Although pesticide spraying benefits agricultural production, pesticide residues and their metabolites may contaminate and affect food products at all stages from production to consumption. These residues in food products may pollute the environment and pose a threat to the health of consumers.1,2 Thiram belongs to the dithiocarbamate pesticide group, which is widely used to control the infestation of vegetables, fruits and pests. Thiram can be easily accumulated in crops.3 Therefore, strengthening the detection of pesticide residues is of pivotal significance for the protection of the environment and human health.Thus far, various techniques have been proposed for the detection of pesticide residues. For example, immunoassays and enzyme inhibition are rapid approaches with low detection cost, ease of operation and portable advantages but with low sensitivity and complex preparation of antibodies.4 Gas chromatography, gas chromatography-mass spectrometry, high-performance liquid chromatography and other precision instrumental methods are also often used to monitor pesticide residues.5–7 Although the reported methods have made great progress, some are time-consuming with complex operation and, more importantly, low sensitivity, which hinder their development and universality. Therefore, it is urgent to develop a novel approach to detect pesticide residue with high sensitivity and universality.
As an emerging spectroscopic technique, SERS is widely used in the fields of food safety, biosensing, and environmental detection owing to its high sensitivity, low damage to the sample, and ability to provide comprehensive fingerprint information.8–10 SERS is a potential tool to detect a variety of pesticide species with the aid of active SERS substrates. To achieve high sensitivity and trace detection, highly active substrates with signal enhancement are required, and noble metal nanoparticles are often employed as active substrates for SERS detection.11,12 It has been reported that the composition, particle size and morphology of the substrate affect the generation and intensity of the Raman signal of the analyte.13,14 Nanoparticles of noble metals, such as Ag, Au, Pt and Cu substrates have a high density of hot-spot regions, especially Ag NPs.15,16 Ag NPs produce the strongest SERS effect, which has great potential for the detection of trace pollutants.17 For example, Vu reported the trace detection of ciprofloxacin using silver nanostars as SERS substrates.18 Dowgiallo used Au nanoparticles as SERS substrates to determine the detection limits of various pesticides.19 However, poor stability and sensitivity are the biggest drawbacks of single precious metal substrates, and they have limitations in practical applications. Nowadays, the preparation of composite substrates has overcome the shortcomings and implemented the concept of sustainable development. Fe3O4 nanoparticles have received widespread attention owing to their ease of separation, simple synthesis and chemical stability.20 Yang's and Han's group prepared core–shell Fe3O4@Ag and Fe3O4@Au composites, respectively, as recyclable magnetic SERS substrates for thiram detection.21,22 As a multifunctional material, graphene has received extensive attention by researchers for its unique structure and excellent electrical and optical properties. Graphene mainly provides chemical enhancement in the Raman enhancement process, and it has a highly conjugated system. Because graphene is enriched in surface molecules through Π–Π stacking effect synergized with non-covalent bonding forces, it enhances the adsorption effect of most SERS substrates and guarantees the sensitivity and accuracy for the detection of target molecules.23,24 A composite substrate overcomes the shortcomings of a single substrate and combines the advantages of its individual components, extending the application of SERS in the field of food safety analysis.
In this study, a three-layer SERS substrate, Fe3O4/GO/Ag, was fabricated. Based on the adsorption of the composite SERS substrate onto the target molecules, quantitative detection of thiram was achieved, as depicted in Scheme 1. The composite nanomaterial substrate Fe3O4/GO/Ag combines the excellent adsorption properties of graphene oxide (GO), the unique magnetic properties of Fe3O4, and the hot spot of Ag, providing new insights and a platform for the detection of pesticide residues.
 | ||
| Scheme 1 Illustration of the fabrication process of Fe3O4/GO/Ag with its application in SERS. | ||
2. Materials and methods
2.1 Chemicals and materials
Graphite powder was obtained from Tianjin Damao Chemical Reagent Factory. Concentrated sulfuric acid, sodium nitrate, hydrogen peroxide, potassium permanganate, ferric chloride, silver nitrate, acetone and ferrous sulfate were purchased from Sinopharm Chemical Reagent Co. 3-Aminopropltriethoxysilane (APTES), tri-hydroxymethyl aminomethane, dopamine hydrochloride were obtained from Aladdin. All chemicals were of analytical grade, and deionized water was prepared using the millipore water purification system.2.2 Preparation of the SERS substrate
Amino-functionalized Fe3O4/GO (0.1 g) was weighed in a beaker, and 25 mL of deionized water was added and ultrasonically dispersed uniformly. Then, 20 mL of Ag sol was added, ultrasonically dispersed for 15 min, and then left undisturbed for 15 min for magnetic separation. To load a large amount of Ag NPs on Fe3O4/GO, the same operation was repeated 10 times. The final products were washed with ethanol and dried in a vacuum drying oven at 60 °C to obtain Fe3O4/Ag.
2.3 Characterization
The X-ray diffraction (XRD) pattern was recorded on a D8 ADVANCE X-ray diffractometer. UV-visible (UV-vis) spectrophotometry (Shimadzu's Uv-2600) and infrared (IR) spectrometry (Bruker's Vertex 70) were used to obtain the UV-visible absorption peaks and infrared characteristic peaks, respectively, of the prepared materials. The structure and morphology were investigated through transmission electron microscopy (TEM) with FEI Tecnai G2 S-Twin. X-ray photoelectron spectroscopy (XPS, Kratos Axis ULTRA) was conducted. Raman spectroscopy was performed using a confocal microscope Raman spectrometer system (Horiba, LabRAMHR). The Raman spectra of GO and thiram were obtained under different experimental parameters.2.4 Application of the active substrate
Preparation of the thiram sample based on the Fe3O4/GO/Ag substrate: 1 mg of Fe3O4/GO/Ag was added to 500 μL of different concentrations of thiram standard solutions with sonication for even dispersion. Next, 30 μL of the test sample was placed on a slide and dried at room temperature for SERS detection. All tests were conducted in at least triplicate.
Test conditions: Raman measurements were conducted under ambient conditions with a 785 nm wavelength as the excitation source. The integration time per spectrum was 10 s, the laser power of the excitation source was 50 mW and the scanning range was from 400 to 1600 cm−1.30
3. Results and discussion
3.1 Characterization of Fe3O4/GO/Ag nanocomposites
The XRD patterns of the prepared Fe3O4, Fe3O4/GO and Fe3O4/GO/Ag NPs are shown in Fig. 1(a). As shown in Fig. 1(a), 2θ = 30.1°, 35.4°, 43.2°, 53.4°, 57.1°, and 62.8° are the diffraction peaks of the green Fe3O4 nanomaterial solution, which correspond to the six crystallographic diffraction peaks of (220), (331), (400), (511), (440), and (531), respectively, of the crystal structure of Fe3O4, and were identical to the diffraction peaks of the standard card (JCPDS no. 1901-0629).32,33 These results indicate that Fe3O4 was successfully synthesized. 2θ = 12° corresponds to the GO in Fe3O4/GO, represented with a red line, which coincides with reported values.34 For Fe3O4/GO/Ag nanomaterials, in addition to the peaks of Fe3O4 and GO, the spectrum exhibited apparent diffraction peaks at 2θ = 38.1°, 44.2°, 64.3°, and 77.5°, given as the blue line, which were attributed to cubic Ag NPs, with reflections from (111), (200), (220), and (311), respectively.35 Compared with the three lines, the intensity of Fe3O4 diffraction peaks was stronger than that of Fe3O4/GO and Fe3O4/GO/Ag composites because, probably, the GO and Ag on the surface of Fe3O4 weaken its magnetism. In combination with other analyses, it can be concluded that GO and Ag were coated on the surface of Fe3O4. The real image of the prepared Fe3O4/GO/Ag powder is presented in the ESI.†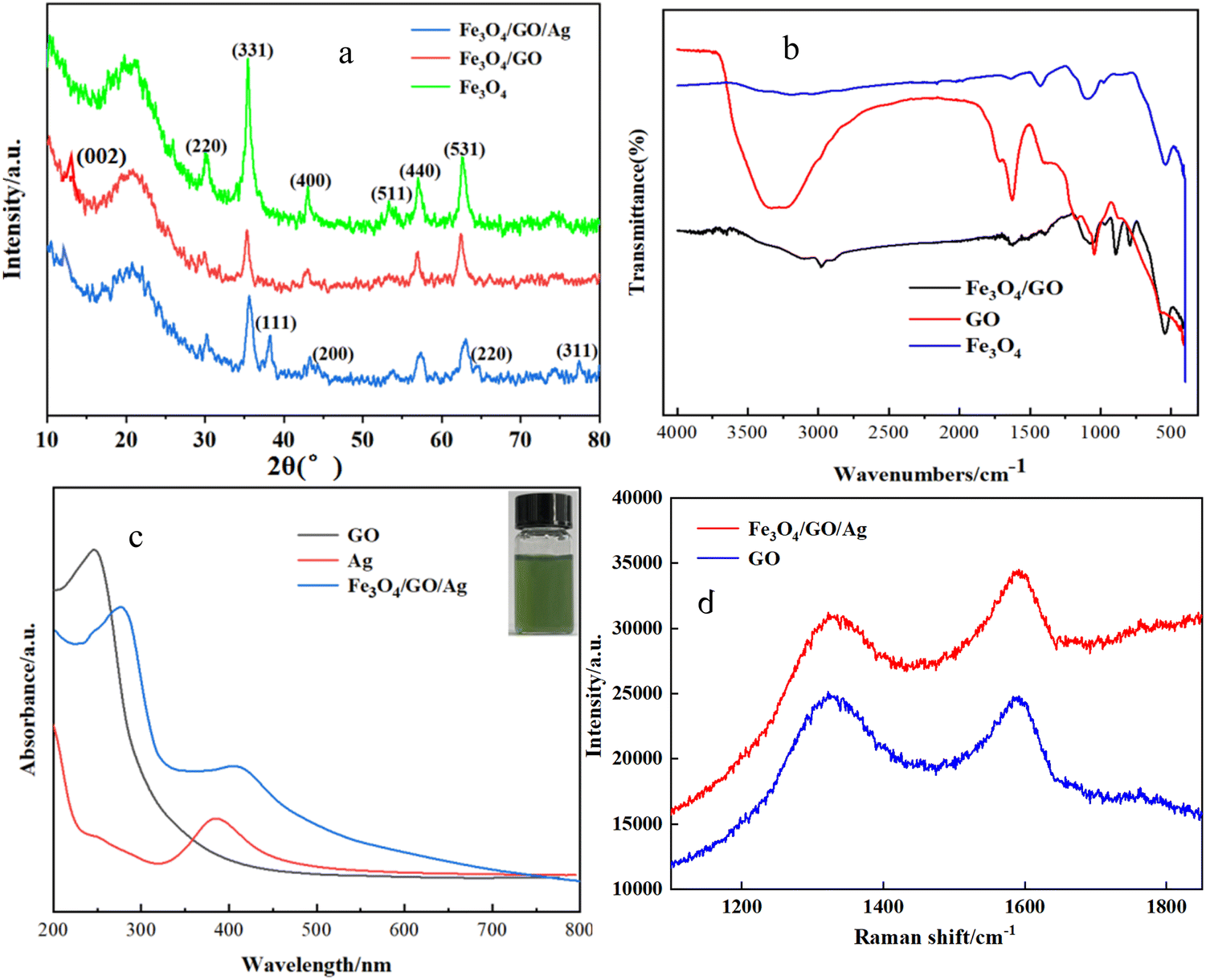 | ||
| Fig. 1 (a) XRD patterns of Fe3O4, GO, and Fe3O4/GO; (b) IR of Fe3O4, GO, and Fe3O4/GO; (c) ultraviolet-visible absorption spectra of each sample (insert: silver nanosol); and (d) Raman spectra of GO in blue line and Raman spectra of Fe3O4/GO/Ag in red line. | ||
The IR of Fe3O4, GO, and Fe3O4/GO are shown in Fig. 1(b). There was an obvious absorption band located at 560 cm−1, which was the characteristic absorption peak of the Fe–O bond on the surface of Fe3O4, given as the blue line. The absorption peaks located at 1041 cm−1 and 897 cm−1 were attributed to the stretching vibration of the epoxy group C–O–C on GO, represented with the red line. The absorption peak located at 3438 cm−1 corresponds to the peak of the stretching vibration of –OH, which indicated an abundance of GO oxygen-containing functional groups. It was beneficial to stabilization of the prepared composites in water and enrichment of the adsorbed material.36 Considering the spectral line of Fe3O4/GO, given in black, in addition to the characteristic absorption band of Fe3O4, the peak at 1638 cm−1 was attributed to the stretching vibration of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the amide bond (–CONH). The wavelength at 1410 cm−1 was the stretching vibration peak of C–N in the amide bond, which indicated that GO was successfully loaded on Fe3O4.37
O in the amide bond (–CONH). The wavelength at 1410 cm−1 was the stretching vibration peak of C–N in the amide bond, which indicated that GO was successfully loaded on Fe3O4.37
Fig. 1(c) shows the UV-visible absorption spectra of Ag, GO, and Fe3O4/GO/Ag. According to literature reports, Fe3O4 has no characteristic absorption peak in the UV-visible region. As shown in Fig. 3, the absorption peak at 240 nm was attributed to the distinct characteristic of GO, which is the π–π transition of conjugated CC on GO. GO was successfully prepared using the Hummers' method. The location of the resonance peaks in the UV-vis absorption spectra of Ag NPs is subject to change with the particle size and morphology. In Fig. 3, a significant absorption peak band around 390 nm, represented as a red line, was observed, which was consistent with the UV-visible characteristic absorption peaks of Ag NP.38,39 Compared with the synthesized pure GO and Ag nanoparticles, the absorption peaks of GO and Ag in Fe3O4/GO/Ag showed little positional shifts, and their absorption peaks appeared as broader peaks at 280 nm and 420 nm. Perhaps the loading of GO and Ag caused a red shift due to plasmonic resonance.
Raman spectroscopy is a powerful tool for characterizing carbon materials. Raman spectra of GO and Fe3O4/GO/Ag are shown in Fig. 1(d). Graphite can be oxidized by potassium permanganate into graphene oxide, and the graphite lamellae were ring-broken after oxidation, with a decrease in the degree of regularity and an increase in the degree of defects. According to the literature, the D and G peaks are Raman characteristic peaks of carbon atom crystals, in which the D peak represents the defects of carbon atom crystals, which usually appear in the range of 1300–1400 cm−1, and the G peak represents the symmetry and orderliness of the graphite structure, which usually appears in the range of 1560–1620 cm−1. The ratio of peak intensity between D and G peaks (ID/IG) is commonly used in Raman analysis to represent the disorder of graphite. The larger the ratio, the greater the disorder, the more the defects, and the higher the degree of oxidation of the material. Fig. 4 shows significant Raman characteristic peaks at 1330 and 1592 cm−1 with (ID/IG) = 0.90, much higher than graphite ID/IG = 0.14, indicating that graphite was fully oxidized and exfoliated to produce GO.40,41 Comparing the two sets of Raman spectra, it was found that graphene oxide and Fe3O4/GO/Ag had clear GO characteristic peaks.
The Fe, O, C and Ag elements were accurately identified using EDS (Fig. 2(a)), as well as SEM and TEM imaging of Fe3O4/GO/Ag (Fig. 2(b) and (d)). Fig. 2(d) shows that the most obvious part was dark black Ag NPs due to their high atomic number, and they were round-shaped, which was consistent with the TEM image of Ag (Fig. 2(c)). Fe3O4 was dark gray, whereas GO was greyish with a thin film. The particle sizes of Ag and Fe3O4 were less than 50 nm. The mapping of individual elements and Fe3O4/GO/Ag are shown in Fig. 3(a) and (b). In summary, we successfully prepared Fe3O4/GO/Ag.
 | ||
| Fig. 2 EDS of Fe3O4/GO/Ag (a); SEM (b) and TEM images of Ag (c); TEM image of Fe3O4/GO/Ag (d). | ||
 | ||
| Fig. 3 (a) Color mapping of individual elements; (b) mapping and (c) SEM image of the same part of Fe3O4/GO/Ag. | ||
The chemical compositions of Fe3O4/GO/Ag were comprehensively analyzed through XPS, as shown in Fig. 4. The presence of iron (Fe), carbon (C), and oxygen (O) elements in the substrate is visible (Fig. 4(a)). The 2p1/2 and 2p3/2 peaks of Fe are observed at binding energies of 724.2 eV and 711.1 eV (Fig. 4(b)), respectively, which distinctly correspond to the characteristics of Fe3O4. For C 1s spectra, binding energies at 286.8 eV and 284.9 eV are attributed to the presence of different chemical states of carbon, namely, the C–O–C and C–C or CC bond, respectively (Fig. 4(c)). Furthermore, C–O at 532.3 eV and H–O at 530.5 eV confirmed the presence of O 1 s in GO (Fig. 4(d)). The double peaks of Ag 3d, including Ag 3d5/2 at 368.2 eV and Ag 3d3/2 at 374.2 eV, were consistent with the standard binding energy (Fig. 4(e)). The combination of the analysis and unique magnetic separability exhibited by the products fully demonstrated the successful fabrication of Fe3O4/GO/Ag NPs.
 | ||
| Fig. 4 (a) XPS spectrum of Fe3O4/GO/Ag and high-resolution XPS of (b) Fe, (c) C, (d) O, and (e) Ag. | ||
3.2 SERS detection of thiram
Thiram as the target was analyzed in detail. Different concentrations of the thiram standard solution were mixed with the prepared active substrate, followed by Raman detection. Fig. 5 shows the Raman spectra of thiram based on Fe3O4/GO/Ag as the substrate. There were obvious characteristic bands at 432 cm−1, 565 cm−1, 745 cm−1, 956 cm−1, 1140 cm−1, 1381 cm−1, 1447 cm−1, and 1536 cm−1, which can be employed as the main Raman characteristic peaks of thiram. Among them, the band at 432 cm−1 was attributed to the bending vibration of CH3CN and the stretching vibration of CS in the molecular structure of thiram. The band at 565 cm−1 was attributed to S–S stretching vibration, and the band at 956 cm−1 was attributed to the vibrational peak of CH3N. The band at 1140 cm−1 belong to C–N stretching vibration. The band at 1381 cm−1 was attributed to C–N stretching vibrations and CH3 symmetric deformation. The peak at 1447 cm−1 was an antisymmetric peak of CH3.42,43 The peak locations were consistent with those reported in the literature. Thiram was completely different from GO in terms of the test conditions and parameters of SERS. Thus, it avoided interference from GO.
 | ||
| Fig. 5 Raman spectra of thiram. | ||
Based on the constructed SERS substrate, the intensities of Raman characteristic peaks of different concentrations are shown in Fig. 6. The intensity of the characteristic peak at 1381 cm−1 is more obvious and enhanced with the increase in the concentration of thiram, ranging from 1 × 10−12 to 1 × 10−3 mol L−1, which was therefore taken as the characteristic peak for thiram detection. To further correct the fluctuation of the Raman signal of thiram due to laser instability and substrate inhomogeneity during the detection process, the intensity of thiram at 1381 cm−1 with different concentrations was used to construct a working curve for the subsequent quantitative detection and analysis of thiram. As shown in Fig. 7, the standard curve showed perfect linearity in the range of 1 × 10−10–1 × 10−4 mol L−1, with a correlation coefficient (R2) of 0.9960, and the optimal fitting equation was Y = 9876![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logX + 115162. Although there were obvious signals for 1 × 10−11 and 1 × 10−12 mol L−1 of thiram, they seem nonlinear (Fig. 8).
logX + 115162. Although there were obvious signals for 1 × 10−11 and 1 × 10−12 mol L−1 of thiram, they seem nonlinear (Fig. 8).
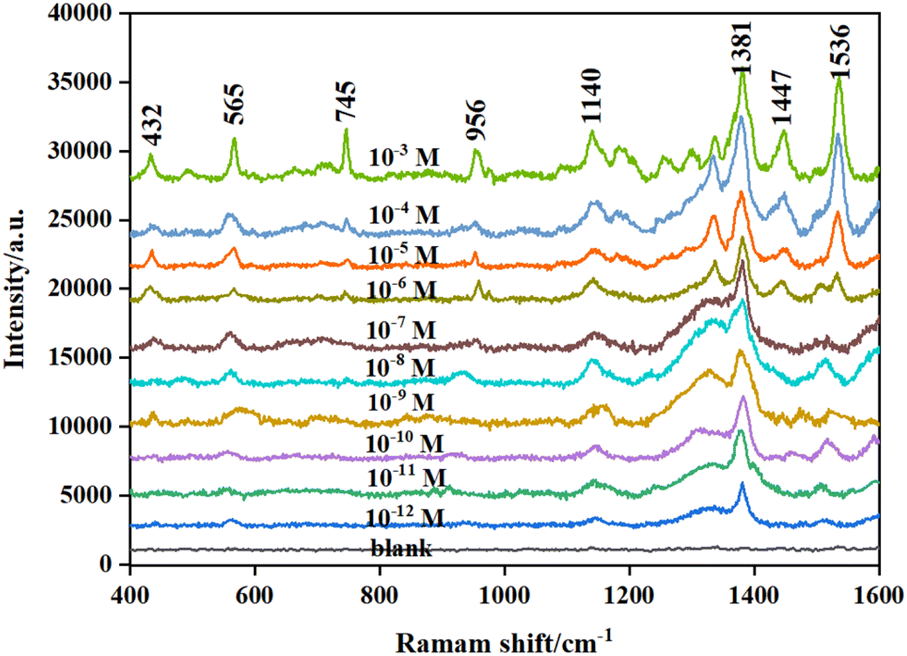 | ||
| Fig. 6 Raman spectra for different concentrations of thiram. | ||
 | ||
| Fig. 7 Corresponding calibration plot of Raman intensity versus concentrations of thiram. | ||
 | ||
| Fig. 8 SERS spectra of 10−4 M thiram using Fe3O4/GO/Ag as substrate that has been stored for various periods. | ||
3.3 Detection of thiram in real samples
We put the proposed methodology into practice. Tap water, apple juice and pear juice as samples were exploited to investigate the spiked recovery experiments. The added amounts of thiram in tap water and fruit juice were 2 × 10−10, 5 × 10−10 and 1 × 10−9 M. As shown in Table 1, the average recovery was 90.2–104.2%. These results indicated that the sensor has great promise for the detection of thiram and other analytes in agriculture, food, medicine and environmental safety.| Samples | Spiked (M) | Recovery (%) |
|---|---|---|
| Pear juice | 1 × 10−9 | 103.8 |
| 5 × 10−10 | 98.7 | |
| 2 × 10−10 | 92.9 | |
| Apple juice | 1 × 10−9 | 103.3 |
| 5 × 10−10 | 104.2 | |
| 2 × 10−10 | 90.2 | |
| Tap water | 1 × 10−9 | 101.9 |
| 5 × 10−10 | 98.4 | |
| 2 × 10−10 | 97.2 |
3.4 Activity, stability and repeatability of Fe3O4/GO/Ag
EF was estimated using the following equation: EF = ISERS × C0/I0 × CSERS; here, I0 and ISERS are the intensities of the normal Raman and intensities of SERS with Fe3O4/GO/Ag as substrate, respectively. CSERS and C0 are the concentrations of the mixed thiram with Fe3O4/GO/Ag and bare thiram. The EF for thiram at 1381 cm−1 was 2.8 × 108. This may be attributed to two types of enhancement mechanisms: electromagnetic enhancement mechanism (EM) and chemical enhancement mechanism (CM).In this study, the minimum visible signal was achieved for 10−12 mol L−1 of thiram. If the concentration of thiram is lower than 10−12 mol L−1, no detectable signal is obtained. Meanwhile, the formulation (LOD = 3δ/k) was employed to calculate the limit of detection (LOD), where δ is the standard deviation of the blank and k is the slope of the calibration curve. At 1381 cm−1, the LOD is 10−11.7 mol L−1. The two results nearly coincide. To verify the stability of Fe3O4/GO/Ag, time-dependent SERS measurements were performed, as shown in Fig. 11 and 9. The intensity of the SERS signal did not substantially change over one month. The results showed that Fe3O4/GO/Ag was stable during preservation. In addition, repeatability plays an important role in SERS. To assess the repeatability of Fe3O4/GO/Ag, three samples were analyzed under the same conditions at 11 random locations on the substrate. High repeatability with a low RSD of approximately 5.1% was obtained, as shown in Fig. 10. The comparison of different materials for detection of thiram by SERS is presented in ESI.†
 | ||
| Fig. 9 SERS spectra of 10−4 M thiram at various periods. | ||
 | ||
| Fig. 10 (a) SERS spectra of 10−4 M thiram from 11 randomly selected Fe3O4/GO/Ag composite microspheres. (b) Raman intensity distribution of 1381 cm−1 peak from the 11 spectra. | ||
3.5 Exploration of the enhancement mechanism
In this study, a high EF of 2.8 × 108 for thiram was obtained at 1381 cm−1. It is necessary to investigate the enhancement mechanism. Generally, it includes electromagnetic enhancement and charge transfer enhancement. However, electromagnetic enhancement is the main contribution. Electromagnetic enhancement means that owing to the local surface plasmon resonance (LSPR) effect of Ag, the electromagnetic field will be enhanced in the region of 10 nm on the surface of Ag.44,45 Specially, when Ag NPs are excited by a specific wavelength of light, LSPR occurs on their surface. The resonance generates highly localized strong electromagnetic fields on Ag NPs, which significantly enhance the Raman signal of thiram adsorbed on the surface of the NPs. In addition, the lightning rod, antenna and mirror image models can explain LSPR enhancement. The tip of Ag NPs was treated as a perfect electrical conductor with a shape needle tip and it is similar to a slender wire. When an incident light was irradiated on the tip of Ag, a strong local electromagnetic field formed near the tip due to the small curvature radius of the tip. This local electromagnetic field interacted with thiram adsorbed near the tip of Ag, resulting in an enhanced Raman signal. The strongly enhanced field in the region of 10 nm is called a hot spot. If thiram is in the hot region, the Raman signal will amplify sharply. In this study, the coordination between Ag and thiram with sulfydryl shortens the distance between the substrate and target molecule, which may be in the hot spot. Furthermore, because of the aggregation of Fe3O4 and the adsorption property of GO, it endows Fe3O4/GO/Ag with enrichment ability. Therefore, the electromagnetic field intensity increased in the region of the hot spot, and the Raman signal amplified.The chemical enhancement mechanism mainly includes resonance Raman enhancement, ground state chemical enhancement and charge transfer enhancement. Among them, charge transfer enhancement is generally approved. Charge transfer enhancement indicates that when incident light irradiates the surface of Ag and the semiconductor (GO), if the Fermi energy level of the surface is between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) of thiram, the energy of incident light matches charge transfer, and resonance Raman scattering occurs.46,47 The free electrons on the surface of Ag and GO resonate and transit to the adsorbed thiram. These changes can significantly increase the polarizability of thiram. Meanwhile, the chemical bonding between thiram and Ag NPs can alter the vibration frequencies of thiram. Thus, the Raman signal of thiram is enhanced. In this study, because of the tunable band gap and distinctive charge transfer capability of GO, GO has been widely used as a SERS substrate. The excitation of electrons from the bands of GO to the LUMO of thiram is shown in Fig. 11. In brief, owing to Ag and GO, as well as Fe3O4, with the aid of electromagnetic fields and charge transfer, the Raman signal was enhanced.
 | ||
| Fig. 11 Schematic of the CM and EM enhancement mechanisms. | ||
4. Conclusion
In this study, a novel and ultra-high sensitive approach for detection of pesticide residues based on SERS was developed. The synthesized magnetic Fe3O4/GO/Ag nanohybrid was used as a SERS active substrate for Raman detection of thiram molecules. Fe3O4 was synthesized using a co-precipitation method, and modified Hummers-prepared graphene was employed to form Fe3O4/GO using a silane coupling agent as a bridge. The Fe3O4/GO/Ag nanohybrid as the active substrate was obtained through amino chemical modification of their surfaces for adsorption of Ag NPs. Its crystalline shape and structure were characterized. Results showed that Fe3O4/GO/Ag composites were successfully fabricated. With its feasible synthesis, Fe3O4/GO/Ag was successfully applied for thiram detection. It showed a notable linear relationship (10−10–10−4 mol L−1) and an R2 of 0.9960. The designed Fe3O4/GO/Ag exhibited excellent enhancement with a high EF. Promising results were obtained for thiram detection in environmental samples. The established method is supersensitive, rapid and convenient with a wide linear range, low detection limit, low consumption, and ease of operation. Therefore, the magnetic nanohybrid as SERS the active substrate can not only improve the sensitivity and accuracy of pesticide residues, but can also have broad application prospects in real samples. The proposed method provides new insights for more rapid, sensitive and accurate detection. As a representative of a precious metal nanohybrid with magnetism, it is a promising active substrate, probe and photocatalyst. Moreover, it is of great significance for applications in the food industry, agriculture, industry, biomedicine and environmental fields.Data availability
All data generated or analyzed during this study are included in this published article (and its ESI).†Author contributions
R. Wu: conceptualization, data curation, funding acquisition, project administration, validation, editing, review, supervision, and draft methodology. X. Song: investigation, writing original. Y. L. Wang and Q. Zhang: formal analysis.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Key Research and Development Plan of Shaanxi Province (2024GX-YBXM-318), Graduate Education and Teaching Reform Project of Shaanxi University of Technology (SLGYKS2401), College Student Innovation and Entrepreneurship Project of Shaanxi University of Technology (202410720020), the Shaanxi Province Education Ministry Research Key Foundation (20JS015) and National Natural Science Foundation of China (22177066).References
- L. Lu, L. Xu and Y. Zhang Y, et al., Appl. Surf. Sci., 2022, 603, 154419–154429 CrossRef CAS.
- K. S. Patel, P. K. Pandey and P. M. Ramos, RSC Adv., 2023, 13, 14914–14929 RSC.
- S. Salam, A. Arif and R. Mahmood, Pestic. Biochem. Physiol., 2020, 164, 14–25 CrossRef CAS PubMed.
- W. Liu, Z. Z. Cong and G. C. Liu, et al., Inorg. Chim. Acta, 2021, 527, 120592–120599 CAS.
- Y. Pico, H. Alfarhan and D. Barcelo, Trends Anal. Chem., 2020, 122, 115720–115753 CAS.
- I. Dominguez, R. R. Gonzalez and F. G. A. Libanas, et al., Trends Environ. Anal. Chem., 2016, 12, 1–12 CAS.
- H. Zhang, Y. Yuan and Y. Sun, et al., Analyst, 2018, 143, 175–181 CAS.
- M. Fan, G. Andrade and A. A. Brolo, Anal. Chim. Acta, 2020, 1097, 1–29 CAS.
- L. Jiang, M. Hassan and S. Ali, et al., Trends Food Sci. Technol., 2021, 112, 225–240 CAS.
- T. N. Wu, S. Yang and W. Feng, Microchem. J., 2024, 198, 110208–110213 CrossRef CAS.
- H. P. Zhou, J. Y. Qiu and Y. H. Zhang, J. Hazard. Mater., 2024, 24, 133868–133874 CrossRef.
- Q. Zhang, R. Wu and C. F. Liu, Rare Met. Mater. Eng., 2024, 53, 546–554 CAS.
- F. Selimoğlu and E. A. Muhammed, Vib. Spectrosc., 2023, 126, 103539–103545 CrossRef.
- N. R. Barveen, J. L. Xu and Y. W. Cheng, J. Environ. Chem. Eng., 2024, 12, 112200–112206 CrossRef CAS.
- J. Y. Li, M. Li and Q. Q. Wang, Spectrochim. Acta, Part A, 2024, 311, 124037–124043 CrossRef CAS PubMed.
- R. Wu, Q. C. Du and H. R. Zhang, et al., J. Saudi Chem. Soc., 2024, 28, 101824–101837 CrossRef CAS.
- R. Wu, F. G. Zhang and X. H. Ji, et al., Arabian J. Chem., 2022, 15, 103519–103528 CAS.
- R. Wu, Q. C. Du and H. R. Zhang, et al., J. Saudi Chem. Soc., 2024, 28, 101824–101837 CAS.
- A. M. Dowgiallo, D. A. Guenther and J. Agric, Food Chem., 2019, 67, 12642–12651 CrossRef CAS PubMed.
- Y. Z. Wei, C. Qi and T. J. Wen, et al., Adv. Chem., 2017, 29, 1366–1394 Search PubMed.
- L. Yang, Z. Bao and Y. Wu, et al., J. Raman Spectrosc., 2022, 43, 848–856 CrossRef.
- D. Han, B. Li and Y. Chen, et al., Nanotechnol, 2019, 30, 2067–2074 Search PubMed.
- L. Y. Wang, H. Chen and S. J. Ma, Vib. Spectrosc., 2023, 129, 103614–103620 CrossRef CAS.
- Z. Wang, S. Wu and L. C. Ciacchi, et al., Analyst, 2018, 143, 5074–5089 RSC.
- J. Z. Yang and C. Y. Zhang, Res. Chem. Intermed., 2020, 50, 50–53 Search PubMed.
- Y. Lin, D. Ding and C. Hu, et al., ACS Omega, 2021, 6, 25996–26003 CAS.
- L. A. Akamine, D. A. V. Medina and F. M. Lanças, Talanta, 2021, 222, 121683–121692 CAS.
- Y. Ai, P. Liang and Y. Wu, et al., Food Chem., 2018, 241, 427–433 CrossRef CAS PubMed.
- Z. Lin, S. Luo and D. Xu, et al., Analyst, 2020, 145, 424–433 RSC.
- A. K. Verma and R. K. Soni, Nanotechnol, 2019, 30, 385502–385516 CrossRef CAS.
- Y. Ma, Y. Wang and Y. Luo, et al., J. Anal. Methods Chem., 2018, 10, 4655–4664 CAS.
- J. He, G. Song and X. Wang X, et al., J. Alloys Compd., 2022, 893, 162226–162235 Search PubMed.
- N. Negin, F. S. Arghavan and D. Nebile, et al., Environ. Sci. Pollut. Res., 2021, 28, 19222–19233 CrossRef.
- R. Wu, S. Song and J. F. Lu, et al., J. Mol. Struct., 2023, 1278, 134908–134914 Search PubMed.
- T. Kavinkumar, K. Varunkumar and V. Ravikumar, et al., J. Colloid Interface Sci., 2017, 4, 1125–1133 CrossRef PubMed.
- M. F. Khan, A. Jama and P. J. Rosy, et al., Inorg. Chem. Commun., 2022, 139, 109422–109432 CrossRef.
- W. Yu, J. Jia and J. Shi, et al., Talanta, 2023, 254, 124181–124193 CrossRef CAS.
- U. T. Khatoon, A. Velidandi and G. S. N. Rao, Mater. Chem. Phys., 2023, 294, 126997–127005 CrossRef CAS.
- A. Mandal, E. Dhineshkumar and T. P. Sastry, Clean Technol. Environ., 2023, 1, 1–18 CrossRef.
- T. Tene, M. Guevara and F. B. Palacios, et al., Front. Chem., 2023, 11, 1–14 Search PubMed.
- B. Das, R. Kundu and S. Chakravarty, Mater. Chem. Phys., 2022, 290, 126597–126602 CrossRef CAS.
- Y. Wang, S. Liu and Y. Hu, et al., Analyst, 2023, 148, 5435–5444 RSC.
- N. Xu, Z. Tang and Y. P. Jiang, et al., J. Environ. Chem. Eng., 2023, 11, 109736–109749 CrossRef CAS.
- M. Liu, X. Hu and C. Zhang C, et al., Sens. Actuators, B, 2024, 398, 134688–134703 CrossRef CAS.
- Y. Luo, L. Niu and Y. Wang, et al., Appl. Surf. Sci., 2023, 607, 155095–155110 CrossRef CAS.
- S. Di, S. Guo and Y. Wang, et al., Mater. Chem. Phys., 2023, 301, 127621–127635 CrossRef CAS.
- R. Mandavkar, S. Lin and S. Pandit, et al., Surf. Interfaces, 2022, 33, 102175–102189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01081k |
| This journal is © The Royal Society of Chemistry 2025 |