
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon-selective uranium capture from seawater by UiO-66 metal–organic framework modified with amidoxime groups†
Lin Ma a,
Chen Huangbc,
Peng Sunb,
Yang Wanga,
Hua Shena,
Hongjuan Mab,
Yuxia Liu*a and
Qingnuan Li*a
a,
Chen Huangbc,
Peng Sunb,
Yang Wanga,
Hua Shena,
Hongjuan Mab,
Yuxia Liu*a and
Qingnuan Li*a
aShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: liuyuxia@sinap.ac.cn; liqingnuan@sinap.ac.cn
bShanghai Applied Radiation Institute, Shanghai University, Shanghai 200444, China
cSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 29th April 2025
Abstract
Seawater is a valuable source of uranium (U) resources, and harnessing it effectively can play a crucial role in promoting nuclear energy. However, current polymer-based adsorbents typically exhibit slow adsorption rates and insufficient selectivity. In this work, the efficient uranyl ion (UO22+) adsorbent (UiO-66-AO) was obtained from UiO-66-NH2 through a simple and mild approach post synthesis modification (PSM). During the PSM process, not only the octahedral morphology (particle size ∼200 nm) but also the crystal structure of UiO-66-NH2 was well maintained. The integration of amidoxime groups (AO) improved the selectivity and adsorption performance towards UO22+. According to the Langmuir model, the maximum adsorption capacity (qmax) of UiO-66-AO was 413.2 mg g−1. In the competitive ion solution containing vanadium(V), iron (Fe), magnesium (Mg), calcium (Ca), etc., UiO-66-AO exhibited a much higher UO22+ adsorption capacity of 12.3 mg g−1 than UiO-66-NH2 (0.9 mg g−1). Furthermore, UiO-66-AO achieved equilibrium for UO22+ adsorption in natural seawater within 3 days, and the adsorption capacity was 3.0 mg g−1. Finally, near-edge X-ray absorption fine structure (NEXAFS) and X-ray photoelectron spectroscopy (XPS) demonstrated the chelation effect of AO on U, proving that the PSM successfully achieved performance enhancement. It is hoped that the simple and mild AO modification approach will provide new insights for the preparation of rapid, highly selective, and efficient UO22+ adsorbents.
1. Introduction
With the rapid increase in global energy demand, nuclear energy has gained significant attention for its high energy density and zero greenhouse gas emissions.1,2 In the long run, the demand for uranium (U) shows a continuous growth trend. However, it is estimated that global conventional U resources are only used by humans for about 100 years.3 Fortunately, the reserves of U in the ocean are abundant, approximately 1000 times that of land, which can compensate for the environmental problems and limited reserves of traditional mining of U from land.4,5 Thanks to its simplicity of operation, high efficiency, and economic feasibility, the adsorption method remains the mainstream approach for uranium extraction from seawater (UES).In recent years, metal–organic frameworks (MOFs) constructed from metal ions or clusters and organic ligands, such as UiO-66-3C4N, UiO-66-neomycin, MIL-101, have attracted significant attention for uranyl ion (UO22+) adsorption due to their high specific surface area, porosity, and structural adjustability.6–9 However, the concentration of UO22+ in the ocean is extremely low (≈3.3 ppb) and is accompanied by various competing ions.10,11 Therefore, effective UES necessitates enhancing the selectivity of MOFs for UO22+ and improving their affinity under trace UO22+ conditions.
Furthermore, achieving MOF adsorbents with targeted performance through simple and efficient methods remains a considerable challenge.12–14 In a recent study, Ghosh and coworkers reported an ionic mesoporous material i-MZIF(90)50 derived from UiO-66-NH2 and ZIF-90 MOFs through precise morphology engineering, which benchmark record of 28.2 mg g−1 UO22+ uptake from natural non-spiked seawater.15 Recent advancements in the synthesis and functionalization of MOFs have highlighted post-synthetic modification (PSM) as an effective and versatile strategy to enhancing the performance of MOFs. This technique involves reactions conducted after the initial synthesis of the frameworks, allowing for tailored modifications.16–19 PSM allows for modifications to the surface environment of MOFs while preserving the original bridging coordination and topological structure.20,21 These modifications can lead to the emergence of new chemical and physical properties, allowing for adjustments in the pores, surface characteristics, stability, hydrophobicity, adsorption capacity, catalytic activity, luminescence, and magnetic properties of MOFs, thus providing new opportunities for applications.22–25 The amidoxime group (AO) is currently one of the most commonly utilized functional groups for UES.26 By introducing AO into MOFs, the selectivity and adsorption efficiency for U can be significantly improved.
To address the low selectivity and complex preparation processes of current MOFs adsorbents for UES. This study selected UiO-66-NH2 as the initial MOFs due to its high framework stability in water. Acetic acid was used as a modulator to affect the crystallinity of MOFs. Zr6 MOFs are ideally 12-coordinated. As acetic acid added, Zr6 MOFs with reduced coordination (8- and 6-coordinated) can be considered as MOFs with defect. Indeed, the modulator facilitates the formation of defects in MOFs, which have an important influence on the stability, reactivity, porosity and thermomechanical behavior of MOFs.16,27 Subsequently, AO was integrated through PSM to obtain UiO-66-AO material (Fig. 1). During the PSM process, the framework structure and morphology of MOFs were well maintained. UiO-66-AO exhibited good selectivity for UO22+ under trace conditions and rapid adsorption rate in seawater, providing reliable insights for MOFs modification and presenting promising materials for UES.
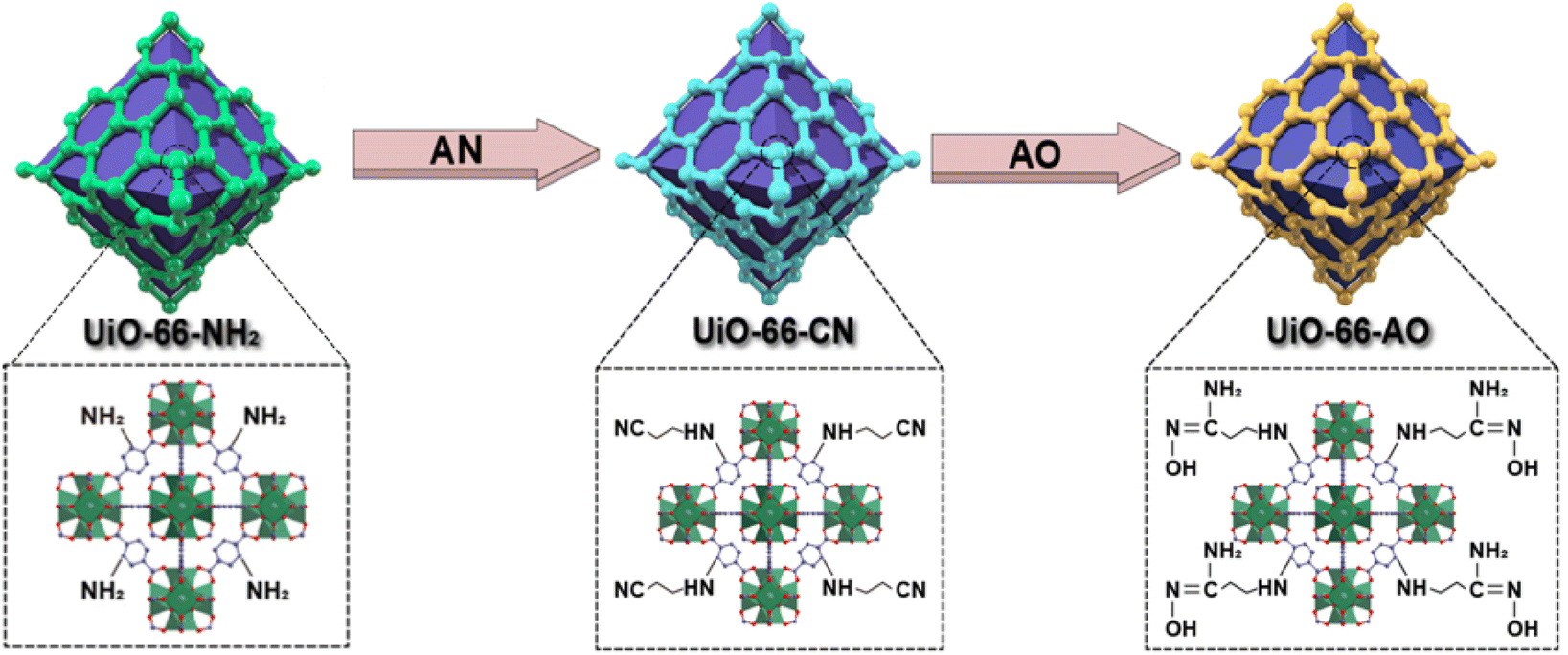 | ||
| Fig. 1 Illustration of the synthesis of UiO-66-AO via PSM. | ||
2. Results and discussion
2.1 Preparation of UiO-66-AO
The desired adsorbent, UiO-66-AO, was synthesized following the route depicted in Fig. 2a. UiO-66-NH2 was synthesized following established methods from previous studies.28 UiO-66-CN was synthesized by PSM in the presence of acrylonitrile (AN) through a Micheal addition reaction.29 The final adsorbent, UiO-66-AO, was synthesized by refluxing UiO-66-CN with hydroxylamine hydrochloride in ethanol for 24 hours, with triethylamine serving as the base. More detailed synthesis information can be found in the supporting literature. | ||
| Fig. 2 (a) UiO-66-AO synthesis process. SEM and TEM images of (b) UiO-66-NH2, (c) UiO-66-CN, and (d) UiO-66-AO. | ||
Acetic acid, as a modulator, can effectively regulate the morphology of MOFs particles, in turn, affect the UO22+ adsorption capacity.16 The effect of acetic acid concentration on the synthesis of UiO-66-NH2 was studied firstly. As shown by SEM in Fig. S1a0†, when without the acetic acid and the concentration of ZrCl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BDC-NH2 was 1.0:1.5, UiO-66-NH2 clusters were successfully formed. With the concentration of acetic acid increased (1.0, 1.7, 2.6, and 4.1 M), the clusters became increasingly dispersed (Fig. S1a1–a4†). When the concentration of acetic acid was 2.6 M, the octahedral particles of UiO-66-NH2 were more uniform with particle sizes around 200 nm (Fig. 2b and S1a3†). As the concentration of acetic acid further increased, at a concentration of 4.1 M (Fig. S1ab†), although the octahedral morphology continued to be maintained, significant defects and damage appeared in the structure. After PSM process with UiO-66-NH2, the UiO-66-CN clusters were reacted, and it could be found their aggregate morphology maintained (Fig. S1b0–b4†). The morphology of UiO-66-AO remained intact after the PSM process with UiO-66-CN (Fig. S1c0–c4†). Fig. 2c and d show that the PSM process had minimal impact on the crystal structure and morphology of the original UiO-66-NH2.
:BDC-NH2 was 1.0:1.5, UiO-66-NH2 clusters were successfully formed. With the concentration of acetic acid increased (1.0, 1.7, 2.6, and 4.1 M), the clusters became increasingly dispersed (Fig. S1a1–a4†). When the concentration of acetic acid was 2.6 M, the octahedral particles of UiO-66-NH2 were more uniform with particle sizes around 200 nm (Fig. 2b and S1a3†). As the concentration of acetic acid further increased, at a concentration of 4.1 M (Fig. S1ab†), although the octahedral morphology continued to be maintained, significant defects and damage appeared in the structure. After PSM process with UiO-66-NH2, the UiO-66-CN clusters were reacted, and it could be found their aggregate morphology maintained (Fig. S1b0–b4†). The morphology of UiO-66-AO remained intact after the PSM process with UiO-66-CN (Fig. S1c0–c4†). Fig. 2c and d show that the PSM process had minimal impact on the crystal structure and morphology of the original UiO-66-NH2.
When the concentration of ZrCl4:BDC-NH2 was 1.0:1.0, the result was consistent with the observation described above (Fig. S2†). With higher concentrations of acetic acid, the MOFs particles became more dispersed, and the octahedral shape became more pronounced. However, at excessively high acetic acid concentrations, defects appeared in the MOFs particles, likely due to acetic acid's effect on the coordination between metal centers and ligands.30 In addition, the PSM process had little influence on the structural integrity of the MOFs.
2.2 Characterization of materials
A comprehensive study was carried out on the physicochemical properties of UiO-66-NH2 during the PSM. As shown in Fourier transform infrared spectroscopy (FT-IR) spectrum (Fig. 3a), UiO-66-NH2 had two signs appeared at 3400–3500 cm−1, corresponding to the stretching vibrations of –NH2, which bonded to the benzene ring. Following the Michael addition of the cyano group, new stretching vibrations of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (–CN) appeared at 2247 cm−1, confirming the successful grafting of AN onto UiO-66-NH2. Ultimately, the disappearance of the –CN group, along with the emergence of stretching vibrations for –C
N (–CN) appeared at 2247 cm−1, confirming the successful grafting of AN onto UiO-66-NH2. Ultimately, the disappearance of the –CN group, along with the emergence of stretching vibrations for –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N− (1642 cm−1) and N–O (916 cm−1), confirmed the conversion of –CN groups to AO groups through amidoximation.31 Analysis of the full X-ray photoelectron spectroscopy (XPS) in Fig. 3b revealed an increase of the N 1s peak (399 eV) after PSM, while the Zr 3d peak (284 eV) remained largely unchanged, which further proved that the –CN and AO groups were successfully obtained.
N− (1642 cm−1) and N–O (916 cm−1), confirmed the conversion of –CN groups to AO groups through amidoximation.31 Analysis of the full X-ray photoelectron spectroscopy (XPS) in Fig. 3b revealed an increase of the N 1s peak (399 eV) after PSM, while the Zr 3d peak (284 eV) remained largely unchanged, which further proved that the –CN and AO groups were successfully obtained.
 | ||
| Fig. 3 Characterization of UiO-66-NH2 before and after modification. (a) FT-IR spectra, (b) XPS spectra, (c) TG curves, (d) PXRD patterns, (e) N2 adsorption/desorption isotherms, and (f) pore size distributions of UiO-66-NH2, UiO-66-CN, and UiO-66-AO. | ||
The thermal stability of MOFs during PSM was examined using thermogravimetric analysis (TG). As shown in Fig. 3c, the thermal stability of MOFs during PSM varied with different functionalities, as indicated by the TG results. The weight loss of MOFs below 120 °C was caused by solvent evaporation.31 The functional groups of UiO-66-CN decomposed after ∼300 °C. After amidoximation, the apparent decomposition temperature of AO group was ∼150 °C, which was consistent with literature reports. All TG curves indicated that the apparent starting temperature for MOFs decomposition to ZrO2 was ∼525 °C.32 As shown in Fig. 3d, the powder X-ray diffraction (PXRD) spectrum of MOFs during the PSM were almost consistent with the peaks of the original UiO-66-NH2, indicating that the crystal structure was formed and maintained in the process.33 It was demonstrated that UiO-66-AO was successfully prepared, and the combination of SEM and TEM (Fig. 2b–d) further showed that the influence of PSM on the crystal structure of the MOFs was not significant. N2 adsorption/desorption isotherms were investigated as shown in Fig. 3e. Apparently, the three isotherms were the combination of type I and type IV isotherms curves according to the IUPAC classification, indicating the presence of microporous and mesoporous in the structures.34 Using the Brunauer–Emmett–Teller method, the specific surface areas were measured as 255.82, 168.75, and 109.66 m2 g−1, respectively (correspond to UiO-66-NH2, UiO-66-CN, and UiO-66-AO). The surface area of MOFs gradually decreased with the PSM process, mainly due to the larger functional groups appeared in the ligand. Micropores contribute a substantial specific surface area and pore volume, boosting the availability of active sites. Mesopores and macropores aid in the quick diffusion of ions and efficient transfer of substances.35 The pore size was primarily centered around ∼2.6 nm (Fig. 3f), supporting effective diffusion of UO22+.36 The number of pores concentrated at 2.6 nm gradually decreased, this was also attributed to pore blockage caused by the large functional groups introduced during the PSM process.
2.3 Uranium adsorption experiments
To optimize the adsorption material, the effects of ligand concentration and acetic acid concentration on UO22+ adsorption were examined. As shown in Fig. S5a,† when the concentration of acetic acid was constant, the effect of the concentration of ligand (molar ratio of ZrCl4:BDC-NH2 = 1.0:1.0 or 1.0:1.5) on UO22+ adsorption was not obvious. However, while the concentration of ligand was constant, as the concentration of acetic acid increased, the adsorption capacity also increased. As the acetic acid concentration was 2.6 M, the adsorption capacity reached its maximum value (10.9 mg g−1). When the molar ratio was increased further, the adsorption capacity decreased because the acetic acid disrupted the MOFs structure (Fig. S1a4–c4†). Acetic acid can regulate adsorption, which may be due to its modulation of the crystallinity of MOFs, affecting the active sites concentration of hydroxo, aquo or monocarboxylic acid on Zr6 coordination.16 UiO-66-AO prepared with ZrCl4:BDC-NH2 = 1.0:1.5 and acetic acid concentration 2.6 M was used as the sample for subsequent adsorption.
Secondly, optimization experiments were conducted on the solid–liquid ratio of the adsorption system. As shown in Fig. 4a, the adsorption capacity gradually decreased (from 97.0 to 6.1 mg g−1) with the m V−1 increased (from 0.002 to 0.05), and the removal rate of UO22+ in the solution gradually increased (from 58.8% to 92.2%). For subsequent adsorption experiments, 0.02 mg mL−1 was chosen for adsorption testing, as its removal rate was moderate and the transition was significant.
 | ||
| Fig. 4 (a) Effect of the mass-to-volume ratio on adsorption performance. (b) Influence of pH on UO22+ adsorption (c0 = 500 ppb, t = 24 h), (c) the effect of anion Cl− concentration on UO22+ adsorption (c0 = 2.0 ppm, t = 24 h). (d) Adsorption in simulated seawater (ion concentrations provided in Table S1,† t = 24 h). (e) SEM and TEM of UiO-66-AO after the adsorption of UO22+ in seawater, and its Zr, and U elemental mapping photographs. | ||
The forms of UO22+ species in solution are highly influenced by the pH level of the solution. Protons not only influence U speciation but also alter the surface charge of the sorbent.37,38 UiO-66-AO could maintain a good octahedral morphology at pH = 3.0–9.0. When pH = 2.0 and 10.0, obvious defects were appeared on the surface of the sample (Fig. S3†). Additionally, to evaluate the impact of pH on UO22+ adsorption and identify the ideal value, adsorption experiments were carried out with pH range from 3.0 to 9.0. As indicated in Fig. 4b, the adsorption capacity of UiO-66-AO was below 1.0 mg g−1 when the pH was ≤ 5.0, but it peaked at 23.8 mg g−1 at pH 6.0. With the increase of pH (pH > 6.0), the adsorption capacity decreased slightly but was still more than 21.0 mg g−1. This characteristic indicates the potential of UiO-66-AO for UES.11
As depicted in Fig. 4c, the influence of counter anions, specifically chloride ions (Cl−), on UO22+ adsorption was investigated. The concentration of Cl− was adjusted by adding NaCl to the U solution. When there was no Cl− in solution, the maximum adsorption capacity of UO22+ was 87.2 mg g−1. When the Cl− concentration was 0.01 and 0.05 M, the UO22+ adsorption capacity decreased slightly to 69.4 and 65.8 mg g−1, respectively. When the Cl− concentration ≥ 0.1 M, as the concentration of Cl− ions increase, the adsorption capacity gradually decreased, and the UO22+ adsorption capacity dropped to 22.3 mg g−1 when the Cl− concentration was 1 M, which meant that the effect on the adsorption of UO22+ was weak when the Cl− concentration was less than 0.05 M.
To study the impact of competing ions on UO22+ adsorption, experiments were conducted using simulated seawater. Competing metal ions were chosen according to prior studies, with their concentrations listed in Table S1.† 39–41 As shown in Fig. 4d, the UO22+ adsorption capacity of UiO-66-AO reached 12.3 mg g−1, greatly surpassing that of UiO-66-NH2 at 0.9 mg g−1. This highlights that introducing AO via the PSM method has significantly improved the UO22+ adsorption performance of the original UiO-66-NH2. In addition, the adsorption capacity of UiO-66-AO for UO22+ was higher than that for V (5.5 mg g−1), Fe (3.7 mg g−1), Co (0.1 mg g−1), Ni (0.6 mg g−1), Cu (2.3 mg g−1), Zn (1.2 mg g−1), Pb (0.7 mg g−1), Ca (3.3 mg g−1), and Mg (2.3 mg g−1), this indicates that UiO-66-AO demonstrates outstanding selectivity for U.42 Homogeneous distribution of UO22+ on the surface of UiO-66-AO (U@UiO-66-AO) could be clearly observed after U adsorption in simulated seawater (Fig. S4†).
Building on the excellent UO22+ adsorption performance of UiO-66-AO observed above, further investigations were conducted to evaluate its ability to UES. As depicted in Fig. S5b,† UiO-66-AO was able to reach adsorption equilibrium for UO22+ in natural seawater within 3 days, demonstrating an adsorption capacity of 3.0 mg g−1, exhibiting good adsorption performance and fast adsorption kinetics (Table S2†), which is related to the number of active sites provided by micropores and rapid ion diffusion promoted by mesopores.35 In addition, the morphology of UiO-66 after adsorbing UO22+ with 16 days remained octahedral morphology (Fig. 4e), indicating good stability of its structure in seawater. Elemental mapping (Fig. 4e and S6b†) revealed the distribution of UO22+ on UiO-66-AO, demonstrating its strong ability to UES.
The adsorption kinetics of UO22+ on the UiO-66-AO at pH 6 is showed in Fig. 5a. A significant increase of the adsorption rate of UO22+ was observed within 400 min, and then a high-level of adsorption capacity (286.8 mg g−1) was observed within 1100 min, which exhibited a faster adsorption rate than UiO-66-NH-(AO) reported previously.29 The adsorption kinetics of UO22+ on the UiO-66-AO was tested by the pseudo-first-order and pseudo-second-order kinetic models.31 And the data of the adsorption kinetics are shown in Table 1. UO22+ adsorption on UiO-66-AO is more accurately described by the pseudo-second-order model, showing a high correlation coefficient (R2 = 0.994, Fig. 5b) compared to the pseudo-first-order model (R2 = 0.981, Fig. 5c). This indicates that chemisorption likely serves as the main rate-determining step.43
 | ||
| Fig. 5 (a) Impact of contact time on UO22+ adsorption (c0 = 8 ppm, t = 24 h), (b) the pseudo-second-order and (c) the pseudo-first-order kinetic model linearized plots for UO22+ adsorption, (d) impact of UO22+ concentration on the adsorption (c0 = 0.1–20.0 ppm, t = 24 h), (e and f) fitting results for the Langmuir and Freundlich isothermal adsorption models of UiO-66-AO. | ||
| Concentration | Pseudo-second-order model | Pseudo-first-order model | ||||
|---|---|---|---|---|---|---|
| R2 | qe (mg g−1) | K2 (g·mg−1·min−1) | R2 | qe (mg g−1) | K1 (g·mg−1·min−1) | |
| 8 ppm | 0.994 | 309.6 | 2.44 × 10−5 | 0.981 | 245.0 | 3.37 × 10−3 |
The influence of UO22+ initial concentration on adsorption was assessed (0.1–20.0 ppm). As illustrated in Fig. 5d, U adsorption continuously increased with higher initial concentrations. At equilibrium, the maximum sorption capacity reached 408.0 mg g−1 under the experimental conditions. The adsorption isotherm data were modeled using the Langmuir and Freundlich isotherms (Fig. 5e and f).44 R2 of Langmuir model was 0.981, with a higher R2 value than the Freundlich model (R2 = 0.787, Table 2), the results suggest that adsorption occurs as a monolayer on the adsorbent's surface.39 The estimated maximum adsorption capacity (qmax) was 413.2 mg g−1, which was significantly greater than the qmax (134.1 mg g−1) of UiO-66-NH-(AO). UiO-66-AO demonstrated a faster adsorption rate and greater adsorption capacity than UiO-66-NH-(AO), attributed to its larger specific surface area (the specific surface areas of UiO-66-NH-(AO) was 59.2 m2 g−1, which was much lower than UiO-66-AO), which offered more active sites and enhanced effective contact with UO22+.35
| Absorbent | Langmuir model | Freundlich model | ||||
|---|---|---|---|---|---|---|
| R2 | qmax (mg g−1) | b (L mg−1) | R2 | kF | n | |
| 2 mg | 0.981 | 413.2 | 1.158 | 0.787 | 160.0 | 2.539 |
2.4 Adsorption mechanism of UiO-66-AO
The possible mechanism of UO22+ adsorption was investigated by XPS and NEXAFS. Through the full spectrum of XPS (Fig. 6a), there was a new sign of U 4f (381.1 eV) appeared in U@UiO-66-AO, this indicates that UiO-66-AO effectively achieved chemical adsorption of uranium (Fig. S6a†). The O 1 s spectrum of UiO-66-AO (Fig. 6b) showed three signs, which were –OH (529.6 eV), CO (531.1 eV), and N–O (531.8 eV). After adsorbing UO22+, the peak of –OH shifted towards lower binding energy fields, while the peaks of N–O shifted towards higher binding energy fields. Additionally, the sign intensities of CO and –OH showed a marked increase, while the intensity of the N–O sign decreased. Likely due to UO22+ adsorption by the AO groups in the MOFs. Analysis of the N 1s spectra of UiO-66-AO and U@UiO-66-AO (Fig. 6c) revealed two distinct characteristic peaks in the N 1s spectra of UiO-66-A1O: CN at 399.0 eV and C–N at 400.6 eV. Upon UO22+ adsorption, the CN and C–N peaks shifted to lower binding energies, and their intensities decreased. This change was attributed to the interaction between the AO and the synergistic –NH– groups in the ligands.45,46 Fig. 6d showed the C 1s spectra of UiO-66-AO and U@UiO-66-AO. The C 1s spectrum of UiO-66-AO was deconvoluted into three bands at 283.6, 284.9, and 287.4 eV, corresponding to C–C, C–O/N, and CO, respectively. After UO22+ adsorption, the C–O/N peak shifted to a higher binding energy, and the intensities of CO and C–O/N peaks decreased. This indicates that nitrogen- and oxygen-containing functional groups played a key role in the effective removal of UO22+. The O k-edge NEXAFS of UiO-66-AO and U@UiO-66-AO was analyzed. As shown in Fig. 6e, the peak of UiO-66-AO at 532.8 eV corresponded to the O of sp2 in CO.47 The π* transition after UO22+ adsorption occurred at 400.9 and 402.6 eV, which shifted towards lower photon energy and may be related to the CN and NH2 in the AO (Fig. 6f).48,49 The coordination effect between AO and UO22+ was more fitted with a cooperative chelating model.10,49 The characterization results confirm that the high adsorption capacity of UiO-66-AO for U is primarily due to the efficient capture of UO22+ by the AO introduced through PSM, along with the synergistic coordination effects of the MOFs ligands.
 | ||
| Fig. 6 (a) XPS spectra, (b) O 1s spectra, (c) N 1s spectra, (d) C 1s spectra and (e) O k-edge of NEXAFS and (f) N k-edge of NEXAFS of UiO-66-AO and U@UiO-66-AO. | ||
3. Conclusions
A novel MOFs, UiO-66-AO was synthesized by grafting AO onto UiO-66-NH2 via the PSM method. The octahedral morphology and crystal structure of the original MOFs UiO-66-NH2 were maintained during the PSM process. But the surface environment of MOFs changed, AO was effectively grafted onto the surface of MOFs, which was conducive to effective contact with UO22+, resulting in UiO-66-AO exhibiting strong adsorption capabilities and selective uptake of UO22+. Adsorption equilibrium was reached within 1100 minutes. The maximum adsorption capacity (qmax) attained 413.2 mg g−1. In simulated seawater, the adsorption capacity was 12.3 mg g−1, much higher than that of the original UiO-66-NH2 (0.9 mg g−1), demonstrating strong affinity and selectivity for UO22+. UiO-66-AO could reach adsorption equilibrium within 3 days in natural seawater, with a fast adsorption rate and an adsorption capacity of 3.0 mg g−1. In addition, XPS and NEXAFS confirmed the chelation between AO and uranium, as well as the synergistic effect of ligands, indicating that the simple PSM method successfully improved the adsorption performance of original MOFs UiO-66-NH2. This study provides a feasible strategy for MOFs modification, and demonstrates the potential of MOFs-based materials for UES.4. Materials and method
4.1 Reagents and materials
All chemicals were purchased from Sinopharm chemical reagent company. (Shanghai, China) and used without purification.4.2 Preparation of UiO-66-AO
UiO-66-NH2 was synthesized according to the previous literature with minor modifications.28 UiO-66-CN was obtained by adding 0.60 g UiO-66-NH2 into the mixed solution of 15 mL AN and 15 mL DMF. The mixture was stirred and heated at 120 °C for 12 h. Then solids were centrifuged and washed with DMF and ethanol. A certain amount of UiO-66-CN (typically 0.20 g) was added into the mixture of 1.29 g NH2OH·HCl, 1.88 g triethylamine and 50 mL ethanol (which was dissolved and mixed in advance). Refluxing for 24 h, the solids were centrifuged and washed by ethanol, and the UiO-66-AO was obtained.4.3 Batch adsorption experiments
Typically, the ratio of adsorbent mass to solution volume (m V−1) was 0.02 mg mL−1, an amount of the adsorbent (2.0 mg) was added to 100 mL UO22+ solution of given concentration and pH value in a plastic bottle. The pH was adjusted by 0.1 M or 1 M HCl and NaOH solution. After being stirred at 100 rpm and 25 °C for 24 h, the sample was taken and filtered by 0.2 μm nylon membrane filter. Adsorption in simulated seawater was carried out according to previous study.10,26,29 Seawater was collected from Qingdao, China and adsorbed under laboratory conditions. UiO-66-AO (37 mg) was stirred in 18.66 L seawater at room temperature for 16 days.Data availability
All data are contained within the manuscript and are available upon request.Author contributions
Data curation and writing – original draft: L. M.; formal analysis: L. M., C. H.; visualization: C. H., P. S.; investigation: C. H., Y. W.; validation: H. M., H. S.; supervision: H. M., Q. L.; writing – review & editing: Q. L., Y. L.; funding acquisition: L. M., Y. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
This work was supported by the Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (No. GZC20232857), the Developing Youth Projects, Shanghai Institute of Applied Physics Chinese Academy of Sciences (SINAP-YXJH-202305), and the Science and Technology Innovation Program, Shanghai Institute of Applied Physics Chinese Academy of Sciences (SINAP-KJCX-202301).References
- W. Lin, Presented in Part at Conference AMER CHEMICAL SOC1155, 16TH ST, Washington, 2017, vol. 3 Search PubMed.
- M. Mirza, R. Abdulaziz, W. C. Maskell, S. Wilcock, A. H. Jones, S. Woodall, A. Jackson, P. R. Shearing and D. J. L. Brett, Energy Environ. Sci., 2023, 16, 952–982 RSC.
- H. Lindner and E. Schneider, Energy Econ., 2015, 49, 9–22 CrossRef.
- W. Cui, F. Li, R. Xu, C. Zhang, X. Chen, R. Yan, R. Liang and J. Qiu, Angew. Chem., Int. Ed., 2020, 59, 17684–17690 CrossRef CAS PubMed.
- J. Kim, C. Tsouris, Y. Oyola, C. J. Janke, R. T. Mayes, S. Dai, G. Gill, L. J. Kuo, J. Wood, K. Y. Choe, E. Schneider and H. Lindner, Ind. Eng. Chem. Res., 2014, 53, 6076–6083 CrossRef CAS.
- H. Liu, T. Fu and Y. Mao, ACS Omega, 2022, 7, 14430–14456 CrossRef CAS PubMed.
- Y. Yuan, S. Feng, L. Feng, Q. Yu, T. Liu and N. Wang, Angew. Chem., Int. Ed., 2020, 59, 4262–4268 CrossRef CAS PubMed.
- J. Xiong, Y. Fan and F. Luo, Dalton Trans., 2020, 49, 12536–12546 RSC.
- Y. Zeng, Y. Ni, S. Liu, J. Xu, A. Zhang, Y. Song, L. Yang, A. Pu, X. Li, L. Lv, L. Yuanli and F. Chi, Microporous Mesoporous Mater., 2022, 343, 112123–112130 CrossRef CAS.
- C. Huang, L. Xu, X. Xu, L. Ma, H. Bao, J. Liao, J. Wang, J. Han, G. Xu, D. Huang, B. Ye, H. Zhang, M. Wu, X. Zhao and H. Ma, Chem. Eng. J., 2022, 443, 136312–136321 CrossRef CAS.
- Y. Yuan, T. Liu, J. Xiao, Q. Yu, L. Feng, B. Niu, S. Feng, J. Zhang and N. Wang, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- C. X. Yu, W. Jiang, M. Lei, M. R. Yao, X. Q. Sun, Y. Wang, W. Liu and L. L. Liu, Small, 2023, 2308910–2308921 Search PubMed.
- P. Sarawade, H. Tan and V. Polshettiwar, ACS Sustain. Chem. Eng., 2013, 1, 66–74 CrossRef CAS.
- Y. D. More, S. Mollick, S. Saurabh, S. Fajal, M. Tricarico, S. Dutta, M. M. Shirolkar, W. Mandal, J. Tan and S. K. Ghosh, Small, 2024, 20, 2302014–2302023 CrossRef CAS PubMed.
- S. Mollick, S. Saurabh, Y. D. More, S. Fajal, M. M. Shirolkar, W. Mandal and S. K. Ghosh, Energy Environ. Sci., 2022, 15, 3462–3470 RSC.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS PubMed.
- O. Karagiaridi, W. Bury, J. E. Mondloch, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 4530–4540 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- A. Tyagi, H. Sharma, A. K. Yadav, P. Singhal, G. Karmakar, D. Bhattacharyya and S. K. Jha, Ind. Eng. Chem. Res., 2024, 63, 10892–10902 CrossRef CAS.
- S. Mandal, S. Natarajan and P. Mani, Adv. Funct. Mater., 2021, 31, 2006291–2006313 CrossRef CAS.
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M. H. Zeng, Coord. Chem. Rev., 2019, 378, 500–512 CrossRef CAS.
- M. Zhang, T. Yang, Z. Wang, X. F. Ma and Y. Zhang, Chem. Sci., 2017, 8, 5356–5361 RSC.
- S. Park, H. Song, N. Ko, C. Kim, K. Kim and E. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 33785–33789 CrossRef CAS PubMed.
- Z. Wang, K. K. Tanabe and S. M. Cohen, Chem.–Eur. J., 2010, 16, 212–217 CrossRef CAS PubMed.
- C. Bi, C. Zhang, C. Wang, L. Zhu, R. Zhu, L. Liu, Y. Wang, F. Ma and H. Dong, Environ. Sci. Pollut. Res., 2024, 31, 16554–16570 CrossRef CAS PubMed.
- X. Xu, H. Zhang, J. AO, L. Xu, X. Liu, X. Guo, J. Li, L. Zhang, Q. Li, X. Zhao, B. Ye, D. Wang, F. Shen and H. Ma, Energy Environ. Sci., 2019, 12, 1979–1988 RSC.
- Z. Zhao, R. Lei, Y. Zhang, T. Cai and B. Han, J. Mol. Liq., 2022, 367, 120514–120522 CrossRef CAS.
- X. Shi, X. Lian, D. Yang, X. Hu, J. Zhang and X. H. Bu, Dalton Trans., 2021, 50, 17953–17959 RSC.
- L. Ma, J. Gao, C. Huang, X. Xu, L. Xu, R. Ding, H. Bao, Z. Wang, G. Xu, Q. Li, P. Deng and H. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 57831–57840 CrossRef CAS PubMed.
- T. L. Ren, M. M. Wang, T. Z. Xiong, X. N. Zhang, Y. Peng, H. R. Wen and S. J. Liu, Cryst. Growth Des., 2023, 24, 696–702 CrossRef.
- L. Chen, Z. Bai, L. Zhu, L. Zhang, Y. Cai, Y. Li, W. Liu, Y. Wang, L. Chen, J. Diwu, J. Wang, Z. Chai and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32446–32451 CrossRef CAS PubMed.
- J. Li, Y. Liu, X. Wang, G. Zhao, Y. Ai, B. Han, T. Wen, T. Hayat, A. Alsaedi and X. Wang, Chem. Eng. J., 2017, 330, 1012–1021 CrossRef CAS.
- J. Li, K. Tuo, C. Fan, G. Liu, S. Pu and Z. Li, Small, 2024, 20, 2306545–2306555 CrossRef CAS PubMed.
- W. Liu, X. Dai, Z. Bai, Y. Wang, Z. Yang, L. Zhang, L. Xu, L. Chen, Y. Li, D. Gui, J. Diwu, J. Wang, R. Zhou, Z. Chai and S. Wang, Environ. Sci. Technol., 2017, 51, 3911–3921 CrossRef CAS PubMed.
- J. Kang, J. Min, S. I. Kim, S. W. Kim and J. H. Jang, Mater. Today Energy, 2020, 18, 100502–100510 CrossRef CAS.
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed.
- F. Endrizzi, C. J. Leggett and L. Rao, Ind. Eng. Chem. Res., 2016, 55, 4249–4256 CrossRef CAS.
- C. J. Leggett, F. Endrizzi and L. Rao, Ind. Eng. Chem. Res., 2016, 55, 4257–4263 CrossRef CAS.
- L. Ma, C. Huang, Y. Yao, M. Fu, F. Han, Q. Li, M. Wu, H. Zhang, L. Xu and H. Ma, Sep. Purif. Technol., 2023, 314, 123526–123538 CrossRef CAS.
- C. Huang, M. Fu, L. Ma, C. Mao, Y. Yao, X. Xu, L. Xu, J. Han, X. Xue, G. Xu, M. Wu, H. Shao and H. Ma, Chem. Eng. J., 2023, 474, 145718–145729 CrossRef CAS.
- P. Sun, L. Ma, J. Liao, J. Wang, C. Huang, L. Xu, M. Fu, J. Wang, Q. Zhou and H. Ma, Sep. Purif. Technol., 2024, 337, 126419–126428 CrossRef CAS.
- S. C. Liu, M. B. Wu, H. Ye, L. Liu, L. L. Ma and J. Yao, Chem. Eng. J., 2021, 426, 131378–131388 CrossRef CAS.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Ǵandara, H. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 127, 11314–11320 CrossRef.
- R. M. Abdelhameed, M. Taha, H. Abdel-Gawad and H. E. Emam, J. Mol. Struct., 2022, 1250, 131914–131924 CrossRef CAS.
- J. Górka, R. T. Mayes, L. Baggetto, G. M. Veith and S. Dai, J. Mater. Chem. A, 2013, 1, 3016–3026 RSC.
- Y. Wang, Z. Gu, J. Yang, J. Liao, Y. Yang, N. Liu and J. Tang, Appl. Surf. Sci., 2014, 320, 10–20 CrossRef CAS.
- R. G. Ryan, A. Stacey, K. M. O'Donnell, T. Ohshima, B. C. Johnson, L. C. L. Hollenberg, P. Mulvaney and D. A. Simpson, ACS Appl. Mater. Interfaces, 2018, 10, 13143–13149 CrossRef CAS PubMed.
- J. Melke, B. Peter, A. Habereder, J. Ziegler, C. Fasel, A. Nefedov, H. Sezen, C. Woll, H. Ehrenberg and C. Roth, ACS Appl. Mater. Interfaces, 2016, 8, 82–90 CrossRef CAS PubMed.
- X. Xu, C. Huang, Y. Wang, X. Chen, Z. Wang, J. Han, M. Wu, G. Liu, L. Li, L. Xu and H. Ma, Chem. Eng. J., 2022, 430, 133159–133170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01079a |
| This journal is © The Royal Society of Chemistry 2025 |