
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzimidazole chemistry in oncology: recent developments in synthesis, activity, and SAR analysis
Basant Farag
 a,
Magdi E. A. Zaki
*b,
Doaa A. Elsayed
a and
Sobhi M. Gomha
*c
a,
Magdi E. A. Zaki
*b,
Doaa A. Elsayed
a and
Sobhi M. Gomha
*c
aDepartment of Chemistry, Faculty of Science, Zagazig University, Zagazig, 44519, Egypt. E-mail: basantfarag@zu.edu.eg; doaaatef641995@gmail.com
bDepartment of Chemistry, Faculty of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia. E-mail: mezaki@imamu.edu.sa
cDepartment of Chemistry, Faculty of Science, Islamic University of Madinah, Madinah, 42351, Saudi Arabia. E-mail: smgomha@iu.edu.sa
First published on 3rd June 2025
Abstract
A six-membered benzene ring is fused with a five-membered imidazole ring at positions four and five, generating benzimidazole, the benzo derivative of imidazole and a bicyclic aromatic chemical compound. Benzimidazole is a significant pharmacophore in a variety of physiologically active heterocyclic compounds due to its distinctive characteristics and structural framework. Because benzimidazole is both aromatic and heterocyclic, it interacts with a range of biological targets via metal ion interactions, π–π stacking, and hydrogen bonding. Its broad range of medicinal chemistry applications, such as anti-inflammatory, antiviral, antifungal, and anticancer therapies, is based on these interactions. Its significance in the development of potentially novel therapeutic pharmaceuticals is highlighted by the fact that its structural flexibility permits the synthesis of derivatives with targeted bioactivity. Derivatives of benzimidazole have garnered significant research interest as potential anticancer medications. These heterocyclic compounds exhibit a wide range of biological activities, such as DNA interaction, enzyme inhibition, and modulation of cellular pathways crucial to cancer development. Thus, to optimize their therapeutic potential, recent studies have focused on evaluating the structure–activity relationships (SAR) of benzimidazole derivatives. The main topics of this review are the current developments in the synthesis, anticancer activity, and SAR studies of benzimidazole derivatives, which will shed light on the increasing role they play in cancer therapies.
 Basant Farag | Dr Basant Farag is a Lecturer of Organic Chemistry in the Faculty of Science at Zagazig University. Most of her publications focus on the synthesis and biological evaluation of various heterocyclic ring systems, utilizing different synthetic routes for the preparation of organic compounds with wide industrial and pharmacological applications. |
 Magdi E. A. Zaki | Prof. Magdi E. A. Zaki is a Professor of Organic Chemistry in the Department of Chemistry at Imam Mohammad Ibn Saud Islamic University, KSA, where he serves as the Scientific Leader of the Advanced Organic Synthesis Group. Prof. Zaki has also been invited as a visiting researcher (1991–2013) to several European institutions in Poland, Germany, and Portugal. Prof. Zaki's research focuses on the design and synthesis of molecules of fundamental importance. He also explores ring transformations, transition metal chelates as potent new anticancer agents, computer-aided drug design, QSAR, cheminformatics, artificial intelligence, molecular dynamics simulations, nanomaterials, environmental research, and organo-catalysis. He has published more than 320 SCI-indexed papers and holds patents in Germany and South Africa. |
 Doaa A. Elsayed | Dr Doaa A. Elsayed is a Lecturer of Organic Chemistry in the Faculty of Science at Zagazig University. Most of her publications focus on the synthesis and biological evaluation of heterocyclic ring systems, as well as the utilization and characterization of nanomaterials for various applications. |
 Sobhi M. Gomha | Prof. Sobhi M. Gomha is a leading expert in Organic Chemistry, renowned for his work on the synthesis of bioactive heterocyclic compounds. He earned his MSc (2002) and PhD (2006) in Organic Synthesis from Cairo University, Egypt, and was promoted to Associate Professor in 2011 and Full Professor in 2016. From May 2019 to September 2024, he served as Head of the Chemistry Department at the Faculty of Science, Islamic University of Madinah, Saudi Arabia. Between 2000 and March 2025, Prof. Gomha published 296 original research articles and 13 books, focusing on heterocyclic synthesis for medicinal, energy, environmental, and industrial applications. His research integrates green chemistry techniques such as microwave irradiation, ultrasound, mechanochemistry, and the use of ionic liquids. As of March 2025, his work has been cited approximately 8150 times, with an h-index of 52. He has supervised 18 MSc and 9 PhD theses, served on 43 journal editorial boards, reviewed for 176 journals, and contributed to faculty promotions and thesis evaluations. Prof. Gomha has received several national and international honors. He was awarded the Best Paper Prize from the Egyptian Heterocyclic Chemistry Society in 2015, and his work was recognized among the most downloaded by the Journal of Heterocyclic Chemistry in 2018–2019. From 2019 to 2024, he was listed among the World's Top 2% Scientists by Stanford University. The AD Scientific Index ranked him 2nd in Organic Chemistry at Cairo University (2020–2023) and 1st in 2024, as well as 1st at the Islamic University of Madinah. ScholarGPS ranked him 3rd globally in heterocyclic compounds and 112th in chemical synthesis. Cairo University honored him annually for research excellence (2008–2024) and named him a Top 10 Publisher (2015–2024). In 2024, he received the Best Researcher in Chemotherapy Award from Pencis for his contributions to sustainable chemotherapy research. |
1 Introduction
According to WHO, deaths due to cancer are predicted to reach 13 million worldwide by 2030.1,2 Most malignancies are characterized by the uncontrolled growth of undifferentiated cells. Current estimates indicate that one in every five people will develop cancer by the age of 75.3 Cancer primarily results from the deregulation of key enzymes and proteins that regulate cell division and proliferation.4,5 However, despite significant advancements in cancer diagnosis and treatment options, many patients do not respond well to therapy, and others relapse after an initial encouraging response. Although chemotherapy is still a crucial component of cancer treatment, the emergence of drug resistance significantly reduces its effectiveness.6 To combat drug resistance, higher dosages of chemotherapeutic drugs are sometimes necessary, which exacerbates drug-induced toxicities.7,8 Therefore, there is a critical need to create and develop novel cancer therapies with potent activity while maintaining a high therapeutic index.9Recently, O- and S-based heterocycles have been attracting increasing attention in the discovery of innovative anticancer drugs, following the extensive investigation of nitrogen-based heterocycles as anticancer agents.10 While heterocyclic compounds with a sulfur atom account for the bulk of FDA-approved drugs, heterocyclic compounds with a nitrogen atom are regarded as the most common type of chemical material utilized in medicinal chemistry.11,12 Benzimidazole and its derivatives are a family of bioactive compounds with important uses in the pharmaceutical sector.13 Imidazole, also known as imidazoline, is a heterocyclic molecule14 classified as an azapyrrole, with two nitrogen atoms separated by a single carbon atom. Historically, this molecule was known as glyoxalin when it was initially synthesized in 1858 by German scientist Heinrich Debus using glyoxal, formaldehyde, and ammonia.15,16 A six-membered benzene ring is fused with a five-membered imidazole ring at positions four and five to form benzimidazole, the benzo-fused imidazole derivative and a bicyclic aromatic molecule. These substances have antiviral, antifungal, antiparasitic, analgesic, anticancer, and antihistaminic properties, among other medicinal applications. Moreover, benzimidazole derivatives have demonstrated potential in the management of numerous conditions associated with neurology, endocrinology, ophthalmology, and cardiovascular disease.17
The benzimidazole nucleus has emerged as a key pharmacophore in cancer research due to its broad cytotoxic potential, adaptive tumor inhibitory mechanisms, and facile synthesis methods for producing a wide range of derivatives. Many bioactive chemicals and anticancer medicines have a benzimidazole motif.18 The benzimidazole scaffold is crucial in the creation of anticancer medications such as bendamustine, carbendazim, nocodazole, and veliparp. It functions in a number of ways.19 Benzimidazoles, a structurally distinct class of Topo I poisons that function as DNA minor groove binders, including Hoechst 33258 and Hoechst 33342.20–23
1.1 Techniques that yield benzimidazoles
The synthesis and chemistry of benzimidazoles have been well reviewed in the literature. In general, the majority of key ingredients mentioned previously may be utilized to develop benzimidazoles, as follows:(1) Phenylenediamines
(2) Acidic compounds
(3) o-(N-arylamino)arylamine
(4) o-Nitro arylamines or o-dinitro arenes
(5) o-Substituted-N-substituted
(6) Imines
(7) Amidines
(8) Heterocyclic scaffolds
(9) Four components
 | ||
| Scheme 1 Primary method for the synthesis of benzimidazole nucleus 2 and its derivative 3. | ||
As shown in Scheme 2, several benzimidazole–chalcone hybrids 10a and b have been produced.25 Refluxing o-phenylenediamine (1) with glycolic acid (4) in HCl produced (1H-benzo[d]imidazol-2-yl)methanol (5). 5 and suitable benzyl bromides 6 were used to produce the substituted (1-benzyl-1H-benzo[d]imidazol-2-yl)methanol (7) in the presence of K2CO3. Subsequently, the corresponding primary alcohols 7 were oxidized using Dess–Martin reagent to provide substituted-1-benzyl-1H-benzo[d]imidazole-2-carbaldehydes 8. Using the Claisen–Schmidt reaction with appropriate acetophenones 9, target compounds 10a and b were subsequently generated from 8.
 | ||
| Scheme 2 Synthetic route for benzimidazole–chalcone hybrids 10a and b. | ||
Zhou et al.25 established a novel class of benzimidazole–chalcone hybrids or BCHs. These BCHs demonstrated anti-proliferative effects in tumor cell lines and significant inhibitory effects in the Topo II-mediated DNA relaxation assay. One of the main enzymes involved in DNA replication, recombination, and repair is type II topoisomerase (Topo II).26,27 Condensation and cyclization of o-phenylenediamine and glycolic acid produced compound 11 and the synthesis pathway for substances 16. The oxidation of the C2-hydroxymethyl of 11 with manganese dioxide produced aldehyde product 12. Intermediate 13 was prepared via the Wittig–Horner reaction of aldehydic compound 12 with methyl diethyl phosphonoacetate. Key intermediate 14 was developed by alkylation of 13 with 3-bromopropionic acid. The appropriate amides were produced by reacting carboxylic acid 14 with amines, and then the amides were treated with lithium hydroxide to produce 16. With an IC50 value of 0.64 μM, compound 16 with a 4-Br substituent had the highest Pin1 inhibitory activity, indicating that the bromine atom is preferred, most likely to react with the residues of amino acid to establish an H-halogen link in this way (Scheme 3).
 | ||
| Scheme 3 Synthesis of a new class of BCHs 15 and 16. | ||
Scheme 4 presents the synthesis of 23. Potassium permanganate oxidizes 17 to yield 1H-benzo[d]imidazole-2-carboxylic acid (18). The carboxylic acid group of 18 was changed to a carbamoyl group by creating the acyl chloride first, and then reacting it with ammonium hydroxide to produce compound 19. Using Lawesson's reagent, the carbonyl in chemical 19 was thiolated to produce compound 20. Compound 20 was condensed and cyclized with ethyl 3-bromo-2-oxopropanoate to produce an intermediate, 2-(1H-benzo[d]imidazol-2-yl)thiazole 21. The use of 3-bromopropionic acid to alkylate 21 produced the desired product 22. Carboxylic acid 22 was combined with amines to form the corresponding amides, which were then reacted with lithium hydroxide to produce the final product 23 (Scheme 4). Wang et al.28 produced derivatives of 1H-benzimidazole and assessed their anticancer potential against the human prostate cancer cell line PC-3. With IC50 values of 0.64 μM and 0.37 μM, respectively, compounds 16 and 23 showed the greatest inhibitory effects. According to the SAR investigations, compounds 23 with thiazole rings as linkers exhibited greater inhibition than 16 with a double bond between the 1H-benzimidazole ring and the terminal carboxyl group.
 | ||
| Scheme 4 Synthesis of derivatives of benzimidazole thiazole 22 and 23. | ||
An effective method for creating novel benzimidazoles 25a and b and 26a–c was accomplished, which began with 2-mercaptomethyl-5-nitro-1H-benzimidazole (24)29 (Scheme 5). El-Gohary et al.30 produced new 1H-benzimidazole compounds and tested them against human cancer cell lines, including HepG-2 (liver), HCT-116 (colon), and MCF-7 (breast), to determine their anticancer potential. The most promising analogues were determined to be compounds 25b, 26b, and 26c. According to the SAR evaluations, compound 25b exhibited a more powerful and broad-spectrum anticancer action when 5-nitrobenzimidazol-2-yl was substituted for benzimidazol-2-yl. Furthermore, the improved lipophilicity of the 4-substituent appeared to enhance its effectiveness against all the examined cell lines. Compound 25b, the 5-nitrobenzimidazol-2-yl homologue, had strong and comprehensive anticancer action. For compounds 26b and c to have anticancer action, the substituent at the 4-position of the phenacyl moiety must be lipophilic. The compounds appeared to be more active against the three examined cell lines as the lipophilicity of the 4-substituent was increased, which the activity following the order of 4-bromophenacyl analog 26c > 4-chlorophenacyl analog 26b.
 | ||
| Scheme 5 Synthesis of novel benzimidazoles 25a and b and 26a–c. | ||
The present study used 2-chloromethyl-1H-benzimidazole 27 (ref. 31) as the starting materials to produce novel benzimidazoles 28 employing a straightforward, effective, and repeatable approach with a simple work-up procedure (Scheme 6). In the following way, the appropriate 27 reacts with 1-(4-fluorophenyl)piperazine in DMF when triethylamine is present to produce 2-(aryl amino)methyl-benzimidazole 28. El-Gohary et al.32 developed and evaluated a variety of 1H-benzimidazole derivatives for their anticancer characteristics. The MTT assay was used to screen the derivatives against the HepG-2 (liver), HCT-116 (colon), and MCF-7 (breast) cancer cell lines. Compound 28 exhibited the strongest activity against all the cell lines, in accordance with the results. The cytotoxic tests of the compounds revealed that they were less active than the reference medication, 5-fluorouracil. According to SAR investigations, the anticancer activity increased with the length of the contact between the aromatic moiety and 5-nitro-1H-benzimidazole.32
 | ||
| Scheme 6 Synthesis of target 2-(aryl amino)methyl-benzimidazole 28. | ||
The proposed mechanistic route for the formation of benzimidazoles by reacting a derivative of o-phenylene diamine with an organic acid has already been studied.24,33 Furthermore, the role of hydrochloric acid in the reaction has also been investigated.24,34 The catalytic action of hydrochloric acid is explained by the protonation of oxygen, which activates the carboxyl group. The reaction intermediate is an addition product, which is produced when the carbonyl group of the protonated acid is attacked by the shared electron pair of a nitrogen. Nevertheless, the researchers concluded that the generation of the monoacyl derivative was a necessary step in the benzimidazole ring building process.24,34 It has been reported that heating aromatic carboxylic acids with o-phenylene diamine in a sealed tube at 180–190 °C produces good yields of 2-arylbenzimidazoles (29, R = Ar). An improved method for producing 2-arylbenzimidazoles 29 from aromatic carboxylic acid uses polyphosphate ester (PPE) or polyphosphoric acid (PPA) as a dehydrator. As an alternative, phosphorus pentoxide has also been documented to function as a dehydrator in the process of producing a derivative of 2-arylbenzimidazole (Scheme 7).34
 | ||
| Scheme 7 Synthetic route for the formation of 2-arylbenzimidazole 29. | ||
Scheme 8 shows the preparation of certain N′-(substituted-benzylidene)-4-(5-methyl-1H-benzimidazol-2-yl)benzohydrazide derivatives 32a and b for the analysis. Three steps were used to get the targeted synthetic substances. Firstly, methyl-4-formylbenzoate and 5-methyl-1,2 phenylenediamine were mixed equimolarly in DMF with Na2S2O5 to yield 30. Compound 31 was produced by refluxing a mixture of ethanolic solution of hydrazine hydrate and 30, which occurred in approximately seven hours. The desired compounds 32a and b were obtained via condensation of hydrazide with different substituted benzaldehydes. Compounds 32a and b were discovered to exhibit significant effectiveness against a number of cancer cell lines, preventing their proliferation by 50–84%.35 SAR studies revealed that the antiproliferative action of compound 32b was enhanced by the presence of tri-methoxy as an electron-donating group on hydrazone, as opposed to the electron-drawing di-chloro substitution in 32a.
 | ||
| Scheme 8 Preparation of certain N′-(substituted-benzylidene)-4-(5-methyl-1H-benzimidazol-2-yl)benzohydrazide derivatives 32a and b. | ||
Scheme 9 shows that commercially produced 4-nitrobenzene-1,2-diamine (33) was employed as the starting material for the synthesis of benzimidazole derivatives 37. In polyphosphoric acid (PPA), 33 first interacted with benzoic acid to produce 34, which then combined with (3-bromo-propyl)-benzene to produce intermediate 35 in the presence of K2CO3. Amino intermediate 36 was produced by reducing intermediate 35 with Pd/C in THF. Essential compounds 37 were obtained by treating amine 36 with suitable acyl chlorides or sulfonyl chlorides and triethylamine in dichloromethane. After various screening rounds, 37j was shown to be an effective G9a antagonist (IC50 = 1.32 μM), which caused the MCF-7 cancer cell line to undergo autophagy (IC50 = 5.73 μM), where increased concentrations caused the MCF-7 cells to undergo apoptosis.36 The antiproliferative action was increased by the addition of a moiety containing 1H-benzimidazole and a potent hydrophobic substituent, phenylpropyl (found in compound 37j), to the N atom of 1H-benzimidazole. Stable binding with G9a was improved by the presence of Br in 37j.
 | ||
| Scheme 9 Synthesis of benzimidazole derivatives 37. | ||
Scheme 10 describes the approach for the synthesis of benzimidazole-thiazolidinediones 46a–c. The Knoevenagel condensation reaction was used to produce the final products 46a–c in a convergent manner. In the presence of sodium metabisulfite, 3,4-diaminobenzoic acid (38) was natively altered to its methyl ester 39, which subsequently interacted with various substituted benzaldehydes 40 to yield 1H-benzo[d]imidazole-5-carboxylates 41. Lithium aluminum hydride was used to decrease the ester functionalities in 41, resulting in alcohols 42, which were subsequently oxidized to their respective aldehydes 43. Compounds 46a–c were created by employing the Knoevenagel reaction to react different thiazolidinediones with 2-phenyl-1H-benzimidazole-5-carbaldehydes 43 in dry toluene containing piperidine. Both conventional and microwave-assisted synthesis were used to carry out the Knoevenagel condensation reaction for the synthesis of compounds 46a–c. Compounds 46a–c exhibited satisfactory cytotoxicity to prostate, cervical, bone, and lung cell lines (IC50 = 0.096–0.63 μM).37 According to SAR evaluations, with the exception of a few that demonstrated selectivity against A549 cells, compounds with IC50 values less than 1.0 μM were generated when heterocyclic rings such as morpholine, pyrrolidine, and piperidine were present via an oxo-ethyl linkage. On the HeLa, A549, and HT1080 cell lines, more effective compounds were generated at 1.0 μM when the oxo-ethyl group was substituted out for a benzyl substituent. On almost all the screened cancer cell lines, derivatives such as 46b that had 3,4,5-trimethoxybenzyl substitution at the tail produced higher active molecules at concentrations below 1.0 μM. By contrast, derivative 46a, which included a 3-methyl benzyl moiety on the thiazolidinedione tail, was more effective against all cancer cell lines at 1.78 μM. The most powerful derivative, 46c, has benzimidazole replaced with 4-isobutoxy-3-methoxy.
 | ||
| Scheme 10 Approach for the synthesis of benzimidazole-thiazolidinediones 46a–c. | ||
Scheme 11 displays the procedure used to synthesize compounds 54a and b. Using sodium bisulphite in DMF, 4-substituted benzaldehyde 48 reacted with 3,4-diaminobenzoic acid (38) to produce 2-(4-substituted-phenyl)-1H-benzo[d]imidazole-6-carboxylic acid (49) derivatives in the first step. A simple esterification procedure transformed molecule 49 into methyl ester 50, which was then reacted with hydrated-hydrazine to yield 2-(4-substituted-phenyl-6-carbohydrazide)-1H-benzo[d]imidazole (51). Hydrazide derivative 51 was converted to 2-(4-substituted-phenyl)-6-(5-mercapto-1,3,4-oxadiazol-2-yl)-1H-benzo[d]imidazole derivative (52) by reacting with carbon disulfide in boiling basic medium of ethanol and NaOH. Compound 52 and acetylated piperazine derivative 53 reacted in acetone at the last reaction step to yield 2-((5-(2-(4-substituted-phenyl)-1H-benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)-1-(4-substituted-piperazin-1-yl) derivatives of ethane-1-on (54a and b). Çevik et al.38 produced a variety of 1H-benzimidazole-oxadiazole compounds and tested them against HeLa, MCF-7, A549, HepG-2, and C6 human cancer cell lines in vitro to determine their anticancer properties. By inhibiting topoisomerase I, compounds 54a (IC50 = 0.224 ± 0.011 μM) and 54b (IC50 = 0.205 ± 0.010 μM) demonstrated the strongest antiproliferative action against the HeLa cancer cell line when doxorubicin (14.280 mM) was used as the standard medication. However, the majority of the derivatives demonstrated effective antiproliferative activity. The above-mentioned MTT experiment results supported our hypothesis that compounds 54a and b would be more potent than Hoechst 33342 (0.306 mM) due to their potential for effective activity and reduced toxicity.
 | ||
| Scheme 11 Synthesis of compounds 54a and b. | ||
Several aryl aldehyde sodium bisulfates adduct 56 were reacted with the benzoic acid derivative to produce the initial document 2-substituted-benzimidzole-5-carboxylic acids 57. Then, benzimidazole derivatives 57 were converted into the ester congeners 58 by a reaction with ethanol in the presence of sulphuric acid. Hydrazide derivatives 59 were generated when 58 reacted with hydrazine hydrate. Lastly, the target hybrids 60 were generated by allowing hydrazides 59 and acid anhydrides to react in acetic acid (Scheme 12).39 Series 60 was produced by adding a carboxylic group to the dioxoisoindoline moiety at position 5, which increased the potency of 60f (% inhibition = 53.54%) and 60g (% inhibition = 60.36%).
 | ||
| Scheme 12 Synthesis of N-(1,3-dioxo-isoindol-2-yl)-2-phenyl-1H-benzo[d]imidazole derivatives 60 and 61. | ||
Derivatives of benzimidazoles were synthesized and reported by Huang et al. Salicylic acid (62) was first esterified by diazomethane to produce methyl salicylate 63, which allowed the production of the desired derivatives. After being treated with benzyl bromide and potassium carbonate, the hydroxyl group of 63 was protected with benzyl ether to yield 64.40 The saponification reaction then changed component 64 into carboxylic acid 65. After treating compound 65 with dicyclohexylcarbodiimide, amide 67 was produced, which was further transformed into benzimidazole 68 through cyclization–dehydration by refluxing in acetic acid.41 Lastly, 5 M NaOH was used to saponify compound 68, yielding compound 69 (Scheme 13). Benzoimidazole-4-metylacetate, 68 (A549, IC50 70 μM), was found to be more effective than benzoimidazole-4-carboxylic acid, 69 (A549, IC50 87 μM) in the anticancer screening tests.42,43
 | ||
| Scheme 13 Synthesis of benzimidazole analogues 68 and 69. | ||
Scheme 14 shows the main procedure for the preparation of 2-arylbenzoimidazole derivatives. The readily available starting material 33 was reacted with substituted benzoic acid 70 in polyphosphoric acid (PPA) (71) at 150 °C for 10–15 h to yield the 2-arylbenzoimidazole intermediate 72.44 The nitro group was reduced to generate an amino intermediate, which was then reacted with substituted aromatic carboxylic acids 73 at 20 °C for 2–4 h with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) and hydroxy benzotriazole (HOBt) as a condensing agent and triethanolamine45 as a base to obtain 74a–d. According to the CAM experiment, compound 74d demonstrated a great level of angiogenesis inhibition (79% inhibition per 10 nM per egg), demonstrating the highest level of VEGFR-2 kinase inhibitory activity (51.4 nM IC50), and effective anti-proliferative potencies against HepG-2 and HUVEC cells (1.47 μM and 2.57 μM, respectively).46 According to the SAR investigations, the phenylacetamide moiety meta- and para-halide substitutes did not increase the kinase inhibitory activity. Furthermore, the imidazole ring and 4-chloroyphenyl together with the 4-methoxyphenylacetamide moiety in compound 74d improved VEGFR-2 kinase binding.
 | ||
| Scheme 14 Procedure for the preparation of 2-arylbenzoimidazole derivatives 74a–d. | ||
Other benzimidazoles were synthesized similarly. Niementowski started by producing 5-methyl benzimidazole (76, X = 5-methyl) by the condensation of equimolar amounts of 75 and ethyl formate at 225 °C in a sealed tube for three hours to investigate the reaction between 4-methyl-o-phenylenediaminehydrochloride (75) and esters (Scheme 15).24,34
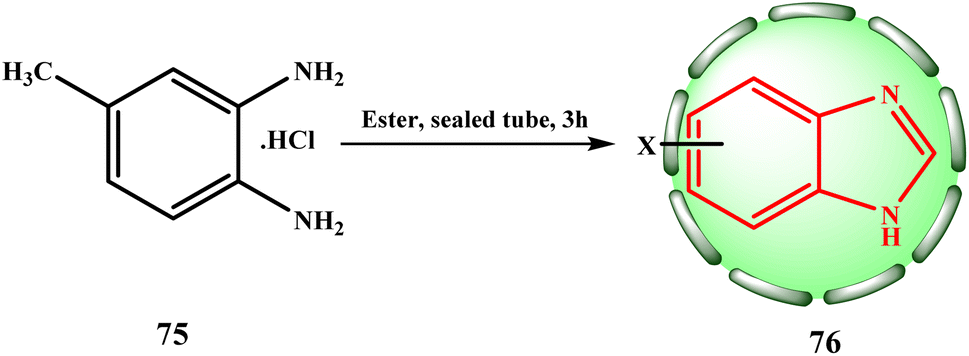 | ||
| Scheme 15 Method for the preparation of 5-methyl benzimidazole (76). | ||
Although a shorter treatment only produced N,N′-diacetyl-phenylenediamine (Scheme 16), the further treatment of 77 with acetic anhydride produced 2-methyl benzimidazole (78, R = CH3). Reinhardt found that the 2-methylbenzimidazole yields from 1% and 2% acetic anhydride were increased using diluted hydrochloric acid.24,34,47
 | ||
| Scheme 16 Synthesis of -2-methylbenzimidazole (78). | ||
With the help of the scientific research by Hoebrecker48 in a six year period, scientists Banerjee49 set the foundation for existing medical chemistry research by discovering a novel heterocyclic molecule (basic structure and synthesis shown in Scheme 17). BZ is the name of this substance.
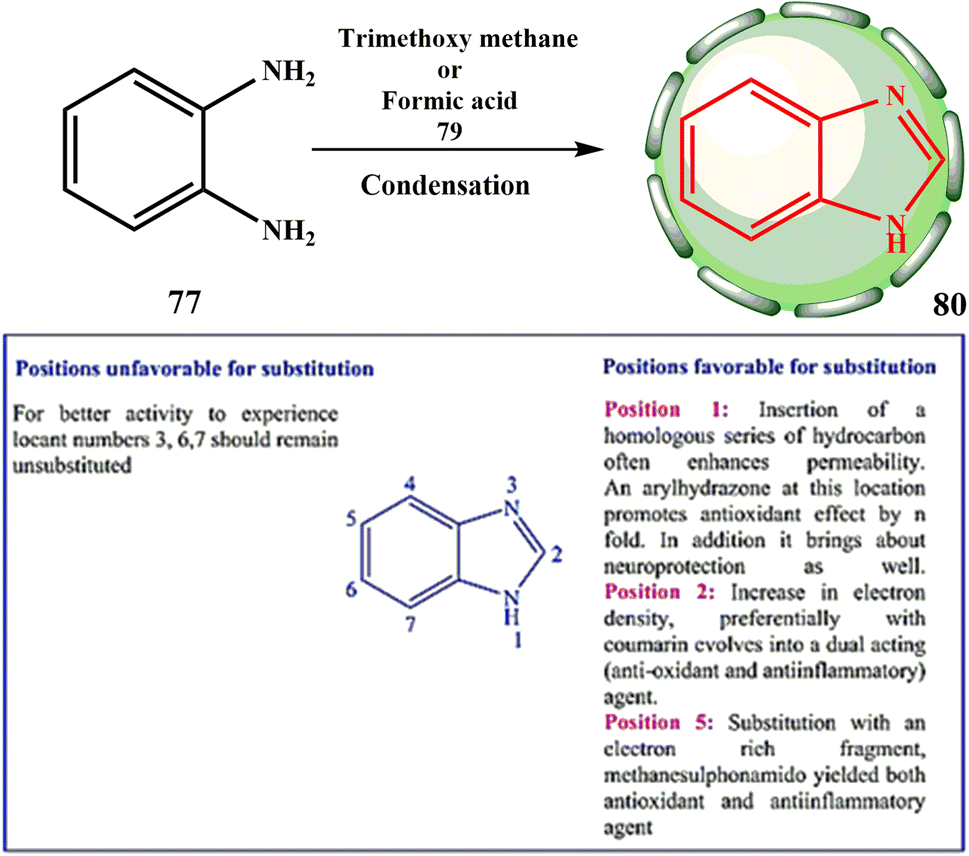 | ||
| Scheme 17 Synthesis of BZ 80. | ||
The synthesis and cytotoxic potential of 1H-benzimidazole scaffolds were examined by Pham et al. N,2,6-trisubstituted 1H-benzimidazole derivatives 83a and b were prepared from benzene-1,2-diamine derivative 77. There are two steps in the synthetic process (Scheme 18). Firstly, using microwave assistance for the heating requirement, benzene-1,2-diamine derivative 77 was condensed with substituted aromatic aldehyde 81 to produce 2,6-disubstituted 1H-benzimidazole derivatives 83a and b. Then, using the non-selective (positive) control involved in paclitaxel (PTX) in the MTT assay, we evaluated the anticancer activity of compounds 83a and b on five cancer cell lines including hepatocellular carcinoma cell line (HepG-2), human breast cancer cell lines (MDA-MB-231 and MCF-7), the aggressive and highly malignant rhabdomyosarcoma cell line (RMS), and colon carcinoma cell line (C-26). Among the synthesized compounds, compounds 83a (3,4-dichloro) and 83b (5-bromo-2-hydroxy) demonstrated the greatest anticancer activity against all the tested cell lines.50
 | ||
| Scheme 18 Synthesis of substituted 1H-benzimidazole derivatives 83a and b. | ||
Given that Na2S2O5 dissolves readily in an EtOH–H2O mixture (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the medium for the manufacture of benzimidazole derivatives, the oxidation reaction to yield 2-phenyl benzimidazole derivatives 86 can occur at room temperature, as illustrated in Scheme 19. Huynh et al.51 produced 2-(substituted-phenyl)benzimidazole derivatives by reacting ortho phenylenediamines with benzaldehydes and using sodium metabisulphite as an oxidant. Three human cancer cell lines, A549, MDA-MB-231, and PC-3, were used to test these substances for their anticancer activities. The functioning of the phenyl ring system at 2-position, the biochemical properties of the cell lines, and electron-withdrawing and -donating groups on the benzimidazole scaffold were shown to be associated with variations in the IC50 values. We established additional benzimidazole derivatives with various substituents on the 2-phenyl ring system, such as –OH, –NO2, –CF3, –I, –OMe, –NMe2, and –OCH2–C6H5, in parallel to provide more information about the impact of the electron-donating and withdrawing groups on the phenyl ring at the 2-position as well as on the benzimidazole frame. Remarkably, the bioactivities of compounds 86 on the A549 and PC-3 cell lines showed that the methyl group at position 5 is crucial in enhancing their bioactivities (86a > 86b > 86c > 86e > 86f > 86g > 86h > 86i).
:1 v/v) as the medium for the manufacture of benzimidazole derivatives, the oxidation reaction to yield 2-phenyl benzimidazole derivatives 86 can occur at room temperature, as illustrated in Scheme 19. Huynh et al.51 produced 2-(substituted-phenyl)benzimidazole derivatives by reacting ortho phenylenediamines with benzaldehydes and using sodium metabisulphite as an oxidant. Three human cancer cell lines, A549, MDA-MB-231, and PC-3, were used to test these substances for their anticancer activities. The functioning of the phenyl ring system at 2-position, the biochemical properties of the cell lines, and electron-withdrawing and -donating groups on the benzimidazole scaffold were shown to be associated with variations in the IC50 values. We established additional benzimidazole derivatives with various substituents on the 2-phenyl ring system, such as –OH, –NO2, –CF3, –I, –OMe, –NMe2, and –OCH2–C6H5, in parallel to provide more information about the impact of the electron-donating and withdrawing groups on the phenyl ring at the 2-position as well as on the benzimidazole frame. Remarkably, the bioactivities of compounds 86 on the A549 and PC-3 cell lines showed that the methyl group at position 5 is crucial in enhancing their bioactivities (86a > 86b > 86c > 86e > 86f > 86g > 86h > 86i).
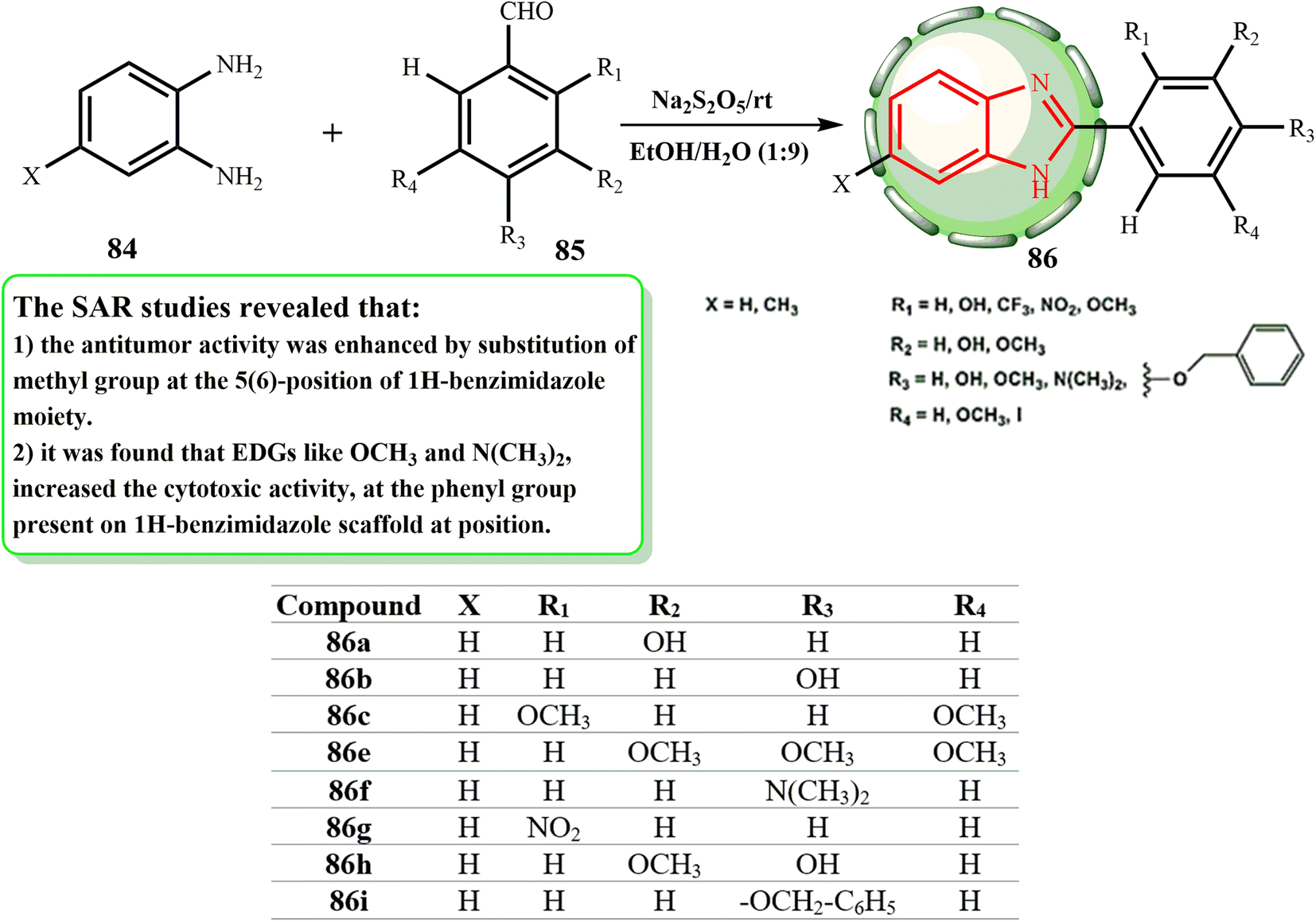 | ||
| Scheme 19 Synthesis of 2-(substituted-phenyl) benzoimidazole derivatives 86. | ||
Scheme 20 illustrates the three-step synthesis of target compounds 92 used in this investigation. Following the procedure outlined in the literature, the substrates, 1-(4-(5-substituted-1H-benzo[d]imidazol-2-yl)phenyl)ethan-1-ones (89) and 4-(5-substituted-1H-benzo[d]imidazol-2-yl)benzohydrazides (90), were created.52 Condensation of the hydrazide with diverse substituted benzaldehydes 91 (ref. 53) led to the related arylidene hydrazides 92. Çevik et al.54 produced a variety of 4-(5-substituted-1H-benzimidazol-2-yl)-N/-((5-substitutedthiophen/furan-2-yl)methylene)benzohydrazides (92) and used the MTT assay to evaluate them for their ability to kill A549 and MCF-7 (breast) cancer cell lines, using cisplatin as a (+ve) control. The normal NIH/3T3 cell line was also employed to check the synthesized synthetic substances. Compound 92g exhibited no effect on the normal cell line, while it had the maximum cytotoxic potential in the A549 cell line (IC50 against A549 = 0.316 μM). Compared to cisplatin, compound 92j (IC50 = 0.0316 μM) exhibited the most potent selective cytotoxicity against the MCF-7 cell line. In comparison to 5-fluoro substitution, SAR investigations demonstrated that 5-chloro substitution at the 1H-benzimidazole ring improved the cytotoxicity against MCF-7 cells.
 | ||
| Scheme 20 Synthesis of target compounds 92. | ||
The appropriate benzyloxybenzaldehyde 94 was produced by reacting 4-hydroxybenzaldehyde (93) with benzyl chloride to synthesize target compounds 95 (Scheme 21). Abdel-Mohsen et al.55 investigated the effects of a novel class of 1,2-disubstituted 1H-benzimidazoles as VEGFR-2 inhibitors on the HepG-2 hepatocellular carcinoma cell line. According to the results, sorafenib (IC50 = 10.99 μM) had lower cytotoxic activity than one of the created hybrids, 95 (IC50 = 1.98 μM). According to the SAR evaluations, the elongated side chains of 1H-benzimidazole at the one position resulted in activity as a VEGFR-2 inhibitor. It was discovered that the presence of a linker and substituents at positions N-1, C-2, C-5, and C-6 is promising in the anticancer action.
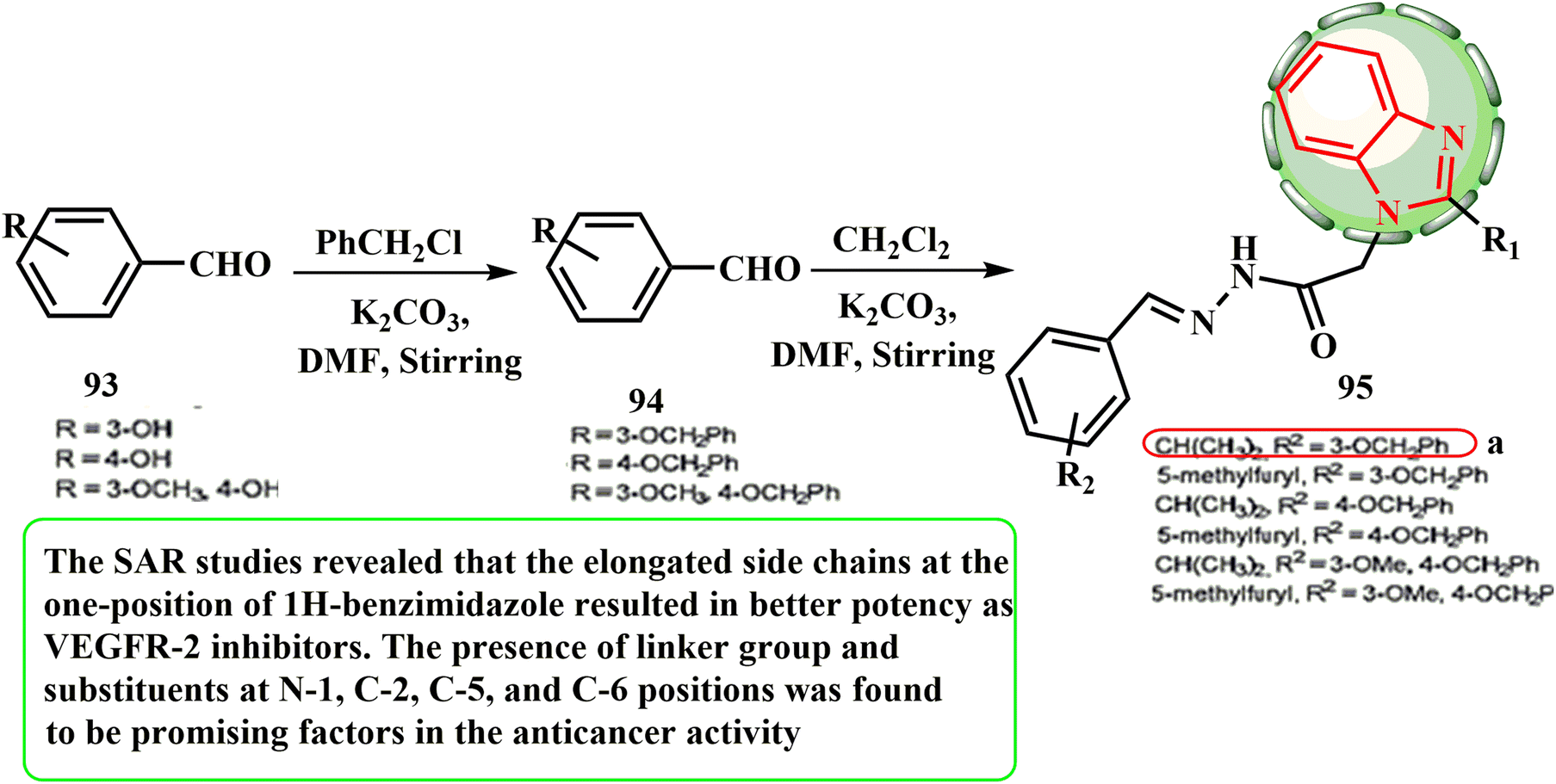 | ||
| Scheme 21 Synthetic protocol for benzimidazole hybrids 95. | ||
 | ||
| Scheme 22 Synthesis of benzimidazole-linked 2-acetylbenzofuran derivatives 99a and b. | ||
Niementowski heated free base 84 with appropriate amides 100 to produce methyl benzimidazole (101, R1 = 5 (6)1-methyl, R = H, CH3, or Ph) (Scheme 23).
 | ||
| Scheme 23 Synthesis route to target compounds 101. | ||
Here, we describe how we produced benzimidazole–urea derivatives 105. The reaction of O-phenylene diamine (77) and cyanogen bromide (102) in methanol at room temperature produced 2-aminobenzimidazole (103) in 88% yield by ammonium hydroxide workup (Scheme 24). Compound 105j (3-chloro-4-fluorophenyl) was the most active compound against a liver cancer cell lines (HepG-2), with an IC50 value of 7.5 μM. The anticancer activity of the other compounds followed the order of 105d > 105a > 105b > 105e > 105c > 105g > 105f. The anticancer activity of the other compounds followed the sequence of 105a > 105b > 105d > 105i > 105f > 105h > 105g with IC50 values in the range of 2.4–38.5 μM, among which compound 105j was the most effective (IC50 = 1.9 μM) against non-small lung cancer (A549).59
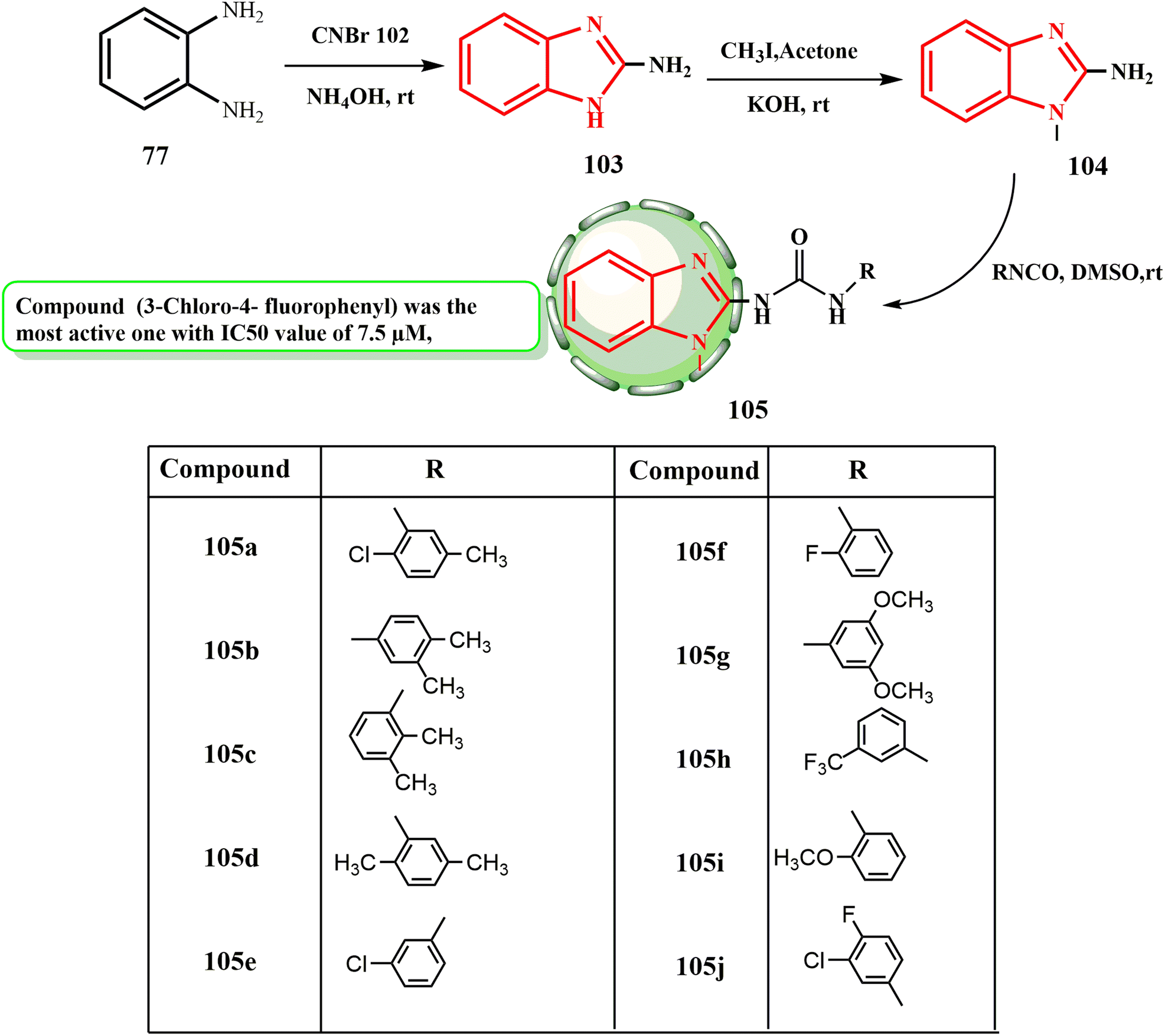 | ||
| Scheme 24 Synthesis of benzimidazole–urea derivatives 105. | ||
 | ||
| Scheme 25 General synthesis of compounds 110a and b. | ||
After being nitrated with sulfuric and nitric acids, the commercially available 1,3-dibromobenzene (111) was converted to 112, which was then selectively replaced with cyclohexylamine in DMF to produce 113. Bis(pinacolato)diboron was used to boronate 113 in the presence of Pd(PPh3)2Cl2 and KOAc, producing 114. Suzuki–Miyaura cross-coupling of 114 with dibromo-imidazo[1,2-a]pyrazine61,62 and Pd(PPh3)4 afforded compound 115 together with traces of disubstituted products. Amines 116 were obtained by reducing the derivatives with Na2S2O4 in ammonia. Then, triethyl orthoformate was cyclized in acetic acid to produce intermediate 117. Using Pd(PPh3)4 and K2CO3, Suzuki reactions of the intermediates with unsubstituted and substituted phenylboronated were performed in CH3CN:H2O, yielding 118a and b (ref. 63) (Scheme 26). Mono benzimidazole derivatives 118a and b showed specific effectiveness against subpanels of melanoma, colon, leukemia, and central nervous system (GI50 = 0.31–0.39 μM). Therefore, 4-methoxyphenyl at the C6 position of imidazo[1,2-a]pyrazine 118b showed superior cytostatic activity compared to phenyl 118a among the mono benzimidazoles.
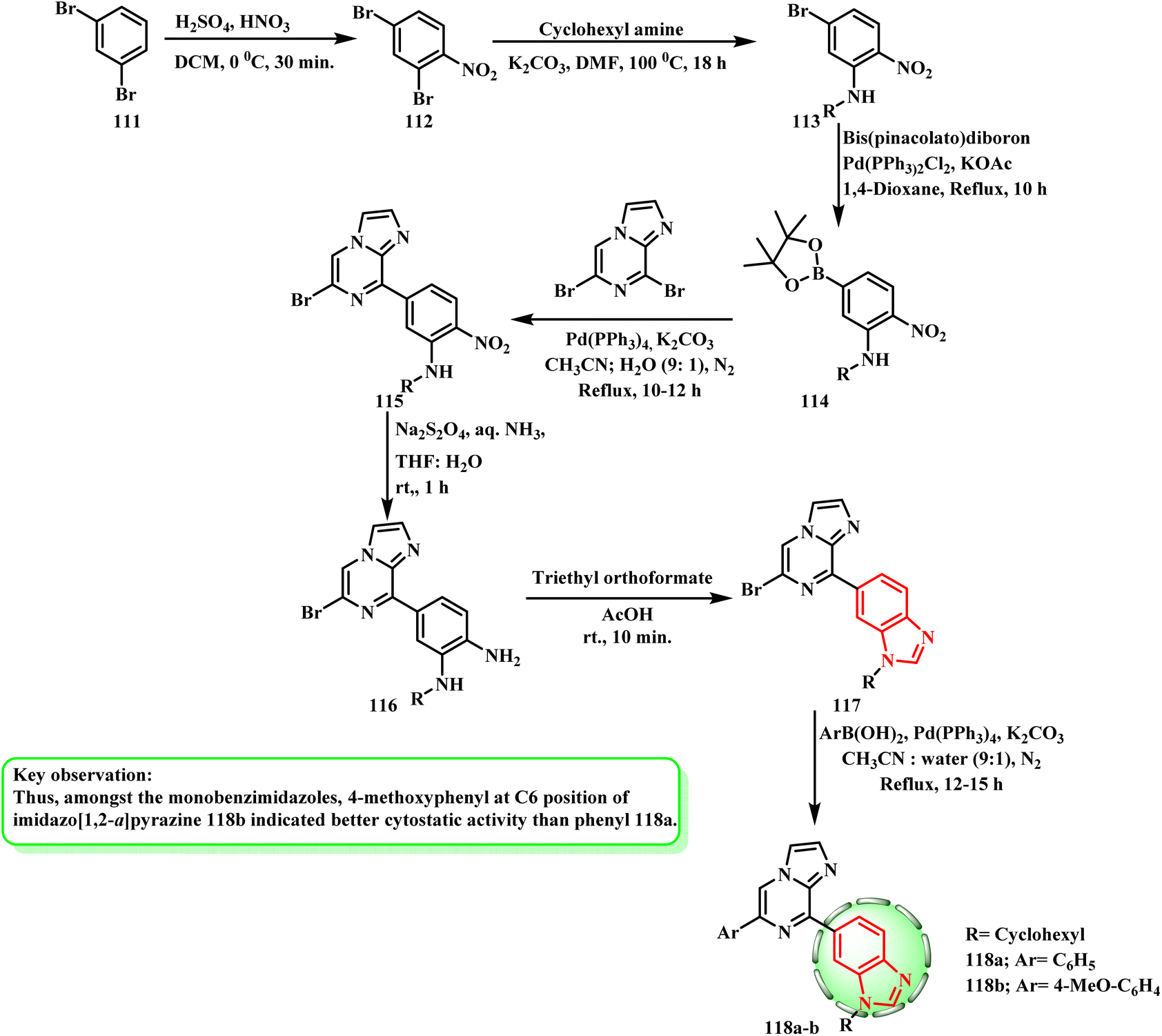 | ||
| Scheme 26 Synthesis of mono benzimidazole derivatives 117 and 118a and b. | ||
The commercially available 1,3-dibromobenzene (111) was nitrated to produce 112, which was then replaced with cyclohexylamine in DMF to produce 113. After reducing 113 with Na2S2O4 in ammonia to yield 119, the corresponding benzimidazole 121 was obtained by cyclization with triethyl orthoformate (120) in acetic acid. Bis(pinacolato)diboron (122) was used to boronate the derivative in the presence of Pd(PPh3)2Cl2 (123) and KOAc, yielding 124. Using Pd(PPh3)4 and K2CO3, Suzuki–Miyaura cross-coupling of benzimidazole boronated 125 with intermediate 124 was carried out to yield 126 (Scheme 27). All the screened cell lines showed the cytotoxicity of compounds 118b and 126, which also displayed significant growth inhibitory concentrations of 2.10 and 2.23 μM, respectively.63 The colon cancer cell line HCC-2998 was discovered to be extremely sensitive to derivative 126 among these tumor-sensitive cell lines, exhibiting a negative growth percentage value (lethal impact). The cancer cell line from the central nervous system (SF-539) was found to be highly susceptible to derivative 118b. Ct-DNA intercalates with imidazo[1,2-a]pyrazine-benzimidazoles 118a and bisbenzimidazole 126,64 exhibiting superior activity than bisbenzimidazole, as a key interaction for basic physiologically noteworthy effects. With cytostatic effects on the cell line, mono benzimidazole derivative 118a was superior to bisbenzimidazole 126; nevertheless, compound 126 subsequently revealed a cytotoxic influence on various cancer cell lines.
 | ||
| Scheme 27 Schematic of the route for the synthesis of bisbenzimidazole 126. | ||
Scheme 28 shows the synthesis of benzimidazole-tethered pyrazole 132. Arylhydrazine and suitable aralkyl ketone 128 are first condensed in glacial acetic acid to produce pyrazole-based carbaldehyde 131. Hydrazone intermediate 129 is then cyclized utilizing Vilsmeier–Haack reaction. Compound 132 had the least toxicity and most promising effects, with IC50 values of 30.9, 32.8, and 80 μM against MRC5 cells, AsPC1, and SW1990.65 Additionally, SAR investigations showed that a 4-fluorophenyl moiety increased its efficacy against all the evaluated cell lines.
 | ||
| Scheme 28 Synthesis of benzimidazole–tethered pyrazole 132. | ||
 | ||
| Scheme 29 Synthesis of benzimidazole derivatives 140a–g. | ||
Methyl(5-(4-(methyl(oxetan-3-yl)amino)benzoyl)-1H-benzo[d]imidazol-2-yl)carbamate (145) was obtained via the microwave67 condensation cyclization of 1,3-bis(methoxycarbonyl)-2-methyl-2-thiopseudourea and intermediate 143 (Scheme 30). Compound 145 with oxetanyl substitution demonstrated significant cytotoxic activity against prostate (PC-3 and PC3MLN4 cell lines), lung (A549 cell lines), and ovarian cancers with considerable activity toward profoundly aggressive carcinogenic cell lines (IC50 = 0.9–3.8 μM).68 The growth of existing tumors was greatly suppressed by compound 145 (30 mg kg−1) without causing any detectable toxicity (T/C: 0.36). Additionally, as seen in compound 145, SAR studies showed that the addition of the para to ketone group in mebendazole and the oxetane group in the phenyl ring on the left side of the compound increased the anticancer activity. A methyl carbamate moiety on the right side of the 1H-benzimidazole ring also increased the cytotoxic activity of the compound.
 | ||
| Scheme 30 Synthesis of (1H-benzo[d]imidazol-2-yl)carbamate analogue 145. | ||
The synthetic route for benzimidazole–quinoline69 hybrid 151 is presented in Scheme 31. Firstly, key intermediate 150,70 obtained initially from core nucleus benzimidazole-5-carboxylate 149, was effectively produced by a ‘one pot’ nitro reductive cyclization process between ethyl 3-nitro-4-(substituted amino)benzoate 148 and 5-bromothiophene-2-carbaldehyde using Na2S2O4 in DMSO.71 When tested against two distinct cell lines, all other drugs showed moderate to good anticancer efficacy. Using cisplatin as a reference medication, compound 151 demonstrated activity against the A37 (IC50 = 34.7 ± 0.9 μg mL−1) and MDA-MB-231 (IC50 = 20.4 ± 1.1 μg mL−1) cell lines using the MTT test. However, the other substances did not show preference for these specific cell lines. Chemical substance 151 also showed cytotoxic properties. The IC50 value of compound 151 (A375) was 34.7 ± 0.9 μg mL−1.42 Compound 151 presented the highest % inhibition and lowest IC50 value of 604.8 μg mL−1.
 | ||
| Scheme 31 Synthetic route for benzimidazole–quinoline hydride 151. | ||
 | ||
| Scheme 32 Synthesis of N-substituted-2-benzimidazolyl acrylonitrile derivatives 156a and b. | ||
Kommi et al. investigated the production and cytotoxic potential of benzimidazole derivatives. Scheme 33 depicts the synthetic method utilized to prepare 1-benzyl-1H-benzimidazole analogues 164a and b. 4-Formyl-N-phenylbenzamide 163 was utilized to condense N1-benzyl-4-chlorobenzene-1,2-diamine (160). Using benzyl bromide (158) and the nitro group of compound 159, the commercially available 4-chloro-2-nitro aniline (157) was first converted to N-benzyl-4-chloro-2-nitroaniline (159). Then, it was further reduced to an amino functional group using stannous chloride dihydrate, which produced N1-benzyl-4-chlorobenzene-1,2-diamine (160).73 Alternatively, the commercially existing 4-formyl benzoic acid (161) and aniline derivative (162) were used to create 4-formyl-N-phenylbenzamide (163).74 Lastly, 4-formyl-N-phenylbenzamide (163) and N1-benzyl-4-chloro orthophenylene diamine (160) were refluxed in ethanol in the presence of Na2S2O5, yielding target compounds 164a and b.75 Among the compounds, with an IC50 value of 7.01 ± 0.20 μM, 164b (4-(1-benzyl-5-chloro-1H-benzo[d]imidazol-2-yl)-N-(4-hydroxyphenyl)benzamide) demonstrated the highest efficacy and stopped the MCF-7 cell cycle in the G2/M phase and S-phase.76 The SAR investigations revealed that larger substituents linked to the benzene ring (164a) reduced the cytotoxic activity of the examined compounds.
 | ||
| Scheme 33 Synthesis of various 1-benzyl-1H-benzimidazole analogues 164a and b. | ||
 | ||
| Scheme 34 General synthesis of compound 169. | ||
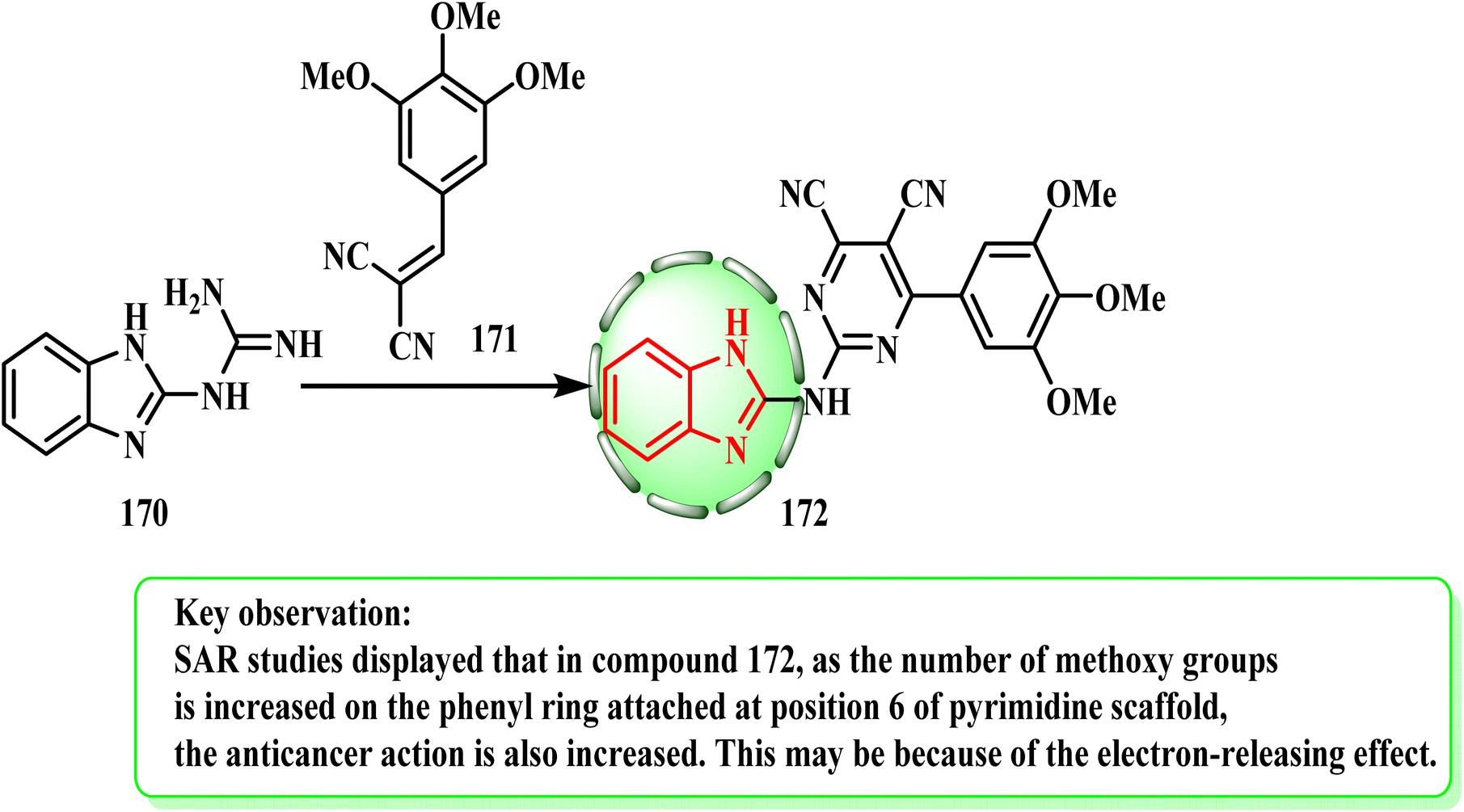 | ||
| Scheme 35 Synthesis of 2-(2-pyrimidinylamino)benzimidazole 172. | ||
 | ||
| Scheme 36 Synthetic scheme for target compound 177. | ||
4-(5-(4-Formylphenoxy)-3-methyl-4-nitro-1H-pyrazol-1-yl)benzonitrile (180b) was obtained by reacting 4-(5-chloro-3-methyl-4-nitro-1H-pyrazol-1-yl)benzonitrile (178b) with 4-hydroxybenzaldehyde (179) in the presence of potassium hydroxide, DMF, and a catalytic amount of copper(I)iodide and triphenylphosphine, respectively. 4-(3-Methyl-4-nitro-5-(4-(5-nitro-1H-benzo[d]imidazol-2-yl)phenoxy)-1H-pyrazol-1-yl)benzonitrile (182b) was produced by the cyclocondensation reaction of compound 180a and b with 4-nitro-1,2-phenylenediamine (181) in the presence of sodium thiosulphate and DMF (Scheme 37). Combining compound (182b) with doxorubicin was shown to greatly boost the anticancer effect of the drug and limit MCF-7 cell cycle growth.84 According to SAR investigations, the anticancer activity of the 1H-benzimidazole moiety decreased when carboxylic or nitro groups were present at position 5, but it increased when a polar cyano-group was substituted for the phenyl group in the pyrazole ring at position 4.
 | ||
| Scheme 37 Synthetic route from pyrazoles 178a and b to pyrazole–benzimidazole conjugates 182a and b, respectively. | ||
The synthesis and cytotoxic potential of imidazo[2,1-b]thiazole-benzimidazole conjugate 188 were examined by Baig et al. As illustrated in Scheme 38, imidazothiazole–benzimidazole conjugate 188 was produced by oxidatively cyclizing imidazo[2,1-b]thiazole-5-carbaldehyde (186) and substituting o-phenylenediamine (187) with sodium metabisulphite in ethanol. By using the Vilsmeier–Haack reaction with the equivalent imidazo[2,1-b]thiazole (185), which was derived from suitable 2-bromo-1-arylethanone 184 and 2-aminothiazole (183), imidazo[2,1-b]thiazole-5-carbaldehyde (186) was produced. Compound 188 (IC50 = 1.08 μM) showed significant cytotoxicity against the A549 cell line.85 The cytotoxicity of conjugates containing electron-withdrawing groups as substituents, such as p-trifluoromethyl and p-methoxy substituents, against A549 cells was demonstrated by SAR evaluations.
 | ||
| Scheme 38 Synthesis of imidazo[2,1-b]thiazole-benzimidazole conjugate 188. | ||
The synthesis and cytotoxic potential of 2-(aminomethyl)benzimidazole derivatives were examined by Al-Sultan et al. Scheme 39 states that all synthesized compounds began with amine derivative 189 and an equivalent quantity of TEA (primary amines did not use TEA). After being dissolved in 25 mL of diethyl ether and allowed to cool to 5 °C, chloroacetyl chloride (190) was gradually added to the mixture until the fumes stopped.86 The antiproliferative features of the developed compounds on human breast cancer (T47D) and human alveolar cell carcinoma (A549) cells were evaluated in vitro using Vero cells (from the kidney of an African green monkey) as a standard control. The morphology of T47D with an inhibitory concentration of the cytotoxic chemicals, gefitinib, and the control was explained by additional research to confirm the antiproliferative effects of the extremely toxic substances (193) on T47D. Lastly, compounds 193 displayed IC50 values that were nearly identical to that of the positive control.
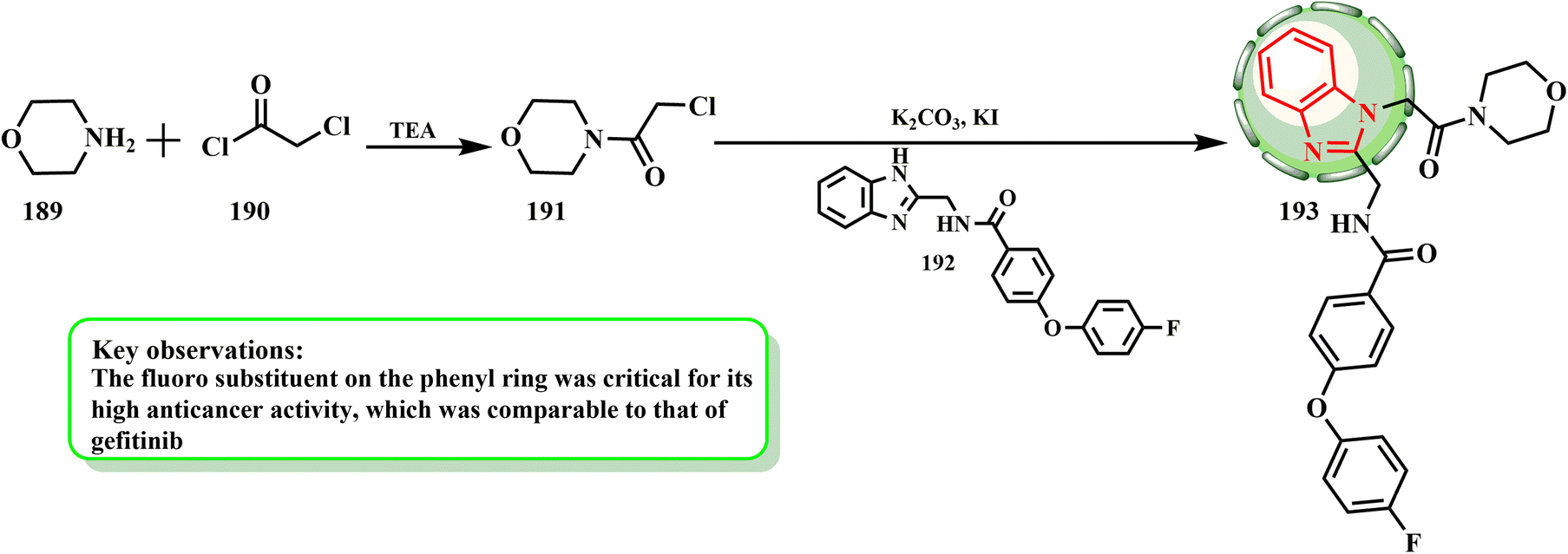 | ||
| Scheme 39 Synthesis of designed compound 193. | ||
In THF and TEA, we used a normal reaction between substituted benzoyl chloride 195 (1.1 mmol) and 2-aminobenzimidazole (194) (1 mmol) (Scheme 40). N-(Benzimidazol-2-yl)-2-substituted benzamide structures 196 were synthesized efficiently in a single step. Interestingly, compound 196 showed the strongest anti-proliferative action against MCF-7 cancer cells (IC50 = 3.84 ± 0.62 μM).87
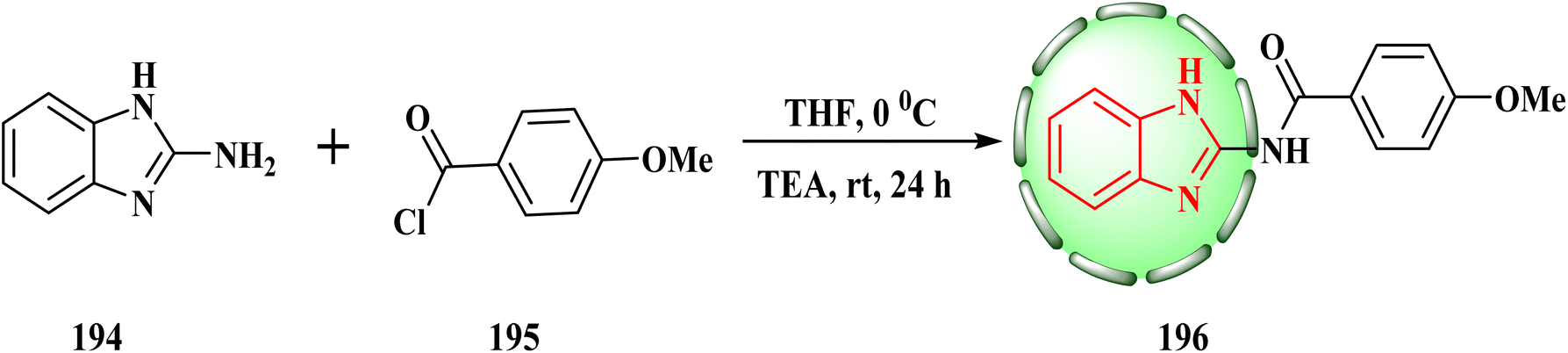 | ||
| Scheme 40 Reaction of substituted benzoyl chloride with 2-aminobenzimidazole. | ||
A series of 3,3′-(1,3-phenylene(methylene))(1-alkyl-benzimidazolium)salts (200) was produced and characterized using the stated approach (Scheme 41).88 We previously employed propyl chloride (197) as the electrophile for 1,2-(bromomethylene)benzene.89 The method adopted for the synthesis of the title compounds was selective for the 1-position heterocycle. Indeed, upon treating alkyl benzimidazole 198 with m-xylyl dibromide (1,3-(bromomethylene)benzene) (199), the respective bisbenzimidazolium salt was observed to give 200. ul Huda et al.90 developed and produced unique 1,10-(1,3-phenylenebis(methylene))bis(3-alkyl/aryl-1H-benzimidazol-3-ium) salt hybrids, and using the SRB assay, evaluated their anticancer potential against the MCF-7 and HCT-116 (CRC) human breast cancer cell lines. Compound 200b was shown to be the most effective anticancer drug overall, inhibiting the growth of HCT-116 cells with an IC50 of 0.1 μg mL−1. According to the SAR evaluations, the notable pharmacological effect of compound 200 was caused by its lengthy N-substituted alkyl chain, which presented it as extremely lipophilic. Compounds with shorter chain lengths, such as isopropyl chain (200a) and nonaromatic replacements, exhibited less cytotoxic action than N-methylene phenyl (200b) substitutions.
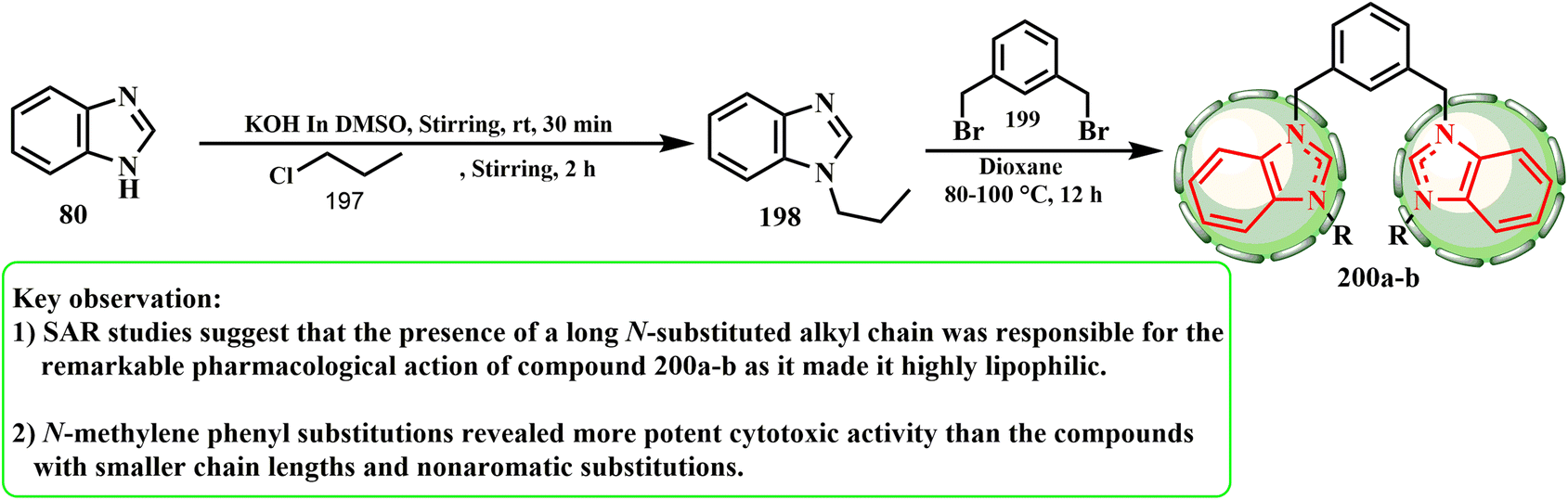 | ||
| Scheme 41 Reaction representation of 1H-benzimidazole with propyl chloride. | ||
The compounds were prepared in accordance with the literature91,92 (Scheme 42). Because of its efficiency in supplying high quantities of hydroxide ions, KOH is a characteristically strong base utilized in a range of laboratory and chemical synthesis applications. For 12 h, the reaction was carried out at 80 °C. According to the results, 202a had relatively antiproliferative efficacy against the A549, DLD-1, and L929 cell lines compared to cisplatin; however, 202b was essentially effective against the A549 cell line. Compound 202b has substituents with the methyl group at the 2-, 3-, 5-, and 6-positions, whereas compound 202a has a chloro group on the benzyl ring at the 3-position. Compared to compound 202b, compound 202a had more advanced, albeit weaker, activity against the cell lines under investigation. The various substituents on the benzyl ring, which may either donate or withdraw electrons, could be the cause of these variations.93
 | ||
| Scheme 42 Reaction of 2-methyl-1H-benzo[d]imidazole with alkyl halide. | ||
Ren et al. researched the production of benzimidazole derivatives and their potential for cytotoxicity. Scheme 43 shows the preparation of compound 207. Benzimidazole intermediates 205 were produced by cyclizing 3-bromo-1,2-benzenediamine (203) with various aldehydes 204. Subsequently, compound 207 was produced by the Suzuki reaction between compound 205 and 3,4,5-trimethoxyphenylboronic acid (206). Compound 207, which is slightly more potent than colchicine, showed the strongest inhibitory effects on the growth of cancer cells (IC50 = 50 nM).94 In melanoma tumors, compound 207 showed a 78.70% inhibition rate. Stronger hydrogen bonds and hydrophobic interactions may be the cause of the significant activity of 207, according to the SAR evaluations.
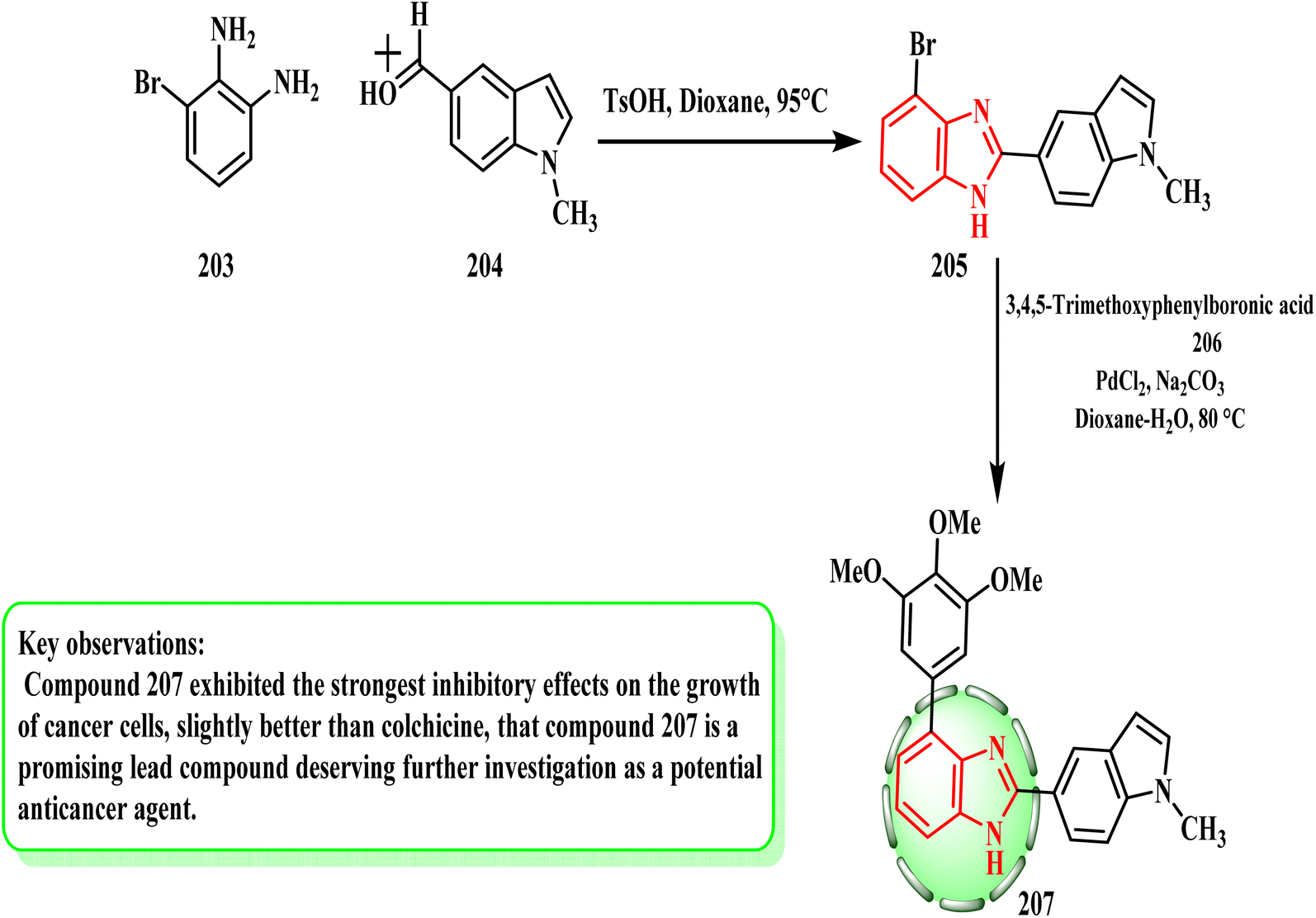 | ||
| Scheme 43 Synthesis of benzimidazole derivatives 205 and 207. | ||
Until the starting materials were exhausted (as determined by TLC), a combination of the proper o-phenylenediamine (1 mmol) (208), related indole derivative (1 mmol) (209), and Na2S2O5 (40%) (2 mL) in EtOH (4 mL) was refluxed95,96 (Scheme 44). Especially compound 210a and b could alter the ER target gene expression, and an integrated stress response was induced in a dose-related manner.97 The MCF-7 transcriptome was shown to be significantly influenced by compounds 210a and b, which resulted in the upregulation and downregulation of an appropriate number of genes. According to the SAR investigations, the presence of an electron-withdrawing group at the indole ring and 4-fluorobenzyl at the 1H-benzimidazole ring demonstrated comparatively stronger anticancer effects. Furthermore, the lipophilic characteristic of the indole moiety was improved by the presence of –Br, which facilitated effective binding.
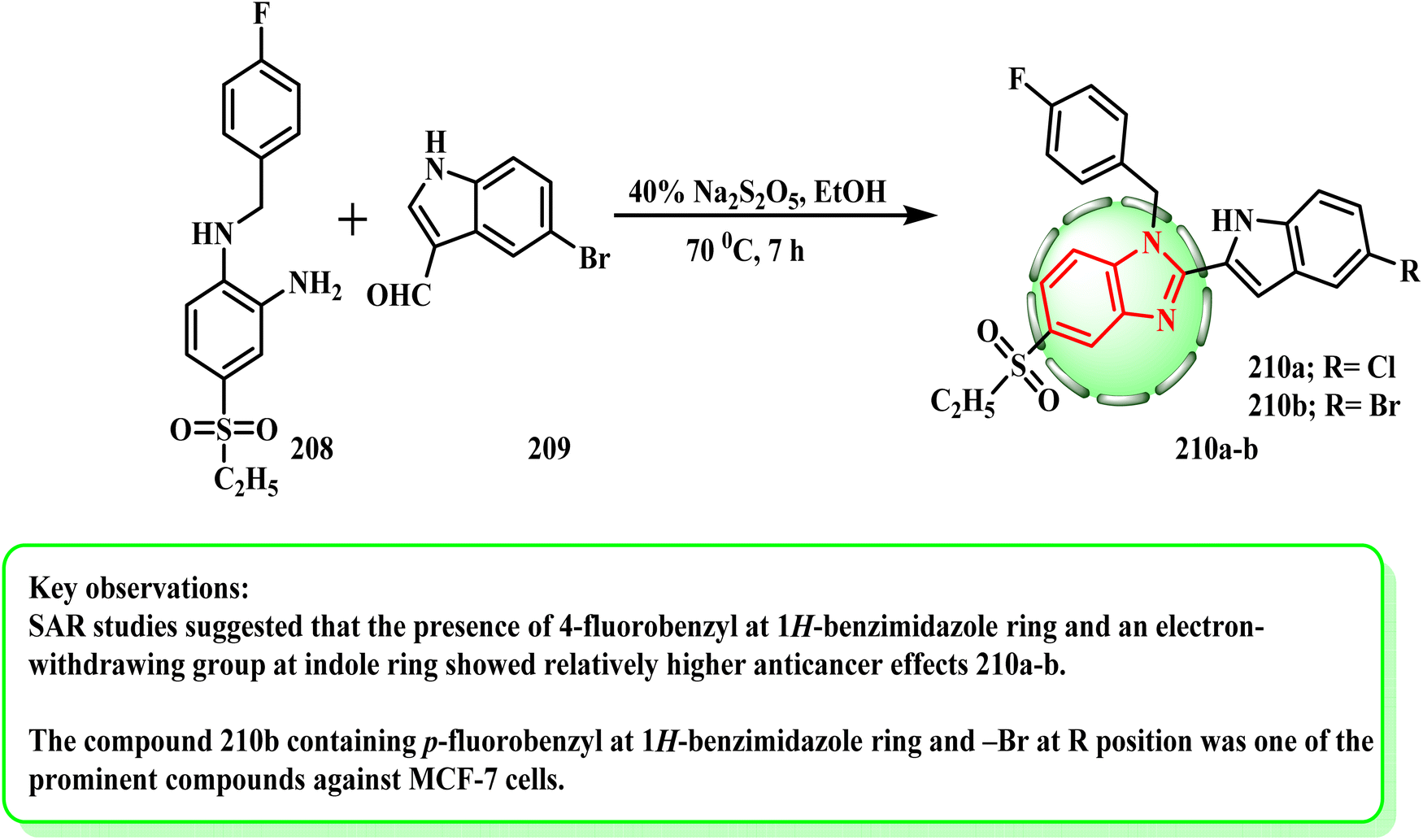 | ||
| Scheme 44 Synthetic route for the preparation of benzimidazoles 210a and b. | ||
The synthesis and cytotoxic potential of tertiary sulfonamide derivatives with a benzimidazole moiety were examined by Gao et al. Benzyl chloride 212 and 3,4,5-trimethoxyaniline (211) interacted in the presence of K2CO3 in acetone to give secondary amine 213, which reacted with different benzene sulfonyl chloride derivatives 37 to obtain tertiary sulfonamide derivatives 214a–d (Scheme 45). MGC-803 cells were more sensitive to compounds 214a–d than PC-3 and MCF-7 cells. Among the compounds that had notable antiproliferative activity, compound 214b demonstrated the strongest anticancer activity against MGC-803 cells (IC50 = 2.19 μM).98 The presence of a 3,4,5-trimethoxy group at the phenyl ring was crucial for the anticancer actions according to the SAR evaluations. The most effective compound against human stomach cancer cell lines was compound 214d, which had a methyl group and a 1H-benzimidazole moiety. 4-Br (214b) > 4-F (214a) > 2-Cl (214c) was the association between halogen substitution and anticancer efficacy. The sulfonyl groups were crucial for their inhibitory effect, which can maintain or improve the antiproliferative activity against the three tested human cancer cells, according to all the reported changes and inhibitory findings.
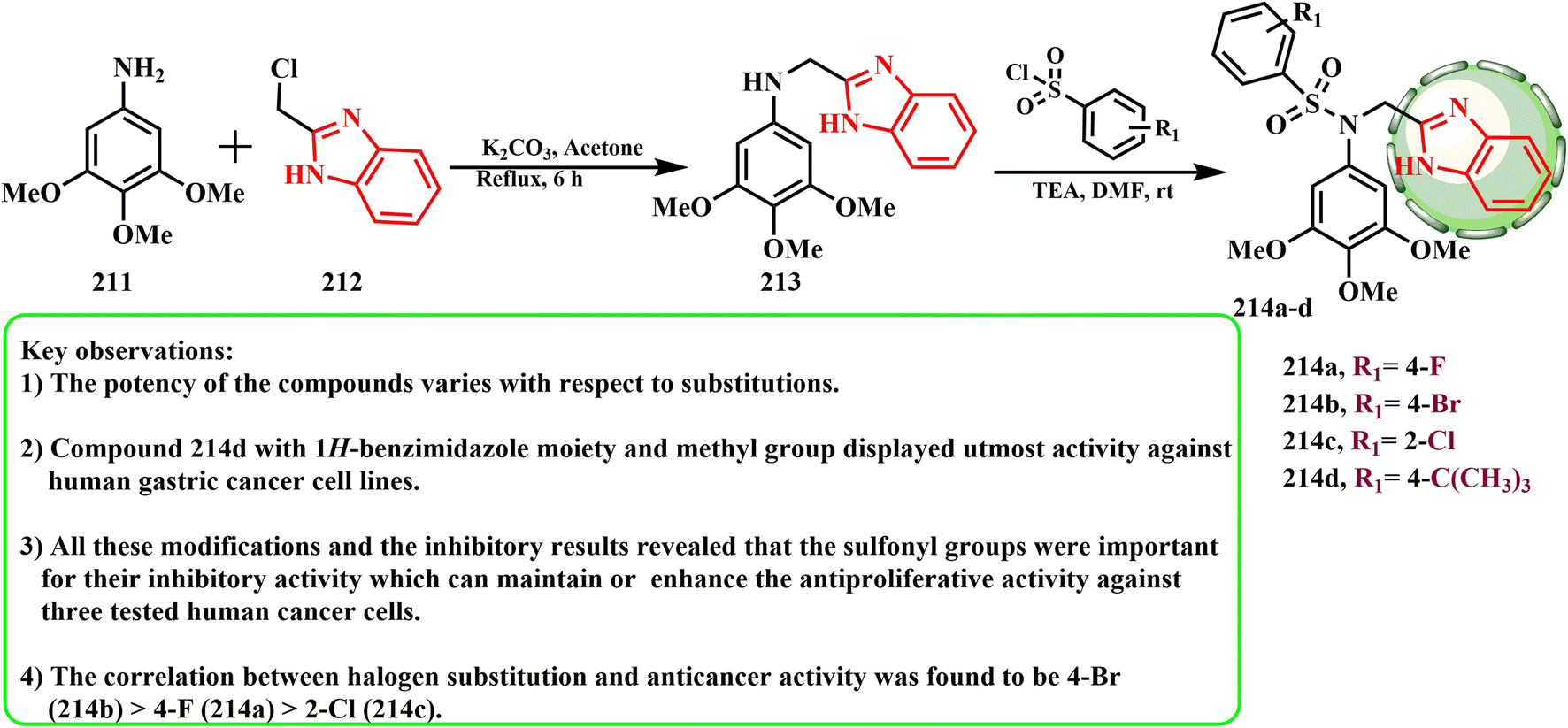 | ||
| Scheme 45 Synthesis of tertiary sulfonamide derivatives containing a benzimidazole moiety 214a–d. | ||
The production and cytotoxic potential of new benzimidazole ligands and their cobalt(II) and zinc(II) complexes were examined by Yılmaz et al. ZnCl2·6H2O or CoCl2 in EtOH was used to produce benzimidazole complexes 216a–d from benzimidazole ligands (215a, 215b, and 215c). Ligands of benzimidazoles 215a (ref. 99) and 215b (ref. 100) were produced using the methods outlined in the literature. In this work, the Mizoroki–Heck reaction was used to synthesize benzimidazole ligand 215d for the first time, similar to the literature technique101,102 (Scheme 46). Compounds 216a–d (logIC50 = −0.97, −1.30, 1.13, and −0.73 μM, respectively) were identified to have higher anticancer potency than the standard docetaxel medication (logIC50 = −0.81 μM) against the A-2780 cell line at 0.1 μM concentration.99
 | ||
| Scheme 46 Synthesis of benzimidazole metal complexes 216a–d. | ||
Scheme 47 shows the route for the preparation of target compound 219 in this work. Acetophenone and 2-mercaptobenzimidazole 217 reacted in refluxing acetic acid with five equivalents of sulphuric acid to produce sulphate salt 218 in a one-pot, two-component heterocyclization process. Phenylthiazolo[3,2-a]benzimidazole 219 was obtained by neutralizing sulphate salt 218 by stirring with an aqueous solution of sodium bicarbonate. HT-29 (colon) and MDA-MB-468 (breast) cancer cell lines were used to investigate the cytotoxic potential of synthetic compound 219. CD133 inhibition in cancer stem cells and the cytotoxicity of specific 3-phenylthiazolo[3,2-a]benzimidazoles including their design, direct synthesis, and in vitro biology were investigated.103 Compound 219 decreased the surface expression of CD133 on cells by 50% and showed strong anticancer activity against both cancer cell lines with IC50 values of 9 and 12 μM, respectively. According to the SAR investigation, the electron-donating group in the phenyl ring of 219 enhanced the suppression of tumor cells.
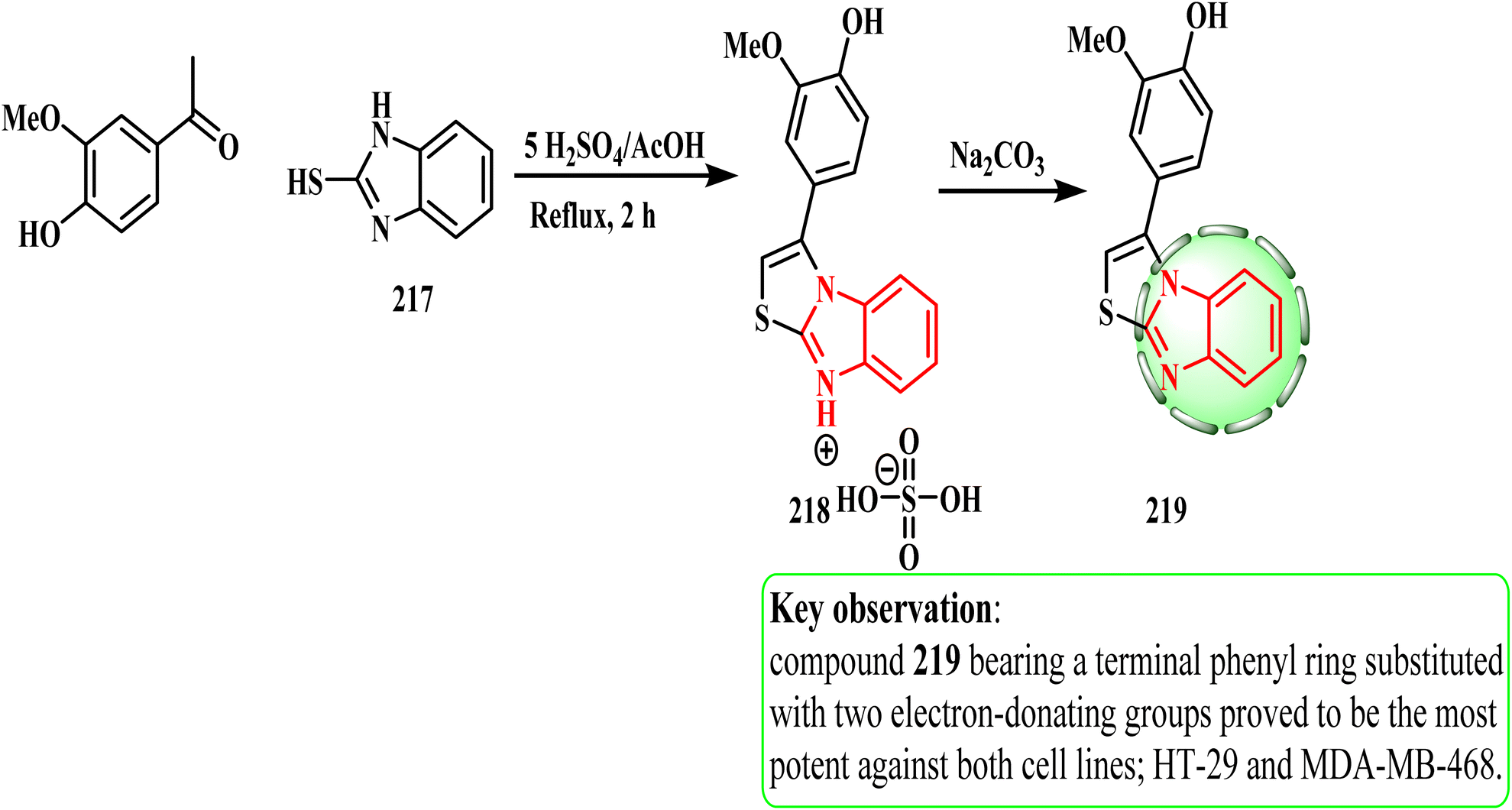 | ||
| Scheme 47 Synthesis of compound phenylthiazolo[3,2-a]benzimidazole 219. | ||
However, employing 3-chloro-2,4-pentanedione, the acetyl thiazolo[3,2-a]benzimidazole derivative (221) was constructed as previously described for the mercaptan alternative 220. The target 223 was obtained, as shown in Scheme 48, by condensing the acetylthiazolo[3,2-a]benzimidazole derivative (221) with 4-(hydrazinecarbonyl)benzene sulfonamide (222) in acetic acid. Compound 223 was found to induce cell cycle arrest and death and exhibit prospective proliferation inhibition against the MCF-7 and MDA-MB231 breast cancer cell lines.104 According to the SAR investigations, the hCA I and II inhibitory activities were improved by moving the sulfamoyl group to the para-position. Compound 223, which has a hydrazide linker and an enaminone spacer, exhibited the highest hCA IX inhibitory activity.
 | ||
| Scheme 48 Synthesis of 3-methylthiazolo[3,2-a]benzimidazole-benzene sulfonamide conjugate 223. | ||
The synthesis and cytotoxic potential of benzimidazole derivatives were examined by Atmaca et al. By first reacting the amino group of 224 with ethylbromo acetate to produce ethyl [5,6-dichloro-2-ethyl-1H-benzimidazol-1-yl]acetate (225), the intended powerful derivative 228 was synthesized. Subsequently, compound 226 was produced by reacting acetate derivative 225 with hydrazine hydrate in ethanol. Finally, the desired hybrid molecule 228 was produced by the successful condensation of the nucleophilic nitrogen with the substituted aromatic aldehyde 227 (Scheme 49). Compound 228 was found to exhibit a potential cytotoxic effect against the MCF-7 (IC50 = 17.8 ± 0.24 μg mL−1), DU-145 (IC50 = 10.2 ± 1.4 μg mL−1), and H69AR (IC50 = 49.9 ± 0.22 μg mL−1) cancer cell lines.105 Compound 228 with a bromo substituent showed the strongest anticancer potential and may be a promising anticancer treatment drug according to the SAR investigations. Furthermore, it was noted that the presence of halogen atoms enhanced its oral absorption and increased its membrane permeability. It was discovered that bromination increased the cytotoxic potential, while also improving the stability and protein binding affinity.
 | ||
| Scheme 49 Synthesis of benzimidazole derivative 228. | ||
The synthesis of benzimidazole derivatives was reported by Yadav et al.106 By first esterifying mercaptan derivative 220 with ethyl chloroacetate (229) to produce acetate 230, the desired derivative 232 was synthesized. After that, 230 was subjected to hydrazinolysis reaction with hydrazine hydrate to yield 231. Lastly, the Schiff base reactionof acetohydrazide 231 with aromatic aldehyde gave 232 (Scheme 50). Yadav et al.106 established a novel series of 1H-benzimidazole compounds and tested their anti-proliferative properties in vitro. When tested against the MCF-7 cell line, compound 232 (IC50 = 0.0013 μM) was found to be a more effective anti-cancer therapeutic candidate than 5-fluorouracil. According to the SAR investigations, the anti-tumor activity increased when an OH group was present on the phenyl ring and decreased when a di- or tri-substituent was present.
 | ||
| Scheme 50 Synthesis of benzimidazole derivative 232. | ||
Using the well-known general techniques, compounds 238a–d were produced in accordance with the reaction sequence shown in Scheme 51. Thus, using established techniques, 2-acetyloxymethylbenzimidazole (233), which was produced by acetylating 2-hydroxymethylbenzimidazole,107 was converted into N-benzylated derivatives 234 by treating it with substituted benzyl halides in the presence of a base. N-Benzyl-2-hydroxymethylbenzmidazoles 235 were produced by hydrolyzing intermediates 234 in aqueous alkali, and they then reacted with thionyl chloride to form alkyl chlorides 236. To obtain the target compounds 238a–d, intermediate alkyl chlorides 236 were finally treated with the relevant phenylpiperazine derivatives 237. Özdemir et al.108 developed and produced a unique range of 1H-benzimidazole-piperazine hybrids, and then tested them against two human cancer cell lines (MCF-7 and A549) to determine whether they have antiproliferative properties. Compound 238b demonstrated the most potent cytotoxicity (IC50 = 11.0 μM against MCF-7 cells and 4.6 μM against A549 cells). A mono-chloro substituent on the N-benzyl ring at the ortho- (238a) or para-positions (238b) increased the potency against the A549 cell line, according to the SAR evaluations. Furthermore, the activity was reduced when large groups such as 3,4,5-trimethoxy and 2,4-dichloro (238c) were present on the N-benzyl ring (238d).
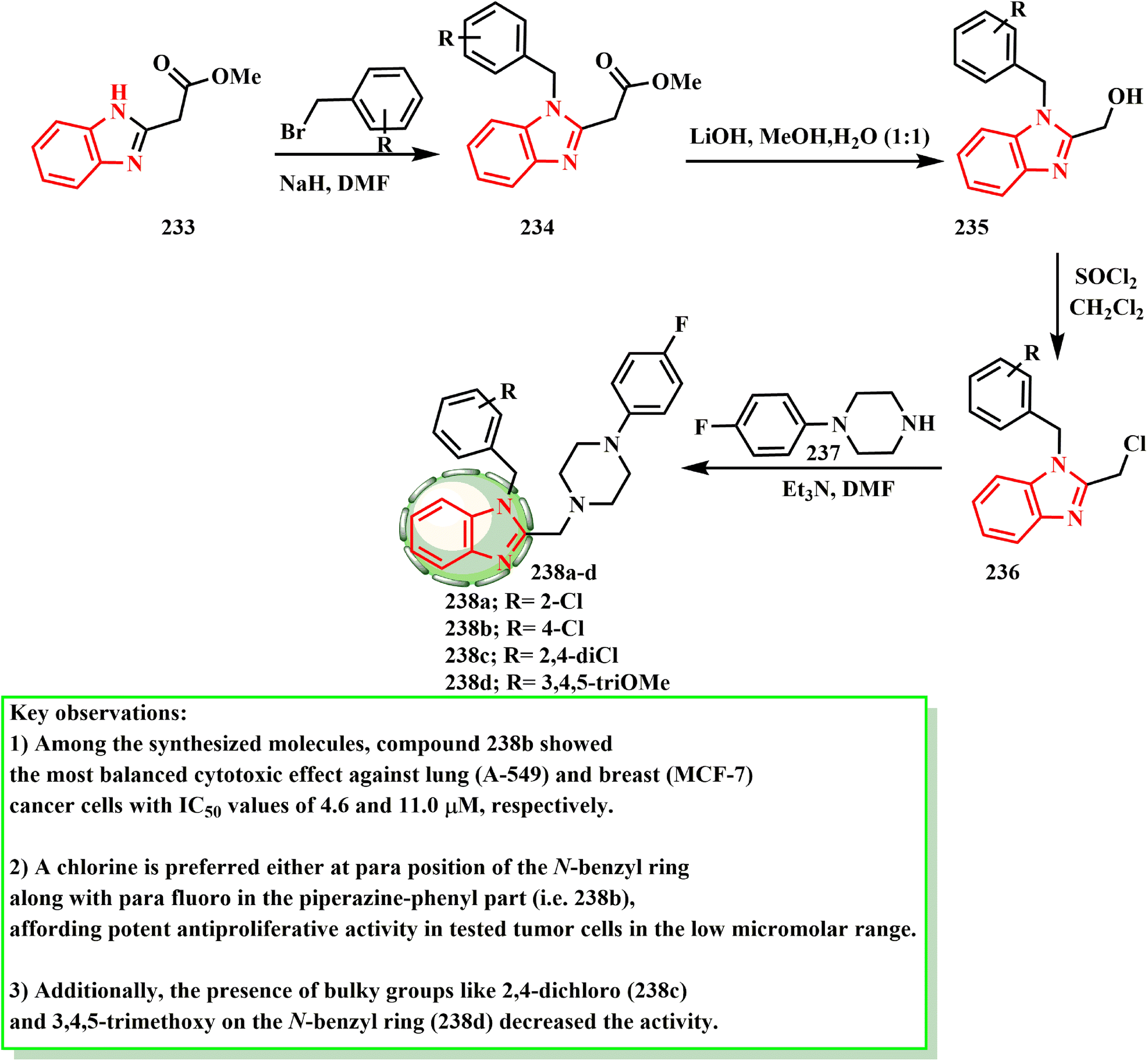 | ||
| Scheme 51 Synthesis of benzimidazole derivatives 238a–d. | ||
Scheme 52 showed the synthesis methods used for constructing the target thiazol–benzimidazole hybrids 241a and b. Thiosemicarbazone 239 (ref. 109–113) with 2-bromo-1-(1-methyl-1H-benzo[d]imidazo-2-yl)ethan-1-one (240) in refluxing ethanol114,115 afforded the corresponding targeted 2-(2-(substituted)hydrazinyl)-4-(1-methyl-1H-benzo [d]imidazol-2-yl)thiazole derivative116 241a and b. Compound 241a revealed strong cytotoxic activity by EGFR TK inhibition117 (IC50 = 109.71 nM)109 as well as an anti-breast cancer agent.118,119 SAR evaluations of this study revealed a notable decrease in potency when 4-nitrophenyl 241b was substituted with cyclopentyl (as in compound 241a). Additionally, it was found that the efficacy of the tested drug was reduced when the polarity of the molecules decreased and the bulkiness of the substituent increased.
 | ||
| Scheme 52 Route for constructing the target thiazolo–benzimidazole hybrids 241a and b. | ||
Subsequently, compound 245 was converted into the intended 2-amino-substituted analogues 246a and b in the reaction with an excess of the appropriate amine microwave irradiation utilizing the previously disclosed optimized reaction conditions120,121 (Scheme 53). Perin et al.122 produced amino-substituted N-methylated-benzimidazo[1,2-a]quinolines and tested them against human cancer cell lines in vitro to determine if they had any anti-proliferative properties. Two of the strongest substances, 246a and 246b, were specifically active against the HCT-116 cancer cell line, causing cell death and a reduction in the proportion of cells in the S phase (IC50 values of 0.2 and 0.4 μM, respectively). Between the acyclic derivatives, compound 246a with the N,N-dimethylaminopropyl substituent demonstrated stronger efficacy against the MCF-7 and HCT-116 cell lines, according to the SAR evaluations.
 | ||
| Scheme 53 Synthesis of new fused benzimidazole analogues 246a and b. | ||
Scheme 54 lists the synthetic pathways for the synthesis of 251. Substituted benzaldehyde 248 and acetophenone 247 were chosen and stirred in a pure form in 30% NaOH to produce chalcone 249. Intermediate compound 250 with compound 249 under reflux in absolute ethanol could be readily converted into the final compound 251.123 By causing cell cycle arrest in the G2/M phase and apoptosis by binding to active pockets of EGFR (IC50 = 0.97 μM), compound 251 demonstrated encouraging growth inhibition on the A549 cell line (IC50 = 2.2 μM).124 Additional research revealed that the effectiveness of compound 251 is attributed to its chloro atom at the 4-position of the phenyl substituent connected to C3 and C5 of the pyrazoline ring.
 | ||
| Scheme 54 Synthesis of benzimidazole–bearing pyrazole derivative 251. | ||
Therefore, the reaction between the pre-synthesized azide derivative 253 and propargyl molecule 252 (ref. 125 and 126) produced a structural alternative of 1,2,3-triazole–benzimidazole–chalcone hybrid 254 (Scheme 55) by using dichloromethane/water (1:1) as the solvent solution, catalyzed by CuSO4·5H2O and sodium ascorbate at room temperature. On all the chosen cancer cell lines, hybrid 254 with the chloro-substituent (2-Cl) demonstrated the strongest cytotoxic effect.127 Compared to doxorubicin, the most active compound, 254, was almost fourfold less active in MDA-MB-231 cells, forty-eight times less active in T47-D cells, and fifteen-fold less active in PC-3 cells. The documented chloro-substituted benzimidazole–triazole hybrids were already demonstrated to have good cytotoxic effects against mouse embryonic fibroblast cell lines NIH/3T3 (IC50 1.63 μM) according to their structure–activity-relationship (SAR).128 Additionally, the SAR showed that the inclusion of chloro-substituents in the chalcone ring increased the cytotoxic effect of the hybrid 1H-benzimidazole derivative against the PC-3, MDA-MB-231, and T47-D cell lines. Furthermore, the antiproliferative properties of the compound were further strengthened by the attachment of a benzyl moiety to the triazole ring.
 | ||
| Scheme 55 Reaction between the azide derivative 253 and propargyl molecule 252. | ||
In the tetrafluoropyridine rings (256), the amines all reacted at the next most electrophilic site, adding to C-2 to create aminopyridine derivatives 257a–c (ref. 129) (Scheme 56), adding to C-2, forming aminopyridine derivatives 257a–c (Scheme 56). Bhambra et al.45 established a library of fluoroaryl benzimidazole derivatives 257a–c, which showed micromolar inhibition against the K-562 and MCF-7 cell lines. The product obtained by adding ethylenediamine 257c was considerably less active against G361 and HOS, but exhibited good activity against two other cell lines. According to reports, compounds 257a and 257c activate caspases, which are crucial for the death of cancer-causing cells.
 | ||
| Scheme 56 Synthesis of different fluoroaryl benzimidazole derivatives 257a–c. | ||
An effective intermediary for the subsequent synthesis of various benzimidazole-heterocyclic compounds was thiosemicarbazone analogue 258. Thus, 2,5-dioxopyrrolidine derivative 259 was formed by the condensation of 258 with various acid anhydrides, such as succinic anhydride in acetic acid. Additionally, thiazolidinone derivative 260 was produced by cyclizing 258 with a haloketone, such as ethyl bromoacetate (Scheme 57). Abd El-Meguid et al.130 established a novel series of 1H-benzimidazole compounds, which were evaluated against the HeLa cell line using doxorubicin as the standard medication. The compounds with the strongest anti-cancer activity against the HeLa cell line were 259 (1.62 ± 0.16 μM) and 260 (1.44 ± 0.06 μM). According to the SAR evaluations, the cytotoxic potential was enhanced by an increase in nitrogen atoms and presence of pyrrolidine or thiazole rings.
 | ||
| Scheme 57 Reaction representative of benzimidazole with different reagents. | ||
The generalized process illustrated in Scheme 58 was used to synthesize each of the ruthenium compounds. The analogue of ruthenium metal complex 262a was obtained by treating benzimidazole ligand 261 with para-cymene ruthenium(II) [(p-cymene)RuCl2]2 and sodium acetate in dichloromethane for 24 h at room temperature. Pentamethylcyclopentadienyl chlorido iridium(III) was the starting point for the preparation of half-sandwich iridium(III) complexes 263. Yellol et al.131 established a variety of novel iridium(III) 263 and ruthenium(II) 262a and b C,N-cyclometalated benzimidazole complexes. The A2780 (ovary), 5637 (bladder), SISO (uterine cervical), and HT29 (rectal) human cancer cell lines were used to investigate their cytotoxicity. In the active hy926 (umbilical vein endothelial) human cell line, certain complexes were anti-angiogenic and triggered apoptosis by enhancing caspase-3 at a concentration of 0.5 μM. In contrast to their respective ligands, the metal complexes exhibited noticeably higher cell growth inhibitory rates. According to the SAR evaluations, ruthenium complex 262a was more potent than its iridium counterpart, compound 263, where a phenyl substitution on the 1H-benzimdazole enhanced the potency in both ligand complexes.
 | ||
| Scheme 58 Generalized process to synthesize benzimidazole complex 262a and b and 263. | ||
The ligand BIGH was produced using a well-known process132 (Scheme 59). By using DOX according to the MTT assay, the produced complexes were tested for their pro-apoptotic and anticancer effectiveness against the normal HEK293 (embryonic kidney), HeLa (cervical), MCF-7 (breast), and A549 human cancer cell lines.133 The produced complexes exhibited a moderate level of activity against the A549, HeLa and MCF-7 cancer cell lines. All the evaluated cancer cell lines were successfully inhibited by complexes 265a (IC50 HeLa = 36.70 ± 0.073 μM; IC50 MCF-7 = 43.20 ± 0.048 μM; and IC50 A-549 = 51.30 ± 0.018 μM) and 265b (IC50 HeLA = 45.85 ± 0.031 μM; IC50 MCF-7 = 51.30 ± 0.083 μM; and IC50 A549 = 32.30 ± 0.052 μM), respectively. The SAR studies indicate that the hydrophobic nature of the methyl-substituted bipyridyl contributed to the higher binding affinity of complex 265b by increasing its intercalation, whereas the electron-donating capacity of the methyl group in complex 265a enhanced the stacking of the substituted molecule.
 | ||
| Scheme 59 Reaction representative of benzimidazole with BIGH ligand. | ||
The synthesis and cytotoxic potential of chrysin-benzimidazole derivatives were examined by Wang et al.134 By first reacting the hydroxy group of 266 with dibromo alkane to yield 267, the most powerful derivatives were synthesized. The essential hybrid molecule 268 was later produced by reacting bromide derivative 267 with benzimidazole in acetone (Scheme 60). Wang et al.134 established a novel variety of chrysin-1H-benzimidazole compounds and tested them against the MFC cell line to see if they may be used as anticarcinogenic agents. Compound 268 was determined to be the most promising antiproliferative drug among the derivatives (IC50 = 25.72 ± 3.95 μM), causing MFC cells to undergo apoptosis and inhibiting the progression of the cell cycle in the G0/G1 phase in a dose-dependent manner. According to the SAR evaluations, the compounds with a methyl substituent at the 1H-benzimidazole ring 2-position had enhanced inhibitory efficacy compared to those with that at the 5- or 6-position of the ring. Additionally, investigations from the previous year revealed that when the OH group was at position 5 and the chrysin derivatives had higher polarity, they showed stronger anti-cancer effects.
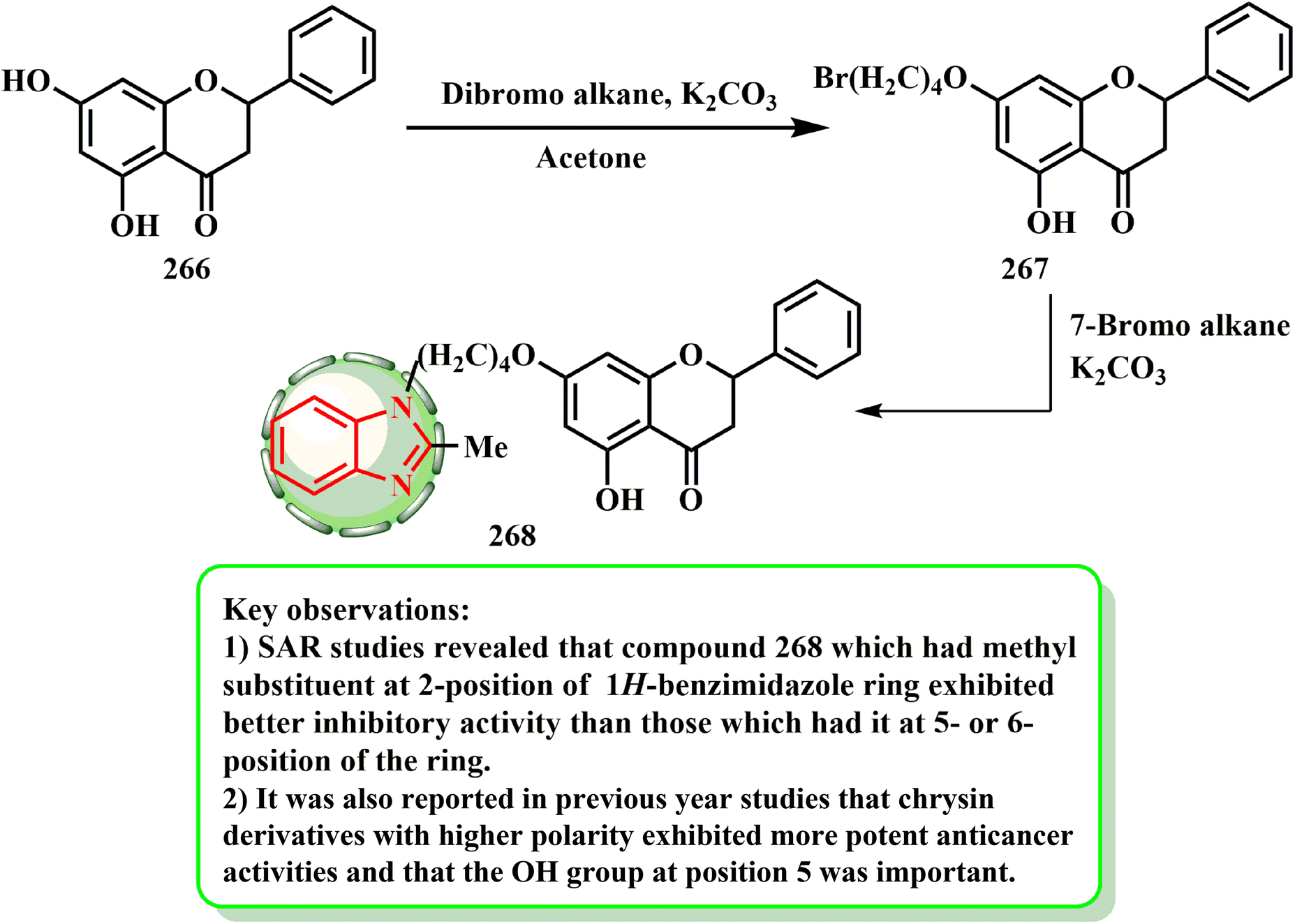 | ||
| Scheme 60 General synthesis of compound 268. | ||
Derivative 272 was prepared according to the route shown in Scheme 61. Trifluoroacetic acid was hydrolyzed to get derivatives 272, and after candesartan and triphenylmethyl bromide reacted to form intermediate 270, which was subsequently condensed with different substituted amines. Compared to candesartan cilexetic, the optimal benzimidazole-derived 272 demonstrated enhanced NAE inhibition, namely CDC (IC50 = 5.51 μM vs. 16.43 μM), together with effective target inhibitory action and selective death of cancerous cells according to research.135 In an enzyme assay, the optimal benzimidazole-derived 272 showed better neddylation inhibition than CDC. It also showed promising target inhibitory action and cancer cell killing selectivity. The results of cellular mechanism investigations and tumor growth suppression in A549 human lung cancer cells in vivo suggest that 272 may be developed as a promising neddylation inhibitor for anticancer treatment. According to SAR investigations, the core structures of tetrazole and 1H-benzimidazole are significant for increasing the neddylation inhibitory activities of the derivatives, whereas changing the substitutions of the heterocyclic cycloalyllic and benzoalylic groups diminished the activity of the compounds.
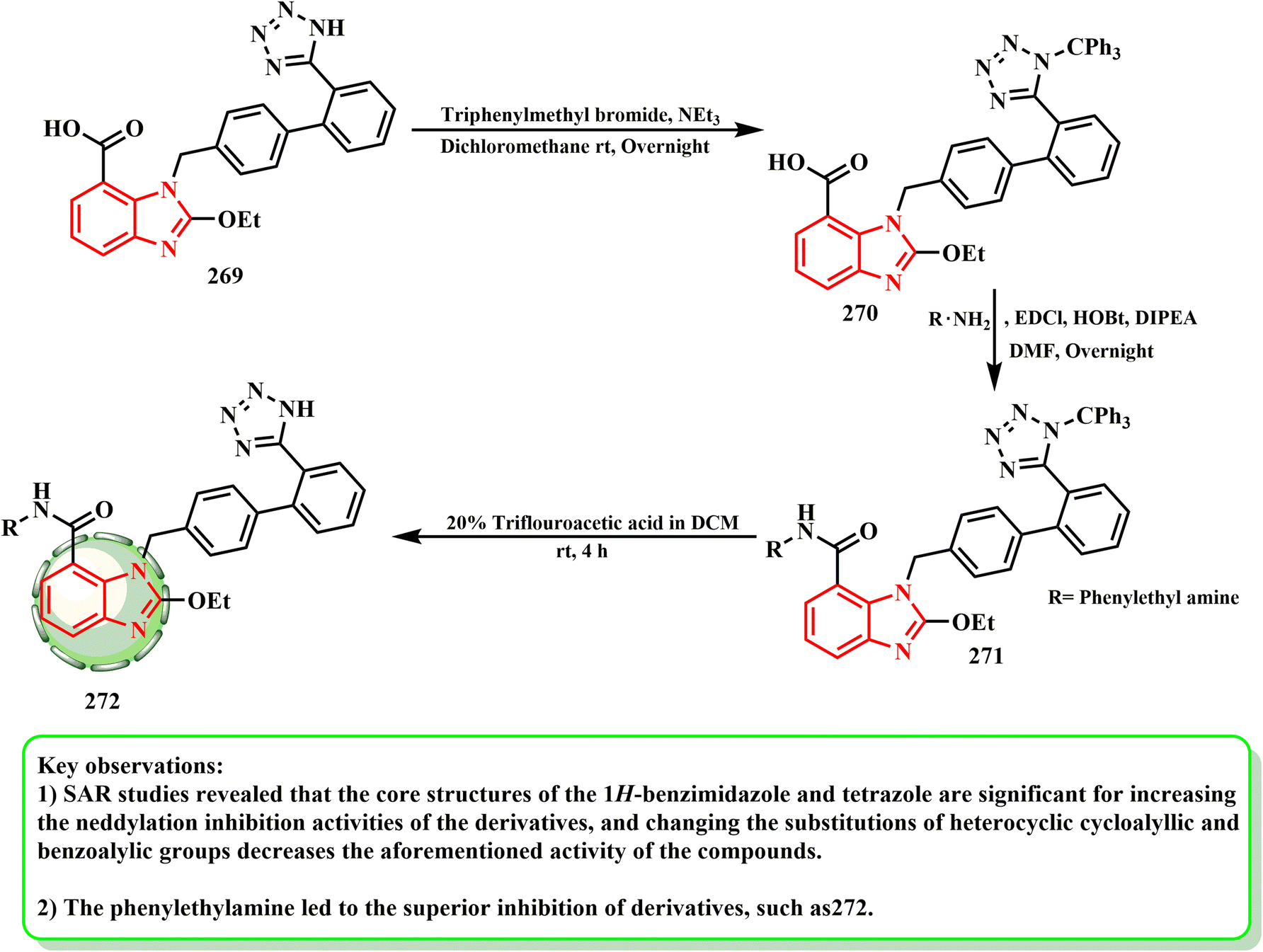 | ||
| Scheme 61 Preparation of derivative 272. | ||
The production and cytotoxic potential of pyrido[1,2-a]benzimidazole hybrids were examined by Samia et al.136 The initial reaction of chloro derivative 273 resulted in the development of the most effective derivative that was investigated,136 and dioxane with appropriate arylamines 274 produced the intended hybrid molecules 275a and b (Scheme 62). The anticancer activity results showed that compound 275b had the most significant inhibition, as indicated by the growth percentage (G%), against the breast cancer BT-549 (19.39%) and melanoma UACC-62 (11.90%) cell lines. Among the compounds tested, compound 275b was found to have promising anticancer activity. This might be because of how the biological activity profile is affected by the lipophilic trifluoromethyl substitution. Compound 275a exhibits weak activity against the breast cancer BT-549 (87.45%) and melanoma UACC-62 (86.58%) cell lines.
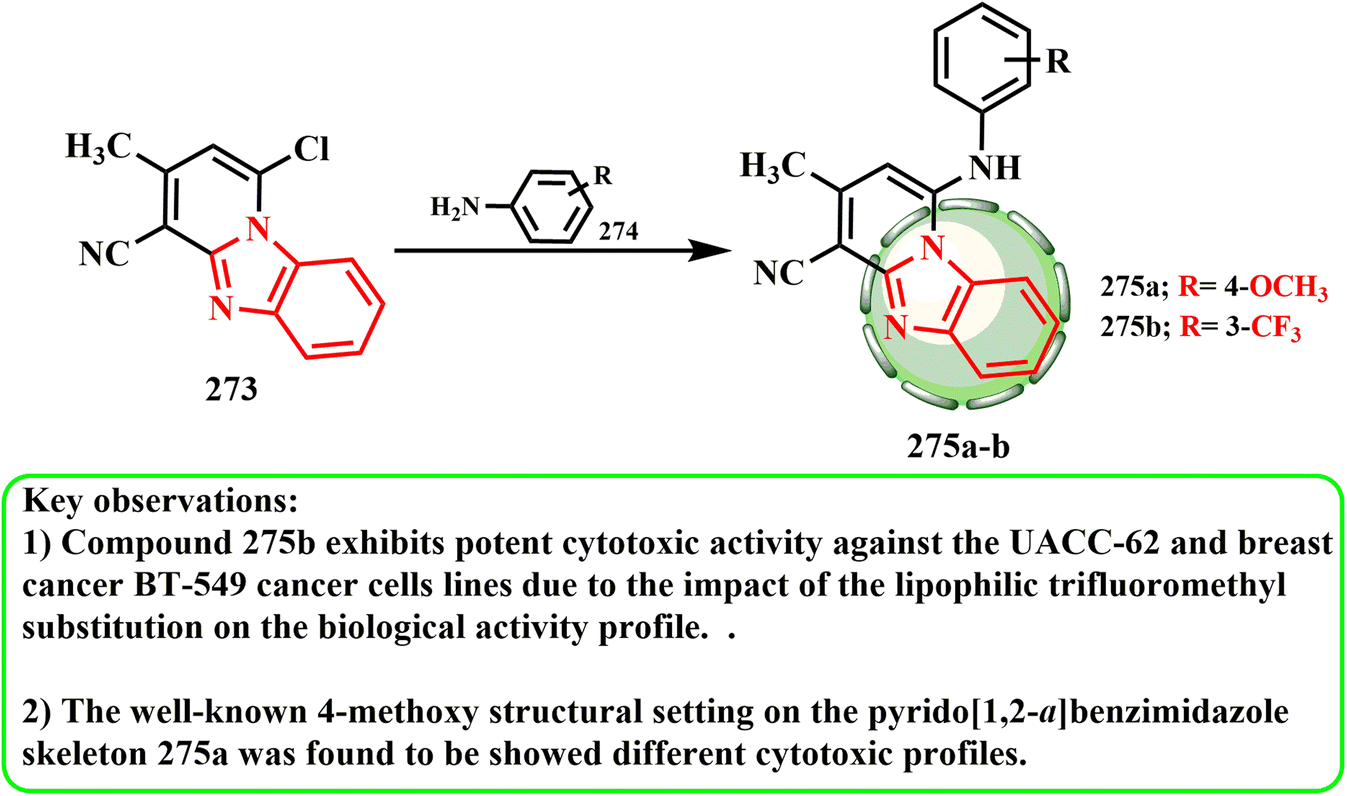 | ||
| Scheme 62 Synthesis of pyrido benzimidazole derivatives 275a and b. | ||
Scheme 63 describes the synthetic procedures used to establish logically constructed 1-(9H-pyrido[3,4-b]indol-1-yl) (281a and b) derivatives that are connected to benzimidazole and benzoxazole. Pure 2-bromo-1-(9H-pyrido[3,4-b]indol-1-yl)ethanone (277) was obtained by reacting the starting compound 1-(9H-pyrido[3,4-b]indol-1-yl)ethanone (276) with N-bromo succinimide (NBS) in dry dichloromethane (DCM) solvent and stirring the reaction mixture for approximately 30 min at room temperature. 1-(2-Methylthiazol-4-yl)-9H-pyrido[3,4-b]indole (278) was obtained by refluxing the resultant ethanone intermediate 277 with ethanethioamide in pure EtOH for approximately 12 h. The obtained thiazole 278 (ref. 137) was converted to pure 4-(9H-pyrido[3,4-b]indol-1-yl)thiazole-2-carbaldehyde (279) by oxidizing it with selenium dioxide in ethanol for 24 h at 100 °C. To produce their respective benzimidazoles/benzoxazoles as pure final compounds 281a and b, aldehyde 279 was finally allowed to react with different substituted 1,2-diamine 280 in the presence of diacetoxyiodobenzene (IBD) in dry 1,4-dioxane by stirring at room temperature for around 15 min. In the study conducted by Sireesha et al.,138 a novel type of 1H-benzimidazole-linked β-carboline was produced, and its anticancer activity against a range of human cancer cell lines was evaluated. Compounds 281a (IC50 = 0.092 ± 0.001 μM) and 281b (IC50 = 0.81 ± 0.062 μM) had the greatest anticarcinogenic properties against the MCF-7 cell line, according to the findings. According to the SAR evaluations, compound 281a was more active than the reference standard due to the presence of 4,5-dimethoxy substitutions (MCF-7 = 0.092 ± 0.001 μM, A549 = 0.72 ± 0.042 μM, Colo-205 = 0.34 ± 0.071 μM, and A-2780 = 1.23 ± 0.55 μM). Alternatively, compound 281b showed good action on four cancer cell lines despite having weak electron-donating 4,5-dimethyl substitutions on the benzimidazole moiety (A-2780 = 1.80 ± 0.59 μM, MCF-7 = 0.81 ± 0.062 μM, A549 = 1.90 ± 0.88 μM, and Colo-205 = 0.41 ± 0.12 μM).
 | ||
| Scheme 63 Synthesis of benzimidazole analogous 281a and b. | ||
 | ||
| Scheme 64 Synthesis of fused benzimidazole–isoquinolinone scaffolds 286a and b. | ||
Tahlan et al.140 employed ((1H-benzo[d]imidazol-2-ylthio)acetamido)benzo hydrazide to establish a novel azomethine of 2-mercapto-1H-benzimidazole. Using 5-fluorouracil as the standard medication, they were screened in vitro using the SRB assay against the HCT-116 (colorectal) human cancer cell line. Compound 287 was found to be the most potent of all the synthesized derivatives and have strong anticancer activity (IC50 = 30 μg mL−1). According to the SAR study findings, the anticancer potential was enhanced by substituting a branched aliphatic aldehyde moiety (compound 287) (Fig. 1).
 | ||
| Fig. 1 2-Mercapto-1H-benzimidazole nucleus with anticancer activity. | ||
However, compound 288 had strong antiproliferative activity against MCF-7 cells, with an IC50 of 5.58 μg mL−1, which was similar to the IC50 of the common therapeutic doxorubicin, which has an IC50 of 4.1 μg mL−1. The fundamental manner of action of compound 288 was to induce cell cycle arrest and apoptosis at the G2/M phase. A thiocarbamate linker was necessary for action, according to the SAR investigation, as shown in Fig. 2.141
 | ||
| Fig. 2 Benzimidazole moiety with its antiproliferative activity. | ||
Compound 289 (ref. 142) B-norcholesteryl benzimidazole demonstrated activity when tested against the HeLa, MCF-7, T-47D, and SKOV3 cell lines, with an IC50 in the range of 7.90 μM to 20.10 μM.143,144 The SAR145 study demonstrated that the electron-donating group enhanced the insertion of this molecule into DNA142 (Fig. 3).
 | ||
| Fig. 3 IH-Benzimidazole scaffold with its in vitro studies. | ||
Hybrid 290 (ref. 146) exhibited strong antiproliferative properties; its GI50 values against the SNB-75 and COLO 205 cell lines were 0.09 μM and 0.35 μM, respectively. Additionally, with a selectivity index of 3.66, it demonstrated moderate selectivity towards prostate cancer cell lines. According to SAR research, the benzimidazole derivatives with a 1,3,4-oxadiazole ring exhibited stronger anticancer properties than those with other heterocyclic rings147,148 (Fig. 4).
 | ||
| Fig. 4 Hybrid 290 with its antiproliferative properties. | ||
Moreover, compound 291 (ref. 149 and 150) had IC50 values of 8.91 μM, 10.93 μM, and 10.67 μM against the MCF-7, HepG-2, and OVCAR-3 cell lines, respectively, indicating its promising antiproliferative action. Alternatively, cisplatin had IC50 values of 11.70 μM, 3.97 μM, and 16.04 μM, respectively.151 According to the SAR investigations, the activities were attributed to the N-substitution of benzimidazole with a nitrogen-linked hydrocarbon spacer149,152 (Fig. 5).
 | ||
| Fig. 5 Benzimidazole–chalcone hybrid 291 with its promising antiproliferative character. | ||
Compounds 292a and b had IC50 values in the range of 6.83 μM to 18.16 μM. Both substances exhibited strong affinity for the tyrosine kinase receptor. P-Methoxy phenyl substituents and unsubstituted phenyl rings are crucial for the anticancer action of the chalcones, according to the SAR studies153 (Fig. 6).
 | ||
| Fig. 6 Benzimidazole–chalcone hybrids 292a and b with their SAR. | ||
When tested against the MDA-MB-231 and HCT-116 cell lines, complexes 293a–c demonstrated greater anticancer activity than their respective benzimidazole ligands, with IC50 values ranging from 4.22 μM to 10.3 μM. Additionally, their ligands showed reduced activity, with IC50 values between 25.51 μM and 34.21 μM, which were similar to that of the reference pharmaceuticals 5-FU (IC50 = 5.5 μM against HCT-116 cells) and tamoxifen (IC50 = 8.20 μM against MDA-MB-231 cells). Increasing the side chain length improved the activity of the complexes, according to a SAR investigation, in which it additionally contributed to making the benzimidazole ring more lipophilic154 (Fig. 7).
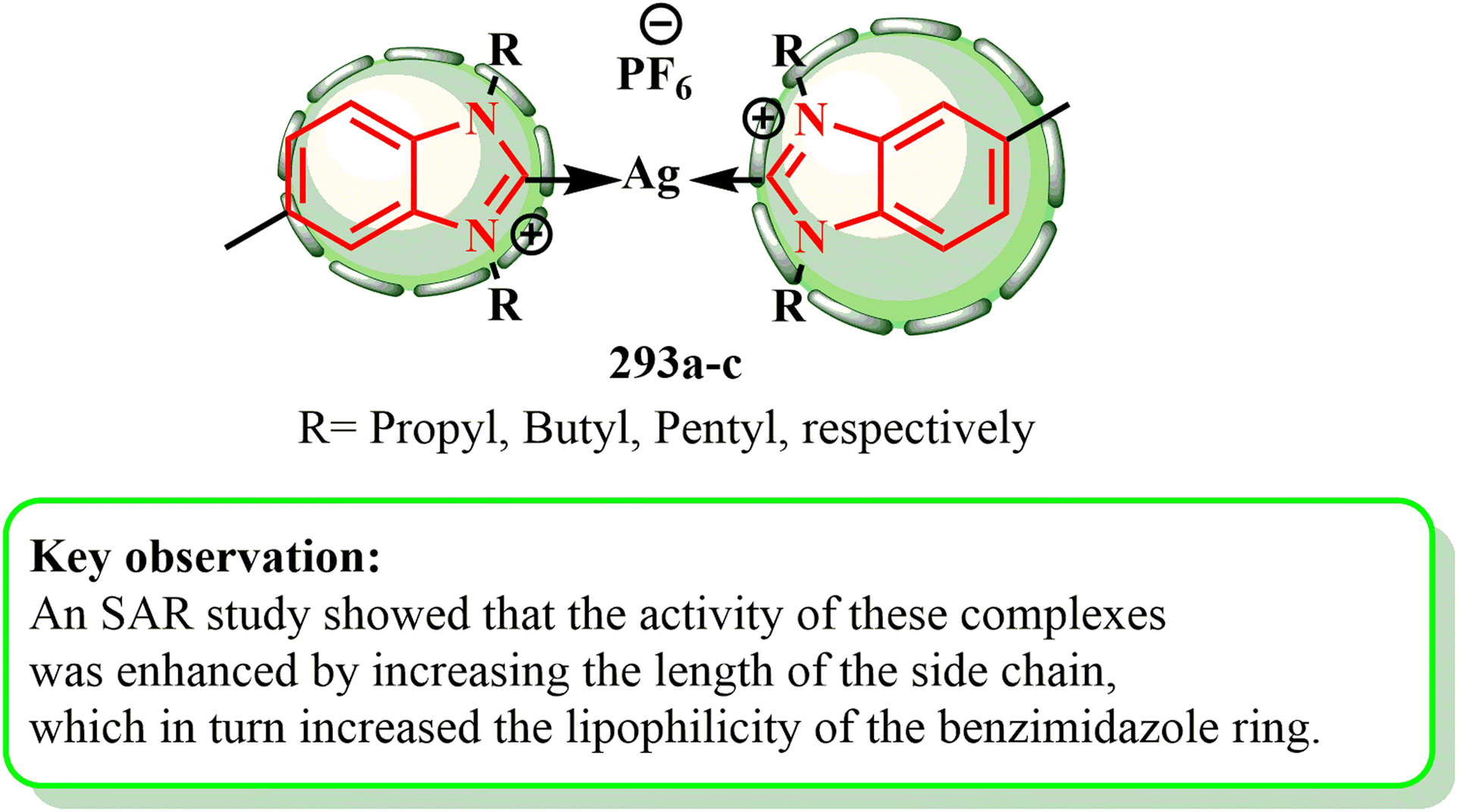 | ||
| Fig. 7 Benzimidazole complexes 293a–c with their SAR. | ||
Methyl 2-(5-fluoro-2-hydroxyphenyl)-1H-benzimidazole-5-carboxylate (MBIC)155 (294) with a methyl ester is substituted with a hydroxyl group at C-2, whereas a fluoro group at the C5 position in the aryl ring inhibits microtubule156 and is very active against breast cancer cells157,158 (Fig. 8).
 | ||
| Fig. 8 1H-Benzimidazole-5-carboxylate 294 with its activity on MCF-7 cell line. | ||
With an IC50 of 10.69 ± 0.14 μM, 13.89 ± 0.74 μM, 7.01 ± 0.20 μM, 14.04 ± 0.62 μM, and 12.91 ± 0.52 μM against the A-549, DU-145, MCF-7, MDA-MB-231, and HCT-116 cancer cell lines, the 4-hydroxy compound 295 showed a noticeable decrease in tumor cell proliferation (Fig. 9). The 4-hydroxy arylamide moiety 295 of the 1H-benzyl benzimidazole derivative had strong anticancer activity, according to the SAR analysis. MCF-7 development was significantly inhibited when aromatic or heterocyclic rings, such as pyrrole, 3-ethoxy-4-hydroxyphen, and 3,5-dimethoxy-4-hydroxyphen, were connected to benzyl benzimidazole scaffolds.159
 | ||
| Fig. 9 Representative SAR studies of 1-benzyl-1H-benzimidazole analogues as anticancer agents. | ||
Further investigation of the distinct 1-benzyl-1H-benzimidazole hybrids for selective gal-1 inhibition resulted in the development of a variety of novel 1-benzyl-1H-benzimidazole-triazole analogues due to the promoting results of 1-benzyl-1H-benzimidazole analogues as possible anticancer agents mediated by gal-1. The 1,3-dipolar cycloaddition of the benzimidazole intermediate with a terminal alkyne group to various benzyl and phenyl azides in the presence of a copper(I) catalyst produced 1-benzyl-1H-benzimidazole-triazole hybrids. 4-Triflourophenyl compound 296 showed no cytotoxicity against normal embryonic kidney cells, but it displayed cytotoxic function against MCF-7, NCI-H460, MDA-MB-231, A-549, and HaCaT cells with IC50 values of 1.3 ± 0.18 μM, 0.99 ± 0.01 μM, 0.94 ± 0.02 μM, 0.63 ± 0.21 μM, and 2.99 ± 0.09 μM, respectively. The SAR investigations demonstrated that in comparison to electron-donating groups and in the absence of any substitution on the aromatic ring, electron-withdrawing groups on the aromatic ring have remarkably greater cytotoxicity (Fig. 10).160
 | ||
| Fig. 10 Representative SAR studies of 1-benzyl-1H-benzimidazole-triazole analogues as anticancer agents. | ||
Hagar et al.161 benzimidazole-1,3,4-oxadiazole–chalcone hybrids were established and synthesized (Fig. 11), and their ability to inhibit EGFR for cell destruction was studied. According to cell line tests, compound 297, which has a paramethoxyphenyl group at the second position of the benzimidazole ring, demonstrated potential potency in inhibiting apoptosis and EGFR inhibition.
 | ||
| Fig. 11 Benzimidazole-containing moiety exhibiting anticancer activity. | ||
The anticancer activities of indole–benzimidazole derivatives were reported.162 Among the evaluated compounds, 298a (ref. 163) (R1 = methyl and R2 = Br) had the highest activity, with an IC50 value of 28.73 μM. As a result, compound 298a may have anticancer properties together with potential modulatory effects on estrogen and the corresponding receptors. The p-fluorophenyl scaffold molecule 298b exhibited less productive anticancer properties, according to the structural activity relationship (SAR). At this point, the anticancer activity was reduced when p-chlorophenyl 298c was substituted for p-fluorophenyl (Fig. 12).
 | ||
| Fig. 12 Indole–benzimidazole derivatives with anticancer properties. | ||
Under efficient metal-free conditions, benzimidazoldiphenyl-2-imino-thiazolidine-4-ol derivatives 299a–c were synthesized.164–166 Doxorubicin was administered as a control for evaluating the targeted compounds against four distinct human cancer cell lines, including breast, colon, prostate, and lung panels. In comparison to doxorubicin (IC50 = 1.75 μM), compounds 299a (IC50 = 3.89 μM), 299b (IC50 = 2.80 μM), and 299c (IC50 = 3.14 μM) had the strongest inhibitory activity against the lung cancer cell line among the published compounds characterized for anticancer activity. Additionally, compared to trifluoromethylphenyl functionalities 299a, the inhibitory effect was enhanced by the fluorophenyl functions 299b–c (Fig. 13).
 | ||
| Fig. 13 Benzimidazoldiphenyl-2-imino-thiazolidine-4-ol 299a–c derivatives with anticancer properties. | ||
In another study, Li et al.167 showed that a panel of benzimidazole–rhodamine conjugates have potent antiproliferative properties against human cervical, breast, lung, and prostate cancer cells, as well as human lymphoma and acute leukemia. As non-intercalative Topo II inhibitors, compounds 300a and b bind to the Topo II enzyme ATP-binding site to inhibit enzymatic activity. The Topo II inhibitory action was significantly influenced by the benzyl and electron donor groups in the compounds (Fig. 14).
 | ||
| Fig. 14 Benzimidazole–rhodanine conjugates 300a and b with anticancer properties. | ||
2 Conclusion
The structural analogy of benzimidazole to nucleosides presents it as a potentially effective anticancer agent. The metal complexes or benzimidazole hybrids with antiproliferative properties were presented in this review. The examples included in this overview demonstrated the variety of synthetic preparation techniques used to achieve benzimidazoles and their antiproliferative properties. Additionally, we focused our attention to the SAR of the various molecular templates based on BZ that have been established by researchers globally. To accomplish our objectives, we collected information from an extensive range of publications to provide researchers, medicinal chemists, and drug designers with an excellent foundation for the development of the next generation of safe and effective BZ-based therapy. This review may shed light on the wide range of cancers that benzimidazoles can target, such as MCF-7, HepG2, MGC-803, HeLa, HCT-116, A-549, PC-3, MDA-MB-231, HUVEC, NIH/3T3, RMS, C-26, HT1080, LNCaP, 22Rv1, C4-2B prostate, DU-145, HEK293, MCF12A, H69AR, A2780, SISO, HT29, HCC2998, SF-539, UACC-62, and breast cancers BT-549, SW620, K-562, A37, SNB-75, COLO 205, and OVCAR-3. Their ability to disrupt important cellular processes such as Topo II-mediated DNA, VGEF, hCA IX, cell cycle progression, and mitosis has been demonstrated by investigations. This can lead to novel possibilities for the development of precision medicine-based benzimidazole anticancer drugs. Furthermore, this is the route forward to develop new medicinal compounds and benzimidazole anticancer drugs.Data availability
The article describes a study that did not utilize any data.Author contributions
Basant Farag, Sobhi M. Gomha, and Doaa A. Elsayed contributed to the conceptualization, supervision, and writing – review and editing. Basant Farag and Doaa A. Elsayed performed formal analysis and data curation. Magdi Zaki and Doaa A. Elsayed were responsible for investigation, methodology, and validation. Basant Farag and Doaa A. Elsayed led the writing – original draft preparation. All authors read and approved the final version of the manuscript.Conflicts of interest
The authors declare that they have no known financial conflicts of interest or personal relationships that could have influenced the work presented in this study.Acknowledgements
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) under grant number IMSIU-DDRSP2501.References
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Global cancer statistics, Ca-Cancer J. Clin., 2011, 61(2), 69–90 Search PubMed.
- V. Careaga and M. Maier, Handbook of Anticancer Drugs from Marine Origin, 2015 Search PubMed.
- J. Ferlay, H. Shin, F. Bray, D. Forman, C. Mathers and D. Parkin, GLOBOCAN V1. 2, Cancer Incidence and Mortality Worldwide: IARC Cancer Base No. 10, International Agency for Research on Cancer, Lyon, France, 2010, vol. 2008 Search PubMed.
- M. Mareel and A. Leroy, Clinical, cellular, and molecular aspects of cancer invasion, Physiol. Rev., 2003, 83(2), 337–376 Search PubMed.
- J. Wesche, K. Haglund and E. M. Haugsten, Fibroblast growth factors and their receptors in cancer, Biochem. J., 2011, 437(2), 199–213 Search PubMed.
- G. Vardatsikos, A. Sahu and A. K. Srivastava, The insulin-like growth factor family: molecular mechanisms, redox regulation, and clinical implications, Antioxid. Redox Signaling, 2009, 11(5), 1165–1190 CrossRef CAS PubMed.
- A. A. Bhat, N. Tandon and R. Tandon, Pyrrolidine Derivatives as Anti-diabetic Agents: Current Status and Future Prospects, ChemistrySelect, 2022, 7(6), e202103757 CrossRef CAS.
- S. Vijayaraghavalu, C. Peetla, S. Lu and V. Labhasetwar, Epigenetic modulation of the biophysical properties of drug-resistant cell lipids to restore drug transport and endocytic functions, Mol. Pharm., 2012, 9(9), 2730–2742 CrossRef CAS PubMed.
- A. A. Bhat, I. Singh, N. Tandon and R. Tandon, Structure activity relationship (SAR) and anticancer activity of pyrrolidine derivatives: Recent developments and future prospects (A review), Eur. J. Med. Chem., 2023, 246, 114954 CrossRef CAS PubMed.
- H. Sachdeva, S. Khaturia, M. Saquib, N. Khatik, A. R. Khandelwal, R. Meena and K. Sharma, Oxygen-and sulphur-containing heterocyclic compounds as potential anticancer agents, Appl. Biochem. Biotechnol., 2022, 194(12), 6438–6467 CrossRef CAS PubMed.
- A. Amin, T. Qadir, P. K. Sharma, I. Jeelani and H. Abe, A review on the medicinal and industrial applications of N-containing heterocycles, Open Med. Chem. J., 2022, 16(1), e187410452209010 CrossRef CAS.
- B. Negi and A. Kwatra, A Review of Recent Progress on the Anticancer Activity of Heterocyclic Compounds, SynOpen, 2024, 8(03), 185–210 CrossRef CAS.
- J. Valdez, R. Cedillo, A. Hernandez-Campos, L. Yepez, F. Hernandez-Luis, G. Navarrete-Vazquez, A. Tapia, R. Cortes, M. Hernández and R. Castillo, Synthesis and antiparasitic activity of 1H-benzimidazole derivatives, Bioorg. Med. Chem. Lett., 2002, 12(16), 2221–2224 CrossRef CAS PubMed.
- F. M. Abdelrazek, M. E. Zaki, S. A. Al-Hussain, B. Farag, A. M. Hebishy, M. S. Abdelfattah, S. M. Hassan, A. F. El-Farargy, L. Iovkova and D. Mross, Facile one-pot synthesis and in silico study of new heterocyclic scaffolds with 4-pyridyl moiety: Mechanistic insights and X-ray crystallographic elucidation, Heliyon, 2024, 10(7), e29221 CrossRef CAS PubMed.
- H. Debus, Ueber die einwirkung des ammoniaks auf glyoxal, Justus Liebigs Ann. Chem., 1858, 107(2), 199–208 CrossRef.
- G. Satija, B. Sharma, A. Madan, A. Iqubal, M. Shaquiquzzaman, M. Akhter, S. Parvez, M. A. Khan and M. M. Alam, Benzimidazole based derivatives as anticancer agents: Structure activity relationship analysis for various targets, J. Heterocycl. Chem., 2022, 59(1), 22–66 CrossRef CAS.
- R. Ranjith, The chemistry and biological significance of imidazole, benzimidazole, benzoxazole, tetrazole and quinazolinone nucleus, J. Chem. Pharm. Res., 2016, 8(5), 505–526 Search PubMed.
- D. I. Othman, A. Hamdi, S. S. Tawfik, A. A. Elgazar and A. S. Mostafa, Identification of new benzimidazole-triazole hybrids as anticancer agents: multi-target recognition, in vitro and in silico studies, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2166037 CrossRef PubMed.
- N. A. Nawareg, A. S. Mostafa, S. M. El-Messery and M. N. Nasr, New benzimidazole based hybrids: Synthesis, molecular modeling study and anticancer evaluation as TopoII inhibitors, Bioorg. Chem., 2022, 127, 106038 CrossRef CAS PubMed.
- I. Sović, S. Jambon, S. K. Pavelić, E. Markova-Car, N. Ilić, S. Depauw, M.-H. David-Cordonnier and G. Karminski-Zamola, Synthesis, antitumor activity and DNA binding features of benzothiazolyl and benzimidazolyl substituted isoindolines, Bioorg. Med. Chem., 2018, 26(8), 1950–1960 CrossRef PubMed.
- C. Sheng, Z. Miao and W. Zhang, New strategies in the discovery of novel non-camptothecin topoisomerase I inhibitors, Curr. Med. Chem., 2011, 18(28), 4389–4409 CrossRef CAS PubMed.
- S. A. Galal, K. H. Hegab, A. M. Hashem and N. S. Youssef, Synthesis and antitumor activity of novel benzimidazole-5-carboxylic acid derivatives and their transition metal complexes as topoisomerease II inhibitors, Eur. J. Med. Chem., 2010, 45(12), 5685–5691 CrossRef CAS PubMed.
- R. M. Noha, M. K. Abdelhameid, M. M. Ismail, M. R. Mohammed and E. Salwa, Design, synthesis and screening of benzimidazole containing compounds with methoxylated aryl radicals as cytotoxic molecules on (HCT-116) colon cancer cells, Eur. J. Med. Chem., 2021, 209, 112870 CrossRef CAS PubMed.
- A. Saini, G. P. Kumar and G. Singh, A review of Benzimidazole derivatives potential activities, Int. J. Pharm. Clin. Res., 2023, 5, 35–40 CrossRef.
- W. Zhou, W. Zhang, Y. Peng, Z.-H. Jiang, L. Zhang and Z. Du, Design, synthesis and anti-tumor activity of novel benzimidazole-chalcone hybrids as non-intercalative topoisomerase II catalytic inhibitors, Molecules, 2020, 25(14), 3180 CrossRef CAS PubMed.
- J. L. Delgado, C.-M. Hsieh, N.-L. Chan and H. Hiasa, Topoisomerases as anticancer targets, Biochem. J., 2018, 475(2), 373–398 CrossRef CAS PubMed.
- W. Hu, X.-S. Huang, J.-F. Wu, L. Yang, Y.-T. Zheng, Y.-M. Shen, Z.-Y. Li and X. Li, Discovery of novel topoisomerase II inhibitors by medicinal chemistry approaches, J. Med. Chem., 2018, 61(20), 8947–8980 Search PubMed.
- S. Wang, L. Guan, J. Zang, K. Xing, J. Zhang, D. Liu and L. Zhao, Structure-based design of novel benzimidazole derivatives as Pin1 inhibitors, Molecules, 2019, 24(7), 1198 CrossRef PubMed.
- J. Gowda, A. Khader, B. Kalluraya and S. Hidayathulla, Synthesis, characterization and antibacterial activity of benzimidazole derivatives carrying quinoline moiety, Indian J. Chem., 2011, 50B, 1491–1495 CAS.
- N. El-Gohary and M. Shaaban, Synthesis, antimicrobial, antiquorum-sensing and antitumor activities of new benzimidazole analogs, Eur. J. Med. Chem., 2017, 137, 439–449 Search PubMed.
- K. Petkar, P. Parekh, P. Mehta, A. Kumari and A. Baro, Synthesis & evaluation of 2-chloromethyl-1H-benzimidazole derivatives as antifungal agents, Int. J. Pharm. Pharm. Sci., 2013, 5(2), 115–119 Search PubMed.
- N. El-Gohary and M. Shaaban, Synthesis and biological evaluation of a new series of benzimidazole derivatives as antimicrobial, antiquorum-sensing and antitumor agents, Eur. J. Med. Chem., 2017, 131, 255–262 CrossRef CAS PubMed.
- G. Sindhu, R. Kholiya, S. Kidwai, P. Singh, R. Singh and D. S. Rawat, Design and synthesis of benzimidazole derivatives as antimycobacterial agents, J. Biochem. Mol. Toxicol., 2022, 36(9), e23123 CrossRef CAS PubMed.
- B. Soni, A short review on potential activities of benzimidazole derivatives, PharmaTutor, 2014, 2(8), 110–118 CAS.
- M. M. Morcoss, E. S. Abdelhafez, H. M. Abdel-Rahman, M. Abdel-Aziz, A. El-Ella and A. Dalal, Novel Benzimidazole/Hydrazone Derivatives as Promising Anticancer Lead Compounds: Design, Synthesis, and Molecular Docking Study, J. Adv. Biomedical Pharm. Sci., 2020, 3(2), 45–52 Search PubMed.
- J. Zhang, D. Yao, Y. Jiang, J. Huang, S. Yang and J. Wang, Synthesis and biological evaluation of benzimidazole derivatives as the G9a Histone Methyltransferase inhibitors that induce autophagy and apoptosis of breast cancer cells, Bioorg. Chem., 2017, 72, 168–181 CrossRef CAS PubMed.
- P. Sharma, T. S. Reddy, N. P. Kumar, K. R. Senwar, S. K. Bhargava and N. Shankaraiah, Conventional and microwave-assisted synthesis of new 1H-benzimidazole-thiazolidinedione derivatives: A potential anticancer scaffold, Eur. J. Med. Chem., 2017, 138, 234–245 CrossRef CAS PubMed.
- U. Acar Çevik, B. N. Sağlık, D. Osmaniye, S. Levent, B. Kaya Çavuşoğlu, A. B. Karaduman, Ö. Atlı Eklioğlu, Y. Özkay and Z. A. Kaplancıklı, Synthesis, anticancer evaluation and molecular docking studies of new benzimidazole-1,3,4-oxadiazole derivatives as human topoisomerase types I poison, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 1657–1673 Search PubMed.
- H. T. Abdel-Mohsen and A. M. Nageeb, Benzimidazole–dioxoisoindoline conjugates as dual VEGFR-2 and FGFR-1 inhibitors: design, synthesis, biological investigation, molecular docking studies and ADME predictions, RSC Adv., 2024, 14(39), 28889–28903 RSC.
- P. Astles, T. Brown, C. Handscombe, M. Harper, N. Harris, R. Lewis, P. Lockey, C. McCarthy, I. McLay and B. Porter, Selective endothelin A receptor ligands. 1. Discovery and structure-activity of 2,4-disubstituted benzoic acid derivatives, Eur. J. Med. Chem., 1997, 32(5), 409–423 Search PubMed.
- D. Kumar, M. R. Jacob, M. B. Reynolds and S. M. Kerwin, Synthesis and evaluation of anticancer benzoxazoles and benzimidazoles related to UK-1, Bioorg. Med. Chem., 2002, 10(12), 3997–4004 CrossRef CAS PubMed.
- S. B. Srinivasa, B. Poojary, B. S. Kalal, U. Brahmavara, D. Vaishali, A. J. Das, T. M. Kalenga, M. Paidikondala and M. K. Shankar, Design, synthesis and anticancer activity of Novel benzimidazole containing quinoline hybrids, Results Chem., 2024, 9, 101631 CrossRef CAS.
- S.-T. Huang, I.-J. Hsei and C. Chen, Synthesis and anticancer evaluation of bis (benzimidazoles), bis(benzoxazoles), and benzothiazoles, Bioorg. Med. Chem., 2006, 14(17), 6106–6119 CrossRef CAS PubMed.
- L. Xi, J.-Q. Zhang, Z.-C. Liu, J.-H. Zhang, J.-F. Yan, Y. Jin and J. Lin, Novel 5-anilinoquinazoline-8-nitro derivatives as inhibitors of VEGFR-2 tyrosine kinase: synthesis, biological evaluation and molecular docking, Org. Biomol. Chem., 2013, 11(26), 4367–4378 RSC.
- A. S. Bhambra, M. Edgar, M. R. Elsegood, L. Horsburgh, V. Kryštof, P. D. Lucas, M. Mojally, S. J. Teat, T. G. Warwick and G. W. Weaver, Novel fluorinated benzimidazole-based scaffolds and their anticancer activity in vitro, J. Fluorine Chem., 2016, 188, 99–109 CrossRef CAS.
- X. Yuan, Q. Yang, T. Liu, K. Li, Y. Liu, C. Zhu, Z. Zhang, L. Li, C. Zhang and M. Xie, Design, synthesis and in vitro evaluation of 6-amide-2-aryl benzoxazole/benzimidazole derivatives against tumor cells by inhibiting VEGFR-2 kinase, Eur. J. Med. Chem., 2019, 179, 147–165 Search PubMed.
- M. Sugumaran and M. Y. Kumar, Synthesis and biological activity of novel 2,5-disubstituted benzimidazole derivatives, Int. J. Pharm. Sci. Drug Res., 2012, 4(1), 80–83 CrossRef CAS.
- F. Hoebrecker, Benzimidazole, Ber, 1872,(5), 920–926 CrossRef.
- S. Banerjee, S. Mukherjee, P. Nath, A. Mukherjee, S. Mukherjee, S. A. Kumar, S. De and S. Banerjee, A critical review of benzimidazole: Sky-high objectives towards the lead molecule to predict the future in medicinal chemistry, Results Chem., 2023, 6, 101013 CrossRef CAS.
- E. C. Pham, T. V. Le Thi, H. H. L. Hong, B. N. V. Thi, L. B. Vong, T. T. Vu, D. D. Vo, N. V. T. Nguyen, K. N. B. Le and T. N. Truong, N,2,6-Trisubstituted 1H-benzimidazole derivatives as a new scaffold of antimicrobial and anticancer agents: design, synthesis, in vitro evaluation, and in silico studies, RSC Adv., 2023, 13(1), 399–420 RSC.
- T.-K.-C. Huynh, T.-H.-A. Nguyen, N.-H.-S. Tran, T.-D. Nguyen and T.-K.-D. Hoang, A facile and efficient synthesis of benzimidazole as potential anticancer agents, J. Chem. Sci., 2020, 132, 1–9 CrossRef.
- U. Acar Cevik, B. Nurpelin Saglik, Y. Ozkay, Z. Canturk, J. Bueno, F. Demirci and A. Savas Koparal, Synthesis of new fluoro-benzimidazole derivatives as an approach towards the discovery of novel Intestinal antiseptic drug candidates, Curr. Pharm. Des., 2017, 23(15), 2276–2286 Search PubMed.
- E. O. Hamed, M. G. Assy, N. H. Ouf, D. A. Elsayed and M. H. Abdellattif, Cyclization of N-acetyl derivative: Novel synthesis–azoles and azines, antimicrobial activities, and computational studies, Heterocycl. Commun., 2022, 28(1), 35–43 CrossRef CAS.
- U. Acar Çevik, B. N. Sağlık, C. M. Ardıç, Y. Özkay and Ö. Atlı, Synthesis and evaluation of new benzimidazole derivatives with hydrazone moiety as anticancer agents, Turk. J. Biochem., 2018, 43(2), 151–158 CrossRef.
- H. T. Abdel-Mohsen, M. A. Abdullaziz, A. M. El Kerdawy, F. A. Ragab, K. J. Flanagan, A. E. Mahmoud, M. M. Ali, H. I. El Diwani and M. O. Senge, Targeting receptor tyrosine kinase VEGFR-2 in hepatocellular cancer: rational design, synthesis and biological evaluation of 1,2-disubstituted benzimidazoles, Molecules, 2020, 25(4), 770 CrossRef CAS PubMed.
- F. Sonmez, B. Zengin Kurt, I. Gazioglu, L. Basile, A. Dag, V. Cappello, T. Ginex, M. Kucukislamoglu and S. Guccione, Design, synthesis and docking study of novel coumarin ligands as potential selective acetylcholinesterase inhibitors, J. Enzyme Inhib. Med. Chem., 2017, 32(1), 285–297 CrossRef CAS PubMed.
- H. A. Abdel-Aziz, H. A. Ghabbour, W. M. Eldehna, S. T. Al-Rashood, K. A. Al-Rashood, H.-K. Fun, M. Al-Tahhan and A. Al-Dhfyan, 2-((Benzimidazol-2-yl)thio)-1-arylethan-1-ones: Synthesis, crystal study and cancer stem cells CD133 targeting potential, Eur. J. Med. Chem., 2015, 104, 1–10 CrossRef CAS PubMed.
- H. S. Ibrahim, M. E. Albakri, W. R. Mahmoud, H. A. Allam, A. M. Reda and H. A. Abdel-Aziz, Synthesis and biological evaluation of some novel thiobenzimidazole derivatives as anti-renal cancer agents through inhibition of c-MET kinase, Bioorg. Chem., 2019, 85, 337–348 CrossRef CAS PubMed.
- S. Rawat and M. Ghate, Benzimidazole-Urea Derivatives as Anti-cancer Agents: In-silico Study, Synthesis and In-vitro Evaluation, Int. J. Health Sci., 2022, 6(S5), 5187–5217 CrossRef.
- Y.-L. Zhang, R. Yang, L.-Y. Xia, R.-J. Man, Y.-C. Chu, A.-Q. Jiang, Z.-C. Wang and H.-L. Zhu, Synthesis, anticancer activity and molecular docking studies on 1,2-diarylbenzimidazole analogues as anti-tubulin agents, Bioorg. Chem., 2019, 92, 103219 CrossRef CAS PubMed.
- R. Goel, V. Luxami and K. Paul, Palladium catalyzed novel monoarylation and symmetrical/unsymmetrical diarylation of imidazo[1,2-a]pyrazines and their in vitro anticancer activities, RSC Adv., 2014, 4(19), 9885–9892 RSC.
- H. Zeng, D. B. Belanger, P. J. Curran, G. W. Shipps Jr, H. Miao, J. B. Bracken, M. A. Siddiqui, M. Malkowski and Y. Wang, Discovery of novel imidazo[1,2-a]pyrazin-8-amines as Brk/PTK6 inhibitors, Bioorg. Med. Chem. Lett., 2011, 21(19), 5870–5875 CrossRef CAS PubMed.
- I. Singh, V. Luxami and K. Paul, Effective synthesis of benzimidazoles-imidazo[1,2-a]pyrazine conjugates: A comparative study of mono-and bis-benzimidazoles for antitumor activity, Eur. J. Med. Chem., 2019, 180, 546–561 CrossRef CAS PubMed.
- W. Shehta, F. Agili, B. Farag, S. A. Said, S. Youssif, M. E. Abdraboh and S. El-Kalyoubi, Design, synthesis and antitumor activity of novel pyran-functionalized uracil derivatives: docking studies, Future Med. Chem., 2023, 15(5), 421–436 CrossRef CAS PubMed.
- R. Sivaramakarthikeyan, S. Iniyaval, V. Saravanan, W.-M. Lim, C.-W. Mai and C. Ramalingan, Molecular hybrids integrated with benzimidazole and pyrazole structural motifs: Design, synthesis, biological evaluation, and molecular docking studies, ACS Omega, 2020, 5(17), 10089–10098 CrossRef CAS PubMed.
- X. Wu, H. Shen, Y. Zhang, C. Wang, Q. Li, C. Zhang, X. Zhuang, C. Li, Y. Shi and Y. Xing, Discovery and characterization of benzimidazole derivative XY123 as a potent, selective, and orally available RORγ inverse agonist, J. Med. Chem., 2021, 64(12), 8775–8797 CrossRef CAS PubMed.
- S. M. Gomha, S. M. Riyadh, R. A. Alharbi, M. E. Zaki, T. Z. Abolibda and B. Farag, Green route synthesis and molecular docking of azines using cellulose sulfuric acid under microwave irradiation, Crystals, 2023, 13(2), 260 Search PubMed.
- J. E. Cheong, M. Zaffagni, I. Chung, Y. Xu, Y. Wang, F. E. Jernigan, B. R. Zetter and L. Sun, Synthesis and anticancer activity of novel water soluble benzimidazole carbamates, Eur. J. Med. Chem., 2018, 144, 372–385 CrossRef CAS PubMed.
- W. S. Shehab, M. M. Amer, D. A. Elsayed, K. K. Yadav and M. H. Abdellattif, Current progress toward synthetic routes and medicinal significance of quinoline, Med. Chem. Res., 2023, 32(12), 2443–2457 CrossRef CAS.
- S. M. Gomha, S. M. Riyadh, B. Farag, S. A. Al-Hussain, M. E. Zaki and M. A. Mohamed, Green synthesis of hydrazono-thiazolones using vitamin B1 and their antibacterial implications, Green Chem. Lett. Rev., 2024, 17(1), 2380746 CrossRef.
- A. Y. Alzahrani, S. M. Gomha, M. E. Zaki, B. Farag, F. E. Abdelgawad and M. A. Mohamed, Chitosan–sulfonic acid-catalyzed green synthesis of naphthalene-based azines as potential anticancer agents, Future Med. Chem., 2024, 16(7), 647–663 CrossRef CAS PubMed.
- N. Perin, L. Hok, A. Beč, L. Persoons, E. Vanstreels, D. Daelemans, R. Vianello and M. Hranjec, N-substituted benzimidazole acrylonitriles as in vitro tubulin polymerization inhibitors: Synthesis, biological activity and computational analysis, Eur. J. Med. Chem., 2021, 211, 113003 CrossRef CAS PubMed.
- D. N. Kommi, D. Kumar, R. Bansal, R. Chebolu and A. K. Chakraborti, “All-water” chemistry of tandem N-alkylation–reduction–condensation for synthesis of N-arylmethyl-2-substituted benzimidazoles, Green Chem., 2012, 14(12), 3329–3335 RSC.
- J. M. Humphrey and A. R. Chamberlin, Chemical synthesis of natural product peptides: coupling methods for the incorporation of noncoded amino acids into peptides, Chem. Rev., 1997, 97(6), 2243–2266 CrossRef CAS PubMed.
- S. I. Alaqeel, Synthetic approaches to benzimidazoles from o-phenylenediamine: A literature review, J. Saudi Chem. Soc., 2017, 21(2), 229–237 CrossRef CAS.
- N. S. Goud, S. M. Ghouse, J. Vishnu, D. Komal, V. Talla, R. Alvala, J. Pranay, J. Kumar, I. A. Qureshi and M. Alvala, Synthesis of 1-benzyl-1H-benzimidazoles as galectin-1 mediated anticancer agents, Bioorg. Chem., 2019, 89, 103016 CrossRef CAS PubMed.
- J. Jang, B. Koh and K. Lee, Discovery of benzimidazole-2-amide BNZ-111 as new tubulin inhibitor, Bioorg. Med. Chem. Lett., 2024, 113, 129953 Search PubMed.
- W. S. Shehab, H. A. Haikal, D. A. Elsayed, A. F. El-Farargy, A.-R. B. El-Gazzar, G. T. El-Bassyouni and S. M. Mousa, Pharmacokinetic and molecular docking studies to pyrimidine drug using Mn3O4 nanoparticles to explore potential anti-Alzheimer activity, Sci. Rep., 2024, 14(1), 15436 CrossRef CAS PubMed.
- F. King, R. Acheson and P. Spensley, 275. Benziminazole analogues of paludrine, J. Chem. Soc., 1948, 1366–1371 RSC.
- S. A. El-Hawash, E.-S. A. Badawey and I. M. El-Ashmawey, Nonsteroidal antiinflammatory agents—part 2 antiinflammatory, analgesic and antipyretic activity of some substituted 3-pyrazolin-5-ones and 1,2,4,5,6,7-3H-hexahydroindazol-3-ones, Eur. J. Med. Chem., 2006, 41(2), 155–165 CrossRef CAS PubMed.
- M. M. Ismail, H. El-Sehrawi, H. S. Elzahabi, T. Shawer and Y. A. Ammar, Synthesis and antitumor activity of novel hybrids of pyrimidine/benzimidazole scaffolds, Polycyclic Aromat. Compd., 2022, 42(5), 2363–2377 CrossRef CAS.
- L. Sun, C. Liang, S. Shirazian, Y. Zhou, T. Miller, J. Cui, J. Y. Fukuda, J.-Y. Chu, A. Nematalla and X. Wang, Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2, 4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase, J. Med. Chem., 2003, 46(7), 1116–1119 CrossRef CAS PubMed.
- N. K. Rasal, R. B. Sonawane and S. V. Jagtap, Potential 2,4-dimethyl-1H-pyrrole-3-carboxamide bearing benzimidazole template: Design, synthesis, in vitro anticancer and in silico ADME study, Bioorg. Chem., 2020, 97, 103660 CrossRef CAS PubMed.
- S. A. Galal, A. S. Abdelsamie, S. A. Shouman, Y. M. Attia, H. I. Ali, A. Tabll, R. El-Shenawy, Y. S. El Abd, M. M. Ali and A. E. Mahmoud, Part I: Design, synthesis and biological evaluation of novel pyrazole-benzimidazole conjugates as checkpoint kinase 2 (Chk2) inhibitors with studying their activities alone and in combination with genotoxic drugs, Eur. J. Med. Chem., 2017, 134, 392–405 CrossRef CAS PubMed.
- M. F. Baig, V. L. Nayak, P. Budaganaboyina, K. Mullagiri, S. Sunkari, J. Gour and A. Kamal, Synthesis and biological evaluation of imidazo[2,1-b]thiazole-benzimidazole conjugates as microtubule-targeting agents, Bioorg. Chem., 2018, 77, 515–526 CrossRef CAS PubMed.
- S. Q. Al-Sultan, M. H. Mohammed and W. H. Talib, Newly designed 2-(aminomethyl) benzimidazole derivatives as possible tyrosine kinase inhibitors: synthesis, characterization, preliminary cytotoxic evaluation and in Silico studies, Chem. Rev. Lett., 2024, 7(3), 545–559 CAS.
- E. Akgoz and S. Colak, Synthesis and antiproliferative activities against breast cancer of N-(benzimidazol-2-yl)-substituted benzamide derivatives, Bioorg. Med. Chem. Rep., 2024, 7(1), 1–9 Search PubMed.
- R. A. Haque, M. A. Iqbal, M. B. Khadeer Ahamed, A. A. Majid and Z. A. Abdul Hameed, Design, synthesis and structural studies of meta-xylyl linked bis-benzimidazolium salts: potential anticancer agents against ‘human colon cancer’, Chem. Cent. J., 2012, 6, 1–14 CrossRef PubMed.
- R. A. Haque and M. A. Iqbal, Synthesis and characterization of ortho-xylyl linked bis-benzimidazolium salts, Asian J. Chem., 2012, 24(6), 2625–2628 CAS.
- N. ul Huda, S. Islam, M. Zia, K. William, F. i Abbas, M. I. Umar, M. A. Iqbal and A. Mannan, Anticancer, antimicrobial and antioxidant potential of sterically tuned bis-N-heterocyclic salts, Z. Naturforsch., C: J. Biosci., 2019, 74(1–2), 17–23 CAS.
- S. Bouchekioua, S. Akkoc and R. Menacer, In Vitro and In Silico Studies on Benzimidazole-Based Compounds, ChemistrySelect, 2024, 9(1), e202304347 CrossRef CAS.
- D. Başat Dereli, S. Akkoc and E. Çubuk Demiralay, Protonation Constant Determination of Some Benzimidazole-Based Drug Candidates by Reverse Phase Liquid Chromatography Method and Cytotoxic Activity Studies, ChemistrySelect, 2023, 8(28), e202301192 CrossRef.
- S. C. Yavuz, Synthesis of new two 1,2-disubstituted benzimidazole compounds: their in vitro anticancer and in silico molecular docking studies, BMC Chem., 2024, 18(1), 146 CrossRef CAS PubMed.
- Y. Ren, Y. Wang, G. Li, Z. Zhang, L. Ma, B. Cheng and J. Chen, Discovery of novel benzimidazole and indazole analogues as tubulin polymerization inhibitors with potent anticancer activities, J. Med. Chem., 2021, 64(8), 4498–4515 CrossRef CAS PubMed.
- M. S. Ibrahim, B. Farag, J. Y. Al-Humaidi, M. E. Zaki, M. Fathalla and S. M. Gomha, Mechanochemical Synthesis and Molecular Docking Studies of New Azines Bearing Indole as Anticancer Agents, Molecules, 2023, 28(9), 3869 CrossRef CAS PubMed.
- Z. ALAGÖZ, C. Kus and T. Coban, Synthesis and antioxidant properties of novel benzimidazoles containing substituted indole or 1,1,4,4-tetramethyl-1,2,3,4-tetrahydro-naphthalene fragments, J. Enzyme Inhib. Med. Chem., 2005, 20(4), 325–331 CrossRef PubMed.
- F. Z. Karadayi, M. Yaman, M. M. Kisla, A. G. Keskus, O. Konu and Z. Ates-Alagoz, Design, synthesis and anticancer/antiestrogenic activities of novel indole-benzimidazoles, Bioorg. Chem., 2020, 100, 103929 CrossRef CAS PubMed.
- Q.-L. Gao, B.-W. Wu, D. Li, L. Shi, T. Zhu, J.-F. Lou, C.-Y. Jin, Y.-B. Zhang, S.-Y. Zhang and H.-M. Liu, Novel tertiary sulfonamide derivatives containing benzimidazole moiety as potent anti-gastric cancer agents: Design, synthesis and SAR studies, Eur. J. Med. Chem., 2019, 183, 111731 CrossRef PubMed.
- Ü. Yılmaz, S. Tekin, N. Buğday, K. Yavuz, H. Küçükbay and S. Sandal, Synthesis and evaluation of anticancer properties of novel benzimidazole ligand and their cobalt (II) and zinc (II) complexes against cancer cell lines A-2780 and DU-145, Inorg. Chim. Acta, 2019, 495, 118977 CrossRef.
- C. Xie, X. Han, J. Gong, D. Li and C. Ma, One-pot strategy of copper-catalyzed synthesis of 1,2-disubstituted benzimidazoles, Org. Biomol. Chem., 2017, 15(27), 5811–5819 RSC.
- H. Küçükbay, N. Şireci, Ü. Yılmaz, M. Akkurt, Ş. P. Yalçın, M. Nawaz Tahir and H. Ott, Synthesis, characterization and microwave-assisted catalytic activity of novel benzimidazole salts bearing piperidine and morpholine moieties in Heck cross-coupling reactions, Appl. Organomet. Chem., 2011, 25(4), 255–261 CrossRef.
- Ü. Yılmaz, H. Küçükbay, S. Deniz and N. Şireci, Synthesis, characterization and microwave-promoted catalytic activity of novel N-phenylbenzimidazolium salts in Heck-Mizoroki and Suzuki-Miyaura cross-coupling reactions under mild conditions, Molecules, 2013, 18(3), 2501–2517 CrossRef.
- G. H. Al-Ansary, W. M. Eldehna, H. A. Ghabbour, S. T. Al-Rashood, K. A. Al-Rashood, R. A. Eladwy, A. Al-Dhfyan, M. M. Kabil and H. A. Abdel-Aziz, Cancer stem cells CD133 inhibition and cytotoxicity of certain 3-phenylthiazolo[3,2-a]benzimidazoles: design, direct synthesis, crystal study and in vitro biological evaluation, J. Enzyme Inhib. Med. Chem., 2017, 32(1), 986–991 CrossRef CAS PubMed.
- A. A. Alkhaldi, M. M. Al-Sanea, A. Nocentini, W. M. Eldehna, Z. M. Elsayed, A. Bonardi, M. F. Abo-Ashour, A. K. El-Damasy, M. S. Abdel-Maksoud and T. Al-Warhi, 3-Methylthiazolo [3,2-a]benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: Design, synthesis, biological and molecular modeling studies, Eur. J. Med. Chem., 2020, 207, 112745 CrossRef CAS PubMed.
- H. Atmaca, S. İlhan, M. B. Batır, Ç. Ç. Pulat, A. Güner and H. Bektaş, Novel benzimidazole derivatives: Synthesis, in vitro cytotoxicity, apoptosis and cell cycle studies, Chem.-Biol. Interact., 2020, 327, 109163 CrossRef CAS PubMed.
- S. Yadav, B. Narasimhan, S. M. Lim, K. Ramasamy, M. Vasudevan, S. A. A. Shah and A. Mathur, Synthesis and evaluation of antimicrobial, antitubercular and anticancer activities of benzimidazole derivatives, Egypt. J. Basic Appl. Sci., 2018, 5(1), 100–109 Search PubMed.
- P. Dubey, P. P. Reddy and K. Srinivas, Alternate Method for the Synthesis of N-Alkyl/aralkyl-2-(α-hydroxy Alkyl/aralkyl) benzimidazoles via Regiospecific Acetylation, Synth. Commun., 2007, 37(10), 1675–1681 CrossRef CAS.
- A. Özdemir, S. Turanli, B. Çalişkan, M. Arka and E. Banoglu, Evaluation of cytotoxic activity of new benzimidazole-piperazine hybrids against human MCF-7 and A549 cancer cells, Pharm. Chem. J., 2020, 53, 1036–1046 CrossRef.
- A. M. Srour, N. S. Ahmed, S. S. Abd El-Karim, M. M. Anwar and S. M. El-Hallouty, Design, synthesis, biological evaluation, QSAR analysis and molecular modelling of new thiazol-benzimidazoles as EGFR inhibitors, Bioorg. Med. Chem., 2020, 28(18), 115657 CrossRef CAS PubMed.
- W. Yi, C. Dubois, S. Yahiaoui, R. Haudecoeur, C. Belle, H. Song, R. Hardré, M. Réglier and A. Boumendjel, Refinement of arylthiosemicarbazone pharmacophore in inhibition of mushroom tyrosinase, Eur. J. Med. Chem., 2011, 46(9), 4330–4335 CrossRef CAS PubMed.
- G. D. Coxon, D. Craig, R. M. Corrales, E. Vialla, L. Gannoun-Zaki and L. Kremer, Synthesis, antitubercular activity and mechanism of resistance of highly effective thiacetazone analogues, PLoS One, 2013, 8(1), e53162 CrossRef CAS PubMed.
- M.-H. Shih, Y.-S. Su and C.-L. Wu, Syntheses of aromatic substituted hydrazino-thiazole derivatives to clarify structural characterization and antioxidant activity between 3-arylsydnonyl and aryl substituted hydrazino-thiazoles, Chem. Pharm. Bull., 2007, 55(8), 1126–1135 CrossRef CAS PubMed.
- R. B. de Oliveira, E. M. de Souza-Fagundes, R. P. Soares, A. A. Andrade, A. U. Krettli and C. L. Zani, Synthesis and antimalarial activity of semicarbazone and thiosemicarbazone derivatives, Eur. J. Med. Chem., 2008, 43(9), 1983–1988 CrossRef CAS PubMed.
- V. M. Reddy and K. R. Reddy, Synthesis and antimicrobial activity of some novel 4-(1H-benz [d]imidazol-2yl)-1,3-thiazol-2-amines, Chem. Pharm. Bull., 2010, 58(7), 953–956 CrossRef CAS PubMed.
- R. Polunin, A. Kozytskiy and S. Kolotilov, Photovoltaic Characteristics of Bis(2-Benzimidazolyl)-Bisthiazole Deposited on TiO2 in the Presence of Zn2+ Ions, Theor. Exp. Chem., 2015, 51, 196–201 CrossRef CAS.
- S. M. Gomha, A.-A. A. El-Sayed, A. Alrehaily, H. M. Elbadawy, B. Farag, A. A. Al-Shahri, S. R. Alsenani, F. E. Abdelgawad and M. E. Zaki, Synthesis, molecular docking, in silico study, and evaluation of bis-thiazole-based curcumin derivatives as potential antimicrobial agents, Results Chem., 2024, 7, 101504 CrossRef CAS.
- J. Y. Al-Humaidi, S. M. Riyadh, B. Farag, M. E. Zaki, T. Z. Abolibda and S. M. Gomha, Synthesis of Bis-thiazoles Tethered 1,4-Dihydropyridine and Pyridine Linkers via Simple Oxidation and their Molecular Docking as VEGFR-TK Inhibitors, Curr. Med. Chem., 2025, 32(9), 1789–1802 CrossRef CAS PubMed.
- T. Z. Abolibda, M. Fathalla, B. Farag, M. E. Zaki and S. M. Gomha, Synthesis and molecular docking of some novel 3-thiazolyl-coumarins as inhibitors of VEGFR-2 kinase, Molecules, 2023, 28(2), 689 CrossRef CAS PubMed.
- W. Shehta, F. Agili, B. Farag, S. Youssif, S. J. Almehmadi, S. M. Elfeky and S. El-Kalyoubi, Synthesis and in vitro study of pyrimidine–phthalimide hybrids as VEGFR2 inhibitors with antiproliferative activity, Future Med. Chem., 2023, 15(8), 661–677 CrossRef CAS PubMed.
- N. Perin, R. Nhili, M. Cindrić, B. Bertoša, D. Vušak, I. Martin-Kleiner, W. Laine, G. Karminski-Zamola, M. Kralj and M.-H. David-Cordonnier, Amino substituted benzimidazo [1, 2-a]quinolines: Antiproliferative potency, 3D QSAR study and DNA binding properties, Eur. J. Med. Chem., 2016, 122, 530–545 CrossRef CAS PubMed.
- N. Perin, K. Bobanović, I. Zlatar, D. Jelić, V. Kelava, S. Koštrun, V. G. Marković, K. Brajša and M. Hranjec, Antiproliferative activity of amino substituted benzo[b]thieno[2,3-b] pyrido[1,2-a]benzimidazoles explored by 2D and 3D cell culture system, Eur. J. Med. Chem., 2017, 125, 722–735 CrossRef CAS PubMed.
- N. Perin, S. Škulj, I. Martin-Kleiner, M. Kralj and M. Hranjec, Synthesis and antiproliferative activity of novel 2-substituted N-methylated benzimidazoles and tetracyclic benzimidazo[1,2-a] quinolines, Polycyclic Aromat. Compd., 2018, 40(2), 343–354 CrossRef.
- M. J. Akhtar, A. A. Siddiqui, A. A. Khan, Z. Ali, R. P. Dewangan, S. Pasha and M. S. Yar, Design, synthesis, docking and QSAR study of substituted benzimidazole linked oxadiazole as cytotoxic agents, EGFR and erbB2 receptor inhibitors, Eur. J. Med. Chem., 2017, 126, 853–869 CrossRef CAS PubMed.
- M. J. Akhtar, A. A. Khan, Z. Ali, R. P. Dewangan, M. Rafi, M. Q. Hassan, M. S. Akhtar, A. A. Siddiqui, S. Partap and S. Pasha, Synthesis of stable benzimidazole derivatives bearing pyrazole as anticancer and EGFR receptor inhibitors, Bioorg. Chem., 2018, 78, 158–169 CrossRef CAS PubMed.
- E. O. Hamed, D. A. Elsayed, M. G. Assy and W. S. Shehab, Design, Synthesis, Docking, 2D-QSAR Modelling, Anticancer and Antioxidant Evaluation of Some New Azo-Compounds Derivatives and Investigation of Their Fluorescence Properties, ChemistrySelect, 2022, 7(41), e202202534 CrossRef CAS.
- D. A. Elsayed, Design, Synthesis, Biological Evaluation, and Molecular Docking Studies of novel Azo-compounds derivatives as Significant Antioxidants, Bull. Fac. Sci. Zagazig Univ., 2023, 2023(1), 1–9 CrossRef.
- A. Djemoui, A. Naouri, M. R. Ouahrani, D. Djemoui, S. Lahcene, M. B. Lahrech, L. Boukenna, H. M. Albuquerque, L. Saher and D. H. Rocha, A step-by-step synthesis of triazole-benzimidazole-chalcone hybrids: Anticancer activity in human cells+, J. Mol. Struct., 2020, 1204, 127487 CrossRef CAS.
- H. Karaca Gençer, U. Acar Çevik, S. Levent, B. N. Sağlık, B. Korkut, Y. Özkay, S. Ilgın and Y. Öztürk, New benzimidazole-1,2,4-triazole hybrid compounds: Synthesis, anticandidal activity and cytotoxicity evaluation, Molecules, 2017, 22(4), 507 CrossRef PubMed.
- S. A. Ouf, S. M. Gomha, B. Farag, M. E. Zaki, M. M. Ewies, I. A. Sharawy, F. O. Khalil and H. K. Mahmoud, Synthesis of novel Bis-1,2,4-Triazolo[3,4-b][1,3,4]Thiadiazines from natural camphoric acid as potential anti-candidal agents, Results Chem., 2024, 7, 101406 CrossRef CAS.
- E. A. Abd El-Meguid, E. M. M. El-Deen, M. A. Nael and M. M. Anwar, Novel benzimidazole derivatives as anti-cervical cancer agents of potential multi-targeting kinase inhibitory activity, Arabian J. Chem., 2020, 13(12), 9179–9195 CrossRef.
- J. Yellol, S. A. Perez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak and A. Espinosa, Novel C, N-cyclometalated benzimidazole ruthenium (II) and iridium (III) complexes as antitumor and antiangiogenic agents: a structure–activity relationship study, J. Med. Chem., 2015, 58(18), 7310–7327 CrossRef CAS PubMed.
- P. Verweij, J. Van der Geest, W. Driessen, J. Reedijk and D. Sherrington, Metal uptake by a novel benzimidazole ligand immobilized onto poly (glycidyl methacrylate-co-ethylene glycol dimethacrylate), React. Polym., 1992, 18(3), 191–201 CrossRef CAS.
- T. Sabithakala and V. R. R. Chittireddy, DNA binding and in vitro anticancer activity of 2-((1H-benzimidazol-2-yl)methylamino)acetic acid and its copper (II) mixed-polypyridyl complexes: Synthesis and crystal structure, Appl. Organomet. Chem., 2018, 32(12), e4550 CrossRef.
- Z. Wang, X. Deng, S. Xiong, R. Xiong, J. Liu, L. Zou, X. Lei, X. Cao, Z. Xie and Y. Chen, Design, synthesis and biological evaluation of chrysin benzimidazole derivatives as potential anticancer agents, Nat. Prod. Res., 2018, 32(24), 2900–2909 Search PubMed.
- X. Chen, X. Yang, F. Mao, J. Wei, Y. Xu, B. Li, J. Zhu, S. Ni, L. Jia and J. Li, Development of novel benzimidazole-derived neddylation inhibitors for suppressing tumor growth in vitro and in vivo, Eur. J. Med. Chem., 2021, 210, 112964 CrossRef CAS PubMed.
- S. M. Rida, F. S. Soliman, E. S. A. Badawey and T. Kappe, Benzimidazole condensed ring systems. 2. New synthesis of substituted 1-oxo-1H,5H-pyrido[1,2-a]benzimidazole-4-carbonitriles and related derivatives, J. Heterocycl. Chem., 1988, 25(6), 1725–1728 CrossRef CAS.
- J. Y. Al-Humaidi, S. M. Gomha, N. A. A. El-Ghany, B. Farag, M. E. Zaki, T. Z. Abolibda and N. A. Mohamed, Green synthesis and molecular docking study of some new thiazoles using terephthalohydrazide chitosan hydrogel as ecofriendly biopolymeric catalyst, Catalysts, 2023, 13(9), 1311 CrossRef CAS.
- R. Sireesha, R. Sreenivasulu, C. Chandrasekhar, S. S. Jadav, Y. Pavani, M. V. B. Rao and M. Subbarao, Design, synthesis, anti-cancer evaluation and binding mode studies of benzimidazole/benzoxazole linked β-carboline derivatives, J. Mol. Struct., 2021, 1226, 129351 Search PubMed.
- L.-J. He, D.-L. Yang, S.-Q. Li, Y.-J. Zhang, Y. Tang, J. Lei, B. Frett, H.-k. Lin, H.-y. Li and Z.-Z. Chen, Facile construction of fused benzimidazole-isoquinolinones that induce cell-cycle arrest and apoptosis in colorectal cancer cells, Bioorg. Med. Chem., 2018, 26(14), 3899–3908 Search PubMed.
- S. Tahlan, B. Narasimhan, S. M. Lim, K. Ramasamy, V. Mani and S. A. Shah, Design, synthesis, SAR study, antimicrobial and anticancer evaluation of novel 2-mercaptobenzimidazole azomethine derivatives, Mini-Rev. Med. Chem., 2020, 20(15), 1559–1571 Search PubMed.
- S. Nashaat, M. A. Henen, S. M. El-Messery and H. Eisa, Synthesis, state-of-the-art NMR-binding and molecular modeling study of new benzimidazole core derivatives as Pin1 inhibitors: Targeting breast cancer, Bioorg. Med. Chem., 2020, 28(11), 115495 CrossRef CAS PubMed.
- C. Gan, X. Huang, J. Zhan, X. Liu, Y. Huang and J. Cui, Study on the interactions between B-norcholesteryl benzimidazole compounds with ct-DNA, Spectrochim. Acta, Part A, 2020, 227, 117525 CrossRef CAS PubMed.
- C. Gan, X. Huang, Y. Wu, J. Zhan, X. Zhang, Q. Liu and Y. Huang, Untargeted metabolomics study and pro-apoptotic properties of B-norcholesteryl benzimidazole compounds in ovarian cancer SKOV3 cells, J. Steroid Biochem. Mol. Biol., 2020, 202, 105709 CrossRef CAS PubMed.
- J. Cui, B. Qi, C. Gan, Z. Liu, H. Huang, Q. Lin, D. Zhao and Y. Huang, Synthesis and in vitro antiproliferative evaluation of some B-norcholesteryl benzimidazole and benzothiazole derivatives, Mar. Drugs, 2015, 13(4), 2488–2504 CrossRef CAS PubMed.
- M. G. Badrey, S. M. Gomha, M. E. Zaki, B. Farag and A. A. El-Reedy, Cyanauric Chloride as a key precursor and a core component for Three-Armed Triazolopyrimidines: Recent finding about SARs and docking analyses, Results Chem., 2024, 7, 101337 CrossRef CAS.
- W. Shehta, N. A. Alsaiari, B. Farag, M. M. Abdel-Aziz, S. Youssif, S. M. Elfeky, S. El-Kalyoubi and N. Awni, Novel phthalimide-pyrimidine hybrids as potent anti-tubercular agents, 2024 Search PubMed.
- M. Rashid, A. Husain, R. Mishra, S. Karim, S. Khan, M. Ahmad, N. Al-Wabel, A. Husain, A. Ahmad and S. A. Khan, Design and synthesis of benzimidazoles containing substituted oxadiazole, thiadiazole and triazolo-thiadiazines as a source of new anticancer agents, Arabian J. Chem., 2019, 12(8), 3202–3224 CrossRef CAS.
- B. Farag, F. Agili, S. El-Kalyoubi, S. A. Said, S. Youssif and W. Shehta, Synthesis, molecular docking and anticancer activity of some 5-aryl-5,10-dihydropyrido[2, 3-d:6,5-d′]dipyrimidine-2, 4,6,8-tetraone derivatives and pyrido[2,3-d]pyrimidines, ChemistrySelect, 2022, 7(7), e202103834 CrossRef CAS.
- C.-Y. Hsieh, P.-W. Ko, Y.-J. Chang, M. Kapoor, Y.-C. Liang, H.-L. Chu, H.-H. Lin, J.-C. Horng and M.-H. Hsu, Design and synthesis of benzimidazole-chalcone derivatives as potential anticancer agents, Molecules, 2019, 24(18), 3259 CrossRef PubMed.
- D. A. Elsayed, M. E. Abdu, M. A. Marzouk, E. M. Mahmoud, W. H. El-Shwiniy, A. M. Spring and W. S. Shehab, Bio-computational modeling, POM analysis and molecular dynamic simulation for novel synthetic quinolone and benzo[d][1,3]oxazine candidates as antimicrobial inhibitors, Sci. Rep., 2024, 14(1), 28709 CrossRef CAS PubMed.
- S. Kaur, J. Kaur, A. Anand, K. Kaur and V. Choudhary, Chalcones: Effective Agents for Fighting Cancer Across Multiple Cell Lines, ChemistrySelect, 2024, 9(9), e202304546 CrossRef CAS.
- K. A. Sheikh, A. Gupta, M. Umar, R. Ali, M. Shaquiquzzaman, M. Akhter, M. A. Khan, M. Kaleem, P. K. Ambast and S. Charan, Advances in chalcone derivatives: Unravelling their anticancer potential through structure-activity studies, J. Mol. Struct., 2024, 1299, 137154 CrossRef CAS.
- S. S. Chhajed, S. S. Sonawane, C. D. Upasani, S. J. Kshirsagar and P. P. Gupta, Design, synthesis and molecular modeling studies of few chalcone analogues of benzimidazole for epidermal growth factor receptor inhibitor in search of useful anticancer agent, Comput. Biol. Chem., 2016, 61, 138–144 Search PubMed.
- A. Habib, M. Nazari, M. A. Iqbal, H. N. Bhatti, M. K. Ahmed and A. A. Majid, Unsymmetrically substituted benzimidazolium based Silver (I)-N-heterocyclic carbene complexes: Synthesis, characterization and in vitro anticancer study against human breast cancer and colon cancer, J. Saudi Chem. Soc., 2019, 23(7), 795–808 Search PubMed.
- M. Hasanpourghadi, N. A. Majid and M. R. Mustafa, The role of miRNAs 34a, 146a, 320a and 542 in the synergistic anticancer effects of methyl 2-(5-fluoro-2-hydroxyphenyl)-1H-benzo[d] imidazole-5-carboxylate (MBIC) with doxorubicin in breast cancer cells, PeerJ, 2018, 6, e5577 CrossRef PubMed.
- Y. T. Lee, Y. J. Tan and C. E. Oon, Benzimidazole and its derivatives as cancer therapeutics: The potential role from traditional to precision medicine, Acta Pharm. Sin. B, 2023, 13(2), 478–497 CrossRef CAS PubMed.
- C. Karthikeyan, V. R. Solomon, H. Lee and P. Trivedi, Synthesis and biological evaluation of 2-(phenyl)-3H-benzo[d]imidazole-5-carboxylic acids and its methyl esters as potent anti-breast cancer agents, Arabian J. Chem., 2017, 10, S1788–S1794 CrossRef CAS.
- M. Hasanpourghadi, A. K. Pandurangan, C. Karthikeyan, P. Trivedi and M. R. Mustafa, Mechanisms of the anti-tumor activity of Methyl 2-(-5-fluoro-2-hydroxyphenyl)-1H-benzo[d] imidazole-5-carboxylate against breast cancer in vitro and in vivo, Oncotarget, 2017, 8(17), 28840 CrossRef PubMed.
- S. G. Nerella, Non-Carbohydrate Galectin-1 Inhibitors as Promising Anticancer Agents: Design Strategies, Structure Activity Relationship and Mechanistic Insights, Eur. J. Med. Chem. Rep., 2024, 100170 CAS.
- N. S. Goud, V. Pooladanda, K. M. Chandra, P. L. Soukya, R. Alvala, P. Kumar, C. Nagaraj, R. D. Bharath, I. A. Qureshi and C. Godugu, Novel benzimidazole-triazole hybrids as apoptosis inducing agents in lung cancer: Design, synthesis, 18F-radiolabeling & galectin-1 inhibition studies, Bioorg. Chem., 2020, 102, 104125 CrossRef PubMed.
- F. F. Hagar, S. H. Abbas, H. A. Gomaa, B. G. Youssif, A. M. Sayed, D. Abdelhamid and M. Abdel-Aziz, Chalcone/1,3,4-Oxadiazole/Benzimidazole hybrids as novel anti-proliferative agents inducing apoptosis and inhibiting EGFR & BRAFV600E, BMC Chem., 2023, 17(1), 116 CrossRef CAS PubMed.
- F. Z. Karadayi, M. Yaman, M. M. Kisla, O. Konu and Z. Ates-Alagoz, Design, synthesis, anticancer activity, molecular docking and ADME studies of novel methylsulfonyl indole-benzimidazoles in comparison with ethylsulfonyl counterparts, New J. Chem., 2021, 45(20), 9010–9019 RSC.
- O. Ebenezer, F. Oyetunde-Joshua, O. D. Omotoso and M. Shapi, Benzimidazole and its derivatives: Recent Advances (2020–2022), Results Chem., 2023, 5, 100925 CrossRef CAS.
- V. Bangade, P. Mali and H. Meshram, Solid-phase reactive chromatography (SPRC): a sustainable method for the synthesis of benzimidazol-diphenyl-2-imino-thiazolidine-4-ols (hemiaminals) which are active against lung cancer, RSC Adv., 2021, 11(4), 2320–2324 RSC.
- J. Y. Al-Humaidi, L. A. Albedair, B. Farag, M. E. Zaki, Y. E. Mukhrish and S. M. Gomha, Design and synthesis of novel hybrids incorporating thiadiazole or thiazole-naphthalene: Anticancer assessment and molecular docking study, Results Chem., 2024, 7, 101475 CrossRef CAS.
- W. A. Mokbel, M. A. Hosny, S. M. Gomha, M. E. Zaki, B. Farag, A. F. El Farargy, A. Al Bahir and Y. H. Zaki, Synthesis, molecular docking study, and biological evaluation and of new thiadiazole and thiazole derivatives incorporating isoindoline-1,3-dione moiety as anticancer and antimicrobial agents, Results Chem., 2024, 7, 101375 CrossRef CAS.
- P. Li, W. Zhang, H. Jiang, Y. Li, C. Dong, H. Chen, K. Zhang and Z. Du, Design, synthesis and biological evaluation of benzimidazole–rhodanine conjugates as potent topoisomerase II inhibitors, MedChemComm, 2018, 9(7), 1194–1205 RSC.
| This journal is © The Royal Society of Chemistry 2025 |